

Abstract
Jaw1 is an endoplasmic reticulum (ER) resident protein representative of a class of proteins post translationally inserted into membranes via a type II membrane anchor (cytosolic NH2 domain, lumenal COOH domain) in a translocon-independent manner. We found that Jaw1 can efficiently deliver a COOH-terminal antigenic peptide to class I molecules in transporter associated with antigen processing (TAP)-deficient cells or cells in which TAP is inactivated by the ICP47 protein. Peptide delivery mediated by Jaw1 to class I molecules was equal or better than that mediated by the adenovirus E3/19K glycoprotein signal sequence, and was sufficient to enable cytofluorographic detection of newly recruited thermostabile class I molecules at the surface of TAP-deficient cells. Deletion of the transmembrane region retargeted Jaw1 from the ER to the cytosol, and severely, although incompletely, abrogated its TAP-independent peptide carrier activity. Use of different protease inhibitors revealed the involvement of a nonproteasomal protease in the TAP-independent activity of cytosolic Jaw1. These findings demonstrate two novel TAP-independent routes of antigen processing; one based on highly efficient peptide liberation from the COOH terminus of membrane proteins in the ER, the other on delivery of a cytosolic protein to the ER by an unknown route.
MHC class I molecules bind peptides of 8–10 residues derived from intracellular proteolytic degradation and present them at the cell surface to CD8+ T lymphocytes (TCD8+) (1, 2). In the absence of high affinity peptide ligands, cell surface class I molecules are unstable at 37°C and rapidly denature (3). Such denaturation can often be detected by mAbs specific for the α1α2 domains: the binding of such mAbs to live cells provides a measure of the capacity of cells to produce class I molecules with stable peptide ligands.
The generation of the majority of class I–associated peptides involves cytosolic proteolysis. Little is known about how proteins are targeted in the cytosol for the production of class I–binding peptides. The nature of the proteases involved is only slightly better defined. The proteasome, an abundant, heterogeneous, macromolecular multicatalytic protease, has been implicated in the generation of a substantial portion of class I–binding peptides (4, 5). Other cytosolic proteases might also contribute to peptide generation, because proteasome inhibitors only partially block class I assembly and antigen presentation (6–9). Peptides of 8–16 or so residues produced by cytosolic proteolysis are transported into the endoplasmic reticulum (ER)1 by the transporters associated with antigen processing (TAP), the MHC-encoded member of the ATP binding cassette transporter family of proteins (10–13). Longer peptides may also be transported, but at much reduced efficiency (14).
Functional TAP is required for the optimal assembly of class I molecules, as shown by the poor cell surface expression of class I molecules by TAP-deficient cells (15–18). This is due to absence of peptides in the ER, because delivery of peptides to the ER by appendage of a hydrophobic signal sequence can restore surface expression of class I molecules (19–22). Such peptides are thought to enter the ER by transiting the translocon, where signal peptidase liberates the class I–binding peptide from the hydrophobic signal sequence.
The ability of TAP to transport peptides longer than those usually recovered from class I molecules raises the possibility of peptide trimming in the ER, with peptide either free or bound to class I as originally proposed (23). Using TAP-deficient cells, it has been shown that class I–binding peptides can be liberated from longer precursors targeted to the ER via the translocon (24, 25). Peptide liberation occurs most readily from short precursors, but under some circumstances, class I–binding peptides can be derived from full-length proteins (26).
In the present study, we explore the capacity of ER-associated proteases to process antigenic peptides from the lumenal domain of Jaw1. Jaw1 is an ER resident protein whose known expression is limited to cells of hematopoietic origin (27). Jaw1 lacks a NH2-terminal signal sequence, and is inserted into the membrane posttranslationally by a hydrophobic transmembrane region at residues 480–503 (28). Jaw1 consists of a large cytosolic domain of several coiled coils, the aforementioned transmembrane region, and a 35-residue lumenal tail (see Fig. 1). The membrane topology of Jaw1 and posttranslational insertion into the ER are representative of a number of integral membrane proteins (29). The membrane insertion of these proteins appears to occur independently of the translocon. In the course of investigating the antigen processing of a form of Jaw1 lacking the membrane anchor/insertion sequence, we unexpectedly encountered a novel route of delivery of antigenic peptides to class I molecules whose generation is dependent on a nonproteasomal activity.
Figure 1.


(A) Schematic representation of rVV expressed Jaw1 chimeric proteins. (B) Biochemical characterization of Jaw1 chimeric proteins. Species reactive with anti-Jaw1 antisera present in detergent extracts of rVV-infected cells pulse radiolabeled and chased for the indicated time in hours were analyzed by SDS-PAGE and visualized using a PhosphorImager. Only the region containing the antibody-reactive species is shown. The far left lane contains molecular weight markers indicated. The asterisk identifies a proteolytic fragment created by the action of Kex2 on Jaw1[NP147–155].
MATERIALS AND METHODS
Biochemical Procedures.
T2 or L929 cells (2 × 106) were infected with recombinant vaccinia virus (rVV) for 3 h at 37°C, incubated for 30 min in 5 ml methionine-free DMEM (Biofluids, Rockville, MD) to deplete intracellular methionine pools. Infected cells were labeled with 50 μCi [35S] methionine (Amersham, Arlington Heights, IL) in 200 μl methionine-free DMEM for 5 min. After washing in PBS containing 2 mg/ml free methionine (PBS/Met), cells were chased in IMDM for the indicated times. Cells were pelletted by centrifugation and suspended in lysis buffer (0.15 M NaCl, 1 mM EDTA, 1% Triton X-100, 10 mM Tris–HCl, pH 7.4, Complete™ protease inhibitor cocktail) (Boehringer Mannheim, Indianapolis, IN). Where indicated, cells were pretreated and proteasome inhibitors added at the concentrations described above. Lysates were collected with affinity-purified rabbit anti-Jaw polyclonal antibodies (28) conjugated to protein A–Sepharose (Pierce, Rockford, IL) for 2 h at 4°C with constant rotation. Beads were washed extensively and boiled in sample buffer containing 0.125 M Tris–HCl, pH 6.8, 4% SDS, 20% glycerol, 10% 2-mercaptoethanol. Samples were analyzed by SDS-PAGE according to Laemmli (30). Gels were dried by vacuum and exposed to phosphorescent screens overnight and for 1 wk. Screens were imaged using a Phosphor Imager (Molecular Dynamics, Sunnyvale, CA). Images were prepared using Adobe Photoshop and printed with a Fujix Pictrography digital printer (Fuji Medical Systems, Stamford, CT).
Cell Lines.
L929 cells (American Type Culture Collection, Rockville, MD) and L929 transfected with genes coding for the MHC molecules Kb (L-Kb) or Db (L-Db cells) were maintained in DMEM supplemented with 7.5% FBS. T2 cells (16) and their class I transfectants T2-Kk, T2-Db were maintained in IMDM supplemented with 7.5% fetal bovine serum (FBS) (vol/vol). T2-Kd cells were maintained in RPMI-1640 supplemented with 7.5% FBS. All cell lines were incubated at 37°C, 91% Air, 9% CO2.
