

Abstract
To determine the role of CD11/CD18 complexes in neutrophil emigration, inflammation was induced in the skin, lungs, or peritoneum of mutant mice deficient in CD18 (CD18−/− mutants). Peripheral blood of CD18−/− mutants contained 11-fold more neutrophils than did blood of wild-type (WT) mice. During irritant dermatitis induced by topical application of croton oil, the number of emigrated neutrophils in histological sections of dermis was 98% less in CD18−/− mutants than in WT mice. During Streptococcus pneumoniae pneumonia, neutrophil emigration in CD18−/− mutants was not reduced. These data are consistent with expectations based on studies using blocking antibodies to inhibit CD11/CD18 complexes, and on observations of humans lacking CD11/CD18 complexes. The number of emigrated neutrophils in lung sections during Escherichia coli pneumonia, or in peritoneal lavage fluid after 4 h of S. pneumoniae peritonitis, was not reduced in CD18−/− mutants, but rather was greater than the WT values (240 ± 30 and 220 ± 30% WT, respectively). Also, there was no inhibition of neutrophil emigration during sterile peritonitis induced by intraperitoneal injection of thioglycollate (90 ± 20% WT). These data contrast with expectations. Whereas CD11/CD18 complexes are essential to the dermal emigration of neutrophils during acute dermatitis, CD18−/− mutant mice demonstrate surprising alternative pathways for neutrophil emigration during pneumonia or peritonitis.
Acute emigration of neutrophils requires CD11/CD18 complexes under most circumstances (for review see reference 1). Antibodies against CD11/CD18 inhibit neutrophil emigration during acute inflammation in animals (2, 3). Leukocytes from human patients with leukocyte adhesion deficiency type 1 (LAD-1),1 a disease arising from mutations in the gene for CD18, lack CD11/CD18 complexes. In LAD-1 patients, neutrophil emigration is not observed using Rebuck skin windows or skin chambers (4–7), and infected peritoneal, laryngeal, esophageal, periodontal, gingival, pharyngeal–glottic, dermal, or umbilical tissues of LAD-1 patients are devoid of emigrated neutrophils (4, 5, 8).
In the lung, neutrophil emigration occurs via CD11/ CD18-dependent, but also via CD11/CD18-independent pathways. Antibodies against CD11 or CD18 inhibit neutrophil emigration in response to Escherichia coli, E. coli LPS, Pseudomonas aeruginosa, phorbol ester, IgG immune complexes, or IL-1 (3, 9–12), but these antibodies do not inhibit pulmonary emigration in response to Streptococcus pneumoniae, hydrochloric acid, or C5a complement fragments (3, 10). In contrast to the other tissues examined, the lungs from an autopsied LAD-1 patient contained emigrated neutrophils (8).
Mice deficient in CD18 (CD18−/− mutants) have now been derived (Scharffetter-Kochanek, K., submitted for publication). Neutrophils from these mice do not express CD11/CD18 adhesion complexes. CD18−/− mutants display a phenotype resembling that seen in humans with LAD-1, including neutrophilia, peripheral lymphadenopathy, splenomegaly, and skin lesions. In this paper, CD18−/− mutant mice were used to determine the roles of CD11/CD18 complexes in neutrophil emigration during inflammation in the skin, lungs, or peritoneum.
Methods
Animals.
The CD18 gene was targeted for disruption in embryonic murine stem cells using a previously published targeting construct (13), blastocysts containing mutant stem cells were transferred to murine foster mothers, and a homozygous mouse line was established by selective breeding, as previously described (14–17). Mouse genotypes were confirmed by Southern blotting. Wild-type mice were from the same genetic background (mixed 129/Sv and C57BL/6). Mice were studied at 8–18 wk of age. All experiments received institutional approval.
CD11/CD18 Expression.
Expression of CD11/CD18 on circulating neutrophils was examined by flow cytometry. Mice were killed by a lethal overdose of halothane. Blood was collected from the inferior vena cava, erythrocytes were hypotonically lysed, and leukocytes were stained with saturating concentrations of the following rat monoclonal antibodies from PharMingen (San Diego, CA): M17/4 (anti–murine CD11a), M1/70 (anti–murine CD11b), or H129.19 (anti–murine CD4, used as a control antibody, nonbinding for neutrophils). Antibodies against CD11a and CD4 were directly conjugated to FITC. Antibodies against CD11b were biotinylated, and CD11b-labeled cells were secondarily labeled by streptavidin conjugated to FITC (PharMingen). Cells were fixed with 1% paraformaldehyde, and then green fluorescence of 5,000 cells in the neutrophil population (identified and gated using scatter profiles) was measured using an Ortho Cytofluorograf 50HTM flow cytometer equipped with a Cicero interface system (Cytomation, Fort Collins, CO).
Expression of CD11/CD18 adhesion complexes by leukocytes in the lungs was examined by immunohistochemistry. After an overdose of halothane, lungs were removed, inflated with a 1:1 mixture of 0.9% NaCl and Tissue-Tek OCT compound embedding medium (Miles Labs., Inc., Elkhart, IN), and snap-frozen in liquid N2. Adhesion molecules were identified in 6-μm sections by antibodies M17/4 (PharMingen) against mouse CD11a, and M1/70 (PharMingen), against mouse CD11b. Antibody control sections were treated with nonspecific rat IgG. After treatment with goat anti–rat secondary antibodies, sites of antibody labeling were stained red using an alkaline phosphatase–based detection system (Kirkegaard and Perry Laboratories, Inc., Gaithersburg, MD). All slides were counterstained with hemotoxylin (Fisher Scientific Co., Fairlawn, NJ).
Dermatitis.
