

Abstract
The chemokine receptor CXCR4 is the major coreceptor used for cellular entry by T cell– tropic human immunodeficiency virus (HIV)-1 strains, whereas CCR5 is used by macrophage (M)-tropic strains. Here we show that a small-molecule inhibitor, ALX40-4C, inhibits HIV-1 envelope (Env)-mediated membrane fusion and viral entry directly at the level of coreceptor use. ALX40-4C inhibited HIV-1 use of the coreceptor CXCR4 by T- and dual-tropic HIV-1 strains, whereas use of CCR5 by M- and dual-tropic strains was not inhibited. Dual-tropic viruses capable of using both CXCR4 and CCR5 were inhibited by ALX40-4C only when cells expressed CXCR4 alone. ALX40-4C blocked stromal-derived factor (SDF)-1α–mediated activation of CXCR4 and binding of the monoclonal antibody 12G5 to cells expressing CXCR4. Overlap of the ALX40-4C binding site with that of 12G5 and SDF implicates direct blocking of Env interactions, rather than downregulation of receptor, as the mechanism of inhibition. Thus, ALX40-4C represents a small-molecule inhibitor of HIV-1 infection that acts directly against a chemokine receptor at the level of Env-mediated membrane fusion.
HIV-1 infection is characterized by massive virus production, calculated to be on the order of 109 virus particles per day (1, 2). New combination chemotherapies have lead to dramatic and sustained reductions of viral load in many individuals, often to undetectable levels (3). However, these therapies require rigorous adherence to a complicated drug regimen, are expensive, and can cause significant side effects. These factors, coupled with the likelihood that resistant viruses may emerge with time, argue for the continued development of compounds that can block HIV-1 replication at multiple levels of the viral life cycle. Recent advances have shown that certain chemokine receptors, in conjunction with CD4, play a critical role in enabling HIV-1 to enter a cell. Macrophage (M)-tropic virus strains use the chemokine receptor CCR5 to enter cells (4–8), whereas the T cell line–tropic viruses that may emerge years after infection use the chemokine receptor CXCR4 (9).
The importance of chemokine receptors for virus infection in vivo is shown by the fact that individuals who lack CCR5 are highly resistant to virus infection (10–12). The central role of CCR5 in viral transmission and the lack of obvious consequences associated with loss of CCR5 function suggests that chemokine receptors may represent invariant cellular targets for antiviral agents. Indeed, the natural ligands to CCR5 and CXCR4 inhibit virus infection in vitro (5, 13–16), and the ligands for CCR5 (RANTES, MIP-1α, MIP-1β) have been identified as major antiretroviral factors secreted by CD8+ T cells (16). Chemically modified forms of RANTES inhibit HIV-1 entry more potently than wild-type RANTES, indicating that more effective chemokines can be developed (17, 18). However, chemokines (8–10-kD proteins) are subject to the limitations of any structurally complex, labile protein in terms of therapeutic potential. Therefore, small molecule inhibitors of chemokine receptor use are desirable. In this study, we demonstrate that a small-molecule inhibitor of HIV-1 infection can be developed that prevents viral entry by directly targeting the chemokine receptor CXCR4.
Materials and Methods
Reagents.
All cells were maintained in DMEM or RPMI-1640 containing 10% FCS. Vaccinia viruses encoding HIV-1 envelopes (Envs) included vSC60 (BH8; reference 19), vCB28 (JR-FL; reference 19), and vBD3 (89.6; reference 7). Primary virus strains were described previously (20), and infection was monitored by p24 production. The development and synthesis of ALX40-4C has been described previously (21).
Infection and Fusion Assays.