Cytofluorography.
T2-Kd cells were infected with rVV at 10 PFU/cell for 16 h at 37°C with gentle rotation. Cells were washed in ice-cold PBS containing 1% rabbit serum, and incubated with SF1.1.1 mAb conjugated to FITC (PharMingen, San Diego, CA) at a 1:10 dilution for 30 min at 0°C, washed extensively in ice-cold PBS, and analyzed with a FACScan® (Becton Dickinson, San Jose, CA). Cells were suspended in ethidium homodimer (Molecular Probes, Eugene, OR) (10 μg/ml), and analysis was restricted to viable cells (nonfluorescent in FL3 photomultiplier tube).
Electron Microscopy.
T2 cells were infected with rVVs for 6 h. Cells were washed twice in PBS and fixed in 0.5% glutaraldehyde for 15 min at room temperature. Cells were washed twice in PBS and fixed in a mixture of 3% paraformaldehyde and 0.5% glutaraldehyde in PBS for 15 min at room temperature. The cells were washed in PBS and further processed for cryomicrotomy and immunolabeling essentially according to methods described by Tokuyashu (31). Frozen–thawed sections were indirectly labeled with a 1:500 dilution of rabbit anti-Jaw antiserum followed by 10-nm colloidal gold coated with protein A (Aurion, Wageningen, Netherlands). The ultrathin sections were examined with a Philips EM400 electron microscope.
Protease Inhibitors.
cbz–LL–CHO, cbz–LLL–CHO, and cbz– LLF–CHO were synthesized as described (32). Lactacystin was purchased from E.C. Corey (Harvard University, Cambridge, MA). N–Ac–LLnL and N–Ac–LLnM were purchased from Calbiochem Novabiochem (La Jolla, CA). Other inhibitors were purchased from Sigma Chemical Co. (St. Louis, MO).
Mice.
6–8-wk-old BALB/cByJ, CBA/J, and C57BL/6J mice were purchased either from the Jackson Laboratories (Bar Harbor, ME) or Taconic Farms (Germantown, NY). Mice were primed intravenously with rVVs (5 × 106 PFU) or intraperitoneally with PR8 (200HAU) in BSS/BSA and spleens taken at least 3 wk after priming.
Microcytoxicity Assays.
Target cells (2 × 106) were infected with rVV (2 × 107 PFU) for 1 h in balanced salt solution, 0.2% BSA (BSS/BSA), followed by an additional 3-h incubation in IMDM. Cells were labeled with 20 μl IMDM with 10 μCi Na51CrO4 (Amersham, Arlington Heights, IL) for 1 h at 37°C and washed in IMDM. Target cells were suspended in IMDM and incubated with splenic effector cells for 6 h at 37°C. Effector cells were generated from splenocytes primed with rVV expressing full-length protein antigen or minigenes and stimulated in vitro by PR8-infected autologous spleen cells. Where indicated, brefeldin A (BFA) (Sigma Chemical Co.) was added to cells at 5 μg/ml and maintained throughout the remaining incubations. In experiments with peptidyl aldehyde inhibitors, target cells were pretreated with either 25 μM inhibitor or as indicated in the Fig. 6 legend for 30 min at 37°C. Inhibitors were present during the 4-h infection.
Figure 6.

Effect of protease inhibitors on generation of the NP147–155 peptide. rVV-infected T2-Kd cells expressing the indicated protein were incubated with the indicated protease inhibitor starting 30 min before infection and ending 4 h after infection. BFA was then added to cells to prevent additional antigen presentation and cells were incubated with NP147–155-specific TCD8+ in a standard microcytotoxicity assay at the effector to target ratios indicated. Inhibitors were used at the following concentrations: NH4Cl, 25 mM; cbz–LL–CH, 12.5 μM; cbz–LLL–CHO 12.5 μM (A and B), 25 μM (C); lactacystin. 10 μM; N–Ac–LLnL, 200 μM; N–Ac–LLnM, 200 μM. The three panels are derived from separate experiments.
Peptide Extraction.
Peptides were extracted in trifluoroacetic acid as previously described (24) with minor modifications. T2 cells (109) were coinfected with rVV expressing Kd or EC15Kd and rVV-expressing Jaw1 constructs at multiplicity of infection (MOI) of 10 for 1 h at 37°C. Cells were incubated at 37°C for an additional 16 h in 100 ml IMDM. Cell pellets were lysed in 10 ml 0.1% trifluoroacetic acid, Dounce homogenized, and sonicated. Lysates were passed through Macrosep filters (Filtron Technology Corp., Northborough, MA) to collect material <3,000 kD. Filtrates were vacuum concentrated and resuspended in 500 μl PBS. Serial twofold dilutions were incubated with 51Cr-labeled P815 cells in 50 μl for 2 h at 26°C. To maximize binding of exogenous peptides, P815 cells were cultured overnight at 26°C with 5 μg/ ml human β2-microglobulin (Sigma). Nucleoprotein (NP)-specific effector cells were added at 10:1 effector to target ratio and incubated an additional 6 h. Percent-specific lysis was calculated as described above.
Viruses.
The influenza virus A/PR8/(H1N1) (PR8) was grown in 10-d-old embryonated chicken eggs and used as infectious allantoic fluid. Recombinant VV were grown in thymidine kinase minus (TK−) human 143B osteosarcoma cells. rVV expressing NP, SNP, cytosolic and ER-targeted peptides have been described (20, 33). VV–ICP47 (34) and VV–Kex2 (35) were provided by B. Rouse (University of Tennessee, Knoxville, TN) and D. Thomas (University of Oregon, Eugene, OR), respectively. Jaw1[NP147–155] and Jaw1 (Lum−)[NP147–155] were engineered by PCR of the full-length Jaw1 mouse cDNA using the 5′ primer CTATTAGGTGACACTATAG A A CAGACACCATGGCTCTCTGTG-TAAAAGGT-CCC with unique 3′ primers GGGGTACCTCATCACACTAGTGCTCGTGTTCGCTGGTATGTAGCCTCCACGGCTGTCTG (Jaw1(Lum−)[NP147–155] and GGGTACCTCATCAC-CTAGTGCTCGTGTTCGCTGGTATGTTCGTTTCGTCGCACTGGCGGTGGTCCATC (Jaw1[NP147–155]). The constructs contain a β-globin leader sequence upstream of the initiator ATG of Jaw1. Jaw1(Lum−) [NP147–155] has the NP147–155 peptide TYQRTRALV fused in-frame after amino acid Ala509 and is followed by two stop codons. Jaw1[NP147–155] has the Kex2/furin protease site RRKR and the peptide TYQRTRALV fused in-frame following the penultimate Val539 of the Jaw1 cDNA. Both PCR fragments were cloned blunt-ended into the SmaI site of pSP72 (Promega), digested with SalI and KpnI, and then subcloned into pSC11. Jaw1(TM−)[NP147–155] was engineered by first amplifying the COOH-terminal half of the Jaw1 cDNA using an internal primer 5′ GGGCTGGTGTCAGGCATG and a 3′ primer GGGGTACCTCATCACACTAGTGCTCGTGTTCGCTGGTATGTGACCCAGGAAGCCACTGA, which placed NP147–155 in-frame with Jaw1 following amino acid Val467 (upstream of the transmembrane domain). This fragment was digested with NaeI and KpnI and then subcloned into the same sites of the Jaw1(Lum−)[NP147–155] plasmid, replacing the COOH-terminal coding region of that plasmid. The [NP147–155]Jaw1 construct used the following primers in PCR of the Jaw1 cDNA: 5′-GATCGTCGACAAACAGACACCATGACATACCAGCGAACACGAGCACTAGTGCTCTGTGTAAAAGGTCCC and 3′-CATTGAGCTGCACGTCAGTCA. This construct fuses the immunogenic peptide in frame immediately following the initiator ATG of full-length Jaw1. The PCR fragment was digested with SalI and BamHI and cloned into pSP72 before shuttling into pSC11 and pSC65. All constructs were sequenced to ensure fidelity of PCR.
