

Abstract
Certain major histocompatibility complex (MHC) class II haplotypes encode elements providing either susceptibility or dominant resistance to the development of spontaneous autoimmune diseases via mechanisms that remain undefined. Here we show that a pancreatic beta cell–reactive, I-Ag7–restricted, transgenic TCR that is highly diabetogenic in nonobese diabetic mice (H-2g7) undergoes thymocyte negative selection in diabetes-resistant H-2g7/b, H-2g7/k, H-2g7/q, and H-2g7/nb1 NOD mice by engaging antidiabetogenic MHC class II molecules on thymic bone marrow–derived cells, independently of endogenous superantigens. Thymocyte deletion is complete in the presence of I-Ab, I-Ak + I-Ek or I-Anb1 + I-Enb1 molecules, partial in the presence of I-Aq or I-Ak molecules alone, and absent in the presence of I-As molecules. Mice that delete the transgenic TCR develop variable degrees of insulitis that correlate with the extent of thymocyte deletion, but are invariably resistant to diabetes development. These results provide an explanation as to how protective MHC class II genes carried on one haplotype can override the genetic susceptibility to an autoimmune disease provided by allelic MHC class II genes carried on a second haplotype.
Insulin-dependent diabetes mellitus (IDDM),1 a prototype of organ-specific autoimmune diseases, results from selective destruction of pancreatic beta cells by a T lymphocyte–dependent autoimmune process in genetically predisposed individuals (1). Genetic susceptibility and/or resistance to most autoimmune disorders, including IDDM, is associated with highly polymorphic genes of the MHC complex and, to a lesser extent, with polygenic modifiers on other chromosomes (2).
Population and animal studies have suggested that the MHC class II–linked susceptibility and resistance to IDDM are inherited as dominant traits with incomplete penetrance (2, 3). In humans, the MHC-associated IDDM susceptibility and resistance are predominantly, but not exclusively, determined by polymorphisms at the human leukocyte antigen (HLA) DQB1 locus. Alleles encoding DQβ chains with serine, alanine, or valine at position 57 provide susceptibility, whereas those encoding DQβ chains with aspartic acid at position 57 provide resistance with differing degrees of dominance (1, 2). In mice, susceptibility and resistance to spontaneous IDDM are also linked to the MHC (H-2). The diabetes-prone nonobese diabetic (NOD) mouse, which spontaneously develops a form of diabetes closely resembling human IDDM, is homozygous for a unique H-2 haplotype (H-2g7). This haplotype carries a nonproductive I-Eα gene and encodes an I-Aαd/I-Aβg7 heterodimer in which the histidine and aspartic acid found at positions 56 and 57 in most murine I-Aβ chains (the counterpart of human DQβ chains) are replaced by proline and serine, respectively (4, 5). Studies of congenic NOD mice expressing non-NOD MHC haplotypes, and of NOD mice expressing I-Eαd, modified I-Aβg7, I-Aαk/I-Aβk, or I-Aβd transgenes, have demonstrated that MHC class II molecules encoded by H-2 haplotypes derived from NOD or IDDM-resistant mice play a direct role in providing either susceptibility or resistance to spontaneous IDDM, respectively (6–18).
The precise mechanisms through which MHC genes provide autoimmune disease susceptibility and resistance, however, remain mysterious. MHC molecules are cell-surface receptors that present short fragments of self and foreign proteins to T lymphocytes and play a pivotal role in instructing T lymphocytes maturing in the thymus how to discriminate between self- and nonself-antigens (19, 20). Thymocytes bearing TCRs capable of recognizing self-peptide–MHC complexes with high affinity/avidity die or are rendered unresponsive to antigenic stimulation (21–27). In contrast, thymocytes bearing TCRs capable of engaging self-peptide–MHC complexes with intermediate affinity/ avidity survive and are exported to the peripheral lymphoid organs as cells capable of recognizing foreign antigens bound to self-MHC molecules (25–30). On the basis of some of this knowledge, a number of authors hypothesized that MHC molecules providing resistance to autoimmune diseases, including IDDM, might do so by mediating the clonal deletion or anergy of autoreactive T cells (8, 11, 31). Studies in MHC-congenic NOD mice and in I-A-, I-E-, and TCR-transgenic NOD mice, however, did not find evidence of T cell tolerance induction, and suggested that the diabetes resistance provided by protective MHC genes likely involved the induction of immunoregulatory functions (10, 13, 16, 18, 32, 33).
The studies presented here were initiated to test the hypothesis that diabetes-resistant genetic backgrounds express non-MHC–linked genetic elements other than endogenous superantigens that are tolerogenic for diabetogenic T cells. To that end, we followed the fate of an NOD islet-derived, beta cell–specific, I-Ag7–restricted transgenic TCR in diabetes-prone NOD mice, and in diabetes-resistant F1 hybrid mice lacking endogenous superantigens binding to the transgenic TCR. In NOD mice, T cells expressing the transgenic TCR underwent positive thymic selection and triggered a dramatic acceleration of the onset of IDDM. In certain F1 hybrid strains, however, the same cells underwent negative selection and the mice did not develop IDDM. Contrary to what we expected, the thymocyte deletion and IDDM resistance observed in these mice cosegregated as a single locus trait with MHC haplotypes known to provide dominant resistance to IDDM in MHC-transgenic and/or -congenic NOD mice (H-2b and H-2k). Thymocyte deletion in these mice was not mediated by endogenous superantigens, since it was absent in single-chain TCR-β–transgenic H-2g7/b and H-2g7/k F1 mice. Studies of bone marrow chimeras, I-Aβb–deficient H-2g7/b mice and I-Ak–transgenic NOD mice demonstrated that the thymocyte deletion and IDDM resistance of these mice resulted from the ability of the transgenic TCR to engage I-Ab, I-Ak, and possibly, I-Ek molecules on thymic bone marrow–derived cells. Additional experiments with H-2g7/q- and H-2g7/nb1-congenic NOD mice revealed that this highly pathogenic TCR was also deleted in the presence of I-Aq and I-A/I-Enb1 molecules, which also provide dominant resistance to autoimmune diabetes in non-TCR–transgenic NOD mice. Our unexpected results provide an explanation as to how protective MHC class II molecules may provide resistance to spontaneous T cell–mediated autoimmune disorders, i.e., by removing certain highly pathogenic autoreactive T cells.
MATERIALS AND METHODS
Generation of TCR Transgenes.
The TCR-α and -β cDNAs of NY4.1 were cloned by anchored PCR and multiple recombinants sequenced as described (34). The cDNAs were then amplified by PCR using primers containing L-V and J-C intron sequences, the corresponding splice donor and acceptor sites and convenient restriction sites, cloned into pBS-SK+ (Stratagene, La Jolla, CA), and sequenced. Inserts with the expected sequences were released from the vector by digestion with ClaI and NotI (TCR-β) or XhoI and NotI (TCR-α). The 4.1-VDJβ sequence was subcloned into a TCR-β shuttle vector carrying the endogenous TCR-β enhancer and 5′ regulatory sequences (3A9β; gift from M. Davis, Stanford University, Stanford, CA). The 4.1-VJα cDNA was subcloned into PRE53α (from M. Davis). A 2.4-kb ClaI–KpnI fragment from 4.1-VJα/PRE53α, containing 5′ regulatory sequences and the 4.1-VJα sequence, was then coligated into pBS-SK+ with a genomic 11.9-kb KpnI–NotI fragment from AN6.2α (provided by S. Hedrick, University of California San Diego, San Diego, CA), containing the four Cα exons and the downstream TCR-α enhancer.
Production of Transgenic Mice.
After removing prokaryotic sequences by digestion with PvuI and SalI (TCR-β) or ClaI and SacII (TCR-α), the constructs (21.5- and 14.5-kb, respectively) were injected into fertilized (SJL × C57BL/6) F2 eggs (DNX, Princeton, NJ). Offspring were screened for integration of the transgenes by Southern blotting using VDJβ and VJα cDNA probes. Transgenic founder mice (4.1-AN6A3–TCR-α/β, 4.1-AN6B3–TCR-β and 4.1-AN6B7–TCR-α) were backcrossed with NOD/Lt mice (I-Ag7, I-E−; Jackson Laboratory, Bar Harbor, ME) for 3–5 generations to generate TCR-α/β–, TCR-β–, and TCR-α–transgenic NOD mice. TCR-α–, TCR-β–, and TCR-α/β–transgenic NOD mice (of the N5 backcross) were crossed with SJL/J (S; I-As, I-E−), C57BL/6 (B; I-Ab, I-E−), C58/J (C; I-Ak, I-Ek), NOD.H-2g7/q (I-Ag7/q, I-E−), or NOD.H-2g7/nb1 (I-Ag7/nb1, I-Enb1) mice (Jackson Laboratory), to generate TCR-transgenic F1 mice or H-2 heterozygous TCR-transgenic NOD mice. TCR-α/β–transgenic F1 (4.1-F1) mice were also backcrossed with NOD, C57BL/6, or C58/J mice, to generate H-2g7, H-2g7/b, H-2g7/k 4.1 mice with 75% NOD genotype, and H-2b or H-2k 4.1 mice with 75% C57BL/6 or C58/J genotypes, respectively. I-Aβb–deficient 4.1-(N × B) F1 mice were generated by crossing I-Aβb− C57BL-6 mice (Taconic, Germantown, NY) with 4.1-NOD mice. β2 microglobulin (β2m)− 4.1-(N × B) F1 mice were obtained by breeding the β2m mutation of β2m− NOD/Lt mice (Jackson Laboratory) into 4.1-NOD mice, to obtain β2m− 4.1-NOD mice, and then by crossing these mice with β2m− C57BL/6 mice (Taconic). CD8-α− H-2g7/b mice were obtained by crossing CD8-α− mice (I-Ab, I-E−) (gift from T. Mak, University of Toronto, Toronto, Canada) with 4.1-NOD mice, followed by crossing CD8-α−/+ 4.1-H-2g7/b mice with CD8-α− H-2b mice. I-Ak–transgenic 4.1-NOD mice were obtained by crossing I-Aαk/I-Aβk–transgenic NOD mice (from G. Morahan and J.F.A.P. Miller, The Walter and Eliza Hall Institute, Victoria, Australia) with 4.1-NOD mice. Mice were screened for inheritance of the transgenes, mutated alleles, and MHC haplotypes by PCR of tail DNA (4.1α, 4.1β, β2m, I-Ag7, I-Ak) and/ or by flow cytometry (4.1β, CD8-α, I-Ab, I-As, Kd, Kk). All mice were housed in a specific pathogen-free facility.
Antibodies and Flow Cytometry.
