

Abstract
Complement (C) is an important component of innate immunity, and was also shown recently to participate in induction of acquired B cell humoral immunity. In this study, we present evidence that C also participates in acquired T cell immunity.
We found that C was involved in early events of the efferent elicitation phase of contact sensitivity (CS), and delayed-type hypersensitivity (DTH). Thus, CS and DTH were inhibited by administration of a C-blocker, soluble recombinant C receptor-1 (sCR1), when given 30 min before, but not 3 h after local antigen challenge. Among C components, local C5 were thought crucial to elicitation of CS, since local administration of anti-C5 monoclonal antibodies or locally injected C-depleting cobra venom factor also inhibited CS and DTH. These findings were consistent with our previous finding of the importance of C5 for CS elicitation, using congenitally C5-deficient mice. To dissect the mechanism of C dependence in CS, we demonstrated that locally increased early macrophage chemotactic activity (probably C5a) in evolving CS skin extracts, as well as late elaboration of IFN-γ, were both inhibited by anti-C treatment. In addition, histological analysis showed that leukocyte recruitment into CS ear sites was similarly C-dependent. Furthermore, an initiating role of B cell–derived C-fixing immunoglobulin was suggested by demonstration of impaired CS responses in B cell–deficient mice.
In summary, these results suggest that C was activated locally, perhaps via a B cell product, in an important early component of the stepwise events necessary to elicit CS, leading to local production of C5-dependent macrophage chemotactic activity and later IFN-γ, and subsequently leading to cell infiltration, for development of T cell–dependent CS.
Complement (C) is a major component of innate immunity, and is involved in early protective immune responses against pathogens, which occur before induction of acquired T and B cell immunity (1). Furthermore, recent findings demonstrate that innate immunity interacts with acquired immunity (1); for example, innate immunity directs Th-1 versus Th-2 development via IFN-γ production from NK cells (2), or via IL-12 from macrophages (3), and IL-4 from NK1.1 CD4+ T cells (4). Furthermore, C participates in acquired augmentation of B cell Ab responses when C3d is conjugated to Ag (5). This was particularly important when the immunizing Ag was limiting (6, 7). Also, C can participate in elaboration of anaphylatoxins (C3a and C5a), (8), which activate various cell types, as well as via formation of the activating terminal C5b-9 complex on target cell surfaces (9). Although a negative regulatory role of C in cellular immunity was suggested recently by demonstrating that cross-linking of membrane cofactor protein (CD46), led to suppressed IL-12 production (10), the role of C in positive regulation of acquired cellular immunity such as T cell responses like contact sensitivity (CS)1 and delayed-type hypersensitivity (DTH) (11, 12) has not been understood fully.
CS is a classical example of a T cell–mediated cutaneous inflammatory response (13). CS and related DTH are mediated generally by Ag/MHC class II–restricted Th-1 cells, which are recruited in mice to the local tissue site via serotonin (5-HT)–mediated processes which occur early after Ag challenge (14). Thus, local Ag challenge causes an early 2-h release of 5-HT from tissue mast cells (14) and platelets (15, 16), leading to endothelial cell activation via their 5-HT receptors. This enables circulating Th-1 cells to extravasate into the local site of Ag challenge, after this early initiating phase of CS and DTH, to constitute the classical 24-h tissue swelling response. Released 5-HT also may costimulate recruited Th-1 cells via their surface 5-HT2 receptors (17, 18). Then, there are late events of the cascade leading to CS elicitation, in which local APC activate the recruited Th-1 cells to produce proinflammatory lymphokines such as IFN-γ (19, 20), TNF-α (20, 21), and migration inhibitory factor (22), to locally recruit and activate nonspecific bone marrow–derived inflammatory leukocytes (neutrophils and monocytes) (13).
In the course of screening for immunomodulators which might specifically affect certain immune responses in vivo, such as Ab production versus DTH (23), we found that Actinomyces produced a DTH-specific immunosuppressant which was identified previously as a C5a antagonist (24–27). C5a is a peptide fragment derived from cleavage of C5 during C activation. C5a is known to be important in local immune inflammation, and in elimination of microbes, via C5a receptors on various cells, especially neutrophils, macrophages, and mast cells (28). Thus, C5a mediates chemotaxis, mast cell degranulation, vascular permeability, smooth muscle contraction (29, 30), and possibly 5-HT release from platelets (31). Since 5-HT release from mast cells (14) and platelets (15, 16), was demonstrated to be important in early events of CS, we previously suggested a role of C5 early in the initiation of CS by using congenitally C5-deficient mice (32).
In this study, we establish a role for C5 in cutaneous T cell CS and DTH responses in normal mice, employing C-depleting soluble recombinant C receptor-1 (sCR1) (33–35), and cobra venom factor (CVF), and also anti-C5 mAb for C5 depletion (36, 37). We demonstrate that local C5 acted in the early initiating phases of elicitation of CS to mediate production of C-derived macrophage chemotactic activity in CS ear extracts, and later IFN-γ, which is the principle Th-1 cytokine of CS/DTH, and also mediates subsequent cell infiltration. Additional studies in B cell– deficient mice suggest that B cell–derived immunoglobulin may be important in early activation of C. Thus, we suggest that C5, possibly derived from Ab fixation of C, played an important role in the early initiating and subsequent late T cell-dependent aspects of these cutaneous immune responses.
MATERIALS AND METHODS
Mice.
Specific pathogen-free female CBA/J, ICR, BDF1, C57Bl/6, and B cell–deficient C57Bl/6-Igh-6 (μMT) mice (6–8 wk old) were obtained from the Jackson Laboratory (Bar Harbor, ME), and were rested at least 1 wk before use.
Reagents.
Picryl chloride (PCl), obtained from Nacalai Tesque, Inc. (Tokyo, Japan) was recrystallized twice as described previously (14), and stored protected from light. Zymosan and CVF were purchased from Sigma Chemical Co. (St. Louis, MO). SRBC and anti-SRBC polyclonal Ab were products of Organon Teknika (Durham, NC). Trinitrobenzene sulfate sodium salt was obtained from Wako Chemicals (Osaka, Japan). Normal mouse serum, drawn freshly from naive ICR mice via cardiac puncture, was stored at −70°C, thawed, and then incubated with sterile 5 mg/ ml zymosan at 37°C for 60 min to activate C, followed by centrifugation at 14,000 rpm for 10 min. Supernatant was used as zymosan activated mouse serum (ZAMS). sCR1 was a gift from T Cell Sciences, Inc. (Needham, MA). Mouse anti–murine C5 mAb (BB5.1) and isotype (IgG1)-matched mouse anti–human C8 mAb (135.8) were purified by protein A column from ascites generated in nude mice injected i.p. with appropriate hybridoma cells.
CS Responses.
CBA/J mice were contact sensitized by topical application of 100 μl of 5% PCl in absolute ethanol and acetone (4:1) applied to the shaved chest and abdomen. 4 d later, CS was elicited by painting both ears via topical application of 10 μl of a low challenge dose of 0.4% PCl in acetone and olive oil (1:1), compared to conventionally employed high dose of 0.8% PCl, unless otherwise noted, since an effect of C alteration on CS usually was observed with a moderate dose of challenge Ag (32). B6 background mice are hyporeactive to PCl CS and thus were contact sensitized twice on days 0 and 1. Resulting thickness of the Ag-challenged ears was measured on days 4 or 7 in the case of B6 background mice with a dial caliper (Ozaki MFG Co., Tokyo, Japan) before challenge and at 2 and 24 h after challenge. Increased ear thickness was expressed as mean ± SE.
TNP-SRBC–induced DTH Responses.
