

Abstract
The lymphotoxin (LT)/tumor necrosis factor (TNF) family has been implicated in the neurologic inflammatory diseases multiple sclerosis (MS) and experimental allergic encephalomyelitis (EAE). To determine the role of individual family members in EAE, C57BL/6 mice, LT-α–deficient (LT-α−/− mice), or LT-β–deficient (LT-β−/− mice), and their wild-type (WT) littermates were immunized with rat myelin oligodendrocyte glycoprotein (MOG) peptide 35-55. C57BL/6 and WT mice developed chronic, sustained paralytic disease with average maximum clinical scores of 3.5 and disease indices (a measure of day of onset and sustained disease scores) ranging from 367 to 663 with central nervous system (CNS) inflammation and demyelination. LT-α−/− mice were primed so that their splenic lymphocytes proliferated in response to MOG 35-55 and the mice produced anti-MOG antibody. However, LT-α−/− mice were quite resistant to EAE with low average clinical scores (<1), an average disease index of 61, and the negligible CNS inflammation and demyelination. WT T cells transferred EAE to LT-α−/− recipients. LT-β−/− mice were susceptible to EAE, though less than WT, with an average maximum clinical score of 1.9 and disease index of 312. These data implicate T cell production of LT-α in MOG EAE and support a major role for LT-α3, a minor role for the LT-α/β complex, and by inference, no role for TNF-α.
Several studies have implicated members of the lymphotoxin(LT)1/TNF family in multiple sclerosis (MS) and its animal model, experimental allergic encephalomyelitis (EAE). LT-α (also known as TNF-β) is a member of the immediate MHC-linked TNF family, which consists of TNF-α, LT-α, and LT-β. LT can be secreted as an LT-α3 homotrimer, which can bind to either the p55 TNFRI or p75 TNFRII, where it emulates many of the activities of TNF-α. It is also present as a cell surface complex in association with a type II transmembrane protein, LT-β. The most common form, LT-α1β2, binds to the LT-βR, and the less common form, LT-α2β1, can bind to p55 TNFRI (1). Studies that implicate members of the family in the pathogenesis of MS and EAE include the presence of LT- and TNF-α in MS plaques (2) and the correlation of T cell clones' encephalitogenicity with their production of LT-α and/or TNF-α (3). Even more compelling evidence for the role of members of the family in EAE includes the ability of antibodies that neutralize LT-α and TNF-α to inhibit passive transfer of both acute (4, 5) and relapsing remitting disease (6). On the other hand, other studies do not support the conclusion that TNF-α is pathogenic in EAE. One study suggests that administration of a rabbit anti-TNF antibody at the time of immunization of mice with myelin basic protein (MBP) does not affect the development of EAE (7) and another indicates that administration of TNF-α, through a vaccinia virus vector delivery system, inhibits the development of EAE (8). Furthermore, a study published while this manuscript was being prepared indicated that mice deficient in both LT-α and TNF-α and backcrossed to SJL mice (though retaining the H-2b knockout chromosome) developed clinical signs of EAE after immunization with MBP or mouse spinal cord homogenate (9). In view of the conflicting data, and the fact that none of the previous studies clearly distinguished between LT and TNF-α, nor did any address the role of secreted LT-α3 as opposed to the membrane associated LT-α/β heterotrimer, we systematically addressed the role of the individual members of the LT family (and by inference TNF-α) in EAE in C57BL/6 H-2b mice.
Recently, mice selectively deficient in LT-α or LT-β have been produced (10–12). LT-α–deficient mice (LT-α−/− mice) have profound defects in lymphoid organ development (10, 11). They are missing all LNs and Peyer's patches (PP), exhibit marked splenic disorganization, and lack germinal centers (13). Despite these anatomic defects, humoral immune responses can be elicited. The mice produce normal levels of anti-nitrophenyl antibody when challenged with a high dose of nitrophenyl-ovalbumin and their immunoglobulin genes undergo somatic hypermutation resulting in antibody affinity maturation (14). LT-α−/− mice produce normal or enhanced levels of IgM to SRBC immunization, although IgG levels are reduced compared to wild-type (WT) littermates (12). Delayed type hypersensitivity to ovalbumin is markedly impaired although LT-α−/− splenic T cells proliferate to that antigen (Bergman, C.M., and N.H. Ruddle, manuscript in preparation; 15). LT-β−/− mice exhibit some, but not all, of the defects of LT-α−/− mice (12). They lack PP and exhibit splenic disorganization with an absence of germinal centers and antibody responses essentially comparable to those of LT-α−/− mice. LT-β−/− mice differ from LT-α−/− mice in that, although most peripheral LNs are absent, the mesenteric and cervical LNs are present and some members of the lumbar group are variably apparent (12).