RESULTS
Characterization of Jaw1-expressing rVVs.
To study the ability of Jaw1 to deliver an antigenic peptide to the ER, we produced rVVs encoding the NP147–155 peptide at the COOH terminus of either intact Jaw1 (Jaw[NP147–155]), or truncated versions lacking the lumenal domain (Jaw1(Lum−)[NP147–155]), or both the lumenal and transmembrane domains (Jaw1 (TM−)[NP147–155]). In designing Jaw1[NP147–155] we inserted an additional tetrapeptide sequence at the peptide– Jaw1 interface that enables cleavage by the yeast Kex2 protease. We also produced a rVV-expressing Jaw1 with the NP147–155 peptide appended to the NH2 terminus ([NP147–155] Jaw1) that serves as a topological control for the COOH-terminal NP147–155-expressing chimeras. The various Jaw1 chimeric molecules are depicted schematically in Fig. 1 A.
Jaw1-containing gene products were initially characterized by SDS-PAGE of material collected with polyclonal rabbit anti-Jaw1 antibodies from [35S]methionine-labeled detergent extracts prepared from rVV-infected cells (Fig. 1 B). Cells were radiolabeled for 5 min at 37°C and chased for up to 3 h at this temperature. In previous studies, we observed that Jaw1 migrates in SDS-PAGE with apparent mobility 10 kD greater than predicted (27). VV-encoded Jaw1 similarly migrates more slowly than predicted. This may be due to posttranslational modifications, but any such modifications would have to be rapid and complete because pulse-labeled material migrated as a single species. Therefore, it is more likely that the protein migrates aberrantly in SDS-PAGE. None of the Jaw1 chimeric proteins are N-glycosylated as determined by unaltered mobility after endoglycosidase H digestion (data not shown).
As anticipated, Jaw1 (TM−)[NP147–155] migrated most rapidly, followed by Jaw1(Lum−)[NP147–155]. Neither of these proteins was detectably modified over the 3-h chase period. Jaw1[NP147–155] migrated with an apparent molecular mass of 4 kD greater than Jaw1(Lum−)[NP147–155], which is consistent with the addition of a 42-residue lumenal domain. [NP147–155] Jaw1 migrated 3 kD more slowly than Jaw1(Lum−)[NP147–155]. Sequencing of the 5′ and 3′ ends of all of the genes revealed the expected sequences, so we again attribute the different mobilities to nonclassical SDS-PAGE behavior. During the chase period, [NP147–155] Jaw1 and Jaw1[NP147–155] were converted into more rapidly migrating forms. We previously showed this was likely due to proteolysis of the COOH terminus (28). By contrast, Jaw1(Lum−)[NP147–155] and Jaw1(TM−)[NP147–155] were not detectably cleaved over the 3-h chase period. This confirms the absence of lumenal domains in these proteins. The function of the Kex2 protease site was confirmed by coinfection of cells with VV–Jaw1[NP147–155] and a rVV that directs the synthesis of Kex2, a secretory protease expressed at limiting levels by most cells (35). This resulted in the presence of a novel species migrating slightly faster than the pulse-labeled material (designated by an asterisk in Fig. 1 B), consistent with the removal of a small terminal peptide.
The intracellular targeting of two of the recombinant Jaw1 constructs was examined by cryoimmuno electron microscopy using colloidal gold-conjugated secondary antibodies to detect binding of anti-Jaw1 antibodies (Fig. 2). Almost all Jaw1(Lum−)[NP147–155] detected was associated with intracellular membranes, whereas Jaw1(TM−)[NP147–155] localized to the cytosol and was not specifically associated with membranes. Immunofluorescence of fixed and permeabilized cells with the anti-Jaw1 antiserum confirmed the expected location of each of the four Jaw1-chimeric proteins (data not shown).
Figure 2.
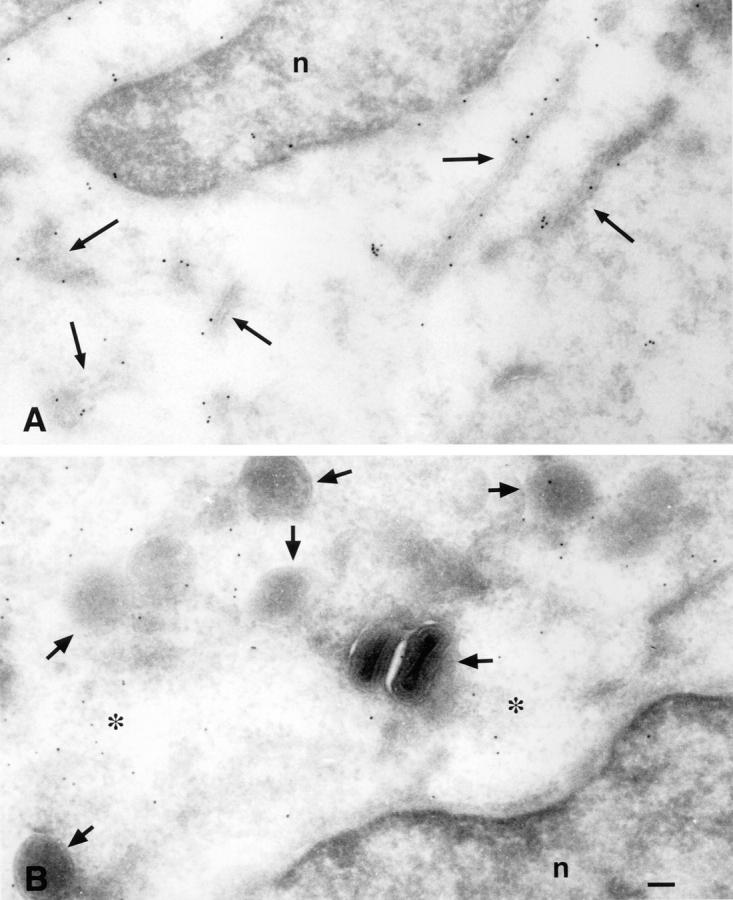
Immunogold localization of Jaw1 proteins. Jaw1 was located in rVV-infected T2 cells by cryoimmunogold labeling. (A) Cells expressing Jaw1 (Lum−)[NP147–155]. Gold particles specifically decorate the nuclear membrane and cytoplasmic membrane structures. (B) Cells expressing Jaw1 (TM−)[NP147–155]. Gold particles are located in the cytosol. In addition to the difference in patterns between A and B, the specificity of Jaw staining was shown by the absence of gold particles on sections prepared from cells infected with a control rVV.