DTH against TNP-SRBC (24, 38) was induced in BDF1 mice with TNP-SRBC conjugated by incubating SRBC (5 × 108 cells/ml) with 10 mM trinitrobenzene sulfate sodium salt in PBS at 37°C for 60 min, and then washing three times with PBS. For immunization, mice were injected i.v. with 105 TNP-SRBC on day 0. On day 5, both footpads were challenged s.c. with TNP-SRBC (2 × 108 cells in 40 μl PBS) to elicit DTH, and footpad thickness was measured before and 24 h after challenge.
C3 and C5 Titration.
C3 and C5 activity were titrated by measuring Ab-dependent C-mediated hemolysis of SRBC by C3- or C5-deficient human serum (39). In brief, SRBC (5 × 108/ml) were coated with Ab in rabbit antiserum to SRBC. 100 μl of each serum sample or ear extract was mixed with 100 μl of human C3- or C5-deficient serum diluted 1:20 with gelatin veronal buffer (pH 7.4), and with 50 μl of Ab-coated SRBC (5 × 108), followed by incubation for 60 min at 37°C. Then hemolyzed supernatants were collected and OD measured at 405 nm.
In Vitro Evaluation of Chemotactic Activity in Ear Extracts.
Ears that were the site of CS reactions were removed at the base and three punch biopsies were collected from the distal portion. Biopsies were 12.5 mm2 and were frozen rapidly in liquid N2. Either the whole ear or three punch biopsies per ear were extracted in 300 or 500 μl cold PBS, with a tissue homogenizer (Biospec Products, Racine, WI) on ice, followed by centrifugation at 14,000 rpm for 15 min to obtain the supernatant of extracts. Protein concentration was determined by BCA-protein determination kit (Pierce, Rockford, IL). C5 content was evaluated by the C5-titration assay.
Chemotactic activity was determined with the J774A.1 murine macrophage cell line that responds to C5a. The cells were maintained in RPMI 1640 supplemented with 10% FBS (GIBCO BRL, Gaithersburg, MD), 25 mM Hepes, 100 U/ml penicillin, and 100 μg/ml streptomycin. The J774A.1 cells were washed three times with RPMI 1640 without FBS, and were suspended in RPMI 1640 with 0.25% gelatin without FBS (RPMI-gelatin) (2–5 × 106 cells/ml). Triplicate ear extract samples were diluted with RPMI gelatin and were placed in lower wells of 96-well chemotaxis chambers (NeuroProbe Inc., Cabin John, MD). Two- or fourfold diluted ear extracts were used according to the previous observation that J774A.1 cells migrated in a dose-dependent fashion, in up to eightfold dilution. No proteinase inhibitors were added due to their inhibitory effect on chemotaxis.
50 μl of J774A.1 cells was added to the upper wells, allowing cells to migrate through a polyvinylpyrrolidone-free polycarbonate filter with 5 μm pores at 37°C for 4 h in a humidified air atmosphere, containing 5% CO2. The J774A.1 cells attached firmly to the other surface of the filter, and then were fixed and stained with Diff Quick solution (Wako Chemicals). Migrated cells were counted at five different filter spots of each well. Among the complement components, J774A.1 chemotaxis was specifically mediated by C5a, since there was no chemotaxis against ZAMS prepared from C5-deficient serum. We also verified that J774A.1 migration was not due to chemokinesis by showing that addition of ear extracts to the upper wells where the cells were loaded caused diminished migration. Thus, the number of migrated J774A.1 cells without ear extracts in the upper wells was 103.0 ± 2.8 versus 27.9 ± 2.7 (P <0.001) with ear extracts added to the upper wells.
In Vitro Quantitative Measurement of IFN-γ in Ear Extracts.
A quantitative sandwich ELISA for IFN-γ used two specific mAbs (PharMingen, San Diego, CA). In brief, wells were coated overnight with 2 μg/ml capture mAb (R4-6A2) in 0.1 M NaHCO3 (pH 8.3) at 4°C. After blocking with 1% BSA in PBS at room temperature for 2 h, samples and dilutions of standard recombinant mouse IFN-γ (Genzyme Corp., Cambridge, MA) were added and incubated for 1 h at room temperature. Since there was no significant difference in IFN-γ content between ears extracted with or without proteinase inhibitor cocktail, which included PMSF, EDTA, leupeptin, E-64, and pepstatin A, cold PBS alone was used to extract ears. Then, 1 μg/ml of the other biotinylated anti–IFN-γ mAb (XMG1.2), and 1:3,000 diluted horseradish peroxidase–conjugated streptavidin (Vector Labs., Burlingame, CA), were added to probe for IFN-γ. Peroxidase substrate (TMB tetramethylbenzidine) and TMB one component stop solution (Kirkegaard & Perry Labs, Inc., Gaithersburg, MD) were used for color development at 450 nm.
Histological Evaluation of Cell Infiltration in CS Ear Responses.
5-μm sections of formalin-fixed, paraffin-embedded ear tissue were stained with hematoxylin and eosin. Semiquantitative evaluation of infiltrating cells per area (1, no infiltration; 2, slight infiltration; 3, modest infiltration; 4, strong infiltration), and also formation of intraepidermal abscesses (1, not seen; 2, few seen; 3, many seen) were determined. Histology in ears was read by an observer blinded as to the experimental design and assigned a numerical grade, from which group means and SE were calculated.
Statistics.
Statistics were performed using the two-tailed Student's t test and P <0.05 was taken as the level of significance. Each experimental group consisted of four to six mice.
RESULTS
C is Involved Early in the Elicitation of CS.
sCR1 is a newly developed C-blocking reagent (33–35) which enabled us to investigate when C acts in the efferent elicitation phase of CS. When sCR1 was injected systematically 30 min before Ag challenge in immunized CBA/J mice, both early (2-h) and late (24-h) components of CS ear swelling responses were reduced significantly (Fig. 1 A, group D vs C). Since both 2- and 24-h ear swelling responses were affected by sCR1 treatment, the decreased 24-h response may have been due to effects of sCR1 on the early 2-h events of CS, which are required for elicitation of the 24-h ear swelling responses (14, 15). Thus, when sCR1 was given 3 h after Ag challenge, and thus after the required early events, there was no effect on 24-h ear swelling responses (Fig. 1 A, group E), suggesting that required C acted early, before 3 h, in the elicitation of CS.
Figure 1.

C is involved in the early phase of eliciting CS and DTH. (A) CBA/J mice (4/group) were actively contact sensitized with PCl applied topically to the shaved chest and abdomen. 4 d later the mice were skin challenged to elicit CS by painting both ears with PCl. The resulting CS ear swelling responses were measured with a dial caliper by comparing thickness before, vs. 2 and 24 h after challenge. Net ear swelling was calculated by subtracting ear thickness before challenge from that at 2 and 24 h. (B) TNP-SRBC (105 RBC) were injected i.v. to immunize BDF1 mice (4/group) which were challenged s.c. 5 d later in both hind footpads with 108 TNP-SRBC. Footpad swelling was measured with a dial caliper before and 24 h after challenge. In both A and B, sCR1 was injected (750 μg/mouse) i.p. 30 min before Ag challenge (groups B and D), or 3 h after challenge (group E). *P <0.05, **P <0.01, compared with PBS injected and immunized and Ag challenged controls (group C).
To determine whether another system of DTH responses was similarly dependent on C, acting in the early phase, BDF1 mice were immunized i.v. with TNP-SRBC and then hind footpads were challenged with TNP-SRBC on day 5. Again, sCR1 treatment 30 min before (Fig. 1 B, group D vs C), but not 3 h after Ag challenge (group E) inhibited DTH. Taken together, these results suggested that classical 24-h CS and 24-h DTH, in two different strains of mice, actually depended on C-mediated early events.
C in the Local Site Is Important for CS Elicitation.