293T cells were transfected with plasmids encoding Env and the NL4-3 luciferase virus backbone (pNL-Luc-E−R−; reference 22). Plasmids encoding the HIV-1 Envs ADA, HXB2, and NL4-3 were provided by John Moore (Aaron Diamond AIDS Research Center, New York), and pNL-Luc-E−R− was provided by Ned Landau (Aaron Diamond AIDS Research Center). Cells were infected with 100 μl of viral supernatant in a total volume of 500 μl with 4 μg/ml polybrene. Cells were lysed 4 d after infection and 50 μl of the resulting lysate was assayed for luciferase activity. ALX40-4C was added to cells 1 h before infection. Virus production from PBMCs and MT2 cell infections was assessed in culture supernatant by p24 content (Coulter Corp., Miami, FL).
To quantitate cell–cell fusion events, we used a luciferase-based gene reporter assay (7, 23). PA317-T4 cells were transfected with T7 luciferase and coreceptor constructs by CaPO4 transfection, and incubated at 37°C overnight. T7 RNA polymerase and Env proteins were introduced into effector 293T cells by infection with recombinant vaccinia viruses and incubation at 32°C overnight in the presence of rifampicin. Target and effector cells were mixed in 24-well plates at 37°C in the presence of ara-C and rifampicin. After 5 h, cells were lysed and assayed for luciferase activity. For inhibition, 10 μM ALX40-4C was preincubated with target cells 30 min before addition of effector cells.
Ca2+ Mobilization Assays.
Response to ligand was determined in the human T cell line SupT1. Cells were incubated in media containing 2.5 μM Fura-2/AM (Molecular Probes Inc., Eurgene, OR) at 37°C in the dark for 1 h. Cells were allowed to efflux for 15 min in PBS before resuspension in Delbecco's PBS containing calcium and magnesium at 2 × 106 cells/ml. Ca2+ mobilization was measured in an Aminco-Bowman luminescence spectrometer.
Flow Cytometry.
Quantitation of surface molecules on SupT1, PM1, and 293T cells was performed by washing with 2% fetal bovine serum (FBS)/PBS, blocking with 10% normal rabbit sera in PBS, staining with 10 μg/ml of the indicated monoclonal antibody in 100 μl for 30 min, and detecting with an FITC-conjugated second antibody. For blocking, ALX40-4C was added after blocking cells with rabbit sera, ∼5 min before addition of antibodies, and in a total volume of 100 μl. Cells were fixed with 1% paraformaldehyde after staining and were kept on ice during entire procedure. Cells were analyzed on a Becton Dickinson (Mountain View, CA) FACScan® using CellQuest software.
Results
ALX40-4C Blocks Virus Infection at the Level of Entry.
ALX40-4C (N-α-acetyl-nona-d-arginine (Arg) amide), a polypeptide of nine Arg residues stabilized by terminal protection and inclusion of d amino acids, inhibits infection by T-tropic virus strains at a step before reverse transcription with an IC50 of 3 nM in the HUT78 T cell line and in PBLs, but does not inhibit infection by M-tropic virus strains (21). The region of HIV-1 responsible for this tropism-specific inhibition localizes to the V3 loop of the Env glycoprotein, but the mechanism of inhibition is undefined (21). To further localize the effects of ALX40-4C, we used luciferase reporter viruses based on a common NL4-3 HIV-1 core and pseudotyped with different Env proteins (22). As a result, all reporter viruses are genetically identical and have identical HIV-1 cores, so that postentry blocks will affect all reporter viruses equally.
Reporter viruses pseudotyped with the T-tropic Envs NL4-3 and HXB2 (that use CXCR4 as a coreceptor), the M-tropic Env ADA (that uses CCR5), or the murine leukemia virus (MLV) Env that enters cells independently of CD4 or chemokine receptors, were used to infect PM1 cells, a T cell line that expresses CD4, CXCR4, and CCR5 (6). As shown in Fig. 1 A, ALX40-4C inhibited infection of viruses containing the NL4-3 and HXB2, but not the ADA or MLV Envs. An antibody to CD4 inhibited all HIV-1 strains tested. Inhibition of HXB2 infection in PM1 cells was achieved with an IC50 of ∼0.3 μM (data not shown). Since ALX40-4C did not prevent infection by ADA, we conclude that it did not block CD4 binding or postentry steps of viral replication, suggesting that its inhibitory effects are at the level of virus entry and might be coreceptor specific, consistent with involvement of the V3 loop in ALX40-4C sensitivity (21). To test this, we transiently transfected feline cells that stably express human CD4 with plasmids encoding the coreceptor of interest. HIV-1 NL4-3 only infected cells expressing CXCR4, whereas HIV-1 ADA only infected cells expressing CCR5 (Fig. 1 B). As with PM1 cells, ALX40-4C inhibited the entry of NL4-3, but did not significantly affect entry of ADA.