Liberation of NP147–155 from Jaw 1 Constructs: Antigen Presentation.
The antigen processing of Jaw1 constructs was studied in T2 cells (a TAP-deficient human lymphoid cell line) or L929 cells (a TAP-expressing mouse cell line). To enable recognition by Kd-restricted, NP-specific TCD8+, cells were coinfected with a rVV expressing Kd in addition to the Jaw1-expressing rVV. As seen in Fig. 3, L929 cells presented NP147–155 from the COOH termini of both Jaw1(Lum−)[NP147–155] and Jaw1(TM−)[NP147–155]. Presentation from the NH2 terminus of Jaw1 occurred at much lower levels. Because a Met-initiated cytosolic minigene product that is identical to the 10 NH4-terminal residues of [NP147–155] Jaw1 is effectively presented by L929 cells (designated in Fig. 3 and [NP147–155]), this indicates that the Jaw1-derived COOH-terminal flanking sequences cannot be efficiently removed from NP147–155 by cytosolic proteases. This compromises the use of this construct as a control in T2 cells, but only partially, because the activities of cytosolic proteases and secretory proteases are expected to be independent.
Figure 3.

Presentation of NP147–155 by rVV-infected cells. rVV-infected cells expressing the Jaw chimeric proteins were tested for lysis by NP-specific secondary TCD8+ at the indicated effector to target ratio. Cells were coinfected with a rVV expressing Kd to enable recognition.
In T2 cells, both Jaw1(Lum−)[NP147–155] (Fig. 3) and Jaw1[NP147–155] (data not shown) were efficiently presented after short VV infections (<4 h). This cannot be attributed to leakiness of the cells for cytosolic peptides, because under the same conditions, the cytosolic minigene, that is produced in enormous amounts relative to the amount of peptide liberated in the cytosol from proteins (36), was not presented above background values obtained with a control VV. [NP147–155] Jaw1-infected cells were recognized by NP-specific TCD8+ at levels observed using cells infected with control rVVs. This is consistent with the predicted topology of Jaw1, but we cannot eliminate the possibility that the secretory pathway, like the cytosol, is incapable of liberating the NP147–155 peptide from the NH2 terminus of Jaw1. NP147–155 was liberated from Jaw1(TM−)[NP147–155], but at far lower efficiency than from the transmembrane domain containing molecules (see Figs. 4 and 5).
Figure 4.

Rescue of Kd cell surface expression. T2-Kd cells were infected with the rVV indicated and the amount of cell surface Kd present on viable cells cytofluorographically determined using a directly conjugated Kd-specific mAb. Data are expressed as mean channel fluorescence × 100. The last three bars on the right represent cells coinfected with Jaw1[NP147–155] and the rVV indicated (ACE, angiotensin-converting enzyme).
Figure 5.

Quantitation of acid-soluble peptides in cell extracts. Extracts prepared from cells expressing ER retained Kd (Kd Ret) and the indicated Jaw1 chimeric protein were tested for their ability to sensitize P815 cells for lysis by NP-specific TCD8+. Cell mix refers to a sample in which ER-retained Kd expressing cells were mixed with Jaw1(Lum−)[NP147–155]- expressing cells before lysis.
Liberation of NP147–155 from Jaw1-constructs: cytofluorography.
We recently found that after infection of T2-Kd cells with rVVs encoding ER-targeted class I–binding peptides, enhanced Kd cell surface expression can be cytofluorographically detected after staining with Kd-specific mAbs (37). This provides a much more quantitative measure of the efficiency of antigen presentation than TCD8+-mediated lysis. As previously observed, infection with a rVV expressing ER-targeted NP147–155 resulted in a clear increase in the amount of mAb-reactive cell surface Kd on the surface of T2-Kd cells (Fig. 4). Remarkably, the greatest increase in Kd expression was observed after infection with VV–Jaw1 (Lum−)[NP147–155]. Kd was rescued slightly less efficiently by expression of Jaw1[NP147–155]. These effects can be attributed to the presence of the NP147–155 peptide at the COOH terminus of Jaw1, because expression of [NP147–155] Jaw1 had no effect on Kd expression. Thus, peptides generated from Jaw1 sequences do not themselves bind Kd and rescue expression. These findings demonstrate that Jaw1 efficiently delivers peptides to the class I processing pathway. In contrast with membrane-targeted Jaw1, expression of Jaw1(TM−)[NP147–155] had no effect on Kd expression. This demonstrates the importance of proper insertion of Jaw1 [NP147–155] into the membrane to enable efficient peptide liberation in the secretory pathway.
In this experiment, we also examined whether liberation of NP147–155 from Jaw1[NP147–155] could be enhanced by coexpression of the yeast Kex2 protease encoded by a rVV. Because this entails coinfection with two rVVs, it was necessary to control for competition between rVVs for gene expression. Coinfection with VV reduced the Jaw1[NP147–155]- mediated rescue of Kd expression. Above this baseline value, Kex2 coexpression clearly increased Kd rescue, whereas a rVV expressing the secretory carboxypeptidase angiotensin-converting enzyme had little effect on Kd expression, demonstrating the specificity of Kex2-mediated enhancement. These findings indicate that it is possible to modify the antigen processing of Jaw1 by coexpressing a protease.
NP147–155 Liberated from Jaw1 Binds Kd in the ER of T2 Cells.
To examine the site of liberation of NP147–155 from Jaw1, we extracted peptides from cells coinfected with the Jaw1 expression rVVs and VV–EC15Kd. EC15Kd is a modified Kd molecule retained in the early secretory pathway by replacement of the normal cytosolic domain with that of E3/19K (38). The low relative molecular mass peptides present in trifluoroacetic acid homogenates prepared from infected cells were used to sensitize P815 cells for lysis by NP-specific TCD8+. As seen in Fig. 5, antigenic peptides were recovered from cells expressing either Jaw1(Lum−) [NP147–155] or Jaw1(TM−)[NP147]. At least eightfold greater quantities of peptide were recovered from Jaw1(Lum−) [NP147–155] expressing cells than from Jaw1 (TM−)[NP147–155]- expressing cells. In an additional experiment in which we reached an endpoint with all of the lysates, we recovered ∼40-fold more NP147–155 activity from cells expressing Jaw1(Lum−)[NP147–155] than those expressing Jaw1(TM−) [NP147–155]. These findings are consistent with data in Fig. 4, in demonstrating that ER-targeted Jaw1 is a much more efficient vehicle for TAP-independent loading of Kd molecules with NP147–155 than cytosolic Jaw1. Peptide recovery from Jaw1(TM−)[NP147]-expressing cells absolutely required coexpression of EC15Kd, whereas recovery from cells expressing Jaw1(Lum−)[NP147–155] was largely, but not entirely dependent on the EC15Kd expression (<5% of peptides were recovered in a Kd-independent manner). This small Kd-independent population of peptides may derive from endogenously expressed human class I molecules by T2 cells that weakly bind the peptides. Alternatively, the peptide (or a precursor that can be processed by cell surface of serum proteases in the course of sensitizing target cells), may be produced/stabilized in a class I–independent manner in the secretory pathway. To demonstrate that peptide association with Kd occurred before lysis of the cells, cells expressing VV–EC15Kd were mixed with VV–Jaw1(Lum−) [NP147–155]-infected cells and then lysed (Fig. 5, Cell Mix). This resulted in only a slight increase in the amount of peptide present in the extracts relative to VV–Jaw1(Lum−) [NP147–155]-infected cell lysates, demonstrating that the vast majority of NP147–155 liberated from Jaw1(Lum−)[NP147–155] associates with Kd intracellularly. Based on these findings, we conclude that NP147–155 is produced from both Jaw1(TM−) [NP147–155] and Jaw1 (Lum−)[NP147–155] in the early secretory pathway of T2 cells, probably the ER itself.
