

Abstract
B cells that mediate normal, T cell–dependent, humoral immune responses must first pass through germinal centers (GCs) within the cortex of antigenically stimulated lymph nodes. As they move through the dark zone and then the light zone in the GC, B cells are subjected to somatic hypermutation and switch recombination within their rearranged immunoglobulin genes and also participate in a number of other processes that control development into memory cells or cells specialized for antibody secretion. To investigate the molecular mechanisms that contribute to B cell development within GCs, we constructed a recombinant DNA library enriched for cDNAs derived from human genes expressed in B cells at this site. This library was found to contain a cDNA structurally and functionally related to genes in bacteria and yeast for the DNA repair enzyme 8-oxoguanine DNA glycosylase. Northern blot analysis indicated that the human gene is expressed as two alternatively spliced messenger RNAs within GC B cells at levels greatly exceeding that found in other tissues. In situ hybridization studies revealed that expression of this gene is most abundant within the dark zones of GCs. Both the function and localized expression of this gene suggest that it may play a role in somatic hypermutation of immunoglobulin genes.
Germinal centers (GCs)1 are sites of B cell development critical to the mounting of normal humoral immune responses (1–4). Anatomically, GCs are well-defined nodular structures that appear in the cortex of lymph nodes after stimulation by T cell–dependent antigens. Each GC contains about 104–105 cells, most of which are B cells, with smaller numbers of scattered T cells, follicular dentritic cells (DCs), and macrophages. Two zones can be distinguished in standard histologic sections of GCs, a dark zone of more densely packed cells located at one pole and a light zone occupying the remaining portion of the GC. Each GC is surrounded by a shell of homogeneously small, resting B cells, constituting a region referred to as the mantle zone, which is usually thickest at the point opposite the dark zone.
A variety of events that occur within GCs are required for the production of antibody with both high affinity for antigen and the proper effector functions necessary for the recognition and elimination of foreign substances in a range of biologic contexts. Naive B cells, which probably first encounter antigen in the interfollicular region of the lymph node cortex, enter the primary lymphoid follicles where they undergo rapid proliferation driven by antigen and cytokines secreted by helper T lymphocytes (5, 6). These B cells assume an altered morphology, with larger overall diameter and vesicular nuclei, and are termed centroblasts. Clonal expansion of these cells establishes the dark zone of the GC. During this phase of B cell development, point mutations accumulate within the DNA of variable (V) gene segments in rearranged immunoglobulin genes, a process referred to as somatic hypermutation (7–13). B cells with mutated V gene segments move out of the dark zone to an adjacent region that becomes the light zone, where the cells take on a morphology with a smaller cellular diameter and irregularly shaped, denser nuclei, in which state they are referred to as centrocytes. Within the light zone, B cells displaying surface immunoglobulin with higher affinity for antigen presented by follicular DCs are selected for further expansion, whereas cells bearing immunoglobulin that fails to bind antigen or does so with lower affinity die through a form of apoptosis and are scavenged by macrophages (14– 16). Together, these events lead to the so-called affinity maturation of the humoral immune response.
Other prominent events in B cell development probably occur only after transit to the light zone. For example, homologous recombination at specific switch regions within the DNA of immunoglobulin heavy chain genes, a process referred to as switch recombination, substitutes different coding sequences for the COOH-terminal part of the heavy chain (3, 5, 17). These sequences determine the class of antibody produced by the B lymphocyte and the ability of that antibody to fix complement, attach to mast cells, or efficiently cross through mucosal barriers. B cells that survive selection in the light zone cease dividing but continue to experience further maturational changes, such as conversion to long-lived memory cells or adaptation for antibody secretion (18), before they finally migrate out of the GC into surrounding areas of the lymph node, undergo further clonal expansion, and eventually enter the general circulation.
Although there has been some progress in recent years toward understanding certain aspects of events in the GC such as switch recombination and the determination of secretory versus memory cell fate, relatively little is known about the molecular mechanisms directly responsible for other processes such as lymphocyte trafficking within GCs and the signals controlling apoptosis of B cells producing low affinity or autoreactive antibody (17, 19–23). In particular, insights into the mutational activities underlying somatic hypermutation, the only known example in biology of a developmentally regulated and sequence-specific mutation, have remained elusive despite intense interest and considerable speculation about a host of hypothetical explanations (24–32). Because of the paucity of information available to explain the many important events that take place within GCs, we constructed a subtractive human cDNA library enriched for reverse transcripts representative of genes expressed in GCs of antigenically stimulated lymph nodes. We describe here two related cDNAs which were isolated from this library and have structural and functional features consistent with a role in DNA repair and possibly in somatic hypermutation.
Materials and Methods
Tissues.
Tonsils from children 2–6 yr of age were collected within 3 h of routine tonsillectomy at the Children's Hospital in Boston (Boston, MA). Fresh frozen lymph node tissues containing small lymphocytic and mantle cell lymphoma were selected from the archives of the Pathology Department of Brigham and Women's Hospital (Boston, MA). By histologic and immunohistochemical evaluation, over 95% of the cells in these tissues were estimated to be malignant. Fetal tissues were dissected from routine abortion specimens of ∼20 wk gestational age which showed no apparent gross abnormalities.
Isolation of GC B Cells from Tonsils.
Single cell suspensions were prepared from tonsils by crushing fragments of tissue between the frosted ends of two microscopic slides. The mononuclear cells were separated by loading the suspensions on top of 15 ml of Ficoll-Hypaque (Pharmacia, Piscataway, NJ) and centrifuging at 1,500 rpm for 30 min at 15°C. Cells at the interface were collected, washed in RPMI tissue culture medium (Sigma Chemical Co., St. Louis, MO) supplemented with 10% heat-inactivated fetal calf serum, and resuspended at a concentration of 107 cells/ml in fresh medium supplemented with serum. The suspensions of mononuclear cells were placed for 1 h at 4°C in 100-mm tissue culture dishes (10 ml/dish) coated with peanut agglutinin (PNA; 160 μg/ml in PBS; Sigma Chemical Co.) to further separate GC B cells used for preparation of cDNA.