Because we wished to study EAE in LT-deficient mice, an immunization regimen that would induce EAE in mice of the H-2b haplotype was necessary. We did not believe it was practical to use the more commonly studied methods of MBP or PLP immunization of H-2s or H-2u mice because the knockout mice were developed from 129 (H-2b) embryonic stem cells, and it was necessary to retain chromosome 17 because the LT/TNF genes map to the MHC. Thus, if the mice were crossed to SJL, WT littermates would of necessity differ at the H-2 locus. Recently, a method of inducing EAE in C57BL/6 (H-2b) mice has been described (16, 17). C57BL/6 mice injected with rat myelin oligodendrocyte glycoprotein (MOG) peptide 35– 55 in CFA and pertussis toxin (Pt) and boosted with peptide in CFA develop a chronic sustained paralysis. These authors reported that the disease could be transferred to naive recipients with MOG peptide–specific T cell lines. MOG peptide immunization also induces EAE in marmosets and rats (18, 19). However, passive transfer of EAE in these models is effected by a combination of specific T cells and antibody, suggesting that the neurologic lesions are initiated by T cells and sustained by antibody. None of the previous studies with MOG in mice, rats, or marmosets have addressed the role of cytokines in this disease, with the exception of a report by Willenborg et al. (20) in IFN-γR−/− mice. However, in that study, WT mice were not susceptible to MOG-induced EAE (although the IFN-γR−/− mice were) probably because human rather than rat MOG peptide was used. In our study, the rat MOG peptide immunization regimen was precisely followed to induce EAE in C57BL/6 and WT controls as we evaluated mice that were selectively deficient in LT-α or LT-β to identify the role of those individual cytokines, and by inference TNF-α, in EAE.
Materials and Methods
Mice.
C57BL/6 (H-2b) mice were obtained from the Jackson Laboratory (Bar Harbor, ME) or from a breeding colony maintained at Yale University School of Medicine (New Haven, CT). A breeding pair of LT-α−/− mice were originally obtained from Dr. David Chaplin (Washington University, St. Louis, MO) and have been previously described (10). They have been maintained as a breeding colony under specific pathogen-free conditions at Yale University for ∼2 yr. LT-β−/− mice were developed in collaboration with Drs. Richard Flavell (Yale University School of Medicine) and Jeffrey Browning (Biogen, Cambridge, MA) as previously described (12) and were also maintained under specific pathogen-free conditions at Yale University School of Medicine. LT-α−/− and LT-β−/− mice were screened by their respective PCR assays as previously described (12). The colonies of LT-α−/− and LT-β−/− mice are of mixed 129 × C57BL/6 origin and were studied in the course of backcrossing to C57BL/6. Each colony was at least at the level of the fourth backcross at the time of these experiments.
MOG Peptide.
MOG peptide 35–55 of rat origin was synthesized by the W.M. Keck Biotechnology Resource Center at Yale University. The sequence was MEVGWYRSPFSRVVHLYRNGK (16, 17). In most experiments, material had been subjected to reverse phase (C18) column HPLC and a TFA/acetonitrile gradient.
Active Induction of EAE with MOG Peptide.
Mice of 8–10 wk of age were injected with 300 μg MOG peptide in CFA (Difco, Detroit, MI) with 500 μg of Mycobacterium tuberculosis. The 0.2-ml emulsion was injected subcutaneously in one flank. Mice received 500 ng Pt (List Biological, Campbell, CA) in 200 μl PBS in the tail vein immediately after the first immunization and 48 h later and were boosted with peptide with CFA subcutaneously in the other flank 1 wk later. The mice were observed daily for clinical signs and scored on a scale of 0–5 with gradations of 0.5 for intermediate scores: 0, no clinical signs; 1, loss of tail tone; 2, wobbly gait; 3, hind limb paralysis; 4, hind and fore limb paralysis; 5, death. The average day of disease onset was calculated by averaging the first day of clinical signs for each mouse in the group. The average maximum disease score was similarly calculated by averaging the highest individual score for each mouse. The disease index was calculated by adding all the daily average disease scores, dividing by the average day of disease onset, and multiplying by 100. The day on which the index was calculated is indicated in individual experiments. For those animals which showed no clinical signs, the onset is arbitrarily calculated as one day after the experiment was terminated.
Histology.
Mice were perfused with 50 ml of cold PBS and with 40 ml of cold Z-fix (formaldehyde, ionized zinc, buffer). The brain and spinal cords were removed. Paraffin sections were prepared and stained with hematoxylin and eosin or Luxol fast blue counterstained with nuclear fast red by the Yale Pathology Services. In other experiments, tissue was embedded in epon, sectioned at 2 microns, and stained with toluidine blue.
ELISA.
Serum samples were prepared from peripheral blood obtained by retroorbital or cardiac puncture immediately before perfusion at days 6 and 30 after the first immunization. ELISA was performed using purified rat MOG 35–55 peptide. The peptide was diluted to 10 μg/ml in carbonate buffer, pH 9.3, and coated onto 96-well microtiter plates (Dynex Technologies, Chantilly, VA). After incubation at 37°C overnight, the plates were washed with TBS, pH 7.5, containing 0.1% Tween 20 and blocked with 5% BSA in TBS (TBS-BSA) for 2 h at 37°C. Twofold serial dilutions of serum were made in TBS-BSA and applied to preblocked wells for 2 h at 37°C. Appropriately diluted alkaline-phosphatase–conjugated goat anti-mouse IgG (H + L) antibody (Pierce, Rockford, IL) was added to the plates and incubated for 1 h at 37°C. The plates were washed and incubated with p-nitrophenyl phosphate substrate for 30 min before optical density was measured at 405 nm. The end-point titer was defined as the dilution at which the OD was 0.05 above background.