NP147–155 Is Liberated from Jaw1(TM−)[NP147–155] by a Novel Protease Activity.
The unexpected presentation of NP147–155 from Jaw1(TM−)[NP147] in T2 cells hinted at an unusual mechanism of peptide entry into the class I presentation pathway. The cellular localization of Jaw1(TM−)/ NP147–155 suggested cytosolic proteases may participate in liberating NP peptide from Jaw1(TM−). Peptide aldehyde inhibitors have been used to characterize protease activities that contribute to the production of class I–binding peptides. We initially studied the effect of the peptide aldehyde inhibitor cbz–LLL–CHO on the liberation of NP147–155 from different precursors in T2 cells. cbz–LLL–CHO blocks all of the known activities of 20S proteasomes in vitro, and causes the accumulation of the ubiquitinated proteins in vivo (4, 32). T2 cells were treated with cbz– LLL–CHO before infection and throughout the 4-h incubation period before the 51Cr release assay. At this time, cbz–LLL–CHO was replaced with BFA to prevent further delivery of class I–peptide complexes produced after the removal of cbz–LLL–CHO.
Using cbz–LLL–CHO at concentrations 12.5 or 25 μM did not detectably affect the liberation of NP147–155 from Jaw1(Lum−)[NP147–155] (Fig. 6). By contrast, recognition of Jaw1(TM−)[NP147] expressing cells was dramatically reduced by cbz–LLL–CHO at either concentration. The effects of cbz–LLL–CHO cannot be attributed to blocking viral gene expression as demonstrated by cytofluorographic analysis of cells infected with a rVV-expressing mouse ICAM 1. Cell surface levels of ICAM detected with saturating amounts of a directly conjugated mAb were not altered by cbz–LLL–CHO under the conditions used for studying antigen presentation (data not shown).
In addition to its effects on proteasomes, cbz–LLL–CHO inhibits other cytosolic and lysosomal proteases. To determine the nature of the target protease, we examined the effects of five additional protease inhibitors on the liberation of NP147–155 in T2 cells (Fig. 6). None of the inhibitors blocked presentation of NP147–155 by Jaw1(Lum−)[NP147–155]- expressing cells, indicating that global effects antigen presentation or viral gene expression are minimal and, therefore, that any effects on presentation of Jaw1(TM−)[NP147]- expressing cells are due to specific blockade of determinant liberation. Two of the additional inhibitors tested, cbz–LLF– CHO (data not shown) and cbz–LL–CHO blocked the liberation of NP147–155 from Jaw1(TM−)[NP147]. cbz–LLF– CHO inhibits both proteasomes and other cellular proteases, whereas cbz–LL–CHO does not detectably affect proteasome activity at the concentration used (39), suggesting that the effects of peptide aldehyde inhibitors on presentation of Jaw1(TM−)[NP147] are due to blockade of nonproteasomal proteases. This was confirmed by the failure of lactacystin to affect presentation of Jaw1(TM−)[NP147]. Lactacystin is the most specific proteasome inhibitor that acts by covalently binding to an enzymatic subunit (40). Like cbz–LLF–CHO, it predominantly blocks the chymotryptic-like, and tryptic-like activities of proteasomes.
It was recently reported that processing of signal peptides in the ER of T2 cells was inhibited by 250 μM N–Ac–LLnL (6). Presentation of Jaw1(TM−)[NP147] was not significantly inhibited by N–Ac–LLnL (or N–Ac–LLnM) at concentrations of 200 μM (Fig. 6) or 300 μM (data not shown), demonstrating that this protease activity is not required for processing of Jaw1(TM−)[NP147]. Presentation was also unaffected by NH4Cl (Fig. 6) or leupeptin (data not shown) indicating that endosomal proteases unlikely to be involved in processing. The sensitivity of Jaw1(TM−)/ NP147–155 to protease inhibitors demonstrates that it and Jaw1(Lum−)[NP147–155] are processed in a fundamentally different manner by TAP-deficient cells.
TAP-independent Processing of Jaw1TM−[NP147] Is Not Lymphoid Restricted.
Because a homolog of mouse Jaw1 is expressed endogenously by T2 cells, it was plausible that a protein with Jaw1 receptor activity might participate in delivering Jaw1(TM−)[NP147] to the ER of T2 cells. We examined the presentation of Jaw1(TM−)[NP147] in HeLa cells, which do not detectably express Jaw1 (27). To block the function of TAP, cells were coinfected with a rVV-expressing herpes simplex virus ICP47, a potent inhibitor of human TAP function (41). Coexpression of ICP47 blocked presentation of VV determinants to polyclonal VV-specific TCD8+, and also full-length NP to NP147–155-specific TCD8+ (Fig. 7). By contrast, the presentation of Jaw1(Lum−) [NP147–155] or Jaw1(TM−)[NP147–155] was unaffected. This demonstrates that the TAP-independent presentation of the two protein occurs in cells that do not naturally express Jaw1.
Figure 7.

Tap-independent presentation in nonlymphoid cells. HeLa cells coinfected with rVVs expressing Kd, a Jaw1 chimerical protein, and either ICP47 or no foreign protein were tested for recognition by TCD8+ populations specific for NP or VV at the indicated effector to target ratio.
DISCUSSION
We recently found that the liberation of peptides from the COOH terminus of Jaw1 is representative of a process that occurs with both soluble and membrane-bound proteins (Snyder, H.L., manuscript submitted for publication). Based on these findings, we have proposed the C-end rule: antigenic peptides are preferentially produced from the COOH terminus of precursor peptides or proteins in the ER. We believe that this reflects the normal NH2-terminal trimming of TAP-transported extended peptides with the proper COOH termini for class I binding.