In later studies, magnetic beads coated with specific antibodies were used for separations of B cells from disaggregated tonsils. T cells were first removed by two rounds of separation using a 1:10 cell/bead ratio of beads coated with anti-CD2 antibody (Dynal, Rochester, NY). The B cells left in suspension were then separated into surface IgD-positive and -negative populations by adding mouse anti–human IgD (1:20 dilution in PBS; Sigma Chemical Co.) plus magnetic beads coated with anti–mouse antibody (beads/cells ratio of 10:1) to cells at a concentration of 107 cells/ml. Flow cytometric analysis of separated cells showed that GC B cells prepared by panning were 75–80% pure (CD19+, CD38+, and surface IgD−) and by magnetic beads were >95%.
Construction of a cDNA Library Using Subtractive Hybridization.
Total RNA was isolated from GC B lymphocytes with Trizol solution (GIBCO BRL, Gaithersburg, MD) following procedures recommended by the manufacturer. PolyA+ RNA was prepared from total RNA with Oligotex (Qiagen, Chatsworth, CA). Conversion of RNA to double-stranded cDNA was performed starting with 5 μg of polyA+ RNA from GC B cells using the LambdaZAP-XR unidirectional cDNA synthesis kit (Stratagene, La Jolla, CA). The resulting cDNAs were then ligated into the DNA of LambdaZAP bacteriophage arms and packaged into phage particles using a Gigagold II packaging kit (Stratagene). The phage were then used to infect Escherichia coli XL1-blue (Stratagene). To generate a subtractive cDNA library, single-stranded DNA circles (the “tracer”) were produced from recombinant LambdaZAP bacteriophage by coinfection with the helper phage R408. Hybridization was performed with pooled, photobiotinylated RNA (the “driver”) isolated from two lymph nodes containing non-Hodgkin's lymphomas considered to be of non-GC origin. A total of 100 μg of nucleic acid at a tracer/driver molar ratio of 1:100 was used in two rounds of hybridization carried out in 50 μl of 50% formamide, 5% dextran sulfate, 5× SSC, 0.1% SDS, and 5× Denhardt's solution at 42°C for 36 h. After hybridization, RNA– DNA hybrids were removed by phenol–chloroform extraction. The remaining single-stranded circles were made double-stranded using the Klenow fragment of DNA polymerase and an M13 reverse primer, and then transformed into competent E. coli, which were grown on agar plates to produce a pBluescript plasmid library of cloned cDNAs enriched for reverse transcripts of genes expressed in GC B cells.
PCR.
With minor modifications, PCR amplifications were performed in 1× PCR buffer (Perkin-Elmer, Foster, CA) with 1.5 mM MgCl2, 10 pmol of each primer, 200 nM dNTPs, and 0.25 U of Taq polymerase (Perkin-Elmer) in a reaction volume of 50 μl. The cycling program consisted of 30 cycles of a three-temperature setup with denaturation at 94°C for 5 s, annealing at 65–55°C for 20 s, and extension at 72°C for 1 min, in a thermocycler (System 9600 PCR; Perkin-Elmer).
Reverse Slot–blot Hybridization and Northern Blot Analysis.
For reverse slot–blot hybridization, cDNAs were amplified by PCR using T3 and T7 primers complementary to pBluescript DNA flanking the insertion site, denatured by boiling in 1N NaOH, and applied to Biotran nylon membranes (Schleicher & Schuell, Keene, NH) in 2× SSC with a blotter (Minifold II; Schleicher & Schuell). The hybridization probes for this study were synthesized by reverse transcription of polyA+ RNA with oligo-dT primers and Superscript II reverse transcriptase (GIBCO BRL, Gaithersberg, MD). RNA from GC B cells was prepared using cells purified by separation with magnetic beads. Hybridization was performed in 5× SSC, 50% formamide, 5× Denhardt's solution, and 0.1% SDS at 42°C for 16 h. After hybridization, blots were washed twice at 65°C for 15 min in 0.5× SSC containing 0.1% SDS and subsequently analyzed by autoradiography against x-ray film.
For Northern blot analysis, total RNA was fractionated by electrophoresis through 1% agarose gels containing 1% (wt/vol) formamide using 1× MOPS running buffer and transferred after electrophoresis onto Biotran nylon membranes. Hybridization probes were prepared by randomly primed synthesis of DNA from individual cDNAs with α-32P-dCTP and the Klenow fragment of DNA polymerase. Hybridization and treatment of membranes was carried out as described for slot blots.
DNA Sequence Analysis.
Sequence analysis was performed by the method of Sanger et al., and the results evaluated on a SUN miniworkstation equipped with a Wisconsin DNA sequence analysis package (GCG release 7). Database searches were performed through the BLAST server at the National Center for Biotechnological Information.
In Situ Hybridization.
Amplified cDNA fragments were recloned in the pBluescipt plasmid vector, and after propagation in Escherichia coli, purified plasmid was linearized to serve as template for probe synthesis. Digoxigenin-labeled riboprobes were synthesized in the presence of digoxigenin-conjugated UTP with T3 and T7 RNA polymerase (Boehringer Mannheim, Indianapolis, IN) following the manufacturer's directions. Tissues were fixed in 10% buffered formalin solution, embedded in paraffin, cut into 5 μm sections, and mounted on glass microscope slides. The mounted sections were deparaffinized, rehydrated, and pretreated with 0.5% acetic anhydride. Hybridization was performed in a solution containing 50% formamide, 5× SSC, 0.1% SDS, 100 μg/ml sonicated salmon sperm DNA, 100 μg/ml yeast tRNA, and 5% dextran sulfate to which probe was added at a final concentration of 0.025 μg/ml. After incubation at 42°C overnight in a humidified chamber, the slides were washed twice at 55°C with 2× SSC, 0.5× SSC, and 0.1× SSC. The washed slides were preblocked with 2% goat serum, incubated with alkaline phosphatase–conjugated antidigoxigenin antibody, washed with PBS, and reacted with substrate solution containing 5-bromo,4-chloro, 3-indolyl phosphate (Sigma Chemical Co.) for 2–16 h at room temperature. The slides were counterstained with methyl green and examined under a standard light transmission microscope.