Figure 1.


(A) Inhibition of virus infection on PM1 cells. The PM1 cell line was infected with single-cycle pseudotyped HIV-1 virions and assayed for luciferase production. (B) Feline CCCS+L-CD4 cells transiently expressing either CXCR4 or CCR5 were infected with pseudotyped luciferase viruses. Results in both panels represent the average of two identical experiments using 10 μM ALX40-4C and 10 μg/ml Leu3A, and error bars represent the range of the two independent experiments. Relative light unit (RLU) values are normalized to 100% for direct infection of permissive cells.
ALX40-4C Blocks Infection by Some Primary Virus Strains.
Previously, ALX40-4C was shown to inhibit infection of PBLs by some primary virus strains (21). To examine this in more detail, we tested the ability of ALX40-4C to inhibit infection of 10 primary virus strains. Of these, seven formed syncytia on MT2 cells (SI isolates) which express CXCR4 but not CCR5, whereas three did not form syncytia (NSI isolates). ALX40-4C inhibited infection of human PBMCs by three of seven SI strains, and did not inhibit the three NSI strains (Table 1). Since a number of primary virus strains have demonstrated dual-tropic properties (5, 7, 24), the failure of ALX40-4C to inhibit all of the SI strains in PBMCs could be due to use of alternative coreceptors such as CCR5. Therefore, we tested the ability of ALX40-4C to inhibit infection of MT2 cells (which do not express CCR5) by the SI strains. In this system, six of seven primary SI strains were inhibited. Whether the single primary SI strain that was not inhibited uses CXCR4 differently from the other viruses or whether it uses an as yet unidentified coreceptor present in MT2 cells is not known. Nevertheless, these results extend earlier findings and show that ALX40-4C can inhibit CXCR4-dependent infection by primary HIV-1 strains.
Table 1.
ALX40-4C Inhibition of HIV-1 Replication
| PBMC | MT-2 | |||||||
|---|---|---|---|---|---|---|---|---|
| Strain | SI/NSI | 1 μM | 10 μM | 10 μM | ||||
| NL4-3 | SI | 85 | >95 | >95 | ||||
| 1 | SI | 42 | 85 | >95 | ||||
| 2 | SI | 7 | 3 | 90 | ||||
| 3 | SI | 73 | >95 | >90 | ||||
| 5 | SI | 2 | 0 | >95 | ||||
| 6 | SI | 57 | >95 | >95 | ||||
| 7 | SI | 10 | 6 | 0 | ||||
| 10 | SI | 0 | 10 | >95 | ||||
| 4 | NSI | 0 | 0 | – | ||||
| 8 | NSI | 0 | 0 | – | ||||
| 9 | NSI | 0 | 0 | – | ||||
Human PBMCs or MT-2 cells were infected with 10 ng of the indicated primary virus strains or with NL4-3 in the presence of either 1 μM or 10 μM ALX40-4C. The amount of viral p24 (60–150 ng/ml for PBMC, 160–450 ng/ml for MT2 cells) released into the supernatant was measured 7 d later. Percentage inhibition is indicated. NSI strains did not grow on MT-2 cells.
ALX40-4C Blocks HIV-1 Usage of CXCR4.