Peptide liberation from the COOH terminus of Jaw occurs much more efficiently than from the other full-length substrates we have tested (an ER-targeted form of NP and CD23). Of these proteins, only Jaw provided sufficient quantities of peptide to detect enhanced expression of Kd in T2 cells (Fig. 4; our unpublished findings). In the case of Jaw1[NP147–155], the relatively high efficiency of peptide liberation is presumably associated with the proteolytic processing of its lumenal domain detectable biochemically. The nature of the endoprotease acting on the lumenal domain of Jaw1 is unknown: we presume that an additional aminopeptidase activity is required for final processing of NP147–155. Endoprotease activity would appear to be limiting inasmuch as peptide liberation was enhanced by coexpression of yeast Kex2.
Peptides were more efficiently liberated form Jaw1 (Lum−)[NP147–155] than from Jaw1[NP147–155]. Jaw1(Lum−) [NP147–155] was produced to take advantage of a potential signal peptidase cleavage site at the junction of the transmembrane and lumenal domains. The efficient liberation of NP147–155 from Jaw1(Lum−)[NP147–155] is consistent with the involvement of signal peptidase. Given the mechanism of Jaw insertion into the ER, this would mean that signal peptidase is operating posttranslationally. There is a precedent for this in antigen processing studies. The limited length of ER-targeted peptides dictates that they also must be posttranslationally inserted into the ER (the leader sequence does not emerge from the ribosome before translation is completed), where it is assumed that signal peptidase liberates the peptide from the leader. However, unlike Jaw1, ER-targeted peptides probably enter the ER via the translocon. The efficient processing of Jaw1 suggests that signal peptidase can act on proteins that are inserted into the membrane in a translocon-independent manner.
ER-targeted peptides have been shown to be more immunogenic than full-length proteins (42). This correlates with their highly efficient loading of class I molecules; we recently found that the NP147–155 determinant processed from full-length VV-encoded NP is present at ∼30 copies/ cell while 55,000 copies are recovered from cells expressing the ER-targeted peptide (36). The data in Fig. 6 suggest that Jaw1 is equally or more adept at delivering peptides to class I molecules than simple signal sequences. It will be of interest to determine how Jaw1 compares with signal sequences in enhancing the immunogenicity of defined peptides.
We unexpectedly found that a control Jaw1 construct lacking a transmembrane region was presented in a TAP-independent manner, albeit much less efficiently than ER-targeted Jaw1. We provide immunocytochemical evidence that the protein remains localized to the cytosol. Although we cannot rule out that a minor fraction of the protein is delivered to ER, examination of its sequence reveals no sequences of sufficient hydrophobicity to account for this activity. Most curiously, presentation of NP147–155 from its COOH terminus was dependent on a protease activity distinct from the proteasome in being unaffected by lactacystin and blocked by cbz–LL–CHO. The same activity was not required for TAP-independent presentation of the NP147–155 peptide liberated from the COOH terminus of other polypeptides directly targeted to the ER. This suggests that the requisite protease is located in the cytosol.
The other major defined cytosolic proteases inhibited by Z–LL–CHO, Z–LLL–CHO, and Z–LLF–CHO are the calpains. However, these are unlikely to be required for the presentation of Jaw1(TM−)[NP147–155], because two additional potent calpain inhibitors failed to affect presentation, even at quite high concentrations. Thus, it appears that we have uncovered a novel cytosolic protease that contributes to antigen processing. Because all of our experiments have used VV to express Jaw1(TM−)[NP147–155], it is possible that the protease is a VV gene product. Similarly, it is possible that transport of the Jaw1(TM−)[NP147–155]–derived peptide into the ER is dependent on an ongoing VV infection.
The putative cytosolic protease may be necessary to maintain the ability of cells to transport the intact Jaw1(TM−)[NP147–155] molecule to the ER. Alternatively, the protease may be involved in producing a fragment from Jaw1(TM−)[NP147–155] that is exported into the ER. This fragment cannot be the naturally processed peptide, because this peptide, when expressed as a minigene product, is not presented by T2 cells under the same conditions. The mechanism by which the fragment gains access to the ER appears to be related to the special properties of the cytosolic domain of Jaw1, because neither NP147–155 nor other peptides are presented in a TAP-independent manner when appended to the COOH terminus of cytosolic NP (Snyder, H.L., I. Bačík, J.W. Yewdell, T.W. Behrens, and J.R. Bennink, manuscript submitted for publication). Because TAP-independent presentation of Jaw1(TM−)[NP147–155] is observed in cells that do not express Jaw1, the mechanism of transport is unlikely to be strictly related to the normal function of Jaw1.
There are a number of examples of TAP-independent peptide presentation that cannot be attributed with reasonable certainty to translocon-mediated transport. RMA/S cells, which lack the TAP2 subunit, are able to present peptides from numerous cytosolic proteins, although at much lower efficiency than parental RMA cells (43–47). However, this has been claimed to result from TAP1 partially functioning (48). By contrast, T2 cells do not detectably present cytosolic antigens presented by RMA/S cells, and human TAP1 has an absolute requirement for TAP2 to transport peptides in T2 cells (13). T2 cells have been reported to present cytosolic peptides produced from transfected minigenes (21). In this case, the levels of direct peptide production are enormous compared with the relatively inefficient liberation of peptides from cytosolic proteins by cellular proteases, and a distinct route into the ER may be used that is less efficient than that used for longer polypeptides. An additional complicating factor in the case of transfectants is the possibility that peptides gain access to the ER due to alterations associated with the process of mitosis, where the nuclear envelope (and the entire secretory pathway) must disassemble and reassemble.
The TAP-independent presentation of Jaw1(TM−) [NP147–155] is most consistent with an antigen-processing pathway that depends on a novel cytosolic protease activity and a novel mechanism for translocation into the ER. Our working hypothesis is that a COOH-terminal cleavage product of Jaw1(TM−)[NP147–155] is translocated into the ER by a ubiquitously expressed transporter that functions to deliver larger polypeptides to the secretory pathway. It seems likely that this pathway is not a major source of class I–binding peptides, and that its detection was facilitated by exploiting the C-end rule liberation of the peptide once the protein was translocated into the ER. Further, the NP147–155-specific TCD8+ used to detect presentation of Jaw1(TM−)[NP147–155] are capable of lysing cells expressing <30 Kd–peptide complexes per cell (36). Together, these factors suggest that the primary function of this pathway is nonimmunological in nature.
Acknowledgments
We acknowledge the excellent technical assistance of B. Buschling, J. Rivard, and T. Bruggmann and U. Luthi for cryoultramicrotomy. We thank P. Cresswell (Yale University, New Haven, CT), J. Sheil (University of West Virginia, Morgantown, WV), and A. Townsend (Oxford University, Oxford, UK) for cell lines, and B. Rouse (University of Tennessee, Knoxville, TN) and D. Thomas (Oregon Health Sciences University, Eugene, OR) for recombinant viruses.
Footnotes
Abbreviations used in this paper: BFA, brefeldin A; ER, endoplasmic reticulum; FBS, fetal bovine serum; MOI, multiplicity of infection; NP, nucleoprotein; rVV, recombinant vaccinia virus; TAP, transporter associated with antigen processing; VV, vaccinia virus.