Production of a Fusion Protein Containing Amino Acid Sequence Encoded by the GCN6 cDNA.
An expression plasmid designed to express a fused protein containing the amino acid sequences of glutathione-S-transferase (GST) and those encoded by the cDNA GCN6 was made by ligating a BamHI–EcoRI fragment which includes all but the first 11 codons of the GCN6 open reading frame into the BamHI–EcoRI sites of the plasmid GST-2TK (Pharmacia). The MC301 mutM− E. coli strain (Yale Genetic Stock Center, New Haven, CT) was transformed with this plasmid, induced to express the composite cDNA by addition of isopropylthio-β-galactoside (IPTG; Sigma Chemical Co.), and the fusion protein purified by binding to and elution from glutathione–Sepharose beads (Pharmacia) according to instructions provided by the manufacturer.
Functional Assays of the GST-GCN6 Protein.
Oligonucleotides contained 8-oxoguanine, ordered from DNAgency (Malvern, PA), had the sequence 5′-CAGTCTCAAGTGTAA[8-oxo-G]TTACATGCACTGGTA-3′ and the reverse complement of this sequence with C inserted opposite the 8-oxo-G. The oligonucleotide was labeled at its 5′ end using T4 DNA kinase and γ-32P-dATP. Unincorporated free nucleotides were removed by centrifugation in a spin column, and the end-labeled oligonucleotides were annealed to their complementary oligonucleotides, precipitated, and resuspended in water. Trapping and cleavage experiments were performed with 1 pmol of oligonucleotide, 10 ng of GST-GCN6 fusion protein in 50 mM Hepes, pH 7.0, and 1 mM EDTA in the presence of either 50 mM freshly prepared NaBH4 (trapping reaction) or NaCl (cleavage reaction). Trapping reactions were incubated for 20 min at 30°C, after which 2× SDS loading buffer containing 1 mM dithiothreitol was added and the reactions boiled for 10 min before loading on a 10% SDS-PAGE gel prepared with standard electrophoresis buffer. Cleavage reactions were handled similarly except for addition of 6× loading buffer before electrophoresis performed on a 12% polyacrylamide-7M urea gel. After electrophoresis, the contents of the gels were detected by autoradiography against x-ray film.
Results
Construction and Screening of a cDNA Library Enriched for Gene Sequences Preferentially Expressed by GC B Cells.
A library of 8 × 105 independent recombinant phage clones containing cDNA inserts averaging 1.2 kb was obtained from five μg of GC B lymphocyte polyA+ RNA. Two rounds of hybridization of single-stranded DNA from the library with a 100-fold excess of RNA isolated from the lymphoma specimens, followed by removal of hybrids and conversion of residual DNA strands to double-stranded plasmids, reduced the number of cDNAs in the library to 740.
To screen the 740 clones in the subtracted library for cDNAs corresponding to genes upregulated in GCs, individual cDNAs immobilized in triplicate on nylon membranes were hybridized with labeled reverse transcription products prepared from the RNA of tonsils and the two lymphoma specimens used to construct the subtracted library. Those cDNAs that produced hybridization signals of about equal intensity with three sets of probes were regarded as unlikely to represent genes specifically involved in GC function and were therefore not further characterized. Those cDNAs that produced stronger signals with the tonsil probes than with either lymphoma probe, as well as cDNAs which gave uniformly weak signals with all probes (and presumably represent rare transcripts), were tested separately as probes on Northern blots prepared with the RNA of GC B cells and non-GC B cells isolated by magnetic beads separation, and with RNA from cells of various other lymphoid, fetal nonlymphoid tissues and several established B and T cell tumor lines.
Among the first 24 cDNAs used as probes, four showed some specificity for RNA in GC B cells. One of these, GCN5, detected two transcripts about 2.0 and 1.8 kb in size predominantly in GC B cells and tonsil, but to a much lesser extent in non-GC B cells, T cells, non–lymphoid human fetal tissues, or lymphoid tumor cell lines (Fig. 1, bands A and B). The larger of these transcripts (band B) is also present at low levels in the RNA from some of the non–lymphoid tissues; however, the smaller transcript (band A) is almost undetectable anywhere other than in the RNA of GC B cells and tonsil. Small amounts of a third transcript, ∼2.3 kb in size (band C), were also identified in RNA of liver, lung, and brain using the GCN5 cDNA as a probe.
Figure 1.

Northern blot analysis of GCN6 and GCN5 expression. 10 μg of total RNA were loaded in each lane. (A) Autoradiogram of the blot after hybridization with radiolabeled GCN5 cDNA. The three bands referred to in the text are labeled A, B, and C. (B) Photograph under ultraviolet light of the agarose gel used in preparation of the Northern blot after staining of the gel with ethidium bromide to indicate the amount and integrity of the RNA loaded on the gel.
Cloning and Structural Analysis of cDNAs Related to GCN5.
Since the size of the GCN5 cDNA in the plasmid clone studied by us was 1.1 kb and the three transcripts identified by Northern blot analysis were in the range of 2 kb, the GCN5 cDNA appeared to fall short of a full-length copy of any identifiable transcript. We therefore attempted to obtain additional cDNAs containing upstream sequences from the same transcript that gave rise to the GCN5 clone by using the 5′ end of the GCN5 cDNA to rescreen the unsubtracted GC B cell cDNA library. Several cDNAs were isolated by this procedure, of which the longest cDNA was termed GCN6. Sequence analysis of each cDNA found indicated that, aside from GCN5 or 6, all others were partial versions of these two.