Although the ability of ALX40-4C to block T-tropic, but not M-tropic viruses at post-CD4 levels of entry implied blocking of coreceptor use, we could not distinguish if ALX40-4C blocked the chemokine receptor directly or blocked regions of Env that may mediate interactions with the chemokine receptor. Therefore, we carried out infections with reporter viruses containing dual-tropic Env proteins from the HIV-1 strains 89.6 and SF2. Both the HIV-1 89.6 and SF2 Env proteins can mediate efficient membrane fusion in either a CXCR4- or a CCR5-dependent manner. We found that ALX40-4C inhibited infection by both viruses only when cells expressed CXCR4 (Fig. 2). CCR5-dependent virus infection was not affected, indicating that the inhibitory effects of ALX40-4C are CXCR4-dependent and are not directed at the Env protein.
Figure 2.

Inhibition of CXCR4 coreceptor usage by dual-tropic Envs. Luciferase virions were pseudotyped with envs from the primary, dual-tropic virus 89.6 and from the lab-adapted strain SF2, both of which can use CXCR4 and CCR5 for entry. Feline CCCS+L-CD4 cells transiently expressing either CXCR4 or CCR5 were infected in the presence or absence of 10 μM ALX40-4C and results are presented as in Fig. 1.
To confirm that the inhibitory effects we observed were due to Env-mediated fusion events, we used a cell–cell fusion assay that depends only on Env-mediated membrane fusion for signal activity (7, 23). In this assay, murine PA317-T4 cells which express human CD4, the coreceptor of interest, and contain luciferase under control of the T7 promoter, are mixed with 293T cells that express the Env protein of interest and T7 polymerase. Fusion of the two cell populations results in cytoplasmic mixing and expression of luciferase. Use of the cell–cell fusion assay confirmed our infection results by demonstrating ALX40-4C inhibition of fusion between cells expressing dual (89.6) and T-tropic (BH8) Env proteins and cells expressing CD4 and CXCR4 (Fig. 3). Fusion between cells expressing dual- and M-tropic (JR-FL) Env proteins and cells expressing CD4 and CCR5 was not significantly affected.
Figure 3.

Inhibition of cell–cell fusion. 293T cells expressing 89.6, JR-FL, or BH8 Envs and T7 polymerase were mixed with PA317-T4 cells expressing CD4, the coreceptor indicated, and a luciferase reporter gene under control of a T7 promoter in the presence or absence of 10 μM ALX40-4C. Fusion of the cell populations results in quantitative luciferase expression. Results are presented as in Fig. 1 with 89.6 normalized to 100% for CXCR4.
ALX40-4C Binds Directly to CXCR4.
Although ALX40-4C inhibited CXCR4-dependent membrane fusion with both T- and dual-tropic Env proteins, chemokine receptors can be used differently by divergent virus strains, raising the possibility that ALX40-4C could interact with a region in a dual-tropic Env protein that mediates interactions with CXCR4 without affecting its ability to use CCR5 (25–28). Thus, we tested its ability to prevent CXCR4 activation by the chemokine stromal-derived factor (SDF)-1α and to prevent binding by an anti-CXCR4 monoclonal antibody (12G5; reference 29). Binding of SDF-1α to CXCR4 results in intracellular G protein–mediated signals that include Ca2+ mobilization (13, 14). We found that ALX40-4C, when incubated with SupT1 cells that express CXCR4 and that normally respond to SDF-1α, was able to eliminate Ca2+ mobilization (Fig. 4 A). We also found that incubation of SupT1 (Fig. 4 B), PM1 (not shown), or 293T cells (not shown) expressing CXCR4 with 10 μM ALX40-4C completely inhibited binding of 12G5. Since binding reactions were performed at 4°C, these results indicate that ALX40-4C inhibited 12G5 binding directly rather than by inducing receptor downregulation.
Figure 4.