Irritant dermatitis was induced by topical application of croton oil. Mice were anesthetized by methoxyflurane inhalation, and each side of one ear was treated with 10 μl of 2% croton oil (Sigma Chemical Co., St. Louis, MO) in 4:1 acetone/ olive oil. After 6 h, mice were killed by an overdose of halothane inhalation. Ear widths were measured using spring-loaded calipers. Peripheral blood was collected from the inferior vena cava. Blood leukocytes were counted with a hemacytometer after erythrocyte lysis, and leukocyte differentials were counted in blood smears stained with LeukoStat (Fisher Scientific Co., Pittsburgh, PA). Each ear was removed, fixed in 10% formalin, embedded in paraffin, sectioned, and stained with hematoxylin and eosin for examination by light microscopy.
Morphometric analysis was used to quantify neutrophil emigration using a drawing tube to reflect a grid onto microscopically viewed histologic sections. For each ear, the volume densities of emigrated neutrophils within four 110-μm-wide cross-sections, separated by 1.5-mm intervals, were assessed by point counting (18). A total of 547–1,677 points/ear were counted. Each point was assessed as falling on (a) epidermis, dermis, or cartilage, and (b) an emigrated neutrophil or not an emigrated neutrophil. The volume fraction of emigrated neutrophils in each ear was standardized to the cartilage volume of that ear, a value not expected to change during acute (6-h) dermatitis, by dividing the neutrophil volume fraction by the cartilage volume fraction.
Cutaneous edema was quantified as the percentage of increase in ear width in the right (croton oil–treated) compared to the left (untreated) ear for each mouse. The width of each ear was measured five times with spring-loaded calipers. Edema (expressed as percentage swelling) was calculated as 100 times the difference in ear widths divided by the width of the untreated ear.
Pneumonia.
Pneumonias were induced by intratracheal instillation of bacteria, as previously described (15, 19). Mice were anesthetized by intramuscular injection of ketamine hydrochloride (100 mg/kg) and acepromazine maleate (5 mg/kg). The tracheas were surgically exposed, and 2.3 μl/g body wt of S. pneumoniae (5 × 109 CFU/ml) or E. coli (107 CFU/ml) were instilled intratracheally. All bacterial suspensions contained 5% colloidal carbon to mark the deposition of the instillate. Radiotracers for measurement of edema formation (see below) were injected into the tail vein 15 min before intratracheal instillation (125I-albumin) and 2 min before euthanasia (51Cr-RBC). After 6 h of infection, mice were killed by overdose of halothane. The peritoneal and thoracic cavities were rapidly opened, the heart vessels were tied off to prevent pulmonary blood loss, peripheral blood samples were drawn from the inferior vena cava, and the lungs were removed and fixed via intratracheal instillation of 6% glutaraldehyde under 22 cm H2O pressure. Circulating leukocytes were counted as above.
Pulmonary neutrophils were quantified by morphometry in histological sections (18). Carbon black–containing lung regions were embedded in paraffin, and 5–7 μm thick sections were cut and stained with hematoxylin and eosin. A counting grid (10 × 10, covering 70,000 μm2 of the magnified field) was reflected onto the field of view using a drawing tube. Randomly selected fields of pneumonic peripheral lung that were largely free of noncapillary blood vessels and bronchioles or larger airways were examined. A total of three grids (300 points) were counted for each lung, and every point was classified as landing on (a) air space or tissue and (b) neutrophil or not a neutrophil. The quantities of neutrophils in air space or tissue were expressed as volume % of the respective compartment (alveolar air space or septal tissue). The volume % of the alveolated region of lung occupied by air space and by tissue did not differ among infected and uninfected WT and CD18−/− mutant mice (data not shown). The ratio of emigrated neutrophils (% alveolated region) to septal neutrophils (% alveolated region) was calculated for pneumonic regions from each mouse.
Pulmonary edema was measured before dissection of lungs for morphometry, as previously described (15, 19). In brief, 125I-albumin and 51Cr-RBC activities of blood and plasma samples and of excised fixed lungs were measured to characterize the pulmonary plasma and blood volumes. The pulmonary intravascular plasma content, calculated from the hematocrit and the lung blood volume, was subtracted from the total lung plasma content to yield extravascular plasma volume, and extravascular plasma volume per lung was expressed as μl pulmonary edema (15, 19).
Peritonitis.
Peritonitis was induced and characterized as previously described (15, 16). After anesthetizing mice by intramuscular injection of ketamine hydrochloride (100 mg/kg) and acepromazine maleate (5 mg/kg), iodinated albumin (0.3 μCi/mouse) was injected into the tail vein. S. pneumoniae (109 CFU/mouse) was injected intraperitoneally 15 min later. Mice were killed by a lethal overdose of halothane after 4 or 24 h of peritonitis, and the peritoneal cavities were lavaged with three aliquots of 5 ml of PBS. Peripheral blood was drawn from the inferior vena cava. Concentrations of total leukocytes in blood and lavage fluid were calculated from hemacytometer counts, and cellular differentials were quantified in smear or cytospin preparations stained with LeukoStat (Fisher Scientific Co.). The 125I-albumin recovered by lavage was expressed as the percentage of 125I-albumin injected and reflects plasma leakage into the peritoneal cavity (15, 16). Bacterial clearance was inferred from the loss of viable bacteria recovered in peritoneal lavage, and CFU recovered 4 or 24 h after instillation were expressed as a percentage of the original number of CFU injected. Sterile peritonitis was induced in separate groups of mice by intraperitoneal injection of 1 ml of sterile-filtered 4% thioglycollate in PBS, and mice were processed as above.
Statistics.