The relationship between GCN5 and 6 is illustrated in Fig. 2. Amplification by PCR of DNA from the unsubtracted GC B cell cDNA library using a 5′ primer complementary to sequence in the middle of GCN6 (Fig. 2, S4) and a 3′ primer complementary to sequence in the first part of GCN5 (Fig. 2, A5) yielded a fragment of the size and sequence expected from a continuous stretch of DNA (data not shown). It therefore appears that GCN5 is an incomplete cDNA that shares a 5′ and coding sequence with GCN6. However, GCN5 and 6 differ from each other in exonic sequences at their 3′ ends, and consequently seem to represent alternatively spliced transcripts of the same gene. This interpretation was supported by the isolation of genomic DNA fragments containing these sequences and the finding within the genomic fragments of apparent intronic sequences adjacent to the exons present in the two cDNAs.
Figure 2.

Diagram of the GCN5 and GCN6 cDNAs. The labeled arrows above each cDNA represent PCR primers used to generate region-specific probes. The two striped regions represent coding exon in the calcium-calmodulin–dependent protein kinase I transcript.
A probe containing sequence common to GCN5 and 6 (the region from S5 to S plus S to A1 in Fig. 2) detected all three transcripts (A–C) identified in the Northern blot (data not shown), whereas a probe specific to GCN5 (the region between S2 and A2) detected only transcript C. Furthermore, a 3′ GCN6-specific probe (the region between S6 and A7) detected both transcript A and B, but not transcript C. We concluded that GCN5 corresponds to transcript C and GCN6 to both transcripts A or B.
Efforts to determine the structural differences between transcripts A and B were unsuccessful. A series of PCR primers constructed from sequences within GCN6 were used to amplify fragments from the GC B cell cDNA library. Amplification with each pair of primers produced only homogeneously sized fragments of lengths expected from the sequence of GCN6. Also, probes prepared from three different regions of the GCN6 (Fig. 2, S5-A3, S-A5, and S6-A7) used on Northern blot analysis all hybridized to the RNA of both bands A and B.
The GCN6 cDNA contains a 1,023-bp open reading frame capable of encoding a protein having a molecular weight of 37 kD. A BLAST homology search against a database of known protein sequences revealed that the putative protein encoded by GCN6 has significant homology to a recently reported yeast base excision DNA repair protein, 8-oxoguanine DNA glycosylase (Ogg)1. Fig. 3 shows a comparison of the GCN6 coding sequence and Ogg1. Several regions of the two sequences are strongly homologous, including one toward the COOH-terminal end of the GCN6 open reading frame (codons 235–310), corresponding to a part of Ogg1 proposed to contain the active site of the enzyme. Within this region, the aspartic acid codon at position 269 of the GCN6 sequence is conserved among many of the DNA glycosylases and is thought to encode the amino acid that acts as the nucleophile during the enzymatic reaction. For as far as it extends, the coding sequence in GCN5 is identical to that in GCN6 before position 307 in the latter, after which GCN5 diverges completely and continues for another 66 codons. Examination of this region of the sequence in the GCN5 cDNA did not reveal significant homology to any protein in the database.
Figure 3.
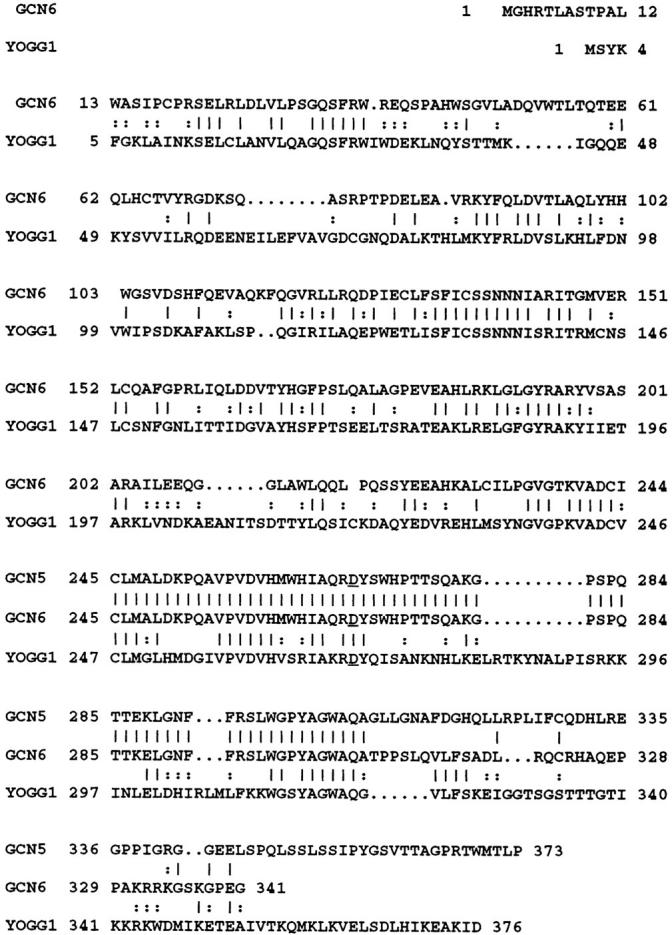
Comparison of amino acid sequences of GCN5, GCN6, and yeast Ogg1 (YOGGI). The alignment was performed by the Bestfit program in the Wisconsin GCG package. Identical amino acids are marked by a vertical line between amino acids in the respective sequences and conserved changes by a colon. Dots represent gaps in the sequence generated by the program to allow better alignment. The overall identity is calculated to be 37%. The GCN5 is a partial cDNA that begins at codon 215 in GCN6. The aspartic acid conserved in the presumed active site among DNA glycosylases is underlined. The DNA sequences reported in the paper are available from EMBL/Genbank/DDBJ under accession number AF 026691.