MOG-specific T Cell Lines.
Two mice were immunized with purified MOG peptide (150 μg) in CFA with Mycobacterium tuberculosis subcutaneously in the tail. After 9 d, the lymph nodes were removed and cultured in the presence of 40 μg/ml MOG peptide. T cell lines were derived and maintained as previously described (4) by feeding MOG peptide (40 μg/ml) with irradiated spleen cells in RPMI media (GIBCO BRL, Gaithersburg, MD) supplemented with 1 mM l-glutamine, penicillin-streptomycin, 10% FCS, fungizone, and 2-mercaptoethanol every 14 d. Cells were fed IL-2 every 7 d. They were incubated in a humidified 37°C, 10% CO2 incubator and routinely tested for antigen specific proliferation.
Proliferative Response of T Cells to MOG Antigen.
The assays were set up in triplicate in 96-well flat-bottomed plates in 250 μl/well with 5 × 104 T cells, 1.5 × 105 irradiated spleen cells, and 30 μg of MOG peptide in RPMI medium supplemented as above. [3H]thymidine, 1 μCi/well, specific activity 5 Ci/mmol (Amersham, Arlington Heights, IL) per well, was added 24 h after culture initiation. Plates were harvested at 48 h using a PHD harvester (Cambridge Technology, Cambridge, MA) and counted by scintillation counter.
Passive Transfer of EAE.
WT MOG–specific T cell lines and spleen cells from MOG immunized mice were used in passive transfer experiments. Cells were injected intraperitoneally into recipient mice which were irradiated (550 rads) or not and treated or not with Pt (500 μg) as indicated in individual experiments. For passive transfer of MOG T cell lines, cells were stimulated with IL-2, irradiated spleen cells, and MOG peptide 4 d before injection, and were fed IL-2 1 d before the transfer. The number of cells injected varied from 2.5–3.8 × 107. For passive transfer of MOG spleen cells, donor mice were immunized with 300 μg of MOG peptide in CFA in both flanks. 14 d later, the spleens were removed and the cells isolated and placed in 20-ml tissue culture dishes at 5 × 106 cells/ml with 20 μg/ml of MOG peptide antigen in RPMI. 4 d later, the cells were washed with HBSS, counted, and 6 × 107 cells were injected into recipient mice.
Results
LT-α−/− Mice Are Primed to MOG.
The protocol of active immunization, virtually identical to that of Mendel and co-workers (16, 17) which induces a chronic EAE in C57BL/6 (H-2b) mice was used. C57BL/6 and LT-α−/− mice were bled at various times after immunization and their sera tested for total IgG by ELISA. There were no detectable titers of IgG against MOG 35–55 6 d after immunization. 30 d after immunization, WT mice (n = 9) had a mean titer of 1420 (200–3,300) and LT-α−/− mice (n = 7) had a mean titer of 380 (130–1,600). Nonimmunized mice (n = 5), mice immunized with adjuvant alone (n = 5), and mice immunized with other antigens (n = 5) did not show any antibody response to MOG 35–55. Spleen cells of MOG immunized LT-α−/− mice proliferated in vitro in response to MOG peptide (Table 1). These results, in agreement with previous studies, indicate that, despite the absence of LNs, LT-α−/− mice can be sensitized to exogenous antigen.
Table 1.
Spleen Cells of LT α−/− Mice Proliferate in Response to MOG
| Source of spleen cells* | No antigen | MOG peptide | SI | |||
|---|---|---|---|---|---|---|
| cpm | ||||||
| C57BL/6 | 74.5 | 616.4 | 8.2 | |||
| LT α−/− | 74 | 374.0 | 5 | |||
Mice were immunized with 300 μg MOG 35–55 in CFA and 14 d later cells cultured in the presence or absence of 30 μg MOG 35–55. [3H]Thymidine incorporation was evaluated after 48 h in culture.
LT-α−/− Mice Show Minimal Clinical Signs after MOG Immunization.