We reexamined whether CVF, which generally is used to deplete C, would inhibit CS. CVF (10 μg) in PBS was injected i.p. twice per day on days 1 and 2 after immunization. This protocol was reported to exert minimal side effects such as decreased platelets (12, 40). This CVF treatment regimen depleted serum C3 and C5, because C3 was undetectable at 24 and 48 h after CVF treatment, and C5 was decreased to 12.5 and 25% of normal, respectively. Platelet counts of saline versus CVF-injected mice were not different 48 h after the final CVF injection. When sensitized mice were challenged with a conventional, high dose of 0.8% PCl, there were no significant differences between saline- and CVF-injected groups (79% of saline control, P = 0.24). However, when the eliciting dose of PCl was decreased to 0.4% to elicit weaker CS, CVF diminished 24-h ear swelling responses (48% of saline control, P <0.05) (data not shown).
We hypothesized that effects on local C levels might explain these observations with CVF. Thus we attempted to deplete C locally by directly injecting CVF into ears. CVF was locally injected into ears 48 h before local Ag challenge, since CVF injected alone s.c. into ears caused no swelling 48 h later. We found that both 2- and 24-h ear swelling responses were decreased significantly by local CVF in CBA mice (Fig. 2 A, group D versus C). In similar DTH experiments which used BDF1 mice, local preinjection of CVF into footpads 48 h before Ag challenge also decreased DTH against TNP-SRBC (Fig. 2 B, group D versus C). Thus, these experiments showed that C depletion with local CVF could inhibit CS and DTH, and pointed to a role of local C activation in CS initiation.
Figure 2.

CS and DTH were inhibited by local depletion of C in ears by CVF injection. (A) CBA/J mice (4/group) were actively contact sensitized with PCl applied topically to the shaved chest and abdomen. 4 d later mice were skin challenged to elicit CS responses by painting both ears with PCl (groups C and D). Subsequent CS ear swelling responses were measured with a dial caliper by comparing thickness before vs. 2 and 24 h after challenge. Net ear swelling was calculated by subtracting ear thickness before challenge from that at 2 and 24 h. PBS or CVF (5 μg/20 μl) was injected directly into the ears s.c. with a 30 gauge, 1/2′′ needle, 48 h before challenge (groups B and D). (B) TNP-SRBC (105 cells) were injected i.v. to immunize BDF1 mice (4/group) which were challenged s.c. 5 d later in both hind footpads with 108 TNP-SRBC. Footpad swelling was measured with a dial caliper before and 24 h after challenge. PBS or CVF (1 μg/40 μl) was injected locally s.c. 48 h before footpad challenge. *P <0.05, **P <0.01, compared to 2- and 24-h positive immune and Ag-challenged controls which were injected with PBS (group C).
Since C depletion by CVF probably was accompanied with generation of anaphylatoxins such as C3a and C5a, which could have confused results, we alternately employed anti-C5 mAb to deplete C5. When anti-C5 mAb was injected i.p. 24 and 4 h before Ag challenge, a negligible amount of C5 remained in serum, as detected by a C5 in vitro titration assay (data not shown). This systemic treatment with anti-C5 mAb significantly inhibited both 2- and 24-h ear swelling responses (Fig. 3 A, group D versus C). Importantly, we also injected anti-C5 mAb locally into ears to deplete C5 at the site of CS. This resulted in decrease of both 2- and 24-h ear swelling responses (Fig. 3 B, group D versus C). Taken together, these results indicated that C and particularly C5, acting early at the local CS site, were important for mediating elicitation of CS responses.
Figure 3.

C5 depletion by anti-C5 mAb–inhibited CS. CBA/J mice (4/group) were actively contact sensitized with PCl applied topically to the shaved chest and abdomen. 4 d later, mice were skin challenged to elicit CS by painting both ears with PCl. CS ear swelling responses were measured with a dial caliper by comparing thickness before, vs. 2 and 24 h after challenge. Net ear swelling was calculated by subtracting ear thickness before challenge from that at 2 and 24 h after challenge. (A) Systemic anti-C5 mAb (1 mg/mouse) was injected i.v. 24 and 4 h before ear challenge with Ag. (B) Local anti-C5 mAb (20 μg/20 μl) was injected s.c. into ears 24 h before ear challenge. *P <0.05, **P <0.01, compared with IgG1 isotype control (group C).
C-dependent Production of Local Chemotactic Activity in CS Ear Extracts.
To determine the mechanism of C participation in CS, we extracted CS ears, which were obtained 24 h after Ag challenge with cold PBS. Total extractable protein was increased in CS, paralleling macroscopic ear swelling measurements 24 h after Ag challenge (Fig. 4 A), and C5 activity in a C-function assay increased significantly in CS responses (Fig. 4 B) but not in controls. We then measured local macrophage chemotactic activity in vitro in CS ear extracts by use of migrating J774A.1 macrophages. We detected increased macrophage chemotactic activity in the CS ear extracts, compared to nonimmune controls (Fig. 5, A and B). To investigate C dependency of this chemotactic activity found in the local ear site, mice were treated systemically with anti-C5 mAb and significant decrease was obtained (Fig. 5 A, right). Furthermore, systemic treatment with sCR1 also diminished macrophage chemotactic activity in CS ears (Fig. 5 B, right), confirming that locally extracted macrophage chemotactic activity was C dependent. These results suggested that either C5a or other C-regulated, locally derived chemotactic factors were responsible for macrophage chemotactic activity in local CS reactions.
Figure 4.

Increased C5-titer in 24-h CS ear extracts. Contact sensitized mice were skin challenged topically on day 4 to elicit CS by painting both ears with PCl. 24 h later the challenged ears were extracted with PBS, as described in Materials and Methods. (A) Total protein concentration (mg/ ear) and (B) C5-titer per ear were determined. Closed and open circles represent two- and four-fold dilution of ear extracts with gelatin-veronal buffer, respectively. Each value per ear was expressed as a separate dot. The number of ear extracts in each group was 12, and was derived from 6 mice.
Figure 5.
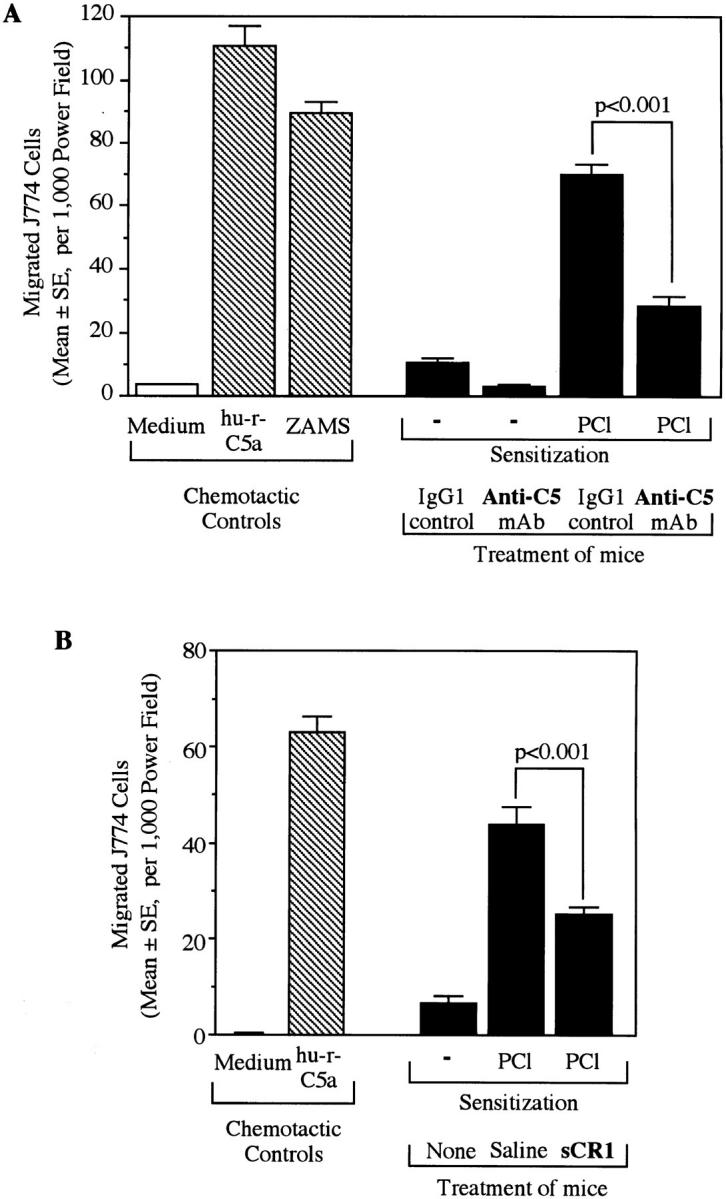
Anti-C treatment resulted in decreased chemotactic activity in 24-h CS ear extracts. Two different C inhibitors, anti-C5 mAb (A) and sCR1 (B), were used. Mice were ear challenged 4 d after PCl contact sensitization. Punch biopsies from each ear were collected 24 h later and then were extracted with PBS. In vitro chemotaxis assay was performed on the ear extracts using target J774A.1 macrophage cells which did not migrate against the RPMI 1640–0.25% gelatin medium. Human rC5a (100 ng/ml) and ZAMS (1:20) prepared from normal mouse serum were used as positive chemoattractant controls. (A) Mice were injected i.v. 24 and 4 h before ear challenge with either anti-C5 (1 mg/mouse) or with isotype (IgG1) matched control anti–human C8 (1 mg/mouse). (B) Mice were injected i.p. with 125 μg/mouse sCR1 30 min before ear challenge.