An unusual feature of both the GCN5 and 6 sequences is the inclusion of short regions of inverted exonic sequence from the calcium-calmodulin–dependent protein kinase I gene (CCDPKI). This finding indicates that the GCN5/6 gene probably overlaps the CCDPKI gene in a reverse orientation within the DNA. Probes containing several regions of the GCN5 and 6 cDNAs hybridize to only a single band in Southern blots of human genomic DNA digested with a number of restriction enzymes (data not shown), suggesting that the gene for GCN5/6 is located at the same chromosomal site as CCDPKI, at band 3p25. The Southern blot results are also consistent with the conclusion that GCN5 and 6 are derived from the same gene.
In Situ Hybridization Analysis of the Expression of GCN6 DNA in Tonsils.
To analyze the distribution of expression of GCN6 DNA in lymph node tissue, in situ hybridization was performed with digoxigenin-labeled sense and antisense riboprobes generated from the 3′ end of GCN6 cDNA (Fig. 2, the region between S6 and A7). As seen in Fig. 4, C and D, hybridization with a GCN6-specific antisense probe was largely localized to the cells in the dark zone of GCs present within sections of tonsil. Examination under high magnification revealed that the cells showing the strongest hybridization had a nuclear morphology compatible with centroblasts. Weaker hybridization was seen in the light zone of GCs, but not in the B lymphocytes making up the mantle zone. In some sections, scattered large cells both outside the GC and between the light and dark zone of the GC displayed intense hybridization. The nature of these cells is not clear.
Figure 4.
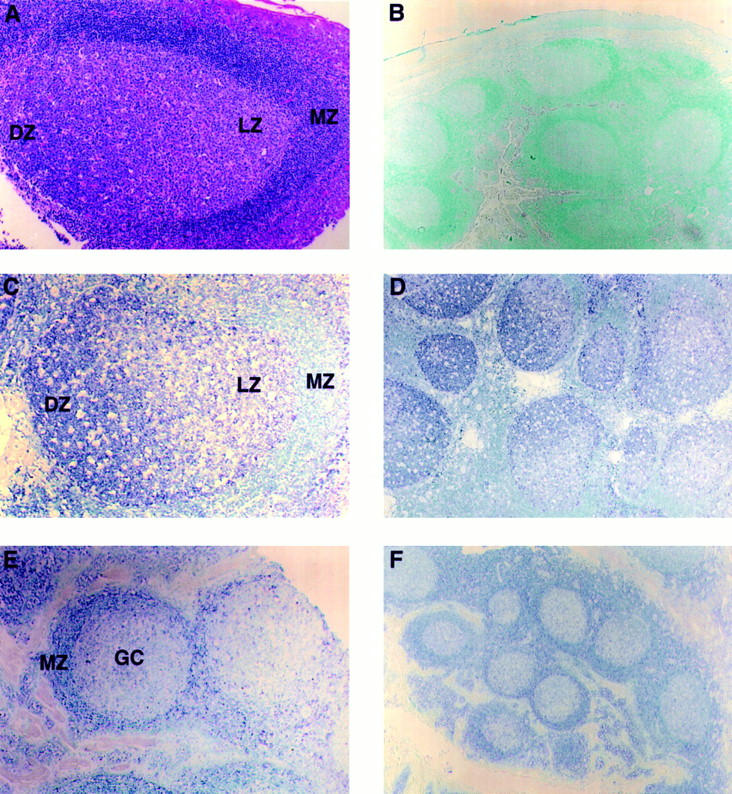
In situ hybridization on paraffin-embedded formalin-fixed sections of human tonsils. (A)A hematoxylin and eosin–stained section showing the dark (DZ), light (LZ), and mantle (MZ) zones of a GC. (B–D) Sections hybridized with GCN6 cDNA into which digoxigenin nucleotides have been incorporated, followed by detection with antidigoxigenin antibody conjugated to alkaline phosphatase and identification of the locations of bound antibody by deposition of dark blue phosphatase reaction product. The nuclei were counterstained with methyl green. (B) Sense strand probe of GCN6. (C and D) Antisense probe of GCN6. (E and F) Antisense probe of GCN7, a cDNA derived from a gene apparently expressed primarily in mantle cells. Original magnification was 40 for B, D, and F and 100 for A, C, and E.
As a control, the sense GCN6 probe uniformly produced only faint background hybridization in all regions of the tonsil (Fig. 4 B). Also, hybridization specific for regions of the tonsil other than GCs was obtained with probes for certain cDNAs that emerged from the subtractive hybridization cloning scheme (Fig. 4, E and F).
Functional Analysis of the GST-GCN6 Fusion Protein.
The yeast Ogg1 protein has been shown to possess an 8-oxoguanine DNA glycosylase activity (33). The glycosylase reaction was demonstrated to proceed through a Schiff base intermediate formed between the enzyme and the DNA at the site of the modified guanine. Furthermore, this intermediate could be trapped by addition of sodium borohydride (NaBH4) to the reaction (34). Given these properties of the yeast enzyme, we tested the ability of the protein encoded by the GCN6 cDNA to form in vitro the same intermediate as the yeast enzyme. A cDNA fusion construct containing all but the first 11 codons of the GCN6 coding sequence fused to sequence encoding GST was expressed in a strain of E. coli mutant for MutM (MC301), the gene for 8-oxoguanine DNA glycosylase in bacteria. Purified GST-GCN6 fusion protein was then incubated under a variety of conditions with end-labeled, double-stranded oligonucleotides with or without an oxidized guanine in one or the other strand at the center of the sequence. As shown in Fig. 5 A, a band representing a covalently linked protein– DNA complex was found only when oxidized guanine was included in the oligonucleotide substrate (Fig. 5 A, lanes 5 and 6) and only in the presence of NaBH4 (Fig. 5 A, lanes 3 and 4). The control GST protein alone produced no complex (Fig. 5 A, lanes 1 and 2). Additionally, the GST-GCN6 fusion protein caused nicking of the oligonucleotides at the position of the oxidized guanine in a manner similar to that observed with the yeast protein (Fig. 5 B).