Hybridomas secreting mAbs H57-597 (anti–TCR-β), 53-6.7 (anti–CD8-α), B220 (anti–B cells), and M1/70 (anti–Mac-1) were obtained from the American Type Culture Collection (ATCC, Rockville, MD). Anti– Lyt-2 (CD8-α)–PE (53-6.7), anti–L3T4–FITC (IM7), or anti– L3T4–biotin (CD4; H129.19), anti–Vβ11–FITC (RR3-15), anti–H-2Kd–FITC (SF1-1.1), and anti–I-Aβb–biotin (25-9-3) were purchased from PharMingen (San Diego, CA). Purified mouse anti–H-2Kk (11-4.1) was from Becton Dickinson (San Jose, CA). Mouse IgG-absorbed FITC-conjugated goat anti–rat IgG and FITC-conjugated goat anti–mouse IgG were from CALTAG Labs. (South San Francisco, CA) and Becton Dickinson, respectively. Streptavidin-PerCP was from Becton Dickinson. Thymi, spleens, and islet-derived T cells were analyzed by three-color flow cytometry using a FACScan® as described (35).
Cloning and Sequencing of TCR-α cDNAs.
The TCR-α cDNA molecules of splenic CD4+ T cells were cloned and sequenced by anchored PCR as described previously (35).
Preparation of CD8+ T Cell–depleted Splenic and Islet-derived T Cells.
Spleens were disrupted into single cell suspensions and red blood cells lysed in 0.87% ammonium chloride. Remaining cells were washed with complete medium (CM: RPMI 1640 media containing 10% heat-inactivated fetal bovine serum [GIBCO BRL, Gaithersburg, MD], 50 U/ml penicillin, 50 μg/ml streptomycin [Flow Labs., McLean, VA], and 50 μM 2-ME [Sigma Chemical Co., St. Louis, MO]) and then depleted of CD8+ T cells using anti-CD8 mAb (53-6.7)–coated magnetic beads as described (35). Islet-infiltrating CD4+ T cells were isolated from acutely diabetic 4.1-NOD mice essentially as described previously (35), analyzed by flow cytometry, depleted of CD8+ T cells by negative selection with anti-CD8 mAb-coated immunobeads, expanded in rIL-2–containing CM for 1–2 wk, and used for in vitro and in vivo studies.
Proliferation Assays.
Pancreatic islets from 5–8-wk-old nontransgenic male NOD mice were dispersed into single cells by incubation in Ca2+- and Mg2+-free PBS containing 0.125% trypsin and 3 mM EGTA at 37°C for 3 min. 2 × 104 splenic or islet-derived CD4+ cells were incubated, in triplicate, with γ-irradiated (3,000 rad) islet cells (3–100 × 103/well) and unfractionated NOD splenocytes (105/well), as a source of antigen and APCs, respectively, in 96-well round-bottomed tissue culture plates for 3 d at 37°C in 5% CO2 in rIL-2–free CM. Cultures were pulsed with 1 μCi of [3H]thymidine during the last 18 h of culture and harvested. The incorporated thymidine was measured by scintillation counting. Specific proliferation was calculated by substracting background proliferation (cpm of cultures containing islet cells plus APCs alone and cpm of cultures of T cells alone) from islet cell–induced proliferation (cpm of cultures containing T cells, APCs, and islet cells).
For anti–TCR-β stimulation, 96-well flat-bottomed plates were precoated overnight at 4°C with serial dilutions of purified anti–TCR-β mAb (H57-597; 0.3–10 μg/ml) in 50 mM Tris-HCl, 150 mM NaCl (pH 9.5), and washed three times in CM. CD4+ T cells (2 × 104) were added to each well in triplicate, incubated for 48 h, pulsed with [3H]thymidine for 18 h, harvested, and analyzed by scintillation counting.
Histology and Immunopathology.
The body-tail of each pancreas was divided into two pieces. One piece was fixed in formalin, embedded in paraffin, sectioned at 4.5 μM, and stained with hematoxylin and eosin. The degree of insulitis was evaluated by scoring 15–30 islets/mouse in a blinded fashion using the following criteria: 0, normal islet; 1, peri-insulitis; 2, mononuclear cell infiltration in <25% of the islet; 3, mononuclear cell infiltration in 25–50% of the islet; 4, >50% of the islet infiltrated. A second piece of tissue was snap frozen, immersed in OCT, sectioned at 6–7 μM, and stored at −80°C for immunopathology. Sections were fixed in cold acetone for 10 min and stained with anti-CD4 (GK1.5), anti-CD8 (53-6.7), anti–Mac-1, and anti-B220 mAbs, followed by anti–rat IgG-FITC, or with anti-rat IgG-FITC alone, as described (35).
Adoptive T cell Transfer.
CD8+ T cell–depleted islet-derived CD4+ T cells from diabetic 4.1-NOD mice (5 × 106 cells/ mouse) were transfused into the tail veins of scid-NOD/Lt (Jackson Laboratory) in 200 μl of PBS, pH 7.2. Transfused mice were followed for development of IDDM by monitoring blood glucose levels with Glucostix and a glucometer (Miles Canada, Etobicoke, Ontario). Mice were killed at IDDM onset for flow cytometry and immunopathological studies.
Bone Marrow Chimeras.
Bone marrow chimeras were generated following standard protocols (36). In brief, bone marrow suspensions (5–10 × 106 cells) from donor mice (transgenic NOD or [N × B] F1 mice) were injected into the tail vein of recipient mice (nontransgenic NOD, [N × B] F1 or [N × S] F1 mice) treated with two doses of 500 rads 3 h apart from a 137Cs source (Gammacell; Atomic Energy of Canada, Ottawa, Ontario). Chimeric mice were killed 5–6 wk after bone marrow transplantation.
Statistical Analyses.
Statistical analyses were performed using Mann-Whitney U and χ2 tests.
RESULTS
Generation of Beta Cell–specific, I-Ag7–restricted TCR-α/β– transgenic NOD Mice.
A CD4+ T cell clone (NY4.1) that was derived from pancreatic islets of a diabetic NOD mouse and that recognized a putative beta cell autoantigen in the context of I-Ag7 (37) was chosen as donor of the TCR transgenes. This T cell clone transcribed one functional TCR-β rearrangement, carrying Vβ11 and Jβ2.4 sequences and one functional TCR-α rearrangement, carrying a novel Vα gene (Vαx4.1) and the Jα33 element (These sequence data are available from EMBL/GenBank/DDBJ under accession numbers U80816 and U80817). These TCR rearrangements were subcloned into genomic TCR-β and -α shuttle vectors carrying endogenous TCR enhancers and 5′-regulatory sequences. The resulting genomic constructs were then used to produce transgenic mice. Founders expressing the transgenes (4.1-AN63A–TCR-α/β, 4.1-AN6B3– TCR-β, and 4.1-AN6B7–TCRα) were crossed with NOD mice for several generations to generate 4.1–TCR-α/β-, 4.1–TCR-β–, and 4.1–TCR-α–transgenic NOD mice, respectively.
Expression of the 4.1-TCR in 4.1–TCR-α/β–transgenic NOD (4.1-NOD) Mice.
Three-color cytofluorometric studies showed that >90% CD4+CD8− thymocytes (Fig. 1 A) and splenic CD4+ T cells (Fig. 1 B) from 4.1-NOD mice expressed Vβ11+ TCRs, compared to ∼6% of the CD4+CD8− thymocytes and splenic CD4+ T cells from nontransgenic littermates, thus indicating TCR-β transgene expression. Although we do not have a transgenic TCR-α chain-specific antibody and thus cannot directly quantitate TCR-α transgene expression, five different lines of evidence suggest that the transgenic TCR-α chain is expressed on a sizeable fraction of thymocytes and peripheral T cells of 4.1-NOD mice. First, CD4+CD8+ thymocytes from 4.1-NOD mice expressed higher levels of total TCR-α/β (as determined by staining with an anti-Cβ mAb) than CD4+CD8+ thymocytes from TCR-β–transgenic NOD mice (mean fluorescence intensities: 59 ± 12 versus 31 ± 3, respectively; P <0.001), suggesting early TCR-α chain expression (23, 38, 39). Second, thymocyte development in 4.1-NOD mice, but not TCR-β–transgenic NOD mice, was skewed towards the CD4+CD8− subset (Fig. 1 A), compatible with TCR-α transgene-dependent positive selection of 4.1-CD4+ thymocytes. Third, skewing of thymocytes into the CD4+CD8− subset occurred in transgenic mice expressing the selecting I-Ag7 molecule, but not in transgenic mice expressing only nonselecting I-A molecules (i.e., I-As, see below), as seen with other MHC class II–restricted TCR-α/β–transgenic models (38, 40). Fourth, 37 out of 37 4.1-NOD TCR-α cDNA sequences, generated from splenic CD4+ T cell–derived RNA by anchored PCR, were TCR-α transgene–derived. Finally, splenic CD4+ T cells from 4.1-NOD, but not TCR-β–transgenic or nontransgenic, NOD mice proliferated in a dose-dependent manner in response to irradiated NOD islet cells (Fig. 1 C). The islet cell–induced proliferation of 4.1- and control CD4+ T cells was quite variable between experiments, perhaps due to variability in the quality of the islet cell preparations; however, the differences within individual experiments were reproducible. Taken together, these results provide strong evidence that in 4.1-NOD mice, the 4.1-TCR specificity is expressed appropriately, and that, in the presence of the selecting I-Ag7 molecule, 4.1–TCR-α/β transgene expression fosters the positive selection of beta cell–reactive CD4+ T cells.
Figure 1.


Expression of the TCR-α/β transgenes in 4.1-NOD mice. (A and B) CD4, CD8, and Vβ11 profiles of thymocytes (A) and splenic cells (B) from transgenic and nontransgenic mice. (Top) CD4 versus CD8 dot plots of cell suspensions stained with anti–CD8–PE, anti–Vβ11– FITC, and anti–CD4–biotin plus Streptavidin-PerCP. (Bottom) Vβ11 fluorescence histograms of each T cell subset after electronic gating. Numbers indicate the average percentage of cells (top) or the average number of Vβ11+ cells (bottom) in each subset. Data correspond to 6–10 3–5-wk-old mice/group. DP, double-positive cells; DN, double-negative cells. (C) In vitro proliferation of splenic CD4+ T cells in response to islet cells. Cultures of 2 × 104 splenic CD4+ T cells from TCR-α/β–transgenic, TCR-β–transgenic, and nontransgenic NOD mice were incubated with γ-irradiated NOD islet cells and splenocytes (105/well) for 3 d, pulsed with [3H]thymidine, and harvested. Bars show the standard error of the means.
Early Onset of IDDM in 4.1-NOD Mice.