C-dependent Production of Local IFN-γ in CS Ear Extracts.
Since IFN-γ is a crucial cytokine in CS responses (19), we also quantified IFN-γ in CS ear extracts. Increased IFN-γ was found in CS ear extracts 24 h after Ag challenge (Fig. 6, A and B). Since little connection is usually recognized between C and cytokines, it was of interest to note that both systemic and local treatment with anti-C5 mAb significantly inhibited IFN-γ production in 24-h CS ears (Fig. 6, A and B). Thus, C5/C5a was also suggested to regulate local production of IFN-γ, which is an important Th-1 cytokine for elicitation of CS responses.
Figure 6.
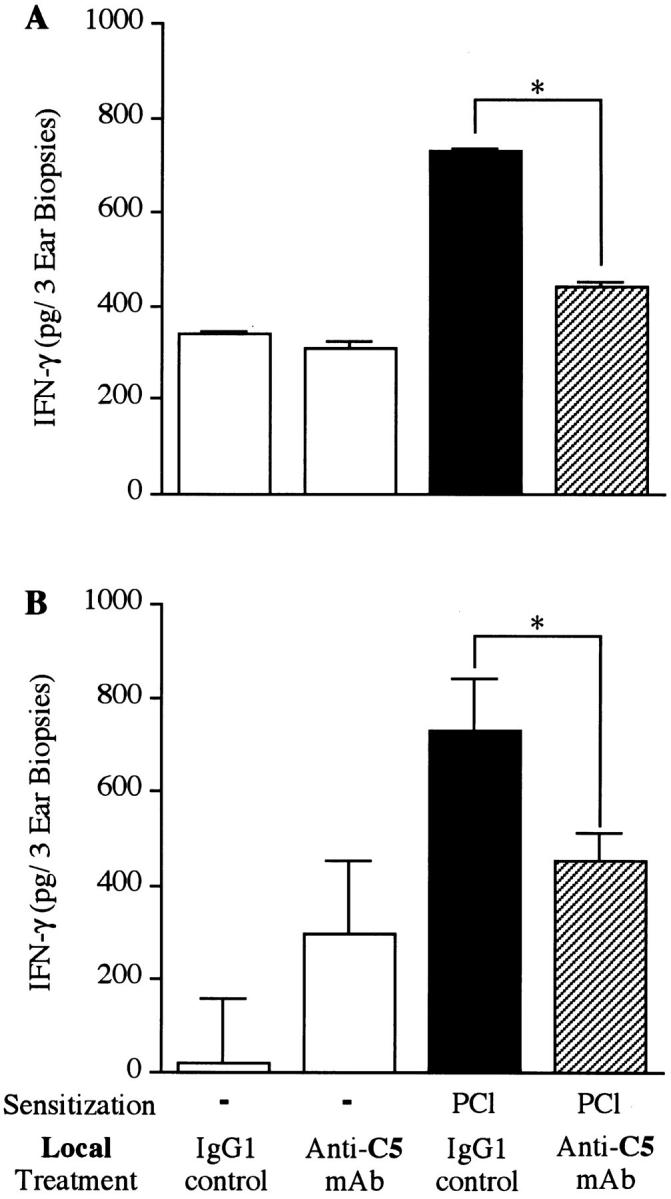
Anti-C5 mAb treatment also decreased IFN-γ in 24-h CS ear extracts. Mice (4/group) were actively contact sensitized with PCl applied topically to the shaved chest and abdomen. 4 d later mice were skin challenged to elicit CS by painting both ears with PCl. Punch biopsies from each ear were collected 24 h later and were extracted with PBS. IFN-γ production in the ear extracts was determined by quantitative sandwich ELISA. (A) Systemic anti-C5 mAb (1 mg/mouse) was injected i.v. 24 and 4 h before ear challenge. (B) Local anti-C5 mAb (20 μg/20 μl) was injected s.c. into the ear 24 h before challenge. The number of mice in each group was four and eight samples of ear extracts in each group were pooled (A) or were assayed separately (B). *P <0.001 compared with isotype (IgG1) controls.
C-dependent Cell Migration into CS Sites.
Finally, to determine whether cell infiltration as well as macrophage chemotactic activity (Fig. 5), and IFN-γ production (Fig. 6), were decreased by C5 depletion, we examined histology of CS ear sections. Decreased local cell infiltration in CS ears (Fig. 7, d versus c) and also inhibited intraepidermal abscesses formation (Fig. 7, f versus e) were noted in mice treated with anti-C5 mAb. Semiquantitative histologic observation was carried out blindly and indicated that C5 depletion resulted in significant decreases in leukocyte infiltration of CS (Fig. 8 A), and in decreased formation of intraepidermal abscesses (Fig. 8 B).
Figure 7.
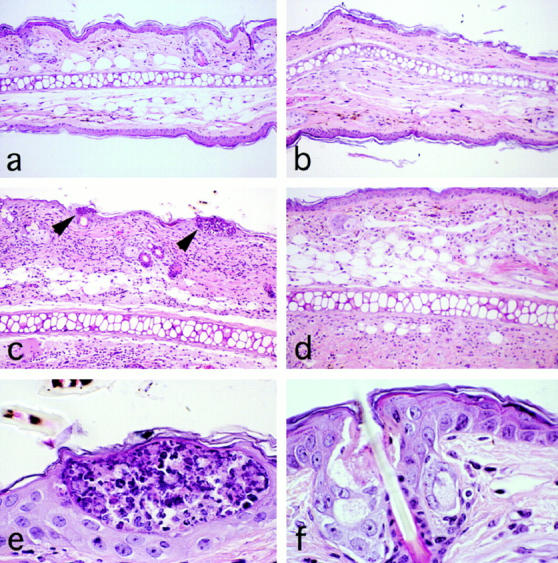
Cell infiltration in 24-h CS ears was decreased by systemic treatment with anti-C5 mAb. Mice were immunized by painting with vehicle (a and b) or 5% PCl (c–f ) on day 0, and challenged on the ears with PCl on day 4. Then, saline (a, c, and e) or 1 mg anti-C5 mAb (b, d, and f ) was injected i.v. at 4 and 24 h before ear challenge. 5-μm sections of ears were obtained 24 h after Ag challenge, and were stained with hematoxylin and eosin. Magnification is 200 (a–d) or 1,000 (e and f ). Leukocyte infiltration in CS era responses (c) was decreased by anti-C5 mAb treatment (d). Intraepidermal abscesses of leukocytes were observed frequently in ears from immune and challenged mice treated with saline (c and e), but not in ears of mice treated with anti-C5 mAb (d and f ). Arrowheads in (c) denote intraepidermal abscess.