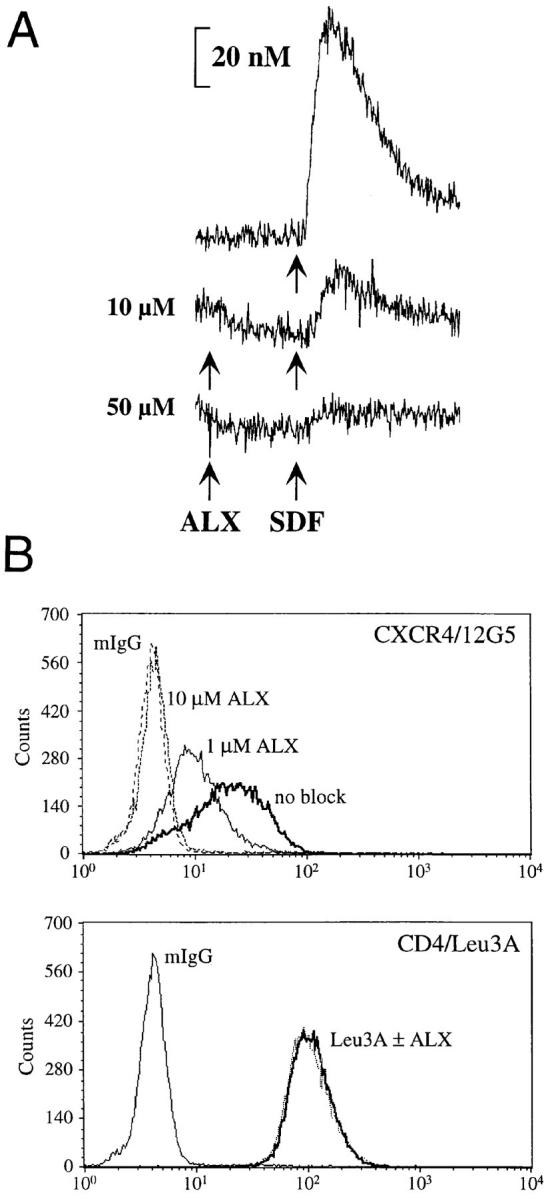
ALX40-4C inhibits interaction of SDF-1α and 12G5 with CXCR4. (A) Addition of 10–50 μM ALX40-4C inhibited the response of SupT1 cells to 500 ng/ml SDF-1α (Peprotech) ∼90 s later. (B) Inhibition of 12G5 binding. SupT1 cells were stained with 12G5, a monoclonal antibody directed to CXCR4, Leu3A, a monoclonal antibody directed to CD4, or mouse IgG. Binding of monoclonal antibodies and inhibition by ALX40-4C was detected by flow cytometry.
Discussion
ALX40-4C, a polypeptide of nine Arg residues stabilized by terminal protection and inclusion of d amino acids, inhibits HIV-1 NL4-3 with an IC50 of 3 nM in the HUT78 T cell line and in PBLs (21). In this study, we found that ALX40-4C inhibited entry of T-tropic, but not M-tropic HIV-1 strains. ALX40-4C also inhibited primary dual-tropic virus strains, but only when cells expressed CXCR4 alone. Infection of cells that express both CCR5 and CXCR4 by dual-tropic virus strains was not inhibited by ALX40-4C, likely reflecting the ability of these viruses to use CCR5 when CXCR4 is blocked.
Addition of ALX40-4C to cells expressing CXCR4 prevented SDF-1α–induced changes in intracellular calcium and prevented binding of a monoclonal antibody, 12G5, to cells expressing CXCR4. The epitope recognized by 12G5 resides in the first and second extracellular loops, the same region of CXCR4 that is used by both dual- and T-tropic HIV-1 strains (26, 30) and that overlaps with the activation site of SDF-1α (our unpublished results). These loops are highly negatively charged, and so represent an obvious site to which the cationic ALX40-4C can bind. It is interesting to note that increasing numbers of basic residues in the V3 loop of Env play an important role in the shift from M to T tropism (31, 32). Since the V3 region is implicated in coreceptor specificity and is the region of Env responsible for sensitivity to ALX40-4C (21), it is tempting to speculate that the V3 region of T- and dual-tropic Envs interacts with the negatively charged first and second extracellular loops of CXCR4, and that this interaction is prevented by the polycationic compound ALX40-4C. Our results with SDF-1α and 12G5 suggest that ALX40-4C interacts directly with CXCR4. However, we cannot rule out the possibility that ALX40-4C may also bind to other cell surface molecules. Of note, ALX40-4C inhibits entry of herpes simplex virus type 1, a virus that is also sensitive to other polycations (33).