To determine whether positive selection of the beta cell–specific TCR in 4.1-NOD mice had any pathogenic significance, we followed 4.1-NOD mice of the N3–N5 backcrosses of the founder mouse onto the NOD background, and nontransgenic NOD mice for development of IDDM. As shown in Fig. 2 A, 4.1-NOD mice developed IDDM much earlier than nontransgenic NOD mice (IDDM onset at 43.6 ± 13 versus 119 ± 26 d in females, and at 56 ± 27 versus 157 ± 28 d in males; P <0.0001) (Fig. 2 A). In males, transgene expression also increased the incidence of IDDM (73.3 versus 45.7%; P <0.05). The kinetics of disease penetrance in the transgenic and nontransgenic populations were, however, remarkably similar (Fig. 2 A). Disease acceleration in 4.1-NOD mice required coexpression of the TCR-α and -β transgenes, since the few 4.1–TCR-β–NOD mice that became diabetic (3/7, 43%) did so significantly later than 4.1-NOD mice (103 ± 20 versus 46 ± 19 d; P <0.01). These results contrast with those obtained with the only other existing beta cell–specific TCR-transgenic NOD mouse model (32), which, when housed under specific pathogen-free conditions, develops IDDM less frequently and significantly later (10–15% at 6 mo) than 4.1-NOD mice (41). Taken together, our results indicate that the 4.1–TCR-α/β is highly diabetogenic in the NOD background, and suggest that the events that trigger accelerated diabetogenesis in 4.1-NOD mice are similar to those that trigger IDDM in nontransgenic NOD mice.
Figure 2.
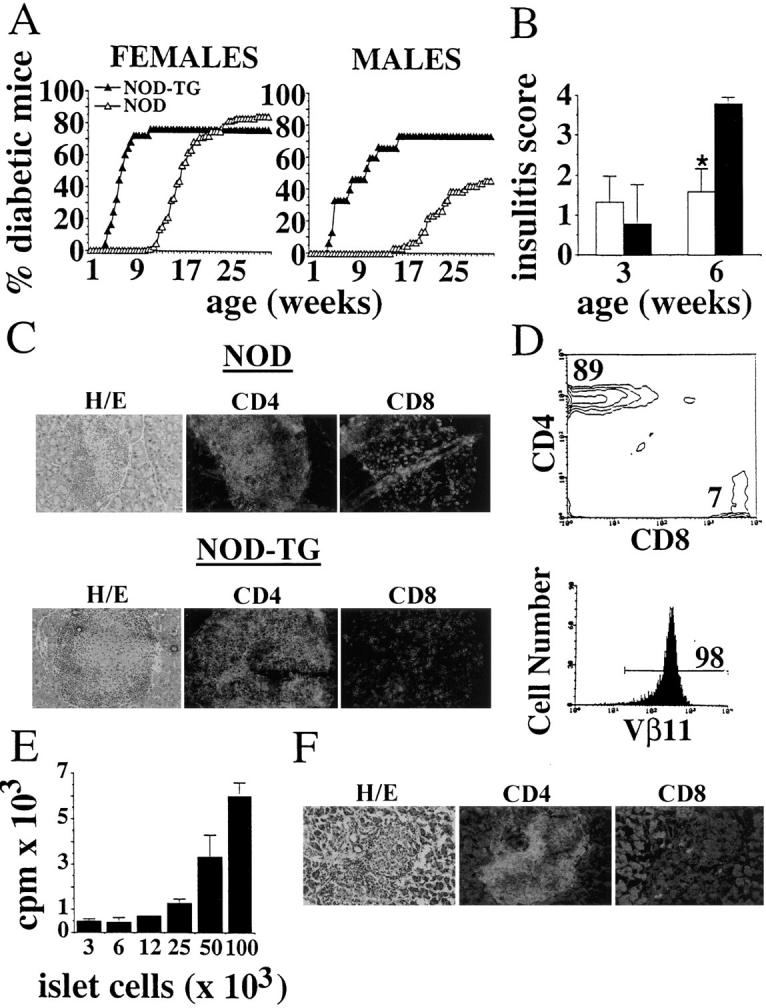
4.1-TCR-α/β–transgene expression and diabetogenesis. (A) Cumulative incidence of IDDM in female (25 transgenic and 114 nontransgenic) and male (15 transgenic and 59 nontransgenic) NOD mice. (B) Progression of insulitis in transgenic and nontransgenic NOD mice. Hematoxylin–eosin stained pancreatic sections of 3- and 6-wk-old mice (4–13 mice/age group) were scored for the degree of insulitis as described in Materials and Methods (4 is the maximum score). Bars show the standard deviation of the means. *P <0.0001 (χ2). (C) Phenotype of islet-infiltrating T cells in nontransgenic and transgenic NOD mice. Pancreas sections were stained with anti-CD8 (53.6-7) or anti-CD4 (GK1.5) mAbs and FITC-labeled anti–rat IgG. Original magnification: 200. (D) Flow cytometric profile of islet-derived T cells from diabetic 4.1-NOD mice. (E) Islet cell reactivity of islet-derived CD4+ T cells from 4.1-NOD mice. See legend to Fig. 1 C for details. (F) Phenotype of islet-infiltrating T cells in a diabetic scid-NOD mice that had been transfused with CD8+ T cell–depleted CD4+ T cells (5 × 106) derived from islets of a diabetic 4.1-NOD mouse.
To investigate the mechanisms underlying disease acceleration in 4.1-NOD mice, we then followed the progression of insulitis in prediabetic and diabetic mice. Histopathological studies of pancreata from 3- and 6-wk-old prediabetic 4.1-NOD mice showed that acceleration of diabetes in these mice was a result of faster progression, but not earlier onset, of islet inflammation (Fig. 2 B). As expected, the insulitis lesions of diabetic 4.1-NOD mice contained more CD4+ and fewer CD8+ T cells (but similar numbers of B cells and macrophages [not shown]), than those of diabetic nontransgenic NOD mice (Fig. 2 C). Islet-derived CD4+ T cells from 4.1-NOD mice expressed high levels of the transgene-encoded Vβ11+ chain (Fig. 2 D), proliferated in response to NOD islet cells in vitro (Fig. 2 E), and transcribed messenger RNA for IL-2 and IFN-γ, but not IL-4 (data not shown). These data indicate that these cells were transgenic, beta cell reactive, and of the Th1 type, as expected. Moreover, purified islet-derived CD4+ T cells from three different diabetic 4.1-NOD mice were able to transfer IDDM into three different scid-NOD mice shortly after transfusion (36 ± 12 d) in the absence of CD8+ T cells in the inflamed islets (Fig. 2 F). We thus conclude that expression of the 4.1-TCR in the NOD background promotes the selection of highly diabetogenic CD4+ Th1 cells and their accelerated recruitment into pancreatic islets, leading to massive beta cell destruction and IDDM within the first few weeks of life.
Thymocyte Deletion, and Insulitis and IDDM Resistance in 4.1-F1 Hybrid Mice.
The exquisite pathogenicity of the 4.1-TCR provided us with a powerful tool with which to test our initial hypothesis that diabetes-resistant backgrounds encode non-MHC–linked elements other than endogenous mouse mammary tumor virus superantigens (vSAgs) that are tolerogenic for diabetogenic T cells. To investigate this, we crossed 4.1-NOD mice (H-2g7) with SJL/J (H-2s), C57BL/6 (H-2b), and C58/J mice (H-2k); F1 mice resulting from crosses of nontransgenic NOD mice with these strains express the diabetogenic I-Ag7 molecule, but are diabetes-resistant, and do not delete Vβ11+ T cells (42). The lack of vSAg-mediated deletion of Vβ11+ or Vαx4.1+ cells in these backgrounds was confirmed by the fact that the thymocyte profiles of single-chain TCR-β– or TCR-α–transgenic F1 mice and those of TCR-β– or TCR-α–transgenic NOD mice, respectively, were indistinguishable; as shown in Fig. 3, the percentages of thymic and splenic CD4+CD8−Vβ11+ cells in TCR-β–transgenic (Fig. 3 A) or nontransgenic F1 mice (Fig. 3 B) were virtually identical to (if not greater than) those seen in TCR-β–transgenic or nontransgenic NOD mice, respectively.
Figure 3.

Absence of deletion of Vβ11+CD4+ T cells in TCRβ–transgenic (A) and nontransgenic (B) F1 hybrid mice. Data correspond to average values from 3–6 mice/group. T, thymocytes; S, splenocytes. *P <0.02.
The flow cytometric profiles of thymocytes (Fig. 4 A) and splenocytes (Fig. 4 B) from 4.1-(N × S) F1 mice (n = 8) were comparable to those seen in 4.1-NOD mice (Fig. 1), indicating that the 4.1-TCR specificity also undergoes positive selection in H-2g7/s mice. In contrast, all 4.1-(N × B) F1 mice (n = 22) and most 4.1-(N × C) F1 mice (n = 16/19) had only one-third to one-half the number of thymocytes seen in 4.1-NOD mice (and 4.1-[N × S] F1 mice), and displayed flow cytometric profiles of thymocytes and splenocytes compatible with negative selection of the 4.1-TCR, as compared with other TCR-α/β–transgenic models (23, 24, 39, 43) (Fig. 4). When compared to 4.1-NOD mice, 4.1-(N × B) F1 and 4.1-(N × C) F1 mice (also referred to as “deleting”) displayed a significant reduction in the percentage of CD4+CD8− thymocytes, a reduction in the percentage of CD4+CD8− thymocytes expressing the transgene-encoded Vβ11 element, and an increase in the percentage of CD4−CD8− thymocytes (Fig. 4 A). In the spleen, 4.1-NOD mice had significantly more CD4+ T cells and more CD4+ T cells expressing Vβ11+ TCRs than deleting mice (Fig. 4 B). The few Vβ11+CD4+ T cells that matured in deleting mice expressed half as many transgenic TCR-β chains on the cell surface (but comparable numbers of total TCR-α/β complexes) as Vβ11+CD4+ T cells of 4.1-NOD mice, both in the thymus and in the spleen (P <0.003; data not shown), suggesting that in deleting mice these cells were selected on endogenous TCR chains that had bypassed allelic exclusion, as seen in other models (23, 24, 39, 43).
Figure 4.

CD4, CD8, and Vβ11 profiles of thymocytes (A) and splenic cells (B) from transgenic F1 hybrid mice. See legend to Fig. 1 for details. Data shown are average values of 7–29 mice/group. In the text, transgenic NOD are referred to as 4.1-NOD; (NOD × SJL) F1-TG as 4.1-(N × S) F1; (NOD × B6) F1-TG as 4.1-(N × B) F1; and (NOD × C58) F1-TG as 4.1-(N × C) F1. When compared to 4.1-NOD mice, 4.1-(N × B) F1 and 4.1- (N × C) F1 mice had fewer CD4+CD8− thymocytes (P <0.0002), fewer Vβ11+CD4+CD8− thymocytes (P <0.0002), and more CD4−CD8− thymocytes (P <0.0002) (A). In the spleen (B), 4.1-NOD mice had more CD4+ T cells (P <0.0001) and more Vβ11+ CD4+ T cells (P <0.002) than 4.1- (N × B) F1 and 4.1-(N × C) F1 mice. All comparisons were done using the Mann-Whitney U test.