Figure 8.
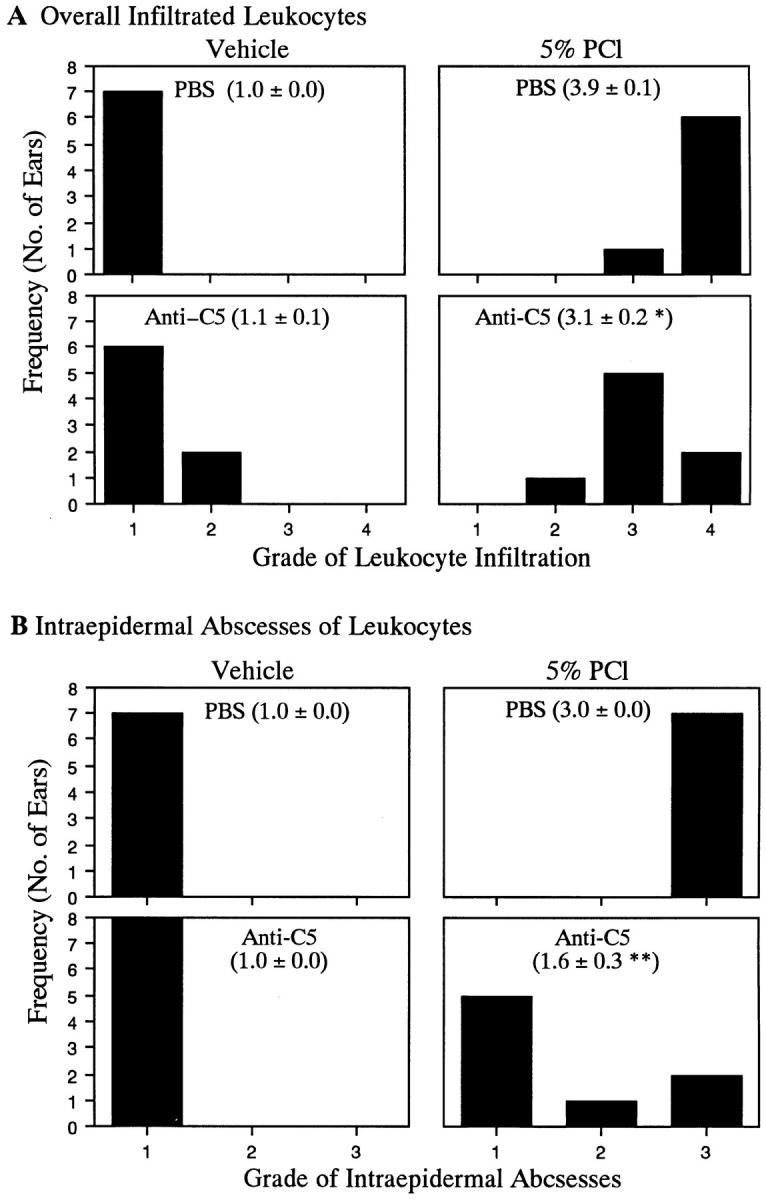
Quantitation of decreased cell infiltration in CS ears of mice treated with anti-C5 mAb. Sections of ears, as in Fig. 4, were histologically evaluated blindly. (A) Infiltration of leukocytes was graded from one to four, where one through four are, respectively, no infiltration, slight infiltration, modest infiltration, and significant infiltration. (B) Formation of intraepidermal abscesses was graded from one to three where they are, respectively, none seen, few seen, and many seen. Numbers in parentheses denote mean ± SE of histologic scores for each portion of the figure. *P <0.02 and **P <0.002, compared to positive controls (5% PCl-immunized, PBS-injected, ear-challenged group).
Impaired 24-h CS in B Cell–deficient Mice.
A central question concerns the mechanism of C activation early in the cascade of events which leads to T cell infiltration, and then local production of chemotactic activity as well as IFN-γ production. We tested CS responses in actively sensitized B cell–deficient μMT mice which have a deletion in the transmembrane portion of surface IgM, and as a consequence display an absence of all mature B cells (41). Fig. 9 shows that 24-h CS was induced in positive control C57Bl/6 mice (group B) versus unsensitized controls (group A). C57Bl/6 are known to have CS hyporeactivity, compared to, e.g., CBA/J mice (for CBA/J see Figs. 1 and 2, group C, at 24 h). Fig. 9 presents similar unsensitized (group C) versus sensitized (group D) groups in C57Bl/6-Igh-6 (μMT) mice, showing no induction of 24-h CS in immune group D versus nonimmune group C. There was a significant decrease in 24-h CS in μMT mice (group D) compared to the positive C57Bl/6 controls (group B). The 2-h macroscopic responses were too small for conclusions about statistical significance.
Figure 9.

Impaired CS responses in B cell–deficient μMT mice. CS was induced in relatively hyporeactive C57Bl/6 control mice (groups A and B), and in C57Bl/6-Igh-6 (μMT) B cell–deficient mice (groups C and D) by topical application of PCl on days 0 and 1. Then, 6 d later the mice were skin challenged to elicit CS by painting both ears with 0.8% PCl. The resulting CS ear swelling responses were measured with a dial caliper by comparing thickness before, versus 2 and 24 h after challenge. Net ear swelling was calculated by subtracting ear thickness before challenge from that at 2 and 24 h after challenge. Number of mice in each group was eight. *P <0.01, compared with C57Bl/6 mice controls (group B).
These results demonstrated a defect in elicitation of CS in B cell–deficient mice. This may signify that antibodies are responsible for C activation early in CS, and for subsequent derivation of C-related chemotactic factors which help recruit T cells, later produce IFN-γ, and generate the late 24-h inflammatory infiltrate.
DISCUSSION
Our results point to a previously unrecognized role of C in CS and DTH which occurs during early required events in the efferent phase of these classical T cell–mediated immune responses. The early events of CS are characterized by processes that are required to recruit Th-1 cells to local sites of Ag challenge to initiate subsequent classical “delayed” aspects of these in vivo immune inflammatory reactions by producing Th-1 cytokines such as IFN-γ. Although these late steps in the Th-1 cell–dependent inflammatory cascade of CS and DTH have been well described (13), the processes that lead to tissue Th-1 cell recruitment are not fully understood. Recombinant sCR1 is a recently developed C-blocker which enabled us to distinguish an early versus late locus of C in elicitation of CS responses by administration 30 min before, compared to 3 h after, local Ag challenge. Thus, only when sCR1 was given 30 min before Ag challenge were CS and DTH responses inhibited, while giving sCR1 3 h after challenge produced no effect (Fig. 1). Thus, it appeared that after Ag challenge, C was involved in 0–3-h early events which were needed to elicit 24-h CS.
In contrast to sCR1, systematic CVF depletion of C failed to demonstrate an involvement of C in CS and DTH, when a high amount of Ag was used for sensitization and challenge (11, 12). However, it was shown previously in studies of Ab production that C participation was more evident when the amount of Ag was limiting (6, 7). Similarly, in congenitally C5-deficient mice (32), we demonstrated previously that CS was impaired when moderate Ag doses were used for sensitization or elicitation. Thus in this study, we reexamined the effect of C depletion by CVF by using a moderate eliciting Ag dose and found impaired CS. We therefore propose that elicitation of CS with a large Ag dose can overcome CVF-induced, or congenital C depletion, whereas C is required to elicit CS when moderate Ag doses are employed. An explanation for this may be that a large Ag dose triggers induction of elicitation of CS via early events that are in part due to nonspecific early irritation and nonspecific inflammation and that probably produce vascular activation and permeability. In fact, it was shown recently that an irritative chemically reactive hapten such as TNP (PCl) contributes nonspecifically to elicitation of Ag-specific CS (42). Thus, specific and nonspecific effects of a given Ag and the dose need to be taken into account in deciding the pathogenesis of the early initiating aspects of CS.