Figure 5.

Functional assays of the GST-GCN6 fusion protein. (A) Trapping of the Schiff base intermediate by the addition of sodium borohydride. Double-stranded, 3′ end–labeled oligonucleotides either with (oG:C, lanes 1–4) or without (G:C, lanes 5 and 6) an 8-oxoguanine at the center of the fragment were incubated with either with GST alone (lanes 1 and 2) or GST-GCN6 fusion protein (lanes 3–6) in the presence (lanes 2, 4, and 6) or absence (lanes 1, 3, and 5) of sodium borohydride. The reaction products were analyzed by 8% SDS-PAGE gel electrophoresis followed by autoradiography. The presence of a protein–DNA conjugate can be detected as a slow mobility band indicated by the arrow. (B) Cleavage of double-stranded oligonucleotides containing 8-oxoguanine. Double-stranded, 3′ end–labeled oligonucleotides with or without an 8-oxoguanine were incubated with GST alone (lanes 1 and 5) or GST-GCN6 fusion protein (lanes 3 and 6), and the reaction products analyzed by electrophoresis in a 12% polyacrylamide-7M urea gel followed by autoradiography.
Discussion
Our decision to use subtractive hybridization for the identification of genes involved in B lymphocyte functions within GCs was based on several key considerations. Many events essential to the normal humoral immune response occur within the GC, but relatively little is known about the molecular mechanisms or genes controlling these events. Although subtractive hybridization of non-GC transcripts from GC transcripts would not define a set of RNAs with discrete functions, we felt that any RNA that we could demonstrate was derived from a gene that showed significantly increased expression in GCs would likely be relevant to one or the other important process within these highly active structures. Although a priori, we had no simple assay to test for any specific function of genes we might identify through construction of a subtractive GC cDNA library, we believed we could probably infer functional information by searching for sequence homologies between a cDNA within the library and entries within the ever-expanding databases of gene sequences from various organisms. We chose a cloning strategy involving subtractive hybridization over competing techniques, such as a candidate gene approach (35) or differential display (36), because we wished to survey both known and previously undiscovered genes and because we did not want to exclude from our study genes expressed as low abundance transcripts, which may happen more easily using methods that rely on PCR.
A major factor in any successful application of subtractive hybridization to cloning of cDNAs is the quality of the positive and negative starting materials. The positive starting material in our study, B lymphocytes isolated from the GCs of tonsils by antigenic selection, provided small but adequate amounts of RNA to begin the procedure. However, these isolated populations of B lymphocytes were not pure, as indicated by flow cytometric analysis of the cells, the difficulty in quantitating gene expression in slot blots prepared with RNA from the isolated cell populations, and the inclusion in the subtracted library of cDNA clones representing genes expressed in regions of the lymph node outside the GC (for example, in the mantle zone; see Fig. 4, E and F). The impurity of the GC B lymphocytes probably also accounts in part for the unexpectedly large size of the subtractive library we obtained.
Pooled lymph nodes containing non–Hodgkin's lymphomas were used as the source of negative cells because the two subtypes of lymphomas present in these specimens are known to be composed of neoplastic B lymphocytes that do not undergo somatic hypermutation or switch recombination (37–39). More generally, B lymphocytes in the two subtypes of lymphoma mimic the stages of normal B cell development preceding passage through the GC and might therefore be more likely to express genes involved in a range of B cell functions exclusive of those that take place only in the GC. Furthermore, the large amounts of fresh lymphoma tissue available to us provided a convenient supply of relatively homogeneous, non-GC B lymphocytes for the preparation of the abundant amounts of RNA required in the subtraction step. Finally, the cells within these tumors are actively dividing, and accordingly, subtraction with RNA extracted from them might be anticipated to remove cDNA clones related only to cell proliferation per se. On the other hand, the tissues containing the two lymphomas are undoubtedly contaminated by nonmalignant B lymphocytes, some of which may have been present in residual GCs. Consequently, it is possible that cDNAs for some rare GC transcripts may have been eliminated from the library by subtraction with RNA from these lymphoma specimens.
Despite all the potential theoretical and practical problems associated with subtractive hybridization, the ultimate justification for the strategy we pursued is the finding within our library of cDNAs that clearly correspond to genes upregulated within GCs. In this report, we have described in detail two such cDNAs, GCN6 and GCN5, which originate from a gene that is over-expressed in GC B lymphocytes relative to other cells and tissues. The two cDNAs apparently represent alternatively spliced transcripts of the same gene, but only the sequences contained within GCN6 are present at increased levels in the RNA of GC B lymphocytes by blot or in situ hybridization. Hybridization of Northern blots with probes from the GCN6 sequence detected, in turn, two transcripts, A and B. The structural differences between these two transcripts is not apparent based on any of the analyses performed by us, but the shorter transcript (A) is virtually limited to GC B lymphocytes in our investigations so far, although the level of the longer transcript B is increased in GC B lymphocytes as well.
Our analyses indicate that the polypeptide encoded by the GCN6 cDNA is both structurally and functionally homologous to Oggs found in lower organisms. In E. coli, this enzyme, encoded by the MutM gene, together with the products of MutY and MutT, comprise the GO repair system (40), which protects against the mutagenic effects of oxidatively damaged guanosine nucleotides. The human homologues of the MutY and MutT genes, and their protein products have been identified and characterized biochemically (41–44). Both perform functions similar to their bacterial counterparts (MutY: excision of adenine incorporated opposite 8-oxoguanine; MutT: removal of 8-oxo-dGTP from the deoxynucleoside triphosphate pool). Enzymatic activity similar to that of MutM in E. coli has also been identified in Hela cell extracts (45, 46). The ability of the polypeptide encoded by GCN6 to bind in vitro to oligonucleotides containing 8-oxoguanine and cause nicking of DNA at this site is compatible with the properties of both the E. coli enzyme and the activity in Hela cell extracts.