We next tested whether MOG-immunized LT-α−/− mice could develop EAE. C57BL/6 and LT-α+/+ littermates exhibited clinical signs by ∼12 d after the initial immunization. The clinical manifestations were similar to those previously reported (16, 17) with a chronic and severe paralysis (Fig. 1) and weight loss. In some cases, an altered gait was apparent a day or two before the tail was affected. However, in general the tail eventually became flaccid. Most mice progressed to hindlimb paralysis, and several progressed to hind- and forelimb paralysis or death. The clinical signs continued for the duration of the observation period and beyond (for >60 d). A similar clinical course was seen in 129 mice (data not shown). In contrast, LT-α−/− mice were markedly resistant to active induction of EAE (Fig. 2; Table 2). In two separate experiments involving a total of nine mice, only one animal attained a score >0.5, and this was much later (at day 23) than the usual peak scores in the other two groups (Table 2). The mean maximum clinical score was significantly less than any of the control groups, including the LT-α+/− mice. Interestingly, LT-α+/− mice, despite their full complement of lymphoid organs, exhibited a sensitivity intermediate between LT-α+/+ and LT-α−/− mice, suggesting a dose related effect of LT.
Figure 1.

C57BL/6 mice develop a sustained chronic disease after immunization with MOG 35–55. Mice were injected with MOG peptide 35–55 in CFA and Pt as indicated in Materials and Methods. Mice that died are indicated and their scores are not included further in the graph.
Figure 2.

LT-α−/− mice manifest minimal clinical signs of EAE after active immunization. The average clinical signs of mice in the individual groups of C57BL/6 (▪), LT-α+/− (•), and LT-α−/− (○) mice are indicated. The scores of the 2 out of 6 C57BL/6 mice that died are included in the graph throughout the experiment. No other mice died.
Table 2.
LT α−/− Mice Are Resistant to MOG 35–55 EAE
| Group* | No. sick/total | Day of onset | Mean max clinical score | Death total | Disease index‡ | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| C57BL/6 | 7/7 | 13.3 | 3.5 | 2/7 | 386 | |||||
| LTα+/+ | 4/4 | 10.3 | 4 | 2/4 | 560 | |||||
| LTα+/− | 4/4 | 14.5 | 2.6 | 0/4 | 234 | |||||
| LTα−/− | 6/9 | 13.5 | 0.91§ | 0/9 | 61 |
Average of two separate experiments.
Disease index calculated on day 30.
P <0.0005 compared to C57BL/6 controls.
LT-α−/− Mice Show Minimal CNS Inflammation and no Demyelination after MOG Immunization.
The brain and cervical, thoracic and lumbar spinal cord of individual C57BL/6, LT-α+/+, LT-α+/−, and LT-α−/− mice were evaluated histologically at various times after MOG immunization. All positive control mice exhibited extensive mononuclear infiltration in all areas examined, including the choroid plexus and all levels of the spinal cord. The mononuclear infiltrate, consisting of T cells, B cells, and macrophages (Suen, W.E., C.M. Bergman, and N.H. Ruddle, manuscript in preparation) was diffuse and not restricted to cuffing around the vessels but extended into the parenchyma, predominantly in the white but also in the gray matter. Demyelination was apparent (Fig. 3). In contrast, there was very little inflammation in any sections obtained from LT-α−/− mice. The minimal inflammation was limited almost exclusively to the meninges. The most extensive inflammation detected is shown in Fig. 3 C. There was no evidence of demyelination. Thus, the minimal clinical signs of LT-α−/− mice were mirrored in the very minor histological involvement.
Figure 3.
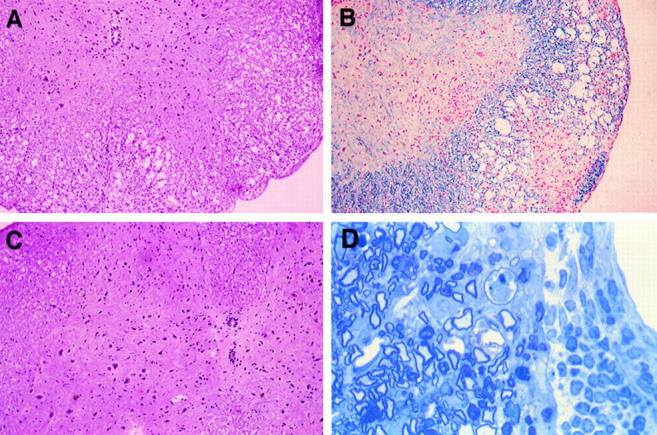
LT-α−/− show minimal CNS inflammation and demyelination after active immunization with MOG peptide. (A and B) Paraffin sections of spinal cord at day 30 of C57BL/6 (score 2.5) and (C) LT-α−/− (score 0.5) mice stained with hematoxylin and eosin (A and C) and Luxol fast blue with neutral red (B). Note extensive infiltration of mononuclear cells and demyelination in sections of the control mice and minimal infiltration and no demyelination in sections from LT-α−/− mice. (Original magnification of A–C × 125). (D) Epon toluidine blue section of infiltrating mononuclear cells in spinal cord of C57BL/6 mouse 35 d after immunization with MOG (Original magnification: × 312.5). Some demyelination is apparent.
LT-α−/− Mice Manifest Clinical Signs after Passive Transfer of WT MOG-specific T Cells.