Groups consisted of four or five mice. Data were presented as mean ± SEM. Data from different groups (WT versus CD18−/− or infected versus uninfected) were compared by t test. Because circulating blood cell counts in CD18−/− mutant mice were not normally distributed and were highly variable, these data were compared by Mann-Whitney U test and expressed as medians in addition to mean ± SEM values. Differences were considered statistically significant when p <0.05.
Results
Expression of CD11/CD18 Adhesion Complexes.
Circulating neutrophils from WT mice expressed both CD11a and CD11b on their surface, as measured by flow cytometry (Fig. 1). Neutrophils from CD18−/− mutants, however, did not express CD11a or CD11b (Fig. 1). Similarly, leukocytes from peripheral lymph nodes or spleens of WT mice expressed CD11a and/or CD11b, whereas lymph node and spleen cells from CD18−/− mutant mice did not express CD11a or CD11b, as measured by flow cytometry (data not shown).
Figure 1.
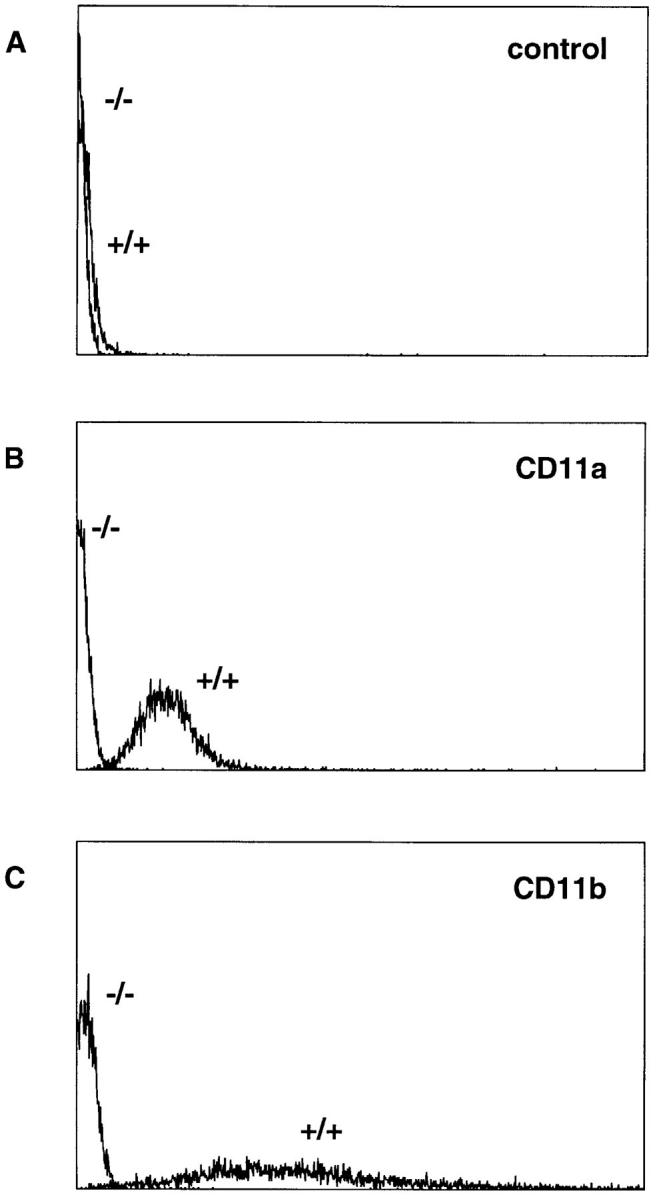
Expression of CD11a–CD18 and CD11b– CD18 by peripheral blood neutrophils from WT and CD18−/− mutant mice. Blood was withdrawn from the inferior vena cava of mice after 6 h of croton oil dermatitis. Shown are representative linear-scale fluorescence histograms from WT (+/ +) or CD18-mutant (−/−) neutrophils labeled with monoclonal antibodies H129.19 against murine CD4 as a nonbinding control (A), M17/4 against murine CD11a (B), or M1/70 against murine CD11b (C).
Expression of CD11/CD18 by leukocytes marginated within lung capillaries was examined by immunohistochemistry. Lungs from WT mice contained leukocytes positive for CD11a or CD11b, but no leukocytes in CD18−/− lungs were positive for CD11a or CD11b. The genetic mutation of CD18 resulted in the complete loss of immunologically recognizable CD11a and CD11b in leukocytes of CD18−/− mutant mice.
Leukocytosis and Splenomegaly.
All CD18−/− mice with or without experimentally induced inflammations had elevated circulating leukocyte and neutrophil counts when compared with WT (Table 1).
Table 1.
Circulating Leukocytes in WT and CD18−/− Mice
| WT | CD18−/− | |||
|---|---|---|---|---|
| No experimental inflammation | ||||
| Total leukocytes | 6.4 ± 0.9 (5.8) | 22.0 ± 3.2 (24.1)* | ||
| Neutrophils | 1.0 ± 0.1 (0.8) | 10.6 ± 2.2 (8.7)* | ||
| Croton oil dermatitis | ||||
| Total leukocytes | 8.2 ± 1.1 (7.9) | 273 ± 232 (29.5)* | ||
| Neutrophils | 1.9 ± 0.3 (1.5) | 235 ± 208 (18.6)* | ||
| E. coli pneumonia | ||||
| Total leukocytes | 1.7± 0.3 (1.6) | 54.2 ± 27.2 (27.1)* | ||
| Neutrophils | 0.4 ± 0.1 (0.4) | 37.8 ± 19.5 (21.1)* | ||
| S. pneumoniae pneumonia | ||||
| Total leukocytes | 2.6 ± 0.4 (2.4) | 29.2 ± 2.9 (26.5)* | ||
| Neutrophils | 0.7 ± 0.1 (0.7) | 21.1 ± 2.2 (18.5)* | ||
| 4-h S. pneumoniae peritonitis | ||||
| Total leukocytes | 3.0 ± 0.6 (3.5) | 13.2 ± 1.7 (13.0)* | ||
| Neutrophils | 1.2 ± 0.3 (1.3) | 9.8 ± 1.7 (9.3)* | ||
| 24-h S. pneumoniae peritonitis | ||||
| Total leukocytes | 3.7 ± 0.8 (3.9) | 19.4 ± 5.7 (15.7)* | ||
| Neutrophils | 0.4 ± 0.04 (0.4) | 15.2 ± 5.2 (11.0)* | ||
| 4-h thioglycollate peritonitis | ||||
| Total leukocytes | 5.6 ± 1.0 (5.2) | 27.4 ± 8.6 (27.2)* | ||
| Neutrophils | 2.7 ± 0.6 (2.6) | 18.0 ± 5.5 (17.4)* | ||
| 24-h thioglycollate peritonitis | ||||
| Total leukocytes | 4.6 ± 0.8 (4.0) | 38.2 ± 1.5 (39.2)* | ||
| Neutrophils | 0.2 ± 0.1 (0.1) | 28.1 ± 1.4 (28.8)* | ||
Blood was drawn from the inferior vena cava. Values represent mean ± SEM × 106 cells/ml. Median values are shown in parentheses.