Proliferation assays using splenic CD4+ T cells as responders and irradiated NOD islet cells and splenocytes as antigen and APCs, respectively, revealed the absence of beta cell–reactive CD4+ T cells in the periphery of deleting, but not nondeleting, 4.1-F1 mice (Fig. 5 A). Lack of proliferation in these assays was the result of deletion, rather than anergy, since peripheral CD4+ T cells from deleting and nondeleting 4.1-F1 mice proliferated similarly in response to plate bound anti–TCR-α/β mAb (Fig. 5 B). The absence of diabetogenic 4.1 T cells in the peripheral lymphoid organs of deleting mice was confirmed by the observation that, like their nontransgenic littermates, 4.1-F1 hybrid mice developed neither diabetes nor insulitis (Table 1).
Figure 5.

In vitro proliferation of naive splenic CD4+ T cells from F1 hybrid mice to islet cells (A) and immobilized anti– TCR-β mAb (B). Proliferation assays in A were done as in Fig. 1 C. For anti–TCR-β–induced proliferation (B), CD4+ T cells (2 × 104) were added in triplicate to wells precoated with serial dilutions of anti–TCR-β mAb (H57-597). Bars show the standard error of the means.
Table 1.
Insulitis and Diabetes in NOD versus F1 Hybrid Mice*
| n | IDDM‖ | Age at Onset¶ | Insulitis Score (n)¶ | |||||
|---|---|---|---|---|---|---|---|---|
| d | x ± SD | |||||||
| NOD-TG | 40 | 30/40a | 48 ± 20g | 3.74 ± 0.15‡i(4) | ||||
| NOD–non-TG | 173 | 123/173b | 138 ± 27h | 1.59 ± 0.58‡j(13) | ||||
| (NOD × B6) F1-TG | 10 | 0/10c | – | 0.42 ± 0.81§k(10) | ||||
| (NOD × B6) F1–non-TG | 12 | 0/12d | – | 0§l(12) | ||||
| (NOD × C58) F1-TG | 15 | 0/15e | – | 0.42 ± 0.70§m(15) | ||||
| (NOD × C58) F1–non-TG | 4 | 0/4f | – | 0§n(4) |
Groups include both males and females.
6-wk-old mice;
15-wk-old mice. Insulitis (15–30 islets/mouse) was scored as described in Materials and Methods. x ± SD, mean ± standard deviation; TG, transgenic. a versus c,e, b versus d,f, g versus h, j versus l: P <0.0001; i versus j: P <0.003; i versus k: P <0.0005; i versus m, j versus n: P <0.0032. Data was compared by χ2 (
) and Mann-Whitney U test(
).
Taken together, these data indicate that: (a) C57BL/6 and C58/J mice carry genes encoding elements capable of mediating the deletion of the diabetogenic 4.1-TCR; (b) the deleting element(s) expressed by these strains has complete or incomplete penetrance, respectively; and (c) these elements are not encoded by vSAgs and target T cells coexpressing both chains of the diabetogenic TCR.
Thymocyte Deletion and Resistance to Insulitis and IDDM Cosegregate with H-2b and H-2k.
Previous studies have suggested that the IDDM resistance provided by certain non-NOD MHC class II genes, including I-Ab and I-Ak, involves the induction of immunoregulatory functions rather than the deletion or anergy of autoreactive T cells (10, 16, 18, 32, 33, 44). Accordingly, we hypothesized that thymocyte deletion in 4.1-F1 hybrid mice would be mediated by elements encoded by non-MHC–linked genes. To test this hypothesis, we backcrossed 4.1-F1 mice with NOD mice and investigated whether thymocyte deletion and IDDM resistance in the transgenic offspring (4.1-F2 mice) segregated away from the H-2 haplotypes of the deleting backgrounds (H-2b and H-2k). All 4.1-F2 mice were killed at IDDM onset, or at 10 wk if nondiabetic, to determine: (a) their H-2 phenotypes, (b) the occurrence of thymocyte deletion; and (c) the degree of insulitis. Unexpectedly, we found that deletion of 4.1 thymocytes segregated as a single-locus trait linked to the MHC; it occurred in H-2g7/b (16/16 mice, 100%) or H-2g7/k mice (9/15 mice, 60%; incidence of deletion comparable to that seen in [N × C] F1 mice), but never in H-2g7/g7 mice (0/24 mice, 0%) (Table 2). Like deleting 4.1-F1 mice, deleting 4.1-F2 mice did not harbor detectable beta cell–reactive CD4+ T cells in the spleen (data not shown). Furthermore, deleting H-2g7/b or H-2g7/k 4.1-F2 mice developed neither IDDM nor insulitis, whereas H-2g7/g7 4.1-F2 mice developed moderate to severe insulitis (100%) and diabetes (∼50%) within 10 wk (Table 2). These data thus indicate that the deleting elements of C57BL/6 and C58/J mice are encoded by genes tightly linked to their H-2b and H-2k complexes, and demonstrate that insulitis and diabetes resistance in 4.1-F2 mice carrying these haplotypes results from deletion of diabetogenic thymocytes.
Table 2.
Cosegregation of Thymocyte Deletion and Resistance to Insulitis and IDDM with H-2b and H-2k Haplotypes in 4.1-transgenic Mice*
| H-2 | Deleting status | Thymocyte profile | Age at onset | Insulitis score ‡,∥ (n) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | CD4+CD8− (Vβ11+) | CD4+CD8+ | CD4−CD8− | CD4−CD8+ | IDDM | |||||||||||||||
| %, x ± SD‖ | n§ | d | x ± SD | |||||||||||||||||
| (NOD × B6) F1 × NOD | H-2g7 | 15 | − | 31 ± 7a | 44 ± 12 | 22 ± 7f | 3 ± 1 | 7/15k | 53 ± 17 | 2.4 ± 1.4 (8)p | ||||||||||
| (92 ± 2)a | ||||||||||||||||||||
| H-2g7/b | 16 | + | 15 ± 3b | 49 ± 12 | 33 ± 10g | 3 ± 1 | 0/16l | – | 0.4 ± 0.6 (16)q | |||||||||||
| (66 ± 5)b | ||||||||||||||||||||
| (NOD × C58) F1 × NOD | H-2g7 | 9 | − | 35 ± 5c | 43 ± 7 | 19 ± 4h | 3 ± 2 | 5/9m | 47 ± 16 | 3.4 ± 0.7 (4)r | ||||||||||
| (93 ± 1)c | ||||||||||||||||||||
| H-2g7/k | 6 | − | 35 ± 4d | 45 ± 8 | 16 ± 6i | 4 ± 2 | 0/6n | – | 1.7 ± 1.2(6)s | |||||||||||
| (89 ± 9)d | ||||||||||||||||||||
| H-2g7/k | 9 | + | 17 ± 3e | 45 ± 6 | 33 ± 6j | 4 ± 3 | 0/9o | – | 0.2 ± 0.2 (8)t | |||||||||||
| (58 ± 9)e | ||||||||||||||||||||
All mice were killed at IDDM onset or at 10 wk if nondiabetic. Flow cytometry was done as described in the legend to Fig. 1.
Nondiabetic mice only. Groups of mice include both male and female mice (∼ 50% each). No differences in the incidence of IDDM nor in the degree of insulitis were noted between female and male mice within groups. Insulitis (15–30 islets/mouse) was scored as described in Materials and Methods. a versus b,c versus e,d versus e,f versus g,h versus j,m versus j: P <0.0002; k versus l: P <0.0001; m versus n, m versus o, s versus t: P <0.002; p versus q: P <0.0006; r versus s: P <0.0007; r versus t: P <0.01 (compared by χ2[
] and Mann-Whitney U test [
]).
I-Ab and I-Ak Molecules as Triggers of 4.1 Thymocyte Deletion.
The data presented above suggested that the deletion of transgenic thymocytes and the IDDM resistance observed in 4.1-F1 and -F2 mice might be mediated by MHC class I and/or class II molecules encoded by the protective H-2 haplotypes. To determine whether 4.1 thymocyte deletion required the engagement of MHC class I molecules, we followed the maturation of 4.1 thymocytes in CD8-α- or β2m-deficient 4.1-(N × B) F1 mice (H-2g7/b), which either do not express the MHC class I-binding CD8 coreceptor on thymocytes, or lack MHC class I molecules, respectively. These mice deleted transgenic thymocytes as efficiently as wild-type 4.1-F1 mice (data not shown), thus indicating that deletion of transgenic thymocytes was not mediated by MHC class I molecules.
Since H-2g7/b mice do not express I-E molecules, we reasoned that deletion in these mice might be mediated by I-Ab. To test this notion, we followed the development of 4.1 thymocytes in I-Aβb–deficient 4.1-(N × B) F1 mice; except for the I-Aβb mutation, I-Aβb–deficient 4.1-(N × B) F1 and 4.1-(N × B) F1 mice are genetically identical. Selective abrogation of I-Aβb expression in 4.1-(N × B) F1 mice restored, at least in part, the positive selection of the transgenic TCR, as evidenced by (a) significant increases in the percentage of Vβ11+ thymocytes (Table 3), (b) significant increases in the ratio of CD4+CD8− to CD4−CD8− T cells in the thymus and in the ratio of CD4+ to CD8+ T cells in the spleen (Table 3), (c) the reappearance of beta cell–reactive CD4+ T cells in the spleen (Fig. 6, left), and (d) the reemergence of insulitis (Table 3). The overall positive selection of the 4.1-TCR in I-Aβb– deficient 4.1-(N × B) F1 mice, however, was less efficient than in 4.1-NOD mice; I-Aβb–deficient 4.1-(N × B) F1 mice had more CD4−CD8− thymocytes than, and half the splenic CD4+ T cells of, age-matched (3–5-wk-old) 4.1-NOD mice (Table 3). Furthermore, the peripheral CD4+ T cells of I-Aβb–deficient 4.1-(N × B) F1 mice proliferated less efficiently in response to NOD islet cells than the peripheral CD4+ T cells of 4.1-NOD mice (Fig. 6, left). Finally, the insulitis lesions of I-Aβb–deficient 4.1-(N × B) F1 mice were milder than those seen in 4.1-NOD mice and did not lead to IDDM in any of the 15 mice that were followed (Table 3). Therefore, the I-Aβb gene is not the only protective element present in (N × B) F1 mice, but its expression is sufficient in and of itself to induce deletion of 4.1 thymocytes.