Several protocols of passive transfer were used to test whether LT-α−/− mice, although resistant to active induction of EAE, could act as recipients of passive transfer of EAE by cells from WT donors. Several experiments were performed using either T cell lines, lymph node cells, or spleen cells from C57BL/6 mice immunized with MOG 35-55 in CFA. Recipients were irradiated or not and/or injected or not with Pt. In five separate experiments (three of which are shown in Table 3), C57BL/6 mice showed rather mild clinical signs after transfer of as many as 4 × 107 T cells. It is likely that a combination of MOG-specific T cells and anti-MOG antibody will result in more dramatic clinical signs. Nevertheless, in three out of three experiments, LT-α−/− mice were susceptible to passive transfer with WT MOG T cells, even more so than the control recipients. The enhanced susceptibility was manifested as a slightly higher maximum clinical score, and especially as a much earlier day of onset. The enhanced susceptibility of LT-α−/− mice to transfer could be due to the fact that their TNF receptors are not occupied by endogenous LT and hence would be more readily triggered.
Table 3.
WT MOG-specific T Cells Transfer EAE to LT-α−/− Mice
| Experiment No. (cells) | Donor | Recipient | No. sick/total | Day of onset | Mean maximum clinical score | Disease index | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 (MOG T cell line) | B6 | Irrad, Pt B6 | 4/4 | 9.3 | 0.88 | 65 | ||||||
| B6 | Irrad, Pt LT-α−/− | 4/4 | 1.8 | 1 | 1,103 | |||||||
| 2 (MOG T cell line) | B6 | Irrad B6 | 1/2 | 8 | 0.5 | 19 | ||||||
| B6 | Irrad LT-α−/− | 5/5 | 3.6 | 1 | 328 | |||||||
| 3 (MOG spleen cells) | B6 | B6 | 1/4 | 11 | 0.25 | 8 | ||||||
| B6 | LT-α−/− | 3/3 | 5.3 | 1 | 157 |
Disease index calculated at 21 d.
LT-β−/−Mice Are Susceptible to EAE.
To determine whether the effects of LT in EAE are mediated by the soluble LT-α3 form or as a membrane-associated LT-α/β complex, we tested whether LT-β−/− mice are susceptible to EAE. These mice were also of interest because they lack PP and most LNs (although, in contrast to LT-α−/− mice, they still have mesenteric and cervical LNs). LT-β−/− mice exhibit the splenic disorganization and minimal defects in immunoglobulin switching that are seen in LT-α−/− mice. LT-β−/− mice were susceptible to EAE in that they showed clinical signs and histologic manifestations, although they were somewhat less susceptible than WT mice (Table 4). These data indicate that peripheral LNs, PP, and an organized spleen are not required for EAE. The data also suggest that the activities of LT in EAE are mediated predominantly as soluble LT-α3, rather than as an LT-α/β membrane–associated complex.
Table 4.
LT β−/− Mice Are Susceptible to MOG 35–55 EAE
| Group* | No. sick/total | Day of onset | Mean max clinical score | Death total | Disease index‡ | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| C57BL/6 | 9/9 | 10.6 | 3.3 | 2/10 | 515 | |||||
| LTβ+/− | 13/13 | 8.9 | 3 | 2/13 | 525 | |||||
| LTβ−/− | 17/17 | 10.1 | 1.9§ | 0/17 | 312 |
Average of three separate experiments.
Disease index calculated at 30 d.
P <0.003 compared to C57BL/6 controls.
Discussion
In this study, we evaluated mice selectively deficient in LT-α or LT-β in order to identify the role of those individual cytokines, and by inference, TNF-α, in MOG-induced EAE. C57BL/6 and WT littermates of LT-deficient mice were susceptible to EAE with a chronic, sustained paralysis and CNS inflammation and demyelination. Despite evidence that LT-α−/− mice were primed to MOG in that they produced antipeptide antibody and their spleen cells proliferated in vitro in response to challenge with MOG 35–55, LT-α−/− mice were quite resistant to EAE with regard to clinical signs and histologic manifestations. LT-α−/− mice were susceptible to passive transfer of EAE with WT MOG T cells. LT-β−/− were susceptible to both the clinical signs and CNS pathology, but less so than WT mice. These data suggest that the reason LT-α−/− mice do not develop EAE is because they lack T cell production of LT which is crucial for pathogenesis. They implicate the secreted form of LT-α3 more strongly than the LT-α/β complex and, by inference, suggest a very little compensatory role for TNF-α in this inflammatory demyelinating disease. The data suggest that the inability of LT-α−/− mice to develop EAE is largely because of the fact that their antigen-specific T cells do not produce LT, rather than because of the anatomic problems of their lack of LN and PP and their splenic disorganization. LT-α+/− mice were less susceptible than LT-α+/+ mice to the clinical signs of EAE despite the fact that they have normal lymphoid organs. An anatomic problem is also less likely since LT-β−/− mice were susceptible to EAE even though they have many of the same lymphoid organ anomalies as LT-α−/− mice, that is absence of PP, disorganized spleen, and the absence of most LNs.