The identification of chemokine receptors as potential new antiviral targets is offset by the limitations of using chemokine ligands therapeutically and by the variation in susceptibility to chemokine inhibition of different strains of HIV-1 (Trkola, A., and J.P. Moore, manuscript submitted). ALX40-4C demonstrates the feasibility of developing small-molecule antagonists to the chemokine receptors that target a broad spectrum of HIV-1 strains that enter via a specific chemokine receptor. The ability to inhibit T-tropic HIV-1 strains may prove critically important in future treatments since T-tropic strains of HIV-1 are associated with CD4 decline and progression to AIDS (34). In addition, under conditions in which CCR5 is blocked, evolution of HIV-1 towards using CXCR4 could potentially be enhanced, thus raising the possibility of hastening disease progression, rather than delaying it. The use of a compound such as ALX40-4C in conjunction with an anti-CCR5 compound may avoid such pressures by targeting both major HIV-1 coreceptors.
Acknowledgments
The authors thank John Moore for sharing unpublished results on chemokine inhibition.
This work was supported by National Institutes of Health grants AI-40880, AI-38225, and NS-35705, Department of Veterans Affairs medical research funds, and the University of California at Los Angeles AIDS Institute. B.J. Doranz was supported by a Howard Hughes Medical Institute predoctoral fellowship, and K. Grovit-Ferbas was supported by National Institutes of Health postdoctoral training grant T32/AI07388.
References
- 1.Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature (Lond) 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 2.Wei X, Ghosh SK, Taylor ME, Johnson VA, Emini EA, Deutsch P, Lifson JD, Bonhoeffer S, Nowak MA, Hahn BH, et al. Viral dynamics in human immunodeficiency virus type 1 infection. Nature (Lond) 1995;373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 3.Gulick RM, Mellors JW, Havlir D, Eron JJ, Gonzalez C, McMahon D, Richman DD, Valentine FT, Jonas L, Meibohm A, et al. Treatment with indinavir, zidovudine and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N Engl J Med. 1997;337:734–739. doi: 10.1056/NEJM199709113371102. [DOI] [PubMed] [Google Scholar]
- 4.Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA. CC CKR5: a RANTES, MIP-1α, MIP-1β receptor as a fusion cofactor for macrophage-tropic HIV-1. Science (Wash DC) 1996;272:1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- 5.Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, Wu L, Mackay CR, LaRosa G, Newman W, et al. The β-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell. 1996;85:1135–1148. doi: 10.1016/s0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]
- 6.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Marzio PD, Marmon S, Sutton RE, Hill CM, et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature (Lond) 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 7.Doranz BJ, Rucker J, Yi Y, Smyth RJ, Samson M, Peiper SC, Parmentier M, Collman RG, Doms RW. A dual-tropic primary HIV-1 isolate that uses fusin and the β-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell. 1996;85:1149–1158. doi: 10.1016/s0092-8674(00)81314-8. [DOI] [PubMed] [Google Scholar]