REFERENCES
- 1.Townsend A, Bodmer H. Antigen recognition by class I–restricted T lymphocytes. Annu Rev Immunol. 1989;7:601–624. doi: 10.1146/annurev.iy.07.040189.003125. [DOI] [PubMed] [Google Scholar]
- 2.Yewdell JW, Bennink JR. Cell biology of antigen processing and presentation to MHC class I molecule-restricted T lymphocytes. Adv Immunol. 1992;52:1–123. doi: 10.1016/s0065-2776(08)60875-5. [DOI] [PubMed] [Google Scholar]
- 3.Germain RN, Margulies DH. The biochemistry and cell biology of antigen processing and presentation. Annu Rev Immunol. 1993;11:403–450. doi: 10.1146/annurev.iy.11.040193.002155. [DOI] [PubMed] [Google Scholar]
- 4.Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg AL. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78:761–771. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 5.Goldberg AL, Rock KL. Proteolysis, proteasomes and antigen processing. Nature (Lond) 1992;357:375–379. doi: 10.1038/357375a0. [DOI] [PubMed] [Google Scholar]
- 6.Hughes EA, Ortmann B, Surman M, Cresswell P. The protease inhibitor, N-Acetyl-l-Leucyl-l-Leucyl-l-Norleucinal, decreases the pool of major histocompatibility complex class I–binding peptides and inhibits peptide trimming in the endoplasmic reticulum. J Exp Med. 1996;183:1569–1578. doi: 10.1084/jem.183.4.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sijts AJAM, Villanueva MS, Pamer EG. CTL epitope generation is tightly linked to cellular proteolysis of a Listeria monocytogenesantigen. J Immunol. 1996;156:1497–1503. [PubMed] [Google Scholar]
- 8.Suh W-K, Mitchell EK, Yang Y, Peterson A, Waneck GL, Williams DB. MHC class I molecules form ternary complexes with calnexin and TAP and undergo peptide-regulated interaction with TAP via their extracellular domains. J Exp Med. 1996;184:337–384. doi: 10.1084/jem.184.2.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vinitsky A, Antón LC, Snyder HL, Orlowski M, Bennink JR, Yewdell JW. The generation of MHC class I associated peptides is only partially inhibited by proteasome inhibitors: involvement of non-proteasomal proteases in antigen processing? . J Immunol. 1997;159:554–564. [PubMed] [Google Scholar]
- 10.Adrolewicz MJ, Anderson KS, Cresswell P. Evidence that transporters associated with antigen processing translocate a major histocompatibility complex class I–binding peptide into the endoplasmic reticulum in an ATP-dependent manner. Proc Natl Acad Sci USA. 1993;90:9130–9134. doi: 10.1073/pnas.90.19.9130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shepherd JC, Schumacher TNM, Ashton-Rickardt PG, Imaeda S, Ploegh HL, Janeway CA, Jr, Tonegawa S. TAP-1–dependent peptide translocation in vitro is ATP dependent and peptide selective. Cell. 1993;74:577–584. doi: 10.1016/0092-8674(93)80058-m. [DOI] [PubMed] [Google Scholar]
- 12.Schumacher TNM, Kantesaria DV, Heemels M-T, Ashton-Rickardt PG, Shepherd JC, Fruh K, Yang Y, Peterson PA, Tonegawa S, Ploegh HL. Peptide length and sequence specificity of the mouse TAP1/TAP2 translocator. J Exp Med. 1994;179:533–540. doi: 10.1084/jem.179.2.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heemels M-T, Ploegh H. Generation, translocation, and presentation of MHC class-I restricted peptides. Annu Rev Biochem. 1995;64:463–491. doi: 10.1146/annurev.bi.64.070195.002335. [DOI] [PubMed] [Google Scholar]
- 14.Urban RG, Chicz RM, Lane WS, Strominger JL, Rehm A, Kenter MJH, UytdeHaag FGCM, Ploegh H, Uchanska-Ziegler B, Ziegler A. A subset of HLA-B27 molecules contains peptides much longer than nonamers. Proc Natl Acad Sci USA. 1994;91:1534–1538. doi: 10.1073/pnas.91.4.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeMars R, Chang CC, Shaw S, Reitnauer PJ, Sondel PM. Homozygous deletions that simultaneously eliminate expression of class I and class II antigens of EBV-transformed B-lymphoblastoid cells. I. Reduced proliferative responses of autologous and allogeneic T cells to mutant cells that have decreased expression of class II antigens. Human Immunol. 1984;11:77–97. doi: 10.1016/0198-8859(84)90047-8. [DOI] [PubMed] [Google Scholar]
- 16.Salter RD, Cresswell P. Impaired assembly and transport of HLA-A and -B antigens in a mutant T×B cell hybrid. EMBO (Eur Mol Biol Organ) J. 1986;5:943–949. doi: 10.1002/j.1460-2075.1986.tb04307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hosken NA, Bevan MJ. Defective presentation of endogenous antigen by a cell line expressing class I molecules. Science (Wash DC) 1990;248:367–370. doi: 10.1126/science.2326647. [DOI] [PubMed] [Google Scholar]
- 18.Townsend A, Öhlen C, Bastin J, Ljunggren H-G, Foster L, Kärre K. Association of class I major histocompatibility heavy and light chains induced by viral peptides. Nature (Lond) 1989;340:443–448. doi: 10.1038/340443a0. [DOI] [PubMed] [Google Scholar]
- 19.Anderson K, Cresswell P, Gammon M, Hermes J, Williamson A, Zweerink H. Endogenously synthesized peptide with an endoplasmic reticulum signal sequence sensitizes antigen processing mutant cells to class I–restricted cell-mediated lysis. J Exp Med. 1991;174:489–492. doi: 10.1084/jem.174.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bacik I, Cox JH, Anderson R, Yewdell JW, Bennink JR. TAP-independent presentation of endogenously synthesized peptides is enhanced by endoplasmic reticulum insertion sequences located at the amino but not carboxy terminus of the peptide. J Immunol. 1994;152:381–387. [PubMed] [Google Scholar]
- 21.Zweerink HJ, Gammon MC, Utz U, Sauma SY, Harrer T, Hawkins JC, Johnson RP, Sirotina A, Hermes JD, Walker BD, Biddison W E. Presentation of endogenous peptides to MHC class I–restricted cytotoxic T lymphocytes in transport deletion mutant T2 cells. J Immunol. 1993;150:1763–1771. [PubMed] [Google Scholar]
- 22.Henderson RA, Michel H, Sakaguchi K, Shabanowitz J, Apella E, Hunt DF, Engelhard VH. HLA-A2.1–associated peptides from a mutant line: a second pathway of antigen presentation. Science (Wash DC) 1992;255:1264–1266. doi: 10.1126/science.1546329. [DOI] [PubMed] [Google Scholar]
- 23.Falk K, Rötzschke O, Rammensee H-G. Cellular peptide composition governed by major histocompatibility complex class I molecules. Nature (Lond) 1990;348:248–251. doi: 10.1038/348248a0. [DOI] [PubMed] [Google Scholar]