Table 3.
Influence of I-Ab,I-Ak, I-Aq, and H2nb1 MHC Class II Molecules on 4.1 Thymocyte Development
| Percentage of cells* | Ratios* | IDDM‡ (n) | Insulitis‡ score (n) | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | Organ | CD4+CD8−(Vβ11+) | CD4+CD8+ | CD4−CD8− | CD4−CD8+ | CD4+/ DN | CD4+/CD8+ | |||||||||||||
| x ± SD | x ± SD | |||||||||||||||||||
| (NOD × B6) F1 | 29 | T | 15 ± 5a (67 ± 7) | 53 ± 10 | 27 ± 9 | 4 ± 3 | 0.6 ± 0.2s | – | 0 (10)ae | 0.42 ± 0.81 (10)ak | ||||||||||
| S | 12 ± 6 (78 ± 12) | – | 84 ± 8 | 4 ± 2 | – | 2.9 ± 1.5y | ||||||||||||||
| (NOD × I-Ab−B6) F1 | 9 | T | 36 ± 5b (93 ± 3) | 37 ± 9 | 23 ± 4o | 4 ± 1 | 1.6 ± 0.2t | – | 0 (15)af | 1.59 ± 0.45 (10)al | ||||||||||
| S | 12 ± 6c (86 ± 11) | – | 86 ± 7 | 2 ± 1 | – | 4.6 ± 1.4z | ||||||||||||||
| NOD | 9 | T | 30 ± 7d (94 ± 2)e | 53 ± 9 | 13 ± 4p | 4 ± 2 | 2.2 ± 0.6u | – | 30 (40)ag | 3.74 ± 0.15 (4)am | ||||||||||
| S | 24 ± 4f (88 ± 4)g | – | 72 ± 5 | 4 ± 1 | – | 8.7 ± 2.9aa | ||||||||||||||
| NOD.H-2g7/I-Ak | 7 | T | 25 ± 3 (93 ± 1) | 49 ± 5 | 21 ± 3q | 5 ± 1 | 1.2 ± 0.2v | – | 0 (9)ah | 2.2 ± 0.8 (7)an | ||||||||||
| S | 6 ± 1h (90 ± 1) | – | 92 ± 1 | 2 ± 1 | – | 2.9 ± 0.3ab | ||||||||||||||
| NOD.H-2g7/q | 7 | T | 19 ± 3i (88 ± 3) | 59 ± 5 | 19 ± 3 | 3 ± 1 | 1.0 ± 0.2w | – | 0 (7)ai | 0.7 ± 0.4 (7)ao | ||||||||||
| S | 11 ± 8j (87 ± 4) | – | 86 ± 10 | 3 ± 2 | – | 2.9 ± 1.4ac | ||||||||||||||
| NOD.H-2g7/nb1 | 11 | T | 11 ± 3k (37 ± 9)l | 52 ± 7 | 34 ± 5r | 3 ± 1 | 0.3 ± 0.1x | – | 0 (11)aj | 0.1 ± 0.1 (7)ap | ||||||||||
| S | 6 ± 2m (51 ± 16)n | – | 91 ± 4 | 2 ± 1 | – | 2.9 ± 0.4ad | ||||||||||||||
Mice were studied at 3–5 (
) or at 10 wk (
) of age. Flow cytometry was done as indicated in the legend to Fig. 1. DN, double-negative cells (CD4−CD8−); T, thymus; S, spleen. Statistics (Mann-Whitney U test): CD4+CD8−: a versus b, P <0.0001; c versus f, P <0.003; h versus f, P <0.0009; i versus d, P <0.0001; j versus f, P <0.005; k versus d, P <0.0002; m versus f, P <0.0002; CD4+CD8−Vβ11+: 1 versus e, P <0.0002; n versus g, P <0.0002; CD4−CD8−: o versus p, P <0.03; q versus p, P <0.015; r versus p, P <0.0002; CD4+/DN: s versus t, P <0.0001; u versus v, P <0.03; w versus u, P <0.002; x versus u, P <0.0002; CD4+/CD8+: y versus z, P <0.015; z versus aa, P <0.002; ab versus aa, P <0.0009; ac versus aa, P <0.0002; ad versus aa, P <0.0002; IDDM: ad,ae,ah,ai,aj versus ag, P <0.0001; Insulitis Score: al versus ak, P <0.0006; ak versus am, P <0.0005; al versus am, P <0.005; an versus am, P <0.03; ao versus am, P <0.008; ap versus am, P <0.006.
Figure 6.

Peripheral beta cell-reactivity in I-Aβb- 4.1-(N × B) F1, 4.1/I-Ak-NOD, 4.1-NOD. H-2g7/q, and 4.1-NOD.H-2g7/nb1 mice. Assays were done as in Fig. 1 C. Values correspond to cultures of 2 × 104 CD4+ T cells with 5 × 104 (left) or 105 NOD islet cells (middle and right).
To investigate whether the H-2k–dependent deletion of thymocytes in 4.1-(N × C) F1 mice was also I-A mediated, we crossed 4.1-NOD mice with I-Aαk/I-Aβk (I-Ak)- transgenic NOD mice, to generate 4.1/I-Ak–NOD mice. Except for the presence of the I-Ak transgenes in 4.1/I-Ak– NOD mice, 4.1-NOD and 4.1/I-Ak–NOD mice have virtually identical genetic backgrounds. We found that 4.1-NOD mice had significantly more thymocytes (data not shown) and greater ratios of CD4+CD8− to CD4−CD8− thymocytes and of CD4+ to CD8+ splenocytes than 4.1/ I-Ak–NOD mice (Table 3), results compatible with deletion of 4.1 thymocytes in 4.1/I-Ak–NOD mice. This deletion, however, was incomplete; unlike the splenic CD4+ T cells of deleting H-2g7/k 4.1-F2 mice, the few CD4+ T cells that matured in 4.1/I-Ak–NOD mice expressed high levels of the transgenic TCR-β chain, and these cells proliferated in response to beta cells in vitro (Tables 2 and 3; and Fig. 6, middle). Furthermore, unlike deleting H-2g7/k 4.1-F2 mice, 4.1/I-Ak–NOD mice developed moderate insulitis, suggesting that these cells were also responsive to antigen stimulation in vivo (Table 3). The insulitis lesions of these mice, however, were less severe than those of 4.1-NOD mice, and did not lead to diabetes in any of the nine mice that were followed (Table 3). It appears, then, that (a) deletion of thymocytes in H-2g7/k 4.1 mice is triggered, at least in part, by I-Ak molecules, and (b) in addition to causing partial deletion of 4.1 thymocytes, I-Ak molecules may somehow abrogate the diabetogenic potential of the 4.1 T cells that escape deletion.
Thymocyte Deletion in 4.1-NOD.H-2 g7/q and 4.1-NOD.H-2g7/nb1 Mice.
We next asked whether the 4.1-TCR could engage additional antidiabetogenic MHC class II molecules during thymocyte development. We therefore crossed 4.1-NOD mice with NOD mice congenic for H-2q or H-2nb1 haplotypes, which provide dominant resistance to diabetes in nontransgenic NOD.H-2g7/q or nb1 mice (7, 15), and followed the fate of the 4.1-TCR in the TCR-transgenic offspring. As shown in Table 3 and Fig. 6 (right) 4.1-NOD.H-2g7/q and 4.1-NOD.H-2g7/nb1 mice had phenotypes compatible with partial or complete deletion of the 4.1-TCR, respectively. When compared to 4.1-NOD mice, 4.1-NOD.H-2g7/q mice had twofold fewer thymocytes (data not shown), increased percentages of CD4− CD8− thymocytes, and reduced percentages of thymic and splenic CD4+CD8− T cells (Table 3). The splenic CD4+ T cells of these mice proliferated in response to NOD islet cells in vitro, but did so less efficiently than those of 4.1-NOD mice (Fig. 6, right). Furthermore, these mice only developed very mild periinsulitis and never became diabetic (Table 3). In contrast, thymocyte deletion in 4.1-NOD.H-2g7/nb1 mice was complete; these mice had a three- to fourfold reduction in the absolute number of thymocytes (data not shown), dramatically increased percentages of CD4−CD8− thymocytes, and reduced percentages of thymic and splenic Vβ11+CD4+CD8− T cells (Table 3). These mice did not contain detectable beta cell–reactive CD4+ T cells in the spleen (Fig. 6, right) and developed neither diabetes nor insulitis (Table 3). Thymocyte deletion in these mice was not mediated by putative NOD vSAgs (i.e., mouse mammary tumor virus superantigen 17) binding to Vβ11 and H-2q or H-2nb1 MHC class II molecules, since the spleens of nontransgenic NOD. H-2g7/q and NOD.H-2g7/nb1 littermates harbored more Vβ11+CD4+ T cells than nontransgenic NOD mice (20 ± 7 and 12 ± 2% versus 6 ± 1%, P <0.008 and P <0.04, respectively).
Thymocyte Deletion in H-2 Heterozygous 4.1 Mice Is Not Restricted by I-Ag7.
To elucidate whether in vivo deletion of 4.1 thymocytes was “restricted” by I-Ag7 molecules, we next studied thymocyte maturation in 4.1 mice homozygous for either nonselecting/nondeleting (H-2s) or deleting (H-2b or H-2k) MHC haplotypes. As expected, 4.1 thymocytes underwent neither positive nor negative selection in H-2s 4.1 mice; these mice had significantly fewer CD4+CD8− thymocytes (but not more CD4−CD8− thymocytes) than H-2g7/s 4.1 mice (compare Fig. 4, left and 7, left; P <0.005). In contrast, the thymocyte profiles of H-2b and H-2k 4.1 mice were compatible with deletion; these mice had fewer thymocytes and greater percentages of CD4−CD8− thymocytes than H-2s 4.1 mice (Fig. 7, middle and right). Therefore, unlike the positive selection of 4.1 T cells in selecting (I-Ag7+) mice, the I-Ab– and I-Ak–mediated deletion of 4.1 thymocytes in 4.1-F1 mice is not I-Ag7– restricted.
Figure 7.

CD4, CD8, and Vβ11 profiles of thymocytes from transgenic mice homozygous for nonselecting or deleting H-2 haplotypes. See legend to Fig. 1 for details. Data are average values corresponding to 7–10 mice/group. H-2b and H-2k 4.1 mice had fewer thymocytes (P <0.006), and more CD4−CD8− thymocytes than H-2s 4.1 mice (P <0.02) (Mann-Whitney U test).
Thymocyte Deletion in 4.1-F1 Mice Is Preceded by Positive Selection and Is Mediated by Hematopoietic Cells.