Significantly different from WT (P <0.05).
CD18−/− mice exhibited splenomegaly (spleen weights of 171.7 ± 11.2 and 62.3 ± 5.3 mg in CD18−/− and WT mice with bacterial peritonitis, respectively; p <0.05). Neither liver weights (1.02 ± 0.04 and 0.94 ± 0.04 g in CD18−/− and WT mice, respectively) nor body weights (18.9 ± 0.6 and 19.2 ± 0.8 g in CD18−/− and WT mice, respectively) differed between WT and mutant mice with bacterial peritonitis.
Neutrophil Emigration During Dermatitis.
Croton oil application induced significant neutrophil emigration in WT mice (Fig. 2). In contrast, there was no increase in emigrated neutrophils in the dermis of CD18−/− mutant mice after 6 h of croton oil dermatitis (Fig. 2).
Figure 2.

Dermal neutrophil emigration in WT and CD18−/− mice 6 h after topical application of 2% croton oil. Emigrated neutrophils were quantitated morphometrically and expressed as mean ± SEM standardized volume fractions for WT (closed bars) or CD18−/− (open bars) mice. *Significant differences from WT; ‡significant differences from control (untreated) ears (P <0.05).
Dermal Edema.
After 6 h, croton oil induced edema accumulation in both WT and CD18−/− ears (Table 2). There was, however, significantly less edema accumulation in the CD18−/− ears than in WT ears (Table 2).
Table 2.
Edema Accumulation in WT and CD18−/− Mice
| WT | CD18−/− | |||
|---|---|---|---|---|
| Dermatitis (percent increase in ear width) | ||||
| Croton oil, 6 h | 41.8 ± 3.4 | 17.8 ± 8.7* | ||
| Pneumonia (μl/lung) | ||||
| E. coli 6 h | 78.1 ± 21.5 | 96.6 ± 15.3 | ||
| S. pneumoniae 6 h | 54.9 ± 12.1 | 129.2 ± 24.0* | ||
| S. pneumoniae peritonitis (%125I-albumin recovered) | ||||
| 4 h | 2.7 ± 0.7 | 1.4 ± 0.3 | ||
| 24 h | 1.2 ± 0.1 | 2.6 ± 0.4* | ||
| Thioglycollate peritonitis (%125I-albumin recovered) | ||||
| 4 h | 4.0 ± 1.1 | 2.5 ± 0.5 | ||
| 24 h | 1.6 ± 0.2 | 5.8 ± 1.2* |
Edema accumulation was quantified during dermatitis, pneumonia, or peritonitis, as described in the text. Values represent mean ± SEM.
Significantly different from WT (P <0.05).
Neutrophil Emigration During Pneumonia.
More neutrophils were present in the alveolar septae of uninfected CD18−/− mutants than in those of uninfected WT mice (Fig. 3). There were almost no neutrophils in the alveolar air spaces of WT and CD18−/− mutant mice (Fig. 4).
Figure 3.

Neutrophils in the alveolar septae of uninfected WT or CD18−/− mice and 6 h after intratracheal instillation of E. coli or S. pneumoniae. Septal neutrophils were quantitated morphometrically and expressed as the mean ± SEM volume percent of septal tissue occupied by neutrophils for WT (closed bars) or CD18−/− (open bars) mice. *Significant differences from WT; ‡significant differences from uninfected mice (P <0.05).
Figure 4.

Neutrophils in the alveolar air spaces of uninfected WT or CD18−/− mice and 6 h after intratracheal instillation of E. coli or S. pneumoniae. Emigrated neutrophils were quantitated morphometrically and expressed as the mean ± SEM volume percent of alveolar air space occupied by neutrophils for WT (closed bars) or CD18−/− (open bars) mice. *Significant differences from WT; ‡significant differences from uninfected mice (P <0.05).
During E. coli and S. pneumoniae pneumonias, the volume fraction of septal tissue occupied by neutrophils increased in both WT and CD18−/− mutant mice (Fig. 3). CD18−/− mutants had more neutrophils in alveolar septae for each type of pneumonia than did WT mice (Fig. 3).
Neutrophils emigrated into the alveolar air spaces during E. coli and S. pneumoniae pneumonias for both WT and CD18−/− mutant mice (Fig. 4). During either pneumonia, CD18−/− mutants had more neutrophils in their air spaces than did WT mice (Fig. 4). The two types of bacteria induced a similar degree of neutrophil emigration in WT mice, but fewer neutrophils (P <0.05) emigrated during E. coli than during S. pneumoniae pneumonia in CD18−/− mutant mice (Fig. 4).