We are currently investigating the role of nonlymphoid cells and antibody in MOG EAE. It is possible that LT production, both by infiltrating T cells and by cells in the CNS such as astrocytes, is necessary for full manifestation of the disease. The data presented here, which demonstrate that LT-α−/− mice make low, but detectable amounts of anti-MOG peptide antibody that do not differ significantly from that made by controls, are consistent with the interpretation that antibody alone is not sufficient for manifestations of this disease, although it may be necessary. This concept is in agreement with the conclusions reached in studies of MOG-induced EAE in rats and marmosets (19, 21) and suggests that LT production by antigen-specific T cells initiates the lesion that is sustained by antibody.
There are several possible avenues through which LT may exert its effects in EAE. The LT-α secreted by T cells may contribute to the initial entrance of inflammatory cells into the CNS. We have recently shown that murine LT induces the expression of E-selectin on murine endothelial cells in vitro (Schwartz, J., C.M. Bergman, K. Russell, J. Bender, and N.H. Ruddle, manuscript in preparation). LT-α could also contribute to recruitment in EAE. It induces expression of vascular cellular adhesion molecule (VCAM) and intracellular adhesion molecule (ICAM) in vitro. VCAM expression correlates with clinical signs in EAE and its upregulation is inhibited by treatment with anti-LT/ TNF antibody (22) and the interaction between VLA4 on antigen-specific T cells and VCAM on endothelial cells is crucial for EAE (23). LTs may also contribute to the pathogenesis of EAE through an effect on oligodendrocytes, since it has been shown that LT-α is even more effective than TNF-α in inducing apoptosis of this myelin-producing cell (24). LTs may contribute to the determinant spreading that is apparent in some forms of EAE. That this could also very well be an aspect of the mouse MOG-induced EAE is supported by the chronic manifestation of clinical signs in this model. LT-α and the LT-α/β complex induce lymphoid organs in embryogenesis, and an LT-α transgene induced inflammation represents a phenomenon we have termed lymphoid neogenesis (25). It is possible that the role of LT in EAE includes this organizational aspect which may contribute to perpetuation through the presentation of new antigens.
The studies presented here should be considered in light of the different but overlapping roles of the members of the LT/TNF family with regard to inflammation and lymphoid organ development. LT-β appears to be primarily concerned with lymphoid organ development as an LT-α/ β complex. Its role in inflammation is less crucial. It does not induce expression of VCAM or ICAM (26) and plays a minor role in EAE (as shown in this paper). LT-α appears to be crucial in lymphoid organ development both as an LT-α3 homotrimer and an LT-α/β heterotrimer LT-α is also a major participant in inflammation, which it induces in transgenic mice (27). It is crucial for delayed type hypersensitivity (Bergman, C.M., and N.H. Ruddle, manuscript in preparation) and for EAE, as demonstrated here. TNF-α plays almost no role in lymphoid organ development in that TNF-α−/− mice have normal LNs and PP, though they exhibit some disorganization of B cell areas in the spleen. TNF-α has generally been thought of as a major participant in the inflammatory process (28) and it induces expression of ICAM and VCAM in vitro. Thus, we were surprised that it did not compensate for the absence of LT-α in LT-α−/− mice and we eagerly await the results of MOG immunization of TNF-α−/− mice. It appears from the data presented here that its role may have been misinterpreted in experiments in which it appeared to be essential for EAE.
While this manuscript was being prepared for publication, a communication was published that indicated that TNF-α and LT-α were not required for EAE induced by proteolipid protein or mouse spinal cord homogenate (9). Mice in that study had a simultaneous deletion of the tnfa and lta genes and the intervening sequences and had been backcrossed to SJL. These animals developed clinical signs of EAE after immunization with antigen. It is difficult to reconcile those observations with the ones reported here, but there are many differences in the systems. They differ with regard to antigen, the H-2 haplotype of the controls, background genes, and the fact that this study analyzed mice defective in single genes, whereas Frei et al. (9) involved a large chromosomal deletion. An additional possibility is that LT and TNF-α play different roles in EAE, i.e., LT-α and TNF-α both induce inflammation, but, at high concentrations, TNF-α downmodulates the activities of LT-α. Perhaps in the model of Frei et al. (9) other cytokines compensate for LT-α in the absence of TNF-α.
The data presented here and elsewhere indicate that the members of the LT/TNF family, through their various physical presentations as soluble or membrane forms and by means of their multiple receptors, can each contribute to many different aspects of inflammation and development. They occasionally compensate for each other, and in that way exhibit some redundancy, and in some transgenic systems LT-α and TNF-α appear to have similar activities (29). Nevertheless, in some instances, it is clear that these cytokines have unique functions that cannot be compensated for by other members of the family. Such appears to be the case described here in which LT-α is crucial for MOG-induced EAE.
Acknowledgments
This work was supported by grant RG 2394 from the National Multiple Sclerosis Society. P. Hjelmström was supported by a fellowship from the Swedish Foundation for International Cooperation in Research and Higher Education.