Previous studies have shown that the factors underlying the MHC-linked resistance to spontaneous IDDM predominantly reside in the bone marrow (17, 33, 44–48). To determine whether deletion of diabetogenic thymocytes in 4.1-F1 mice was mediated by hematopoietic cells or by thymic epithelial cells, we transfused bone marrow from deleting H-2g7/b– or selecting H-2g7-4.1 mice into lethally-irradiated nontransgenic NOD (H-2g7) or (N × B) F1 mice (H-2g7/b), respectively, and followed the fate of 4.1 thymocytes in the chimeras. As shown in Fig. 8, the mice that received marrow from H-2g7/b–4.1 mice (expressing I-Ab only on hematopoietic cells), but not those that received marrow from H-2g7-4.1 mice (expressing I-Ab only on thymic epithelial cells), had a phenotype compatible with deletion (low thymocyte CD4+CD8−/CD4−CD8− ratios and small percentages of Vβ11+CD4+CD8− thymocytes). It thus appears that deletion of thymocytes in 4.1-F1 hybrid mice is preceded by their positive selection on radioresistant thymic epithelial cells, and is mediated by hematopoietic cells.
Figure 8.

Bone marrow chimeras. Bone marrow cells (5–10 × 106) from donor mice (4.1-NOD or 4.1-[N × B] F1 mice) were injected into the tail veins of lethally irradiated recipient mice (nontransgenic NOD, [N × B] F1 or [B × S] F1 mice). Thymi of chimeric mice were analyzed by flow cytometry (as in Fig. 1), 5–6 wk after transplantation. Data represent average values of 2–4 mice/group. The mice that received marrow from H-2g7/b-4.1 mice (left) had lower thymocyte CD4+CD8−/CD4−CD8− ratios and fewer Vβ11+CD4+CD8− thymocytes than mice which received marrow from H-2g7-4.1 mice (middle) (P <0.05).
This finding raised one final question: if deletion is preceded by positive selection, why does it also occur in mice homozygous for the deleting H-2 haplotypes, which lack the selecting I-Ag7 molecule? We reasoned that the 4.1-TCR might actually perceive the deleting I-A molecules (i.e., I-Ab) expressed on thymic epithelial cells as selecting. To test this hypothesis, we followed the fate of 4.1 thymocytes arising from marrow of H-2g7–4.1 mice in lethally irradiated H-2s/b mice, which express deleting (I-Ab) and nonselecting/nondeleting (I-As), but not selecting (I-Ag7), MHC class II molecules on thymic epithelial cells. As shown in Fig. 8 (right), the thymocyte profiles of recipient mice were indistinguishable from those of H-2g7/b mice transplanted with marrow from H-2g7–4.1 mice, which were nondeleting (Fig. 8, middle). It thus appears that in heterozygotes, the 4.1-TCR senses the I-Ab molecules expressed by thymic epithelial cells as selecting, rather than as deleting.
DISCUSSION
The first striking observation of this study was the finding that positive selection of the 4.1-TCR in NOD mice caused a dramatic acceleration of the onset of IDDM (by ∼3 mo). This was surprising to us considering the observations of Katz et al. in transgenic NOD mice expressing another beta cell–specific and I-Ag7–restricted transgenic TCR (BDC-2.5) (32, 41). BDC-2.5 mice do not develop an accelerated onset of diabetes (32, 41) and, when housed under specific pathogen-free conditions (41), develop diabetes less frequently than 4.1-NOD mice (15 versus 74%). We are confident that beta cell destruction in 4.1-NOD mice was triggered and/or effected by CD4+ T cells expressing the 4.1-TCR since recombination activating gene 2–deficient 4.1-NOD mice, which cannot rearrange endogenous TCR genes, develop IDDM as early, and as frequently, as recombination activating gene-2+ 4.1-NOD mice (Verdaguer, J., D. Schmidt, B. Anderson, A. Amrani, and P. Santamaria, manuscript in preparation). Most surprising, however, was the observation that 4.1 thymocytes undergo deletion in diabetes-resistant H-2g7/b–, H-2g7/k–, H-2g7/q–, and H-2g7/nb1–4.1-NOD mice by engaging antidiabetogenic I-A (I-Ab, I-Ak, I-Aq) and, possibly, I-E molecules (I-Ek, I-Enb1) on thymic APCs, particularly since previous studies did not find evidence for deletion of autoreactive T cells in congenic or transgenic NOD mice expressing these MHC class II molecules (7, 15, 16, 32, 33, 44). This deletion was clearly not mediated by endogenous superantigens binding to Vβ11, since it was absent in TCR-β–transgenic and nontransgenic mice expressing the deleting MHC haplotypes, and segregated as a single locus trait (MHC linked) in backcross studies. Thus, the 4.1-TCR is unique in that it is both highly diabetogenic and promiscuous.
Abrogation of I-Aβb expression in TCR-transgenic I-Aβb− (N × B) F1 mice clearly restored the positive selection of the 4.1-TCR; however, it did not completely eliminate its negative selection, as suggested by the greater percentage of CD4−CD8− thymocytes in these mice versus age-matched 4.1-NOD mice. Partial deletion of transgenic thymocytes in I-Aβb- (N × B) F1 mice, which express the C57BL/6-derived I-Aαb gene, may be a result of engagement of I-Aβg7/I-Aαb complexes by the 4.1-TCR with an affinity/ avidity approaching the threshold for deletion, or perhaps, to other tolerogenic factors unique to the C57BL/6 background. Similarly, thymocyte deletion in H-2g7/k 4.1-F1 and -F2 mice, which express both I-Ak and I-Ek molecules, was clearly more efficient than that observed in I-Ak–transgenic 4.1-NOD mice, raising the possibility that thymocyte deletion in the former also involves the engagement of I-Ek and/or I-Eαk/I-Eβg7 molecules by the 4.1-TCR. An alternative, but not mutually exclusive, possibility is that the timing and levels of I-Ak expression in I-Ak–transgenic 4.1-NOD mice are different than the timing and levels of expression of endogenous I-Ak molecules in H-2g7/k 4.1-F1 or -F2 mice. In this respect, it is worth noting that, unlike wild-type H-2k mice, I-Ak-transgenic NOD mice do not express I-Ak molecules on bone marrow cells (they do, however, express them on bone marrow–derived APCs), and that the thymic cortical epithelial cells of I-Ak–transgenic NOD mice express lower levels of I-Ak than those of H-2k mice (13). Whatever the explanation for these differences, the MHC-induced T cell tolerance in H-2g7/b and H-2g7/k 4.1-F1 and -F2 mice was complete. Deletion of the 4.1-TCR was also observed in 4.1-NOD.H-2g7/nb1 mice (complete deletion), which express I-Anb1 and I-Enb1 molecules, and in 4.1-NOD.H-2g7/q mice (partial deletion), which express I-Aq, but not I-E, molecules. It thus appears that, unlike many other MHC class II–restricted TCR specificities (32, 38, 40), the highly diabetogenic 4.1-TCR can engage several distinct MHC class II molecules in the thymus with totally different consequences, i.e., positive selection or dominant negative selection.
The extensive promiscuity of the 4.1-TCR raises a series of important questions. How can 4.1 thymocytes recognize so many different MHC class II molecules? Does the 4.1-TCR recognize each of these different molecules bound to specific peptides? Or to a common peptide? Or, does it bind to all of them regardless of the molecular nature of the bound peptides? Is there anything unique about the molecular structure of the deleting I-A molecules that makes them function as such? We do not yet have answers to these questions, but we have some clues. For example, we know that the diabetogenic 4.1-TCR is not a classic alloreactive TCR; the MHC molecules that mediated deletion of the 4.1-TCR when presented by thymic APCs did not trigger proliferation of naive and preactivated CD4+ T cells from 4.1-NOD mice when presented by peripheral APCs from deleting F1 backgrounds (our unpublished data). Since thymocyte tolerance is a more sensitive response than peripheral T cell activation (43), these results need not imply that the putative peptide–MHC class II complexes that mediate thymic deletion in our system are expressed solely by a specialized thymic APC subpopulation; they may also be present on peripheral APCs, but may not be able to trigger mature T cell proliferation. We also know that the I-Aβ chains of all the deleting I-A molecules that we have tested so far (I-Ab, I-Ak, and I-Aq) share residues at positions 57 (aspartic acid; Asp) and 61 (tryptophan). Interestingly, I-Ek and I-Enb1 molecules, which are encoded on the deleting H-2k and H-2nb1 haplotypes, respectively, have the same residues at these two positions (49). I-Anb1, which is encoded on the deleting H-2nb1 haplotype (but may or may not be engaged by the 4.1-TCR), is also Asp-57+ and, like I-Ab, I-Ak, and I-Aq, has a bulky hydrophobic residue at position 61 (phenylalanine). We are not certain as to whether the presence of aspartic acid at I-Eβ and/or I-Aβ position 57, which is negatively associated with human and murine IDDM (1, 2), is neccessary to trigger deletion of the 4.1-TCR. However, we know it is not sufficient, since I-As is also Asp-57+, but it is nondeleting.
Whatever the specific residues involved, the unexpected promiscuity of the highly pathogenic 4.1-TCR raises the intriguing possibility that pathogenicity of autoreactive TCRs and their ability to cross-react with different MHC class II molecules are related phenomena. At the present time, it is difficult to envision how promiscuity may lead to pathogenicity. It is possible, however, that promiscuous autoreactive TCRs, like the 4.1-TCR, are primarily selected (positively and/or negatively) by reaction with MHC residues rather than with peptide residues, as recently proposed for alloreactive TCRs (50). Like the latter (50), these promiscuous autoreactive TCRs may then be able to engage a larger range of peripheral self-peptide–selecting MHC complexes above the affinity threshold required for mature T cell activation than nonpromiscuous autoreactive TCRs, resulting in increased chances for pathogenicity. This would predict that promiscuity would not be unusual among those autoreactive TCRs with the highest pathogenic potential (i.e., those that trigger diabetes), and that MHC molecules providing dominant resistance to a given autoimmune disease would do so predominantly by removing the most pathogenic autoreactive T cells rather than all autoreactive T cells, regardless of their pathogenicity (i.e., those recruited during amplification of the autoimmune response). This postulate would provide an explanation as to why the beta cell–reactive and I-Ag7–restricted BDC-2.5-TCR of Katz et al. (32), which does not accelerate IDDM onset in NOD mice (32, 41), did not undergo tolerance in H-2g7/b F1 mice or in I-E–trangenic NOD mice (32). It would also account for the presence of mildly insulitogenic, but not diabetogenic, T cells in some congenic NOD.H-2g7/b, NOD.H-2g7/q, or NOD.H-2g7/nb1 mice (7, 15, 16), I-Ad–transgenic NOD mice (18), NOD mice reconstituted with bone marrow from I-Eα–transgenic NOD mice (33), and I-Ak-transgenic NOD mice (12, 13).