- 8.Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP, Paxton WA. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature (Lond) 1996;381:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 9.Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane domain, G-protein coupled receptor. Science (Wash DC) 1996;272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 10.Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber C-M, Saragosti S, Lapoumèroulie C, Cogniaux J, Forceille C, et al. Resistance to HIV-1 infection of Caucasian individuals bearing mutant alleles of the CCR5 chemokine receptor gene. Nature (Lond) 1996;382:722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- 11.Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, MacDonald ME, Stuhlmann H, Koup RA, Landau NR. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86:367–377. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- 12.Dean M, Carrington M, Winkler C, Huttley GA, Smith MW, Allikmets R, Goedert JJ, Buchbinder SP, Vittinghoff E, Gomperts E, et al. Genetic resistance of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Science (Wash DC) 1996;273:1856–1862. doi: 10.1126/science.273.5283.1856. [DOI] [PubMed] [Google Scholar]
- 13.Bleul CC, Farazan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, Springer TA. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature (Lond) 1996;382:829–833. doi: 10.1038/382829a0. [DOI] [PubMed] [Google Scholar]
- 14.Oberlin E, Amara A, Bachelerie F, Bessia C, Virelizier J-L, Arenzana-Seisdedos F, Schwartz O, Heard J-M, Clark-Lewis I, Legler DF, et al. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell line-adapted HIV-1. Nature (Lond) 1996;382:833–835. doi: 10.1038/382833a0. [DOI] [PubMed] [Google Scholar]
- 15.Paxton WA, Martin SR, Tse D, O'Brien TR, Skurnick J, VanDevanter NL, Padian N, Braun JF, Kotler DP, Wolinsky SM, Koup RA. Relative resistance to HIV-1 infection of CD4 lymphocytes from persons who remain uninfected despite multiple high-risk sexual exposures. Nat Med. 1996;2:412–417. doi: 10.1038/nm0496-412. [DOI] [PubMed] [Google Scholar]
- 16.Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. Identification of RANTES, MIP-1α, and MIP-1β as the major HIV suppressive factors produced by CD8+T cells. Science (Wash DC) 1995;270:1811–1815. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- 17.Arenzanaseisdos F, Virelizier JL, Rousset D, ClarkLewis I, Loetscher P, Moser B, Baggiolini M. HIV blocked by chemokine antagonist. Nature (Lond) 1996;383:400. doi: 10.1038/383400a0. [DOI] [PubMed] [Google Scholar]
- 18.Simmons G, Clapham PR, Picard L, Offord RE, Rosenkilde MM, Schwartz TW, Buser R, Wells TNC, Proudfoot WEI. Potent inhibition of HIV-1 infectivity in macrophages and lymphocytes by a novel CCR5 antagonist. Science (Wash DC) 1997;276:276–279. doi: 10.1126/science.276.5310.276. [DOI] [PubMed] [Google Scholar]
- 19.Broder CC, Berger EA. Fusogenic selectivity of the envelope glycoprotein is a major determinant of human immunodeficiency virus type 1 tropism for CD4+ T-cell lines vs. primary macrophages. Proc Natl Acad Sci USA. 1995;92:9004–9008. doi: 10.1073/pnas.92.19.9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daar ES, Chernyavskiy T, Zhao JQ, Krogstad P, Chen IS, Zack JA. Sequential determination of viral load and phenotype in human immunodeficiency virus type 1 infection. AIDS Res Hum Retroviruses. 1995;11:3–9. doi: 10.1089/aid.1995.11.3. [DOI] [PubMed] [Google Scholar]