The ratio of alveolar air space neutrophils to septal neutrophils in pneumonic regions is an index of the ability of neutrophils to extravasate. In WT mice, the ratio of air space to septal neutrophils was 1.0 ± 0.2 and 1.1 ± 0.1 during E. coli and S. pneumoniae pneumonias, respectively. In CD18−/− mutants, this ratio was 1.7 ± 0.2 and 2.3 ± 0.3 for E. coli and S. pneumoniae, respectively, significantly greater than WT for each type of pneumonia (P <0.05).
Pulmonary Edema.
Pulmonary edema during E. coli pneumonia did not differ between WT and CD18−/− mice (Table 2). Edema accumulation was significantly greater in CD18−/− mutants than in WT mice during S. pneumoniae pneumonia (Table 2).
Neutrophil Emigration During Peritonitis.
There were few neutrophils in the uninfected peritoneal cavities of WT and CD18−/− mice (Fig. 5). After intraperitoneal injection of S. pneumoniae, neutrophil accumulation was apparent 4 h later, and was greatly increased after 24 h of peritonitis in both WT and CD18−/− mutant mice (Fig. 5). More neutrophils were recovered in the peritoneal lavage fluids of CD18−/− mutants than of WT mice after 4 or 24 h of peritonitis (220 ± 30 and 500 ± 50% WT, respectively; Fig. 5). After intraperitoneal injection of thioglycollate, there was no significant difference between CD18−/− mutants and WT mice in the number of neutrophils recovered by peritoneal lavage after 4 h (110 ± 30% WT; Fig. 6). By 24 h, more neutrophils were recovered from CD18−/− mutants than from WT mice (360 ± 80% WT; Fig. 6).
Figure 5.

Peritoneal neutrophil emigration in WT and CD18−/− mice 0, 4, or 24 h after intraperitoneal injection of S. pneumoniae. Emigrated neutrophils were quantitated in peritoneal lavage fluids and expressed as mean ± SEM neutrophils/lavage for WT (closed bars) or CD18−/− (open bars) mice. *Significant differences from WT; ‡significant differences from 0-h (uninfected) mice (P <0.05).
Figure 6.

Peritoneal neutrophil emigration in WT and CD18−/− mice 0, 4, or 24 h after intraperitoneal injection of thioglycollate. Emigrated neutrophils were quantitated in peritoneal lavage fluids and expressed as mean ± SEM neutrophils/lavage for WT (closed bars) or CD18−/− (open bars) mice. The data from 0 h of peritonitis (no thioglycollate) are the same as shown in Fig. 5. *Significant differences from WT; ‡significant differences from 0-h (uninfected) mice (P <0.05).
Peritoneal Edema.
Peritoneal edema did not significantly differ between mutant and WT mice after 4 h of bacterial or thioglycollate peritonitis (Table 2). After 24 h of either peritonitis, there was more peritoneal edema in CD18−/− mutants than in WT mice (Table 2).
Bacterial Clearance During Peritonitis.
Bacterial clearance did not significantly differ between WT and CD18−/− mutant mice. After 4 h, 24 ± 10% of injected CFU were recovered in the peritoneal lavage from WT mice, and 45 ± 9% of injected CFU from CD18−/− mice. After 24 h, <0.1% of injected CFU were recovered from either WT or CD18−/− mice.
Discussion
Neutrophil emigration was reduced and edema accumulation compromised in CD18−/− mutant mice with croton oil dermatitis. In contrast, neither neutrophil emigration nor edema accumulation were compromised in CD18−/− mutants during S. pneumoniae or thioglycollate peritonitis or during E. coli or S. pneumoniae pneumonia. These data confirm that CD11/CD18 complexes are essential to neutrophil emigration under at least some circumstances, but they definitively demonstrate that CD11/CD18-independent pathways for neutrophil emigration can be used during acute inflammation in the peritoneum and the lung.
These results may be compared and contrasted with (a) studies using blocking antibodies to inhibit CD11/CD18 interactions with ligands, (b) observations with human LAD-1 patients, (c) studies with CD18 hypomorphic mutant mice with decreased but not eliminated expression of CD18, and (d) mutant mice deficient in CD11a or in CD11b.
Antibodies against CD18 inhibit cutaneous emigration of neutrophils in response to diverse stimuli (2), and in this study cutaneous emigration of neutrophils was inhibited in CD18−/− mutant mice during croton oil dermatitis. Whereas antibodies against CD18 inhibit acute peritoneal emigration of neutrophils during S. pneumoniae or thioglycollate peritonitis (3, 20) and acute pulmonary emigration of neutrophils during E. coli pneumonia (3), however, neutrophil emigration was not inhibited during any of these inflammatory reactions in mice completely deficient in CD11/CD18 adhesion complexes due to a CD18 null mutation. This discrepancy suggests that the murine regulation of adhesion pathways differs during lifelong deficiency of CD11/CD18 compared with acute inhibition of CD11/ CD18 by antibodies.
Tissues from human LAD-1 patients with a lifelong deficiency of CD11/CD18 complexes are generally lacking in neutrophils, even when infected by bacteria or fungi. Biopsies from diseased periodontal, gingival, pharyngeal–glottic, dermal, or umbilical tissues from LAD-1 patients show a lack of neutrophils in infected or inflamed sites (4, 5). Autopsy of a LAD-1 patient revealed infection without neutrophil emigration in the peritoneum, larynx, and esophagus (8). The lungs of the autopsied patient were also infected, but in contrast to the other sites examined, emigrated neutrophils were observed (8). Thus, humans can recruit neutrophils via CD11/CD18-independent pathways under at least some circumstances. During acute inflammations in this study, the CD18−/− mutant mice recruited neutrophils into the lungs and the peritoneum, but not into the skin.