In evaluating the relevance of our findings in 4.1 mice with respect to the MHC-linked susceptibility and resistance to spontaneous IDDM in non-TCR–transgenic mice, one should consider two additional aspects. The first aspect has to do with the pathophysiological consequences of thymocyte selection in 4.1 mice. There was an absolute correlation between deletion of thymocytes in H-2g7/b and H-2g7/k 4.1-F2 mice and resistance to insulitis and diabetes; deleting offspring of the second backcross of 4.1-F1 mice to NOD mice never developed insulitis, whereas all their nondeleting littermates developed moderate to severe insulitis, and ∼50% of those which inherited two H-2g7 haplotypes became diabetic (a significant percentage given the polygenic nature of murine IDDM; reference 2). The same was true for 4.1-NOD.H-2g7/q and 4.1-NOD.H-2g7/nb1 mice, which developed mild periinsulitis or no insulitis, respectively, and did not become diabetic. It is noteworthy that these observations are highly reminiscent of the resistance of NOD.H-2g7/b, NOD.H-2g7/q, or NOD.H-2g7/nb1-congenic mice, and of I-E–, I-Ad–, and I-Ak–transgenic NOD mice to insulitis and/or IDDM (6–14, 16–18). The second aspect relates to the geography and timing of negative selection of diabetogenic thymocytes in 4.1 mice. As in other models of thymocyte selection (51, 52), negative selection of thymocytes in 4.1-F1 mice was preceded by positive selection and was mediated by hematopoietic cells. This observation also accords with the widely accepted notion that the factors underlying the MHC-linked resistance to spontaneous IDDM predominantly reside in the bone marrow (17, 33, 44–48). Taken together, then, these data lend strong support to the hypothesis that the MHC-associated resistance to insulitis and IDDM of non-TCR–transgenic mice (and perhaps humans) may also result from negative selection of highly diabetogenic thymocytes.
Nonetheless, deletion of diabetogenic thymocytes does not account for all the protection afforded by MHC class II molecules. It appears, also, that in the absence of additional susceptibility factors and/or in the presence of additional resistance factors, positive selection of autoreactive T cells bearing pathogenic TCRs does not automatically imply autoreactivity. These interpretations stem from three observations. First, I-Ak/4.1-NOD mice developed insulitis but not diabetes, suggesting that engagement of the partially deleting I-Ak molecules by the few 4.1 T cells that escaped deletion/anergy might have fostered their differentiation into nondiabetogenic Th-cells (perhaps of the Th2 type, or of the Th1 type, but incapable of effecting beta cell damage). Second, unlike their H-2g7 littermates, the H-2g7/k 4.1-F2 mice that did not tolerate the 4.1-TCR (40%) only developed nondiabetogenic insulitis. The reasons behind the diabetes resistance of these mice may be similar to those argued above for I-Ak/4.1-NOD mice. Alternatively, progression from moderate to severe insulitis and overt diabetes may require a double dose of the beta cell antigen–I-Ag7 complex(es) targeted by diabetogenic T cells, as proposed in nontransgenic systems (16). Finally, I-Aβb–deficient 4.1- (N × B) F1 mice developed insulitis but remained diabetes-resistant, and 4.1-(N × S) F1 mice, which also exported functional 4.1 T cells to the periphery, developed neither diabetes nor insulitis. Whether the diabetes resistance of these mice is due to I-Ag7 hemizygosity, to the presence of additional C57BL/6- or SJL-derived resistance elements, or to “dilution” of NOD-derived recessive susceptibility factors, is currently under investigation.
Whatever the nature and function of these additional protective elements turn out to be, our observations in 4.1-TCR–transgenic mice have unearthed what, at the outset of this study, seemed to be an unlikely proposition, namely, the existence of a relationship between thymocyte selection of highly pathogenic T cells and the MHC-linked susceptibility and resistance to one spontaneous autoimmune disease, IDDM. Our results do not imply that deletion is the only mechanism for autoimmune disease protection afforded by MHC class II molecules, but explain how protective MHC genes carried on one haplotype (e.g., I-Ab in mice and DQA1*0102/DQB1*0602 in humans) can override the genetic susceptibility provided by MHC genes carried on a second haplotype (e.g., I-Ag7 in mice and DQA1*0301/DQB1*0302 in humans).
Acknowledgments
We thank T. Mak for providing CD8-α− mice; G. Morahan and J.F.A.P. Miller for providing I-Ak–transgenic NOD mice; M. Davis and S. Hedrick for TCR shuttle vectors; M. Nagata and J.-W. Yoon for providing NY4.1 cells and for exciting discussions; T. Utsugi for advice on histopathology, for providing rIL-2, and for helpful suggestions; R. Sangha and R. Sparkes for help with histology; R. Dawson and L. Mock for excellent animal care; L. Bryant for excellent assistance with flow cytometry; H. Kominek for editorial assistance.
Footnotes
J. Verdaguer was supported by a postdoctoral fellowship from CIRIT (Comissió Interdepartamental de Reçerca i Innovació Tecnològica, Generalitat de Catalunya, Barcelona, Spain). P. Santamaria is a scholar of the Medical Research Council of Canada. This research was supported by a grant from the Medical Research Council of Canada.
Abbreviations used in this paper: 4.1-F1, TCR-α/β–transgenic F1; β2m, β2 microglobulin; Asp, aspartic acid; B, C57BL/6; C, C58/J; CM, complete medium; IDDM, insulin-dependent diabetes mellitus; N or NOD, nonobese diabetic; S, SJL/J; vSAg, virus superantigen.
D. Schmidt and J. Verdaguer contributed equally to this work.
REFERENCES
- 1.Tisch R, McDevitt HO. Insulin-dependent diabetes mellitus. Cell. 1996;85:291–297. doi: 10.1016/s0092-8674(00)81106-x. [DOI] [PubMed] [Google Scholar]
- 2.Vyse TJ, Todd JA. Genetic analysis of autoimmune disease. Cell. 1996;85:311–318. doi: 10.1016/s0092-8674(00)81110-1. [DOI] [PubMed] [Google Scholar]
- 3.Serreze DV, Leiter EH. Genetic and pathogenic basis of autoimmune diabetes in NOD mice. Curr Opin Immunol. 1994;6:900–906. doi: 10.1016/0952-7915(94)90011-6. [DOI] [PubMed] [Google Scholar]
- 4.Hattori M, Buse JB, Jackson RA, Glimcher L, Dorf ME, Minami M, Makino S, Moriwaki K, Kuzuya H, Imura H, et al. The NOD mouse: recessive diabetogenic gene in the major histocompatibility complex. Science (Wash DC) 1986;231:733–735. doi: 10.1126/science.3003909. [DOI] [PubMed] [Google Scholar]
- 5.Acha-Orbea H, McDevitt HO. The first external domain of the non-obese diabetic mouse class II I-Aβ chain is unique. Proc Natl Acad Sci USA. 1987;84:2435–2439. doi: 10.1073/pnas.84.8.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishimoto H, Kikutani H, Yamamura K-I, Kishimoto T. Prevention of autoimmune insulitis by expression of I-E molecules in NOD mice. Nature (Lond) 1989;328:432–434. doi: 10.1038/328432a0. [DOI] [PubMed] [Google Scholar]
- 7.Prochazka M, Serreze DM, Worthen SM, Leiter EH. Genetic control of diabetogenesis in NOD/Lt mice. Development and analysis of congenic stocks. Diabetes. 1989;38:1446–1455. doi: 10.2337/diab.38.11.1446. [DOI] [PubMed] [Google Scholar]
- 8.Uehira M, Uno M, Kürner T, Kikutani H, Mori K-I, Inomoto T, Uede T, Miyazaki J-I, Nishimoto H, Kishimoto T, Yamamura K-I. Development of autoimmune insulitis is prevented in Eαd but not in AβkNOD transgenic mice. Int Immunol. 1989;1:210–213. doi: 10.1093/intimm/1.2.209. [DOI] [PubMed] [Google Scholar]
- 9.Wicker LS, Miller BJ, Fisher PA, Pressey A, Peterson LB. Genetic control of diabetes and insulitis in the nonobese diabetic mouse. Pedigree analysis of a diabetic H-2nod/bheterozygote. J Immunol. 1989;142:781–788. [PubMed] [Google Scholar]
- 10.Böhme J, Schuhbaur B, Kanagawa O, Benoist C, Mathis D. MHC-linked protection from diabetes dissociated from clonal deletion of T-cells. Science (Wash DC) 1990;249:293–295. doi: 10.1126/science.2115690. [DOI] [PubMed] [Google Scholar]
- 11.Lund T, O'Reilly L, Hutchings P, Kanagawa O, Simpson E, Gravely R, Chandler P, Dyson J, Picard JK, Edwards AK, et al. Prevention of insulin-dependent diabetes mellitus in non-obese diabetic mice by transgenes encoding modified I-Aβ-chain or normal I-Eα-chain. Nature (Lond) 1990;345:727–729. doi: 10.1038/345727a0. [DOI] [PubMed] [Google Scholar]
- 12.Miyazaki T, Uno M, Uehira M, Kikutani H, Kishimoto T, Kimoto M, Hishimoto H, Miyazaki J-I, Yamamura K-I. Direct evidence for the contribution of the unique I-Anodto the development of insulitis in non-obese diabetic mice. Nature (Lond) 1990;345:722–723. doi: 10.1038/345722a0. [DOI] [PubMed] [Google Scholar]
- 13.Slattery RM, Kjer-Nielsen L, Allison J, Charlton B, Mandel TE, Miller JFAP. Prevention of diabetes in non-obese diabetic I-Aktransgenic mice. Nature (Lond) 1990;345:724–726. doi: 10.1038/345724a0. [DOI] [PubMed] [Google Scholar]
- 14.Uno M, Miyazaki T, Uehira M, Nishimoto H, Kimoto M, Miyazaki J-I, Yamamura K-I. Complete prevention of diabetes in transgenic NOD mice expressing I-E molecules. Immunol Lett. 1991;31:47–52. doi: 10.1016/0165-2478(92)90009-d. [DOI] [PubMed] [Google Scholar]
- 15.Livingstone A, Edwards CT, Shizuru JA, Fathman CG. Genetic analysis of diabetes in the nonobese diabetic mouse. I. MHC and T cell receptor β gene expression. J Immunol. 1991;146:429–537. [PubMed] [Google Scholar]