C components are produced by multiple cell types (43– 46). Local tissue macrophages were proposed as the C source in a DTH model associated with Listeria protection (47). Thus, C5-deficient mice which were locally reconstituted with normal C5-producing tissue macrophages were protected against Listeria but were not protected with macrophages from C5-deficient donors (47). Therefore in this study, we treated mice with CVF locally into the ears to examine the role of local C in CS. We found that both 2- and 24-h CS responses were decreased significantly by this local depletion of C (Fig. 2), suggesting that locally produced C was involved in the crucial early events required in CS and DTH.
There are limitations to the use of CVF due to its intrinsic generation of C3a and C5a anaphylatoxins and possible alteration in platelets which are known to be involved in CS (15, 16). Thus, we also used anti-C5 mAb to deplete C5 (36, 37), but without these complications. Systemic anti-C5 mAb treatment strongly inhibited both 2- and 24-h CS ear swelling responses (Fig. 3 A, group D versus group C). Further, local injection of anti-C5 mAb directly into the ears, also profoundly inhibited both 2- and 24-h CS responses (Fig. 3 B, group D versus group C), similar to local depletion of C produced by CVF (Fig. 2). These results strongly indicated that local C, possibly C5, is required early for eliciting CS, and that the trigger for activation of C in CS occurs locally.
To directly analyze events in CS, we developed a technique to extract macromolecular components from the ears of CS ear reactions. We found an increase in chemotactic activity for macrophages at the local CS sites, that was decreased by systemic administration of anti-C5 mAb, or by sCR1 (Fig. 5). C5-derived C5a itself, or possible other C-induced chemotactic factors, may be responsible for the observed chemotactic activity in CS ear extracts. Since migration inhibitory factor (22) and IP-10 (48), both of which are chemotactic, have been reported to be upregulated in sites of DTH reactions, further study is necessary to discriminate C5a from these other macrophage chemotactic factors. However, since we detected increased C5 in 24-h CS ear extracts (Fig. 4 B), potent chemotactic C5a may have been generated by newly arrived C5, in addition to locally generated C5a which was suggested by treatment with local CVF, and also local anti-C5 mAb.
Important additional findings were made regarding IFN-γ in CS ear extracts. Since IFN-γ is a crucial Th-1 cytokine in DTH (19), we also measured IFN-γ in local CS ear sites and as expected found increased local levels at 24 h. Since little connection generally is recognized between C and cytokines, we were surprised to find that the increased 24-h IFN-γ production in CS ears was decreased significantly by C5 depletion, either systemically or locally, following treatment with anti-C5 mAb, which has a locus early in elicitation of CS (Fig. 6). In addition, we also performed time course experiments to quantitate IFN-γ by sandwich ELISA and semiquantitative reverse transcription–PCR for IFN-γ mRNA in CS ears. These studies showed that neither IFN-γ nor its mRNA was detected at CS sites for up to 4 h after Ag challenge, when the early CS-initiating events were completed (Tsuji, R.F., unpublished observations). At this time, Th-1 cells probably were not yet fully recruited and locally activated, but C activation was complete. In further studies, we noted that immunodeficient TCR-α knockout mice showed no IFN-γ production in CS ear extracts at any time. Taken together, these observations strongly suggested that IFN-γ production in local CS ear sites was due to late arriving Ag/MHC-specific α/β TCR+ CS-effector Th-1 cells. Therefore, anti-C treatment via anti-C5 probably impaired the early phase, C-dependent recruitment of Th-1 cells into the local CS site, resulting in decreased later production of IFN-γ at these local CS sites.
Since Th-1 cell recruitment and activation for subsequent IFN-γ production should lead to local recruitment of inflammatory leukocytes, we performed histology to determine the possible C dependency of the characteristic cell infiltration in CS. Accordingly, we found that anti-C5 mAb treatment reduced the numbers of infiltrating leukocytes (Figs. 7, d versus c, and 8 A). In particular, anti-C5 mAb treatment led to drastic decreases in characteristic intraepidermal abscesses in 24-h CS ears (Figs. 7, e versus f, and 8 B). This apparent C5 dependency of leukocyte infiltration was thus a histology analogue which was consistent with our finding of decreased macrophage chemotactic activity in 24-h CS ear extracts (Fig. 5). Although we did not directly study the effect of C on Th-1 cell recruitment, decreased local production of IFN-γ in CS ears of anti-C5 mAb–treated mice (Fig. 6) suggested that recruitment of Th-1 cells was decreased. Thus, the inhibited leukocyte infiltration may have been due to decreased C-dependent Th-1 recruitment into CS ears. We noted previously that treatment with specific antiplatelet Ab led to depletion of platelets, a potential source of local 5-HT able to activate endothelium for Th-1 recruitment, and also led to marked decrease in formation of intraepidermal abscesses in CS (15). Thus, the action of C in CS might involve intermediate platelet and mast cell activation, perhaps via C5a acting on their C5a receptors, with subsequent 5-HT release, contributing to Th-1 cell recruitment and then leukocyte arrival.
The exact function of C5 acting early in elicitation of CS responses is not known. There are two potential C5-derived mediators such as anaphylatoxin C5a, and the terminal C5b-9 complex, generated after C5 cleavage, and also a potent cell activator in sublytic amounts (9). One likely possibility is that either or both of these C-mediators trigger local mast cells and/or platelets via C5a receptors early in CS, to release 5-HT which is a key mediator of vascular permeability, and perhaps vasoactivity in the initiation of CS (29, 31, 49). Alternately, or in addition, these C5- derived mediators may directly activate the local vasculature to enhance permeability and expression of adhesion molecules (50, 51), contributing to local T cell and/or leukocyte recruitment. In fact, it was reported that C augments immune inflammation in the lung by upregulating expression of vascular ICAM-1 (52). Also C5a was shown recently to be responsible for upregulation of lung vascular P-selectin (53). In other CS studies, we noted prolonged C5a activity in vivo when we gave an inhibitor of the C5a-inactivating enzyme, which converts active C5a to inactive C5a desArg (54). There were also augmented CS ear swelling responses, suggesting that endogenous released C5a is involved in CS elicitation. In addition, experiments with C5a receptor (−/−) mice (55) suggest a role of C5a in CS (Tsuji, R.F., C. Gerard, and P.W. Askenase, manuscript in preparation).
It was of interest to find what causes the crucial early C activation in CS responses. We investigated this by using B cell–deficient μMT mice and found that their 24-h CS was decreased (Fig. 9). Taken together, an involvement of B cells and therefore probably of immunoglobulins in CS responses is suggested, but awaits further detailed demonstration. These findings suggest a heretofore unappreciated crucial role of B cell–derived specific Ab in the early initiating phase of T cell–mediated immunity in vivo, as exemplified by CS and DTH. Current studies suggest that IgM is an important involved immunoglobulin isotype and may be produced by the B-1 subset of B cells. (Tsuji, R.F., and P.W. Askenase, manuscript in preparation).
In summary, this study shows that local C activation is involved in crucial early initiating events which are required to elicit CS and DTH. This may be due to C activation triggered via B cell–derived immunoglobulin, and subsequent local generation of C5a and/or the terminal C5b-9 complex, leading to local production of C-related chemotactic factors such as C5a, which facilitate local recruitment of Th-1 cells to then interact with APC to produce IFN-γ at the local site of CS/DTH to recruit an inflammatory infiltrate. These data provide a new example of an important augmenting role of C, which is usually regarded as a mediator of innate immunity but here acts as an initiator in an early required phase of acquired T cell immunity in vivo.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health to P.W. Askenase (AI-12211, AI-26689, and AI-07174).