Mutant mice that express 2–16% of WT levels of CD11/ CD18 on their leukocyte surfaces (CD18 hypomorphs) have been previously characterized (13). The numbers of neutrophils recovered by peritoneal lavage 4 h after intraperitoneal injection of thioglycollate were not significantly different in CD18 hypomorphs when compared with WT mice (13). Thus, either the remaining CD11/CD18 mediates neutrophil emigration in the hypomorphs, or alternative pathways for neutrophil emigration are used in the hypomorphs as in the CD18−/− mutants.
Mutant mice lacking CD11a–CD18 secondary to a CD11a mutation have been generated, and their neutrophils are compromised in their ability to emigrate during thioglycollate peritonitis (21). In contrast, CD18−/− mutants lack CD11a–CD18 as well as the other members of the CD11/CD18 family, yet neutrophil emigration is not compromised in these mice during peritonitis. Thus, the CD18−/− mutants, but not the CD11a–CD18 mutants, induce CD11/CD18-independent neutrophil emigration during thioglycollate peritonitis. Mutant mice deficient in CD11b do not demonstrate reduced neutrophil emigration during thioglycollate peritonitis (22, 23). CD11a–CD18 function, however, remains critical to neutrophil emigration in CD11b-deficient mice as indicated by CD11a-blocking antibodies (23). It is unclear why mutant mice with single deficiencies in CD11a–CD18 or CD11b–CD18 require CD11a–CD18 for neutrophil emigration under circumstances in which CD18−/− mutants do not demonstrate impaired emigration. CD18−/− mutants are deficient in CD11a–CD18, CD11b–CD18, CD11c–CD18, and CD11d–CD18, whereas the CD11a−/− and CD11b−/− mutant mice each express three of the four adhesion complexes. Furthermore, the CD18−/− mutants have extremely high circulating neutrophil counts, which has not been reported for the CD11a−/− or CD11b−/− mutants. These factors may contribute to the differing recruitment of CD11a-independent pathways for the emigration of neutrophils in the peritoneal cavities of these mutant mice.
To our knowledge, this is the first evidence that a CD11/CD18-independent pathway can be used by neutrophils in nonpulmonary tissues during the first hours of acute inflammation. CD11/CD18-independent neutrophil emigration in the peritoneum has been observed over prolonged (12–24-h) periods of inflammation (20, 24). Macrophages recovered after 72 h of protease peptone-induced peritonitis can elicit CD11/CD18-independent neutrophil emigration when transferred to naïve peritoneal cavities before eliciting acute (4 h) streptococcal peritonitis (25). CD11/CD18-independent neutrophil emigration also occurs in the joints during chronic inflammation (26). In CD18−/− mutants, the peritoneal emigration of neutrophils occurred in the absence of CD11/CD18 after only 4 h of peritonitis.
These data suggest that neutrophils can use different adhesion molecule pathways in emigrating from the systemic vasculature in the skin (during croton oil dermatitis) and in the peritoneum (during S. pneumoniae or thioglycollate peritonitis). These tissues may regulate the emigration of neutrophils differently. Whereas neutrophil emigration into the skin was quantified in situ by morphometric analysis of histologic sections, neutrophil emigration into the peritoneum was quantified in lavage fluid. Studying the roles of adhesion molecules in neutrophil emigration by examination of lavage fluid during peritonitis is complicated by the fact that leukocytes use adhesion molecules, including CD11/CD18 and ICAM-1, to adhere to the peritoneal mesothelium (27, 28). Thus, leukocytes may be less likely to adhere to the mesothelium and may be more likely to be lavaged in situations in which CD11/CD18-ICAM-1 interactions are disrupted. Despite these confounding factors in interpreting lavage data, it remains clear that neutrophils emigrated during peritonitis in CD18−/− mutant mice. Neutrophil emigration was observed in situ during both E. coli and S. pneumoniae pneumonias in CD18−/− mutant mice as well.
The observation of increased emigration of neutrophils in the lungs and peritoneal cavities of CD18−/− mutants when compared with WT mice likely resulted from the increased numbers of circulating neutrophils in CD18−/− mutant mice, although other explanations such as decreased resolution of inflammation (22) are also possible. Animals rendered neutrophilic by administration of G-CSF demonstrate increased neutrophil numbers in bronchoalveolar lavage fluid during pneumonia induced by S. pneumoniae, Klebsiella pneumoniae, or E. coli (29–31), and increased neutrophil numbers in peritoneal lavage fluid during peritonitis induced by Listeria monocytogenes, fecal flora, or E. coli (32– 34). The number of circulating neutrophils in CD18−/− mutants with pneumonia or peritonitis was increased 8–fold or more when compared with WT mice. Thus, the increased neutrophil emigration observed in CD18−/− mutants with pneumonia or peritonitis likely resulted from their peripheral neutrophilia.
In conclusion, CD18−/− mutant mice displayed the expected phenotype of reduced acute neutrophil emigration when studied during croton oil dermatitis. CD18−/− mutants, however, deviated surprisingly from what was expected when studied during acute S. pneumoniae or thioglycollate peritonitis or during acute E. coli pneumonia, in that neutrophil emigration was not reduced, but was in fact increased during these types of inflammation. These genetically engineered mice now highlight and help to define two important questions to be studied: (a) which signals induce neutrophils to emigrate via the CD11/CD18-dependent or -independent pathways?, and (b) which adhesion molecules mediate CD11/CD18-independent neutrophil emigration?
Acknowledgments
The authors thank Ervin Melulini for preparation of histological slides, and Amy Imrich for assistance with flow cytometry.