- 21.O'Brien WA, Sumner-Smith M, Mao S-H, Sadeghi S, Zhao J-Q, Chen ISY. Anti-human immunodeficiency virus type 1 activity of an oligocationic compound mediated via gp120 V3 interactions. J Virol. 1996;70:2825–2831. doi: 10.1128/jvi.70.5.2825-2831.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Connor RI, Chen BK, Choe S, Landau NR. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology. 1995;206:935–944. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- 23.Nussbaum O, Broder CC, Berger EA. Fusogenic mechanisms of enveloped-virus glycoproteins analyzed by a novel recombinant vaccinia virus–based assay quantitating cell fusion-dependent reporter gene activation. J Virol. 1994;68:5411–5422. doi: 10.1128/jvi.68.9.5411-5422.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simmons G, Wilkinson D, Reeves JD, Dittman MT, Beddows S, Weber J, Carnegis G, Gesselberger U, Gray PW, Weiss RA, Clapham PR. Primary, syncytium-inducing human immunodeficiency virus type 1 isolates are dual-tropic and most can use either Lestr or CCR5 as coreceptors for virus entry. J Virol. 1996;70:8355–8360. doi: 10.1128/jvi.70.12.8355-8360.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doranz BJ, Lu Z, Rucker J, Zhang T, Sharron M, Cen Y, Wang Z, Guo H, Du J, Accavitti MA, et al. Two distinct CCR5 domains can mediate coreceptor usage by human immunodeficiency virus type 1. J Virol. 1997;71:6305–6314. doi: 10.1128/jvi.71.9.6305-6314.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu Z, Berson JF, Chen Y, Turner JD, Zhang T, Sharron M, Jenks MH, Wang Z, Kim J, Rucker J, et al. Evolution of HIV-1 coreceptor usage through interactions with distinct CCR5 and CXCR4 domains. Proc Natl Acad Sci USA. 1997;94:6426–6431. doi: 10.1073/pnas.94.12.6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Atchison RE, Gosling J, Monteclaro FS, Franci C, Digilio L, Charo IF, Goldsmith M. Multiple extracellular elements of CCR5 and HIV-1 entry: dissociation from response to chemokines. Science (Wash DC) 1996;274:1924–1926. doi: 10.1126/science.274.5294.1924. [DOI] [PubMed] [Google Scholar]
- 28.Bieniasz PD, Fridell RA, Aramori I, Ferguson SSG, Caron MG, Cullen BR. HIV-1 induced cell fusion is mediated by multiple regions within both the viral envelope and the CCR-5 coreceptor. EMBO (Eur Mol Biol Organ) J. 1997;16:2599–2609. doi: 10.1093/emboj/16.10.2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Endres MJ, Clapham PR, Marsh M, Ahuja M, Turner JD, McKnight A, Thomas JF, Stoebenau-Haggarty B, Choe PJVS, Wells TNC, et al. CD4-independent infection by HIV-2 is mediated by fusin/CXCR4. Cell. 1996;87:745–765. doi: 10.1016/s0092-8674(00)81393-8. [DOI] [PubMed] [Google Scholar]
- 30.Brelot A, Heveker N, Pleskoff O, Sol N, Alizon M. Role of the first and third extracellular domains of CXCR-4 in human immunodeficiency virus coreceptor activity. J Virol. 1997;71:4744–4751. doi: 10.1128/jvi.71.6.4744-4751.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeJong J-J, Goudsmit J, Keulen W, Klaver B, Krone W, Tersmette M, deRonde A. Human immunodeficiency virus type 1 clones chimeric for the envelope V3 domain differ in syncytium formation and replication capacity. J Virol. 1992;66:757–765. doi: 10.1128/jvi.66.2.757-765.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fouchier RAM, Groenink M, Kootstra NA, Tersmette M, Huisman HG, Miedema F, Schuitemaker H. Phenotype-associated sequence variation in the third variable domain of the human immunodeficiency virus type 1 gp120 molecule. J Virol. 1992;66:3183–3187. doi: 10.1128/jvi.66.5.3183-3187.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sumner-Smith M, Zheng Y, Zhang YP, Twist EM, Climie SC. Antiherpetic activities of N-α–acetyl-nona-d-arginine amide acetate. Drugs Exp Clin Res. 1995;21:1–6. [PubMed] [Google Scholar]
- 34.Connor RI, Mohri H, Cao Y, Ho DD. Increased viral burden and cytopathicity correlate temporally with CD4+ T-lymphocyte decline and clinical progression in human immunodeficiency virus type 1-infected individuals. J Virol. 1993;67:1772–1777. doi: 10.1128/jvi.67.4.1772-1777.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]