Footnotes
The authors are indebted to Dr. Lindsay N. Donald of T Cell Sciences, Inc. for the kind gift of sCR1. We thank Drs. Peter J. Lachmann, Tony Hugli, Vipin Paliwal, and Rajani Ramabhadran for their valuable advice. We are also grateful to Marilyn Avallone for her superb secretarial skills.
Abbreviations used in this paper: CS, contact sensitivity; CVF, cobra venom factor; DTH, delayed-type hypersensitivity; PCl, picryl chloride (TNP-chloride); sCR1, soluble complement receptor type 1; ZAMS, zymosan activated mouse serum; 5-HT, serotonin (5-hydroxy-tryptamine).
REFERENCES
- 1.Fearon DT, Locksley RM. The instructive role of innate immunity in the acquired immune response. Science (Wash DC) 1996;272:50–54. doi: 10.1126/science.272.5258.50. [DOI] [PubMed] [Google Scholar]
- 2.Scharton-Kersten T, Scott P. The role of the innate immune response in Th1 cell development following Leishmania majorinfection. J Leukocyte Biol. 1995;57:515–522. doi: 10.1002/jlb.57.4.515. [DOI] [PubMed] [Google Scholar]
- 3.Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O'Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science (Wash DC) 1993;260:547–549. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- 4.Bendelac A. Mouse NK1+T cells. Curr Opin Immunol. 1995;7:367–374. doi: 10.1016/0952-7915(95)80112-x. [DOI] [PubMed] [Google Scholar]
- 5.Dempsey PW, Allison MED, Akkaraju S, Goodnow CC, Fearon DT. C3d of complement as molecular adjuvant: bridging innate and acquired immunity. Science (Wash DC) 1996;271:348–350. doi: 10.1126/science.271.5247.348. [DOI] [PubMed] [Google Scholar]
- 6.Colten HR. Drawing a double-edged sword. Nature (Lond) 1994;371:474–475. doi: 10.1038/371474a0. [DOI] [PubMed] [Google Scholar]
- 7.Heyman B, Wiersma EJ, Kinoshita T. In vivo inhibition of the antibody response by a monoclonal complement receptor specific antibody. J Exp Med. 1990;172:665–668. doi: 10.1084/jem.172.2.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Egwang TG, Befus AD. The role of complement in the induction and regulation of immune responses. Immunology. 1984;51:207–224. [PMC free article] [PubMed] [Google Scholar]
- 9.Nicholson-Weller A, Halperin JA. Membrane signaling by complement C5b-9, the membrane attack complex. Immunol Res. 1993;12:244–257. doi: 10.1007/BF02918256. [DOI] [PubMed] [Google Scholar]
- 10.Karp CL, Wysocka M, Wahl LM, Ahearn JM, Cuomo PJ, Sherry B, Trinchieri G, Griffin DE. Mechanism of suppression of cell-mediated immunity by measles virus. Science (Wash DC) 1996;273:228–231. doi: 10.1126/science.273.5272.228. [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto S, Dunn CJ, Capasso F, Deporter DA, Willoughby DA. Quantitative studies on cell-mediated immunity in the pleural cavity of guinea-pigs. J Pathol. 1974;117:65–73. doi: 10.1002/path.1711170202. [DOI] [PubMed] [Google Scholar]
- 12.Cochrane CG, Muller-Eberhard HJ, Aikin BS. Depletion of plasma complement in vivoby a protein of cobra venom: its effect on various immunologic reactions. J Immunol. 1970;105:55–69. [PubMed] [Google Scholar]
- 13.Askenase, P.W. 1993. Effector and regulatory mechanisms in delayed-type hypersensitivity (DTH). In Allergy: Principles and Practice, 4th edition. E. Middleton, Jr., C.E. Reed, E.F. Ellis, and N.F. Atkinson, editors. C.V. Mosby Co., St. Louis, MO. 362–389.
- 14.Van Loveren H, Meade R, Askenase PW. An early component of delayed-type hypersensitivity mediated by T cells and mast cells. J Exp Med. 1983;157:1604–1617. doi: 10.1084/jem.157.5.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geba GP, Ptak W, Anderson GM, Paliwal V, Ratzlaff RE, Levin J, Askenase PW. Delayed-type hypersensitivity in mast cell deficient mice: dependence on platelets for expression of contact sensitivity. J Immunol. 1996;157:557–565. [PubMed] [Google Scholar]
- 16.Matsuda H, Geba GP, Askenase PW. Human platelets can initiate contact sensitivity through serotonin release mediated by IgE. J Immunol. 1997;158:2891–2897. [PubMed] [Google Scholar]
- 17.Ameisen J-C, Meade R, Askenase PW. A new interpretation of the involvement of serotonin in delayed-type hypersensitivity: serotonin-2 receptor antagonists inhibit contact sensitivity by an effect on T cells. J Immunol. 1989;142:3171–3179. [PubMed] [Google Scholar]
- 18.Aune TM, Golden HW, McGrath KM. Inhibitors of serotonin synthesis and antagonists of serotonin 1A receptors inhibit T lymphocyte function in vitro and cell-mediated immunity in vivo. J Immunol. 1994;153:489–498. [PubMed] [Google Scholar]
- 19.Fong TAT, Mosmann TR. The role of IFN-γ in delayed-type hypersensitivity mediated by Th1 clones. J Immunol. 1989;143:2887–2893. [PubMed] [Google Scholar]
- 20.Piguet PF, Grau GE, Hauser C, Vassalli P. Tumor necrosis factor is a critical mediator in hapten-induced irritant and contact hypersensitivity reactions. J Exp Med. 1991;173:673–679. doi: 10.1084/jem.173.3.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwartz A, Kronke C, Trautinger F, Aragane Y, Neuner P, Luger TA, Schwartz T. Pentoxifylline suppresses irritant and contact hypersensitivity reactions. J Investig Dermatol. 1993;101:549–552. doi: 10.1111/1523-1747.ep12365951. [DOI] [PubMed] [Google Scholar]
- 22.Bernhagen J, Bacher M, Calandra T, Metz CN, Doty SB, Donnelly T, Bucala R. An essential role for macrophage migration inhibitory factor in the tuberculin delayed-type hypersensitivity reaction. J Exp Med. 1996;183:277–282. doi: 10.1084/jem.183.1.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsuji RF, Magae J, Nagai K, Yamasaki M. A novel in vivo screening method for immunomodulating substances: development of an assay system. Biosci Biotechnol Biochem. 1992;56:1497–1498. doi: 10.1271/bbb.56.1497. [DOI] [PubMed] [Google Scholar]
- 24.Tsuji RF, Uramoto M, Koshino H, Tsuji NM, Magae J, Nagai K, Yamasaki M. Preferential suppression of delayed-type hypersensitivity by L-156,602, a C5a receptor antagonist. Biosci Biotechnol Biochem. 1992;56:1686–1689. doi: 10.1271/bbb.56.1686. [DOI] [PubMed] [Google Scholar]
- 25.Tsuji RF, Magae J, Nagai K, Yamasaki M. Effects of L-156,602, a C5a receptor antagonist, on mouse experimental models of inflammation. Biosci Biotechnol Biochem. 1992;56:2034–2036. doi: 10.1271/bbb.56.2034. [DOI] [PubMed] [Google Scholar]
- 26.Tsuji RF, Yamakoshi J, Uramoto M, Koshino H, Saito M, Kikuchi M, Masuda T. Anti-inflammatory effects and specificity of L-156,602: comparison of effects on concanavalin A and zymosan-induced footpad edema, and contact sensitivity response. Immunopharmacology. 1995;29:79–87. doi: 10.1016/0162-3109(95)00047-w. [DOI] [PubMed] [Google Scholar]
- 27.Hensens OD, Borris RP, Koupal LR, Caldwell CG, Currie SA, Haidri AA, Homnick CF, Honeycutt SS, Lindenmayer SM, Schwartz CD, et al. L-156,602, a C5a antagonist with a novel cyclic hexadepsipeptide structure from Streptomycessp. MA6348. J Antibiot (Tokyo) 1991;44:249–254. doi: 10.7164/antibiotics.44.249. [DOI] [PubMed] [Google Scholar]