This work was supported by United States Public Health Service grants HL 48160, HL 52466, and AI 32177. Karin Scharffetter-Kochanek was supported by a Heisenberg grant from the Deutsche Forschungsgemeinschaft.
Footnotes
Abbreviations used in this paper: LAD-1, leukocyte adhesion deficiency type 1; WT, wild-type.
References
- 1.Carlos TM, Harlan JM. Leukocyte-endothelial adhesion molecules. Blood. 1994;84:2068–2101. [PubMed] [Google Scholar]
- 2.Arfors K, Lundberg C, Lindbom L, Lundberg K, Beatty PG, Harlan JM. A monoclonal antibody to the membrane glycoprotein complex CD18 inhibits polymorphonuclear leukocyte accumulation and plasma leakage in vivo. Blood. 1987;69:338–340. [PubMed] [Google Scholar]
- 3.Doerschuk CM, Winn RK, Coxson HO, Harlan JM. CD18-dependent and -independent mechanisms of neutrophil adherence in the pulmonary and systemic microvasculature of rabbits. J Immunol. 1990;114:2327–2333. [PubMed] [Google Scholar]
- 4.Bowen TJ, Ochs HD, Altman LC, Price TH, Van Epps DE, Brautigan DL, Rosin RE, Perkins WD, Babior BM, Klebanoff SJ, Wedgwood RJ. Severe recurrent bacterial infections associated with defective adherence and chemotaxis in two patients with neutrophils deficient in a cell-associated glycoprotein. J Pediatr. 1982;101:932–940. doi: 10.1016/s0022-3476(82)80013-9. [DOI] [PubMed] [Google Scholar]
- 5.Anderson DC, Schmalsteig FC, Finegold MJ, Hughes BJ, Rothlein R, Miller LJ, Kohl S, Tosi MF, Jacobs RL, Waldrop TC, et al. The severe and moderate phenotypes of heritable Mac-1, LFA-1 deficiency: their quantitative definition and relation to leukocyte dysfunction and clinical features. J Infect Dis. 1985;152:668–689. doi: 10.1093/infdis/152.4.668. [DOI] [PubMed] [Google Scholar]
- 6.Buescher ES, Gaither T, Nath J, Gallin JI. Abnormal adherence-related functions of neutrophils, monocytes, and Epstein-Barr virus-transformed B cells in a patient with C3bi receptor deficiency. Blood. 1985;65:1382–1390. [PubMed] [Google Scholar]
- 7.Weisman SJ, Berkow RL, Plautz G, Torres M, McGuire WA, Coates TD, Haak RA, Floyd A, Jersild R, Baehner RL. Glycoprotein-180 deficiency: genetics and abnormal neutrophil activation. Blood. 1985;65:696–704. [PubMed] [Google Scholar]
- 8.Hawkins HK, Heffelfinger SC, Anderson DC. Leukocyte adhesion deficiency: clinical and postmortem observations. Pediatr Pathol. 1992;12:119–130. doi: 10.3109/15513819209023288. [DOI] [PubMed] [Google Scholar]
- 9.Mulligan MS, Wilson GP, Todd RF, Smith CW, Anderson DC, Varani J, Issekutz TB, Miyasaka M, Tamatani T, Miyasaka M, et al. Role of β1, β2 integrins, and ICAM-1 in lung injury after deposition of IgG and IgA immune complexes. J Immunol. 1993;150:2407–2417. [PubMed] [Google Scholar]
- 10.Hellewell PG, Young SK, Henson PM, Worthen GS. Disparate roles of the β2-integrin CD18 in the local accumulation of neutrophils in pulmonary and cutaneous inflammation in the rabbit. Am J Respir Cell Mol Biol. 1994;10:391–398. doi: 10.1165/ajrcmb.10.4.7510985. [DOI] [PubMed] [Google Scholar]
- 11.Kumasaka T, Doyle NA, Quinlan WM, Graham L, Doerschuk CM. Role of CD11/CD18 in neutrophil emigration during acute and recurrent Pseudomonas aeruginosa-induced pneumonia in rabbits. Am J Pathol. 1996;148:1297–1305. [PMC free article] [PubMed] [Google Scholar]
- 12.Qin L, Quinlan WM, Doyle NA, Graham L, Sligh JE, Takei F, Beaudet AL, Doerschuk CM. The roles of CD11/CD18 and ICAM-1 in acute Pseudomonas aeruginosa-induced pneumonia in mice. J Immunol. 1996;157:5016–5021. [PubMed] [Google Scholar]
- 13.Wilson RW, Ballantyne CM, Smith CW, Montgomery C, Bradley A, O'Brien WE, Beaudet AL. Gene targeting yields a CD18-mutant mouse for study of inflammation. J Immunol. 1993;151:1571–1578. [PubMed] [Google Scholar]
- 14.Sligh JE, Jr, Ballantyne CM, Rich SS, Hawkins HK, Smith CW, Bradley A, Beaudet AL. Inflammatory and immune responses are impaired in mice deficient in intercellular adhesion molecule 1. Proc Nat Acad Sci USA. 1993;90:8529–8533. doi: 10.1073/pnas.90.18.8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bullard DC, Qin L, Lorenzo I, Quinlin WM, Doyle NA, Bosse R, Vestweber D, Doerschuk CM, Beaudet AL. P-selectin/ICAM-1 double mutant mice: acute emigration of neutrophils into the peritoneum is completely absent but is normal into pulmonary alveoli. J Clin Invest. 1995;95:1782–1788. doi: 10.1172/JCI117856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bullard DC, Kunkel EJ, Kubo H, Hicks MJ, Lorenzo I, Doyle NA, Doerschuk CM, Ley K, Beaudet AL. Infections and deficiency of leukocyte rolling and recruitment in E-/P-selectin mutant mice. J Exp Med. 1996;183:2329–2336. doi: 10.1084/jem.183.5.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scharffetter-Kochanek K, Norman K, Bullard DC, Lorenzo I, van Nood NP, Rich S, Smith W, Beaudet AL. Generation and characterization of a CD18 complete knock-out mouse—new insights in the function of the CD18 molecule? . J Investig Dermatol. 1996;106:809. [Google Scholar]
- 18.Weibel, E.R. 1990. Morphometry: stereological theory and practical methods. In Models of Lung Disease: microscopy and Structural Methods, Volume 47. J. Gil, editor. Marcel Dekker, Inc., New York. 199–252.