We thank Dr. Matthew Hanson and Irene Visintin for instruction and assistance in the perfusion experiments, Dr. Jeffrey Kocsis for assistance in epon embedding and Toluidine blue staining, and Dr. Kocsis and Dr. Jung Kim for assistance in histological evaluation.
Footnotes
Abbreviations used in this paper: CNS, central nervous system; EAE, experimental allergic encephalomyelitis; ICAM, intracellular adhesion molecule; LT, lymphotoxin; MBP, myelin basic protein; MOG, myelin oligodendrocyte glycoprotein; PP, Peyer's patches; Pt, pertussis toxin; VCAM, vascular cell adhesion molecule; WT, wild-type.
References
- 1.Crowe PD, VanArsdale TL, Walter BN, Ware CF, Hession C, Ehrenfels B, Browning JL, Din WS, Goodwin RG, Smith CA. A lymphotoxin-beta specific receptor. Science (Wash DC) 1994;264:707–710. doi: 10.1126/science.8171323. [DOI] [PubMed] [Google Scholar]
- 2.Selmaj K, Raine CS, Cannella B, Brosnan CF. Identification of lymphotoxin and tumor necrosis factor in multiple sclerosis lesions. J Clin Invest. 1991;87:949–954. doi: 10.1172/JCI115102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Powell MB, Mitchell D, Lederman J, Buckmeier J, Zamvil SS, Graham M, Ruddle NH, Steinman L. Lymphotoxin and tumor necrosis factor-alpha production by myelin basic protein–specific T cell clones correlates with encephalitogenicity. Int Immunol. 1990;2:539–544. doi: 10.1093/intimm/2.6.539. [DOI] [PubMed] [Google Scholar]
- 4.Ruddle NH, Bergman CM, McGrath KM, Lingenheld EG, Grunnet ML, Padula SJ, Clark RB. An antibody to lymphotoxin and tumor necrosis factor prevents transfer of experimental allergic encephalomyelitis. J Exp Med. 1990;172:1193–1200. doi: 10.1084/jem.172.4.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Selmaj K, Raine CS, Cross AH. Anti–tumor necrosis factor therapy abrogates autoimmune demyelination. Ann Neurol. 1991;30:694–700. doi: 10.1002/ana.410300510. [DOI] [PubMed] [Google Scholar]
- 6.Santambrogio L, Hochwald GM, Saxena B, Leu CH, Martz JE, Carlino JA, Ruddle NH, Palladino MA, Gold LI, Thorbecke GJ. Studies on the mechanisms by which TGF-β protects against allergic encephalomyelitis: antagonism between TGF-β and TNF. J Immunol. 1993;151:1116–1127. [PubMed] [Google Scholar]
- 7.Teuscher C, Hickey WF, Korngold R. An analysis of the role of tumor necrosis factor in the phenotypic expression of actively induced experimental allergic orchitis and experimental allergic encephalomyelitis. Clin Immunol Immunopathol. 1990;54:442–453. doi: 10.1016/0090-1229(90)90057-w. [DOI] [PubMed] [Google Scholar]
- 8.Willenborg DO, Fordham SA, Cowden WB, Ramshaw IA. Cytokines and murine autoimmune encephalomyelitis: inhibition or enhancement of disease with antibodies to select cytokines, or by delivery of exogenous cytokines using a recombinant vaccinia virus system. Scand J Immunol. 1995;41:31–41. doi: 10.1111/j.1365-3083.1995.tb03530.x. [DOI] [PubMed] [Google Scholar]
- 9.Frei K, Eugster H-P, Bopst M, Constantinescue CS, Lavi E, Fontana A. Tumor necrosis factor α and lymphotoxin α are not required for induction of acute experimental autoimmune encephalomyelitis. J Exp Med. 1997;185:2177–2182. doi: 10.1084/jem.185.12.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Togni P, Goellner J, Ruddle NH, Streeter PR, Fick A, Mariathasan S, Smith SC, Carlson R, Shornick LP, Strauss-Schoenberger J, et al. Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science (Wash DC) 1994;264:703–707. doi: 10.1126/science.8171322. [DOI] [PubMed] [Google Scholar]
- 11.Banks TA, Rouse BT, Kerley MK, Blair PJ, Godfrey VL, Kuklin NA, Bouley DM, Thomas J, Kanangat S, Mucenski ML. Lymphotoxin-alpha–deficient mice. Effects on secondary lymphoid organ development and humoral immune responsiveness. J Immunol. 1995;144:1685–1693. [PubMed] [Google Scholar]
- 12.Koni PA, Sacca R, Lawton P, Browning JL, Ruddle NH, Flavell RA. Distinct roles in lymphoid organogenesis for lymphotoxins alpha and beta revealed in lymphotoxin beta–deficient mice. Immunity. 1997;6:491–500. doi: 10.1016/s1074-7613(00)80292-7. [DOI] [PubMed] [Google Scholar]