- 28.Frank MM, Fries LF. The role of complement in inflammation and phagocytosis. Immunol Today. 1991;12:322–326. doi: 10.1016/0167-5699(91)90009-I. [DOI] [PubMed] [Google Scholar]
- 29.Hugli TE, Müller-Eberhard J. Anaphylatoxins: C3 and C5a. Adv Immunol. 1978;26:1–53. doi: 10.1016/s0065-2776(08)60228-x. [DOI] [PubMed] [Google Scholar]
- 30.Hugli TE. Structural basis for anaphylatoxin and chemotactic functions of C3a, C4a and C5a. Crit Rev Immunol. 1981;1:321–366. [PubMed] [Google Scholar]
- 31.Meuer S, Ecker U, Hadding U, Bitter-Suermann D. Platelet-serotonin release by C3a and C5a: two independent pathways of activation. J Immunol. 1981;126:1506–1509. [PubMed] [Google Scholar]
- 32.Tsuji RF, Kikuchi M, Askenase PW. Possible involvement of C5/C5a in the efferent and elicitation phases of contact sensitivity. J Immunol. 1996;156:4644–4650. [PubMed] [Google Scholar]
- 33.Piddlesden SJ, Storch MK, Hibbs M, Freeman AM, Lassmann H, Morgan BP. Soluble recombinant complement receptor 1 inhibits inflammation and demyelination in antibody-mediated demyelinating experimental allergic encephalomyelitis. J Immunol. 1994;152:5477–5484. [PubMed] [Google Scholar]
- 34.Mulligan MS, Yeh CG, Rudolph AR, Ward PA. Protective effects of soluble CR1 in complement- and neutrophil-mediated tissue injury. J Immunol. 1992;148:1479–1485. [PubMed] [Google Scholar]
- 35.Weisman HF, Bartow T, Leppo MK, Marsh HC, Carson GR, Concino MF, Boyle MP, Roux KH, Weisfeldt ML, Fearon DT. Soluble human complement receptor type I: in vivo inhibitor of complement suppressing post-ischemia myocardial inflammation and necrosis. Science (Wash DC) 1990;249:146–151. doi: 10.1126/science.2371562. [DOI] [PubMed] [Google Scholar]
- 36.Wang Y, Rollins SA, Madri JA, Matis LA. Anti-C5 monoclonal antibody therapy prevents collagen- induced arthritis and ameliorates established disease. Proc Natl Acad Sci USA. 1995;92:8955–8959. doi: 10.1073/pnas.92.19.8955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Hu Q, Madri JA, Rollins SA, Chodera A, Matis LA. Amelioration of lupus-like autoimmune disease in NZB/W F1 mice after treatment with a blocking monoclonal antibody specific for complement component C5. Proc Natl Acad Sci USA. 1996;93:8563–8568. doi: 10.1073/pnas.93.16.8563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Askenase PW, Hayden BJ, Gershon RK. Augmentation of delayed-type hypersensitivity by dose of cytoxan which do not affect antibody responses. J Exp Med. 1975;141:697–702. doi: 10.1084/jem.141.3.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Churchill WH, Jr, Weintraub RM, Borsos T, Rapp HJ. Mouse complement: the effect of sex hormones and castration on two of the late-acting components. J Exp Med. 1967;125:657–672. doi: 10.1084/jem.125.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lewis E, Turk JL. Comparison of the effect of various antisera and cobra venom factor on inflammatory reactions in guinea-pig skin. I. Non-specific inflammation due to the intradermal injection of turpentine. J Pathol. 1975;115:97–109. doi: 10.1002/path.1711150206. [DOI] [PubMed] [Google Scholar]
- 41.Kitamura D, Roes J, Kühn R, Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobin μ chain gene. Nature (Lond) 1991;350:423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- 42.Grabbe S, Steinert M, Mahnke K, Schwartz A, Luger TA, Schwarz T. Dissection of antigenic and irritative effects of epicutaneously applied haptens in mice. Evidence that not the antigenic component but nonspecific proinflammatory effects of haptens determine the concentration-dependent elicitation of allergic contact dermatitis. J Clin Invest. 1996;98:1158–1164. doi: 10.1172/JCI118899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Colten HR, Strunk RC, Perlmutter DH, Cole FS. Regulation of complement protein biosynthesis in mononuclear phagocytes. Ciba Found Symp. 1986;118:141–154. doi: 10.1002/9780470720998.ch10. [DOI] [PubMed] [Google Scholar]
- 44.Johnson E, Hetland G. Mononuclear phagocytes have the potential to synthesize the complete functional complement system. Scand J Immunol. 1988;27:489–493. doi: 10.1111/j.1365-3083.1988.tb02375.x. [DOI] [PubMed] [Google Scholar]
- 45.Garred P, Hetland G, Mollnes TE, Stoervold G. Synthesis of C3, C5, C6, C7, C8 and C9 human fibroblasts. Scand J Immunol. 1990;32:555–560. doi: 10.1111/j.1365-3083.1990.tb03196.x. [DOI] [PubMed] [Google Scholar]
- 46.Johnson E, Hetland G. Human umbilical vein endothelial cells synthesize functional C3, C5, C6, C8 and C9 in vitro. . Scand J Immunol. 1991;33:667–671. doi: 10.1111/j.1365-3083.1991.tb02539.x. [DOI] [PubMed] [Google Scholar]
- 47.Petit J-C. Resistance to listeriosis in mice that are deficient in the fifth component of complement. Infect Immun. 1980;27:61–67. doi: 10.1128/iai.27.1.61-67.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abe M, Kondo T, Xu H, Fairchild RL. Interferon-γ inducible protein (IP-10) expression is mediated by CD8+ T cells and is regulated by CD4+T cells during the elicitation of contact hypersensitivity. J Investig Dermatol. 1996;107:360–366. doi: 10.1111/1523-1747.ep12363337. [DOI] [PubMed] [Google Scholar]
- 49.Sims PJ, Wiedmer T. The response of human platelets to activated components of the complement system. Immunol Today. 1991;12:338–342. doi: 10.1016/0167-5699(91)90012-I. [DOI] [PubMed] [Google Scholar]
- 50.Foreman KE, Vaporciyan AA, Bonish BK, Jones ML, Johnson KL, Glovsky MM, Eddy SM, Ward PA. C5a-induced expression of P-selectin in endothelial cells. J Clin Invest. 1994;94:1147–1155. doi: 10.1172/JCI117430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kilgore KS, Shen JP, Miller BF, Ward PA, Warren JS. Enhancement by complement membrane attack complex of tumor necrosis factor-α-induced endothelial cell expression of E-selectin and ICAM-1. J Immunol. 1995;155:1434–1441. [PubMed] [Google Scholar]
- 52.Vaporciyan AA, Mulligan MS, Warren JS, Barton PA, Miyasaka M, Ward PA. Up-regulation of lung vascular ICAM-1 in rats is complement dependent. J Immunol. 1995;155:1442–1449. [PubMed] [Google Scholar]
- 53.Mulligan MS, Schmid E, Till GO, Hugli TE, Friedl HP, Roth RA, Ward PA. C5a-dependent up-regulation in vivo of lung vascular P-selectin. J Immunol. 1997;158:1857–1861. [PubMed] [Google Scholar]
- 54.Gerard CG, Chenoweth DE, Hugli TE. Molecular aspects of the serum chemotactic factors. J Reticuloendothel Soc. 1979;26:711–718. [PubMed] [Google Scholar]
- 55.Hopken UE, Lu B, Gerard NP, Gerard C. The C5a chemoattractant receptor mediates mucosal defence to infection. Nature (Lond) 1996;383:86–89. doi: 10.1038/383086a0. [DOI] [PubMed] [Google Scholar]