Oxidation of guanine has been proposed to be the most important lesion of DNA leading to spontaneous mutations seen in somatic cells (47–49). The almost certain requirement by all cells for a means to repair this lesion probably explains the low amounts of GCN6 transcripts B and C found in most tissues. The abundance of transcript B and the appearance of large amounts of the distinct transcript A in tissues containing GCs would seem to imply some special function or need for Ogg at these sites. The high concentration of GCN6 RNA in the dark zone of the GC, as demonstrated by in situ hybridization, suggests that the reason for changes in expression of the GCN5/6 gene relate to processes specifically associated with this region of the GC. The two most plausible candidates for these processes are cell proliferation and somatic hypermutation, although, of course, other processes yet to be described in the dark zone could also account for the upregulation of this gene. Since GCN5/6 transcripts were not detected in increased numbers either in rapidly dividing cells of fetal tissues or lymphoid tumor cell lines (Fig. 1), altered expression of the GCN5/6 gene probably does not serve a role connected solely to proliferation. Therefore, based only on those processes known to occur within the dark zone of the GC, we surmise that the GCN5/6 gene is most likely somehow involved in somatic hypermutation.
In view of the function of Ogg in DNA repair, the increased expression of the GCN5/6 gene at a site of enhanced mutation initially seems paradoxical. A variety of explanations for this paradox is possible, but the most cogent one may be the following. Oxidative damage to DNA within cells of the dark zone of the GC occurs at an unusually high rate, due perhaps in part to large amounts of reactive oxygen free radicals released from macrophages during the phagocytosis of cell debris generated by the exuberant apoptosis that takes place in the GC. This damage affects the entire genome, requiring increased levels of Ogg. At the same time, the V segments of rearranged immunoglobulin genes within B cells of the dark zone are protected from repair, possibly by a protein that shields DNA at these loci from Ogg action. In this regard, it may be noteworthy that in one B cell line reported to show some degree of somatic hypermutation, the base substitutions affect G:C more frequently than other basepairs within the immunoglobulin DNA (50). Furthermore, experiments have indicated that after treatment with ultraviolet light, the DNA of rearranged V gene segments in GC B cells are repaired less efficiently than other regions of immunoglobulin genes or similar segments of non-GC B cells (51). In any case, besides conforming with observations previously reported on somatic hypermutation (8, 52, 53), the above model makes a number of specific predictions which should be testable in the near future.
Acknowledgments
We thank Dr. Deborah Schofield for assistance in acquisition of human tonsils and Dr. Geraldine Pinkus for consultation and assistance with the lymphoma specimens.
This work was supported by National Institutes of Health grants CA 38621 and K08-AI01375.
Footnotes
Abbreviations used in this paper: CCDPKI, calcium-calmodulin–dependent protein kinase I gene; GC, germinal center; GST, glutathione-S-transferase; Ogg, 8-oxoguanine DNA glycosylase; V, variable.
References
- 1.Kelsoe G. The germinal center reaction. Immunol Today. 1995;16:324–326. doi: 10.1016/0167-5699(95)80146-4. [DOI] [PubMed] [Google Scholar]
- 2.Maclennan ICM. Germinal centers. Annu Rev Immunol. 1994;112:117–139. doi: 10.1146/annurev.iy.12.040194.001001. [DOI] [PubMed] [Google Scholar]
- 3.Tsiagbe VK, Inghirami G, Thorbecke GJ. The physiology of germinal centers. Crit Rev Immunol. 1996;16:381–421. [PubMed] [Google Scholar]
- 4.Thorbecke GJ, Amin AR, Tsiagbe VK. Biology of germinal centers in lymphoid tissue. FASEB J. 1994;8:832–840. doi: 10.1096/fasebj.8.11.8070632. [DOI] [PubMed] [Google Scholar]
- 5.Brandtzaeg P. The B-cell development in tonsillar lymphoid follicles. Acta Oto-Laryngologica - Suppl. 1996;523:55–59. [PubMed] [Google Scholar]
- 6.Itoh K, Hirohata S. The role of Il-10 in human B cell activation, proliferation, and differentiation. J Immunol. 1995;154:4341–4350. [PubMed] [Google Scholar]
- 7.Kuppers R, Zhao M, Hansmann ML, Rajewsky K. Tracing B cell development in human germinal centres by molecular analysis of single cells picked from histological sections. EMBO (Eur Mol Biol Org) J. 1993;12:4955–4967. doi: 10.1002/j.1460-2075.1993.tb06189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neuberger MS, Milstein C. Somatic hypermutation. Curr Opin Immunol. 1995;7:248–254. doi: 10.1016/0952-7915(95)80010-7. [DOI] [PubMed] [Google Scholar]
- 9.Wagner SD, Neuberger MS. Somatic hypermutation of immunoglobulin genes. Annu Rev Immunol. 1996;14:441–457. doi: 10.1146/annurev.immunol.14.1.441. [DOI] [PubMed] [Google Scholar]
- 10.French DL, Laskov R, Scharff MD. The role of somatic hypermutation in the generation of antibody diversity. Science (Wash DC) 1989;244:1152–1157. doi: 10.1126/science.2658060. [DOI] [PubMed] [Google Scholar]