- 16.Wicker LS, Appel MC, Dotta F, Pressey A, Miller BJ, DeLarato NH, Fischer PA, Boltz RC, Jr, Peterson LB. Autoimmune syndromes in major histocompatibility complex (MHC) congenic strains of nonobese diabetic (NOD) mice. The NOD MHC is dominant for insulitis and cyclophosphamide-induced diabetes. J Exp Med. 1992;176:67–77. doi: 10.1084/jem.176.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Podolin PL, Pressey A, DeLarato NH, Fischer PA, Peterson LB, Wicker LS. I-E+nonobese diabetic mice develop insulitis and diabetes. J Exp Med. 1993;178:793–803. doi: 10.1084/jem.178.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singer SM, Tisch R, Yang X-D, McDevitt HO. An Aβdtransgene prevents diabetes in nonobese diabetic mice by inducing regulatory T-cells. Proc Natl Acad Sci USA. 1993;90:9566–9570. doi: 10.1073/pnas.90.20.9566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zinkernagel RM, Doherty P. H-2 compatibility requirement for T cell–mediated lysis of target cells infected with lymphocytic choriomeningitis virus: different cytotoxic T cell specificities are associated with structures from H-2K or H-2D. J Exp Med. 1975;141:1427–1436. doi: 10.1084/jem.141.6.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kappler JW, Marrack P. Helper T-cells recognize antigen and macrophage surface components simultaneously. Nature (Lond) 1976;262:797–799. doi: 10.1038/262797a0. [DOI] [PubMed] [Google Scholar]
- 21.Kappler JW, Roehm N, Marrack P. T-cell tolerance by clonal elimination in the thymus. Cell. 1987;49:273–280. doi: 10.1016/0092-8674(87)90568-x. [DOI] [PubMed] [Google Scholar]
- 22.MacDonald HR, Schneider R, Lees RK, Howe RC, Acha-Orbea H, Festenstein H, Zinkernagel RM, Hengartner H. T cell receptor Vβ use predicts reactivity and tolerance to Mlsa-encoded antigens. Nature (Lond) 1988;332:40–45. doi: 10.1038/332040a0. [DOI] [PubMed] [Google Scholar]
- 23.Kisielow P, Bluthmann H, Staerz UD, Steinmetz M, von Boehmer H. Tolerance in T-cell receptor transgenic mice involves deletion of nonmature CD4+CD8+thymocytes. Nature (Lond) 1988;333:742–746. doi: 10.1038/333742a0. [DOI] [PubMed] [Google Scholar]
- 24.Sha W, Nelson C, Newberry R, Kranz D, Russell J, Loh D. Positive and negative selection of an antigen receptor on T cells in transgenic mice. Nature (Lond) 1988;336:73–76. doi: 10.1038/336073a0. [DOI] [PubMed] [Google Scholar]
- 25.Hogquist KA, Jameson CS, Heath WR, Howard JL, Bevan MJ, Carbone FR. T-cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 26.Ashton-Rickardt PG, Bandiera A, Delaney JR, van Kaer L, Pircher HP, Zinkernagel RM, Tonegawa S. Evidence for a differential avidity model of T-cell selection in the thymus. Cell. 1994;76:651–663. doi: 10.1016/0092-8674(94)90505-3. [DOI] [PubMed] [Google Scholar]
- 27.Sebzda E, Wallace VA, Mayer J, Yeung RSM, Mak TW, Ohashi PS. Positive and negative thymocyte selection induced by different concentrations of a single peptide. Science (Wash DC) 1994;263:1615–1618. doi: 10.1126/science.8128249. [DOI] [PubMed] [Google Scholar]
- 28.Bevan MJ. In a radiation chimera host H-2 antigens determine the immune responsiveness of donor cytotoxic cells. Nature (Lond) 1977;169:417–418. doi: 10.1038/269417a0. [DOI] [PubMed] [Google Scholar]
- 29.Zinkernagel RM, Callahan GN, Althage A, Cooper S, Klein PA, Klein J. On the thymus in the differentiation of “H-2 self recognition” by T-cells: evidence for dual recognition? . J Exp Med. 1978;147:882–896. doi: 10.1084/jem.147.3.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nikolic-Zugic J, Bevan M. Role of self peptides in positively selecting the T-cell repertoire. Nature (Lond) 1990;344:65–67. doi: 10.1038/344065a0. [DOI] [PubMed] [Google Scholar]
- 31.Reich EP, Sherwin RS, Kanagawa O, Janeway CA., Jr An explanation for the protective effect of the MHC class II I-E molecule in murine diabetes. Nature (Lond) 1989;341:326–328. doi: 10.1038/341326a0. [DOI] [PubMed] [Google Scholar]
- 32.Katz JD, Wang B, Haskins K, Benoist C, Mathis D. Following a diabetogenic T-cell from genesis through pathogenesis. Cell. 1993;74:1089–1100. doi: 10.1016/0092-8674(93)90730-e. [DOI] [PubMed] [Google Scholar]
- 33.Parish NM, Chandler P, Quartey-Papafio R, Simpson E, Cooke A. The effect of bone marrow and thymus chimerism between nonobese diabetic (NOD) and NOD-E transgenic mice, on the expression and prevention of diabetes. Eur J Immunol. 1993;23:2667–2675. doi: 10.1002/eji.1830231042. [DOI] [PubMed] [Google Scholar]
- 34.Santamaria P, Utsugi T, Park B-J, Averill N, Yoon J-W. Beta cell-cytotoxic CD8+T-cells from nonobese diabetic mice use highly homologous T-cell receptor α-chain CDR3 sequences. J Immunol. 1995;154:2494–2503. [PubMed] [Google Scholar]
- 35.Verdaguer J, Yoon J-W, Anderson B, Averill N, Utsugi T, Park B-J, Santamaria P. Acceleration of spontaneous diabetes in TCRβ-transgenic nonobese diabetic mice by beta cell-cytotoxic CD8+T-cells expressing identical endogenous TCRα chains. J Immunol. 1996;157:4726–4735. [PubMed] [Google Scholar]
- 36.Coligan, J.E., A.M. Kruisbeek, D.H. Margulies, E.H. Shevack, and W. Strober. 1995. Assessment of lymphocyte development in radiation bone marrow chimeras. In Current Protocols in Immunology. John Wiley and Sons, Inc., New York. 4.6.1–4.6.8.
- 37.Nagata M, Yoon J-W. Studies on autoimmunity for T cell–mediated beta cell destruction. Diabetes. 1992;41:998–1008. doi: 10.2337/diab.41.8.998. [DOI] [PubMed] [Google Scholar]
- 38.Berg LJ, Fazekas de St B, Groth, Pullen AM, Davis MM. Phenotypic differences between αβ vs. β T-cell receptor transgenic mice undergoing negative selection. Nature (Lond) 1989;340:559–562. doi: 10.1038/340559a0. [DOI] [PubMed] [Google Scholar]
- 39.Pircher H, Burki K, Lang R, Hengartner H, Zinkernagel RM. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature (Lond) 1989;342:559–561. doi: 10.1038/342559a0. [DOI] [PubMed] [Google Scholar]
- 40.Kaye J, Hsu ML, Sauron ME, Jameson SC, Gascoine NR, Hedrick SM. Selective development of CD4+ T-cells in transgenic mice expressing a class II MHC-restricted antigen receptor. Nature (Lond) 1989;341:746–749. doi: 10.1038/341746a0. [DOI] [PubMed] [Google Scholar]
- 41.Kurrer MO, Pakala SM, Hanson H, Katz JD. β cell apoptosis in T-cell mediated autoimmune diabetes. Proc Natl Acad Sci USA. 1997;94:213–218. doi: 10.1073/pnas.94.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tomonari K, Fairchild S, Rosenwasser OA. Influence of viral superantigens on Vβ- and Vα-specific positive and negative selection. Immunol Rev. 1993;131:131–168. doi: 10.1111/j.1600-065x.1993.tb01534.x. [DOI] [PubMed] [Google Scholar]
- 43.Pircher H, Rohrer UH, Moskophidis D, Zinkernagel RM, Hengartner H. Lower receptor avidity required for thymic clonal deletion than for effector T-cell function. Nature (Lond) 1991;351:482–485. doi: 10.1038/351482a0. [DOI] [PubMed] [Google Scholar]
- 44.Slattery RM, Miller JFAP, Heath WR, Charlton B. Failure of a protective major histocompatibility class II molecule to delete autoreactive T-cells in autoimmune diabetes. Proc Natl Acad Sci USA. 1993;90:10808–10810. doi: 10.1073/pnas.90.22.10808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ikehara S, Ohtsuki H, Good RA, Asamoto H, Nakamura T, Sekita K, Muso E, Tochino Y, Ida H, Kuzuya H, Imura H, Hamashima Y. Prevention of type I diabetes in nonobese diabetic mice by allogeneic bone marrow transplantation. Proc Natl Acad Sci USA. 1985;82:7743–7746. doi: 10.1073/pnas.82.22.7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wicker LS, Miller BJ, Chai A, Terada M, Mullen Y. Expression of genetically determined diabetes and insulitis in the nonobese diabetic (NOD) mouse at the level of bone marrow derived cells. Transfer of diabetes and insulitis to non-diabetic (NOD × B10)F1 mice with bone marrow cells from NOD mice. J Exp Med. 1988;167:1801–1808. doi: 10.1084/jem.167.6.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.LaFace DM, Peck AB. Reciprocal allogeneic bone marrow transplantation between NOD mice and diabetes non-susceptible mice associated with transfer and prevention of autoimmune diabetes. Diabetes. 1989;38:894–898. doi: 10.2337/diab.38.7.894. [DOI] [PubMed] [Google Scholar]
- 48.Serreze DV, Leiter EH. Development of diabetogenic T-cells from NOD/Lt marrow is blocked when an allo-H-2 haplotype is expressed on cells of hematopoietic origin, but not on thymic epithelium. J Immunol. 1991;147:1222–1229. [PubMed] [Google Scholar]
- 49.Acha-Orbea H, Scarpellino L. Nonobese diabetic and nonobese nondiabetic mice have unique MHC class II haplotypes. Immunogenetics. 1991;34:57–59. doi: 10.1007/BF00212313. [DOI] [PubMed] [Google Scholar]
- 50.Ignatowicz L, Kappler J, Marrack P. The repertoire of T-cells shaped by a single MHC/peptide ligand. Cell. 1996;84:521–529. doi: 10.1016/s0092-8674(00)81028-4. [DOI] [PubMed] [Google Scholar]
- 51.Surh CD, Sprent J. T-cell apoptosis detected in situ during positive and negative selection in the thymus. Nature (Lond) 1994;372:100–103. doi: 10.1038/372100a0. [DOI] [PubMed] [Google Scholar]
- 52.Laufer TM, DeKoning J, Markowitz JS, Lo D, Glimcher LH. Unopposed positive selection and autoreactivity in mice expressing class II MHC only on thymic cortex. Nature (Lond) 1996;383:81–85. doi: 10.1038/383081a0. [DOI] [PubMed] [Google Scholar]