- 13.Matsumoto M, Mariathasan S, Nahm MH, Baranyay F, Peschon JJ, Chaplin DD. Role of lymphotoxin and the type I TNF receptor in the formation of germinal centers. Science (Wash DC) 1996;271:1289–1291. doi: 10.1126/science.271.5253.1289. [DOI] [PubMed] [Google Scholar]
- 14.Matsumoto M, Lo SF, Carruthers CJL, Min J, Mariathasan S, Huang G, Plas DR, Martin SM, Geha RS, Nahm MN, Chaplin DD. Affinity maturation without germinal centres in lymphotoxin-α–deficient mice. Nature (Lond) 1996;382:462–466. doi: 10.1038/382462a0. [DOI] [PubMed] [Google Scholar]
- 15.Sacca, R., S. Turley, L. Soong, I. Mellman, and N.H. Ruddle. 1997. Transgenic expression of lymphotoxin restores lymph nodes to lymphotoxin α–deficient mice. J. Immunol. In press. [PubMed]
- 16.Kerlero de Rosbo N, Mendel I, Ben-Nun A. Chronic relapsing experimental autoimmune encephalomyelitis with a delayed onset and an atypical clinical course, induced in PL/J mice by myelin oligodendrocyte glycoprotein (MOG)–derived peptide: preliminary analysis of MOG T cell epitopes. Eur J Immunol. 1995;25:985–993. doi: 10.1002/eji.1830250419. [DOI] [PubMed] [Google Scholar]
- 17.Mendel I, Kerlero de Rosbo N, Ben-Nun A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2bmice: fine specificity and T cell receptor Vb expression of encephalitogenic T cells. Eur J Immunol. 1995;25:1951–1959. doi: 10.1002/eji.1830250723. [DOI] [PubMed] [Google Scholar]
- 18.Linington C, Bradl M, Lassmann H, Brunner C, Vass K. Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Am J Pathol. 1988;130:443–454. [PMC free article] [PubMed] [Google Scholar]
- 19.Genain CP, Nguyen M-H, Letvin NL, Pearl R, Davis RL, Adelman M, Lees M, Linington C, Hauser SL. Antibody facilitation of muliple sclerosis–like lesions in a nonhuman primate. J Clin Invest. 1995;96:2966–2974. doi: 10.1172/JCI118368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Willenborg, D.O., S. Fordham, C.C.A. Bernard, W.B. Cowden, and I.A. Ramshaw. 1996. IFN-γ plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein–induced autoimmune encephalomyelitis. J. Immunol. 157:3223–3227. [PubMed]
- 21.Linington C, Berger T, Perry L, Weerth S, Hinze-Selch D, Zhang Y, Lu H, Lassmann H, Wekerle H. T cells specific for the myelin oligodendrocyte glycoprotein mediate an unusual autoimmune inflammatory response in the central nervous system. Eur J Immunol. 1993;23:1364–1372. doi: 10.1002/eji.1830230627. [DOI] [PubMed] [Google Scholar]
- 22.Barten DM, Ruddle NH. Vascular cell adhesion molecule–1 modulation by TNF in experimental allergic encephalomyelitis. J Neuroimmunol. 1994;51:123–133. doi: 10.1016/0165-5728(94)90074-4. [DOI] [PubMed] [Google Scholar]
- 23.Baron JL, Madri JA, Ruddle NH, Hashim G, Janeway CA. Surface expression of α4 integrin by CD4 T cells is required for their entry into brain parenchyma. J Exp Med. 1993;177:57–68. doi: 10.1084/jem.177.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Selmaj K, Raine CF, Farooq M, Norton WT, Brosnan CF. Cytokine cytotoxicity against oligodendrocytes: apoptosis induced by lymphotoxin. J Immunol. 1991;147:1522–1529. [PubMed] [Google Scholar]
- 25.Kratz A, Campos-Neto A, Hanson MS, Ruddle NH. Chronic inflammation caused by lymphotoxin is lymphoid neogenesis. J Exp Med. 1996;183:1461–1472. doi: 10.1084/jem.183.4.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hochman PS, Majeau GR, Mackay F, Browning JL. Proinflammatory responses are efficiently induced by homotrimeric but not heterotrimeric lymphotoxin ligands. J Inflamm. 1996;46:220–234. [PubMed] [Google Scholar]
- 27.Picarella DE, Kratz A, Li C-B, Ruddle NH, Flavell RA. Insulitis in transgenic mice expressing TNF-β (lymphotoxin) in the pancreas. Proc Natl Acad Sci USA. 1992;89:10036–10040. doi: 10.1073/pnas.89.21.10036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paul NL, Ruddle NH. Lymphotoxin. Annu Rev Immunol. 1988;6:407–438. doi: 10.1146/annurev.iy.06.040188.002203. [DOI] [PubMed] [Google Scholar]
- 29.Picarella DE, Kratz A, Li C-B, Ruddle NH, Flavell RA. Transgenic TNF-α production in islets leads to insulitis, not diabetes: distinct patterns of inflammation in TNF-α and TNF-β transgenic mice. J Immunol. 1993;149:4136–4150. [PubMed] [Google Scholar]