- 24.Snyder HL, Yewdell JW, Bennink JR. Trimming of antigenic peptides in an early secretory compartment. J Exp Med. 1994;180:2389–2394. doi: 10.1084/jem.180.6.2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elliot T, Willis A, Cerundolo V, Townsend A. Processing of major histocompatibility class I–restricted antigens in the endoplasmic reticulum. J Exp Med. 1995;181:1481–1491. doi: 10.1084/jem.181.4.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hammond SA, Johnson RP, Kalams SA, Walker BD, Takiguchi M, Safrit JT, Koup RA, Siliciano RF. An epitope-selective, transporter associated with antigen presentation (TAP)-1/2–independent pathway and a more general TAP-1/2–dependent antigen-processing pathway allow recognition of the HIV-1 envelope glycoprotein by CD8+ CTL. J Immunol. 1995;154:6140–6156. [PubMed] [Google Scholar]
- 27.Behrens TW, Jagadeesh J, Scherle P, Kearns G, Yewdell J, Staudt LM. Jaw1, a lymphoid-restricted membrane protein localized to the endoplasmic reticulum. J Immunol. 1994;153:682–690. [PubMed] [Google Scholar]
- 28.Behrens TW, Kearns GM, Rivard JJ, Bernstein HD, Yewdell JW, Staudt LM. Carboxyl-terminal targeting and novel post-translational processing of JAW1, a lymphoid protein of the endoplasmic reticulum. J Biol Chem. 1996;271:23528–23534. doi: 10.1074/jbc.271.38.23528. [DOI] [PubMed] [Google Scholar]
- 29.Kutay U, Hartmann E, Rapoport TA. A class of membrane proteins with a C-terminus anchor. Trends Cell Biol. 1993;3:72–75. doi: 10.1016/0962-8924(93)90066-a. [DOI] [PubMed] [Google Scholar]
- 30.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (Lond) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 31.Tokuyashu KT. Application of cryoultramicrotomy to immunocytochemistry. J Micros. 1986;143:139–149. doi: 10.1111/j.1365-2818.1986.tb02772.x. [DOI] [PubMed] [Google Scholar]
- 32.Vinitsky A, Michaud C, Powers JC, Orlowski M. Inhibition of the chymotrypsin-like activity of the pituitary multicatalytic proteinase complex. Biochemistry. 1992;31:9421–9428. doi: 10.1021/bi00154a014. [DOI] [PubMed] [Google Scholar]
- 33.Eisenlohr LC, Bacik I, Bennink JR, Bernstein K, Yewdell JW. Expression of a membrane protease enhances presentation of endogenous antigens to MHC class I–restricted T lymphocytes. Cell. 1992;71:963–972. doi: 10.1016/0092-8674(92)90392-p. [DOI] [PubMed] [Google Scholar]
- 34.Banks TA, Jenkins FJ, Kanangat S, Nair S, Dasgupta S, Foster CM, Rouse BT. Vaccination with the immediate–early protein ICP47 of herpes simplex virus–type 1 (HSV-1) induces virus-specific lymphoproliferation, but fails to protect against lethal challenge. Virology. 1994;200:236–245. doi: 10.1006/viro.1994.1181. [DOI] [PubMed] [Google Scholar]
- 35.Thomas G, Thorne BA, Thomas R, Allen RG, Hruby DE, Fuller R, Thorner J. Yeast KEX2 endopeptidase correctly cleaves a neuroendocrine prohormone in mammalian cells. Science (Wash DC) 1988;241:226–230. doi: 10.1126/science.3291117. [DOI] [PubMed] [Google Scholar]
- 36.Antón LC, Yewdell JW, Bennink JR. MHC class I–associated peptides produced from endogenous gene products with vastly different efficiencies. J Immunol. 1997;158:2535–2542. [PubMed] [Google Scholar]
- 37.Deng Y, Yewdell JW, Eisenlohr LC, Bennink JR. MHC affinity, peptide liberation, T cell repertoire, and immunodominance all contribute to the paucity of MHC class I–restricted peptides recognized by antiviral CTL. J Immunol. 1997;158:1507–1515. [PubMed] [Google Scholar]
- 38.Lapham CK, Bacik I, Yewdell JW, Kane KP, Bennink JR. Class I molecules retained in the endoplasmic reticulum bind antigenic peptides. J Exp Med. 1993;177:1633–1641. doi: 10.1084/jem.177.6.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vinitsky A, Cardozo C, Sepp-Lorenzino L, Michaud C, Orlowski M. Inhibition of the proteolytic activity of the multicatalytic proteinase complex (proteasome) by substrate-related peptidyl aldehydes. J Biol Chem. 1994;269:29860–29866. [PubMed] [Google Scholar]
- 40.Fenteany G, Standaert R F, Lane WS, Choi S, Corey EJ, Schreiber SL. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science (Wash DC) 1995;268:726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- 41.Hill A, Jugovic P, York I, Russ G, Bennink J, Yewdell JW, Ploegh H, Johnson D. Herpes simplex virus turns off the TAP to evade host immunity. Nature (Lond) 1995;375:411–415. doi: 10.1038/375411a0. [DOI] [PubMed] [Google Scholar]
- 42.Restifo NP, Bacík I, Irvine KR, Yewdell JW, McCabe B, Anderson RW, Eisenlohr LC, Rosenberg SA, Bennink JR. Antigen processing in vivo and the elicitation of primary CTL responses. J Immunol. 1995;154:4414–4422. [PMC free article] [PubMed] [Google Scholar]
- 43.Esquivel F, Yewdell JW, Bennink JR. RMA/S cells present endogenously synthesized cytosolic proteins to class I–restricted cytotoxic T lymphocytes. J Exp Med. 1992;175:163–168. doi: 10.1084/jem.175.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hosken NA, Bevan MJ. An endogenous antigenic peptide bypasses the class I antigen presentation defect in RMA-S. J Exp Med. 1992;175:719–729. doi: 10.1084/jem.175.3.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hermel E, Grigorenko E, Lindahl KF. Expression of medial class I histocompatibility antigens on RMA-S mutant cells. Int Immunol. 1993;3:407–412. doi: 10.1093/intimm/3.4.407. [DOI] [PubMed] [Google Scholar]
- 46.Zhou X, Glas R, Momburg F, Hämmerling GJ, Jondal M, Ljunggren H-G. TAP2-defective RMA-S cells present Sendai virus antigen to cytotoxic T lymphocytes. Eur J Immunol. 1993;23:1796–1801. doi: 10.1002/eji.1830230810. [DOI] [PubMed] [Google Scholar]
- 47.Ossevoort MA, Sijts AJAM, van Veen KJH, Momburg F, Hämmerling GJ, Seelig A, Butcher GW, Howard JC, Kast WM, Melief CJM. Differential effect of transporter TAP-2 gene introduction into RMA-S cells on viral antigen processing. Eur J Immunol. 1993;23:3082–3088. doi: 10.1002/eji.1830231206. [DOI] [PubMed] [Google Scholar]
- 48.Gabathuler R, Reid G, Kolaitis G, Driscoll J, Jeffries WA. Comparison of cell lines deficient in antigen presentation reveals a functional role for TAP-1 alone in antigen processing. J Exp Med. 1994;180:1415–1425. doi: 10.1084/jem.180.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]