- 19.Mizgerd JP, Meek BB, Kutkoski GJ, Bullard DC, Beaudet AL, Doerschuk CM. Selectins and neutrophil traffic: margination and Streptococcus pneumoniae-induced emigration in murine lungs. J Exp Med. 1996;184:639–645. doi: 10.1084/jem.184.2.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Conlan JW, North RJ. Listeria monocytogenes, but not Salmonella typhimurium, elicits a CD18-independent mechanism of neutrophil extravasation into the murine peritoneal cavity. Infect Immunol. 1994;62:2702–2706. doi: 10.1128/iai.62.7.2702-2706.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmits R, Kundig TM, Baker DM, Shumaker G, Simard JJL, Duncan G, Wakeham A, Shahinian A, van der Heiden A, Bachmann MF, et al. LFA-1–deficient mice show normal CTL responses to virus, but fail to reject immunogenic tumor. J Exp Med. 1996;183:1415–1426. doi: 10.1084/jem.183.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, von Andrian UH, Arnaout MA, Mayadas TN. A novel role for the β2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity. 1996;5:653–666. doi: 10.1016/s1074-7613(00)80278-2. [DOI] [PubMed] [Google Scholar]
- 23.Lu H, Smith CW, Perrard J, Bullard D, Tang L, Shappell SB, Entman ML, Beaudet AL, Ballantyne CM. LFA-1 is sufficient in mediating neutrophil emigration in Mac-1 deficient mice. J Clin Invest. 1997;99:1340–1350. doi: 10.1172/JCI119293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winn RK, Harlan JM. CD18-independent neutrophil and mononuclear leukocyte emigration into the peritoneum of rabbits. J Clin Invest. 1993;92:1168–1173. doi: 10.1172/JCI116686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mileski W, Harlan J, Rice C, Winn R. Streptococcus pneumoniae-stimulated macrophages induce neutrophils to emigrate by a CD18-independent mechanism of adherence. Circ Shock. 1990;31:259–267. [PubMed] [Google Scholar]
- 26.Issekutz TB, Miyasaka M, Issekutz AC. Rat blood neutrophils express very late antigen 4 and it mediates migration to arthritic joint and dermal inflammation. J Exp Med. 1996;183:2175–2184. doi: 10.1084/jem.183.5.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andreoli SP, Mallett C, Williams K, McAteer JA, Rothlein R, Doerschuk CM. Mechanisms of polymorphonuclear leukocyte mediated peritoneal mesothelial cell injury. Kidney Int. 1994;46:1100–1109. doi: 10.1038/ki.1994.372. [DOI] [PubMed] [Google Scholar]
- 28.Liberek T, Topley N, Luttman W, Williams JD. Adherence of neutrophils to human peritoneal mesothelial cells- role of intercellular adhesion molecule-1. J Am Soc Nephrol. 1996;7:208–217. doi: 10.1681/ASN.V72208. [DOI] [PubMed] [Google Scholar]
- 29.Nelson S, Summer W, Bagby G, Nakamura C, Stewart L, Lipscomb G, Andresen J. Granulocyte colony-stimulating factor enhances pulmonary host defenses in normal and ethanol-treated rats. J Infect Dis. 1991;164:901–906. doi: 10.1093/infdis/164.5.901. [DOI] [PubMed] [Google Scholar]
- 30.Lister PD, Gentry MJ, Preheim LC. Granulocyte colony-stimulating factor protects control rats but not ethanol-fed rats from fatal pneumococcal pneumonia. J Infect Dis. 1993;168:922–926. doi: 10.1093/infdis/168.4.922. [DOI] [PubMed] [Google Scholar]
- 31.Freeman BD, Correa R, Karzai W, Natanson C, Patterson M, Banks S, Fitz Y, Danner RL, Wilson L, Eichacker PQ. Controlled trials of rG-CSF and CD11b-directed MAb during hyperoxia and E. colipneumonia in rats. J Appl Physiol. 1996;80:2066–2076. doi: 10.1152/jappl.1996.80.6.2066. [DOI] [PubMed] [Google Scholar]
- 32.Serushago BA, Yoshikai Y, Handa T, Mitsuyama M, Muramori K, Nomoto K. Effect of recombinant human granulocyte colony-stimulating factor (rhG-CSF) on murine resistance against Listeria monocytogenes. . Immunology. 1992;75:475–480. [PMC free article] [PubMed] [Google Scholar]
- 33.Lundblad R, Wang MY, Kvalheim G, Lingaas E, Giercksky K. Granulocyte colony-stimulating factor improves myelopoiesis and host defense in fulminant intra-abdominal sepsis in rats. Shock. 1995;4:68–73. doi: 10.1097/00024382-199507000-00011. [DOI] [PubMed] [Google Scholar]
- 34.Dunne JR, Dunkin BJ, Nelson S, White JC. Effects of granulocyte colony stimulating factor in a nonneutropenic rodent model of Escherichia coliperitonitis. J Surg Res. 1996;61:348–354. doi: 10.1006/jsre.1996.0128. [DOI] [PubMed] [Google Scholar]