- 11.Jacob J, Kelsoe G, Rajewsky K, Weiss U. Intraclonal generation of antibody mutants in germinal centres. Nature (Lond) 1991;354:389–392. doi: 10.1038/354389a0. [DOI] [PubMed] [Google Scholar]
- 12.Jacob J, Miller C, Kelsoe G. In situ studies of the antigen-driven somatic hypermutation of immunoglobulin genes. Immunol Cell Biol. 1992;70:145–152. doi: 10.1038/icb.1992.19. [DOI] [PubMed] [Google Scholar]
- 13.Kim S, Davis M, Sinn E, Patten P, Hood L. Antibody diversity: somatic hypermutation of rearranged VH genes. Cell. 1981;27:573–581. doi: 10.1016/0092-8674(81)90399-8. [DOI] [PubMed] [Google Scholar]
- 14.Nakamura M, Yagi H, Kayaba S, Ishii T, Gotoh T, Ohtsu S, Itoh T. Death of germinal center B cells without DNA fragmentation. Eur J Immunol. 1996;26:1211–1216. doi: 10.1002/eji.1830260604. [DOI] [PubMed] [Google Scholar]
- 15.Choe JS, Kim HS, Zhang XH, Armitage RJ, Choi YS. Cellular and molecular factors that regulate the differentiation and apoptosis of germinal center B cells–anti-Ig down-regulates fas expression on Cd40 ligand–stimulated germinal center B cells and inhibits fas-mediated apoptosis. J Immunol. 1996;157:1006–1016. [PubMed] [Google Scholar]
- 16.Billian G, Mondiere P, Berard M, Bella C, Defrance T. Antigen receptor–induced apoptosis of human germinal center B cells is targeted to a centrocytic subset. Eur J Immunol. 1997;27:405–414. doi: 10.1002/eji.1830270210. [DOI] [PubMed] [Google Scholar]
- 17.Hengstschlager M, Maizels N, Leung H. Targeting and regulation of immunoglobulin gene somatic hypermutation and isotype switch recombination. Prog Nucl Acid Res Mol Biol. 1995;50:67–99. doi: 10.1016/s0079-6603(08)60811-9. [DOI] [PubMed] [Google Scholar]
- 18.George J, Claflin L. Selection of B cell clones and memory B cells. Sem Immunol. 1992;4:11–17. [PubMed] [Google Scholar]
- 19.Arpin C, Dechanet J, Vankooten C, Merville P, Grouard G, Briere F, Banchereau J, Liu YJ. Generation of memory B cells and plasma cells in vitro. Science (Wash DC) 1995;268:720–722. doi: 10.1126/science.7537388. [DOI] [PubMed] [Google Scholar]
- 20.Goldfarb AN, Flores JP, Lewandowska K. Involvement of the E2a basic helix-loop-helix protein in immunoglobulin heavy chain class switching. Mol Immunol. 1996;33:947–956. doi: 10.1016/s0161-5890(96)00047-8. [DOI] [PubMed] [Google Scholar]
- 21.Cleary AM, Fortune SM, Yellin MJ, Chess L, Lederman S. Opposing roles of Cd95 (Fas/Apo-1) and Cd40 in the death and rescue of human low density tonsillar B cells. J Immunol. 1995;155:3329–3337. [PubMed] [Google Scholar]
- 22.Lederman S, Yellin MJ, Cleary AM, Pernis A, Inghirami G, Cohn LE, Covey LR, Lee JJ, Rothman P, Chess L. T-Bam/Cd40-L on helper T lymphocytes augments lymphokine-induced B cell Ig isotype switch recombination and rescues B cells from programmed cell death. J Immunol. 1994;152:2163–2171. [PubMed] [Google Scholar]
- 23.Li MJ, Peakman MC, Golub EI, Reddy G, Ward DC, Radding CM, Maizels N. Rad51 expression and localization in B cells carrying out class switch recombination. Proc Natl Acad Sci USA. 1996;93:10222–10227. doi: 10.1073/pnas.93.19.10222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kepler TB, Perelson AS. Modeling and optimization of populations subject to time-dependent mutation. Proc Natl Acad Sci USA. 1995;92:8219–8223. doi: 10.1073/pnas.92.18.8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kepler TB, Perelson AS. Cyclic re-entry of germinal center B cells and the efficiency of affinity maturation. Immunol Today. 1993;14:412–415. doi: 10.1016/0167-5699(93)90145-B. [DOI] [PubMed] [Google Scholar]
- 26.Klotz EL, Storb U. Somatic hypermutation of a lambda 2 transgene under the control of the lambda enhancer or the heavy chain intron enhancer. J Immunol. 1996;157:4458–4463. [PubMed] [Google Scholar]
- 27.Storb U, Peters A, Klotz E, Rogerson B, Hackett J., Jr The mechanism of somatic hypermutation studied with transgenic and transfected target genes. Sem Immunol. 1996;8:131–140. doi: 10.1006/smim.1996.0017. [DOI] [PubMed] [Google Scholar]
- 28.Knight KL, Becker RS. Molecular basis of the allelic inheritance of rabbit immunoglobulin VH allotypes: implications for the generation of antibody diversity. Cell. 1990;60:963–970. doi: 10.1016/0092-8674(90)90344-e. [DOI] [PubMed] [Google Scholar]
- 29.Maizels N. Might gene conversion be the mechanism of somatic hypermutation of mammalian immunoglobulin genes? TIG ( Trends Genet) 1989;5:4–8. doi: 10.1016/0168-9525(89)90004-8. [DOI] [PubMed] [Google Scholar]
- 30.Maizels N. Somatic hypermutation: how many mechanisms diversify V region sequences? . Cell. 1995;83:9–12. doi: 10.1016/0092-8674(95)90227-9. [DOI] [PubMed] [Google Scholar]
- 31.Peters A, Storb U. Somatic hypermutation of immunoglobulin genes is linked to transcription initiation. Immunity. 1996;4:57–65. doi: 10.1016/s1074-7613(00)80298-8. [DOI] [PubMed] [Google Scholar]
- 32.Steele EJ, Pollard JW. Hypothesis: somatic hypermutation by gene conversion via the error prone DNA— RNA—DNA information loop. Mol Immunol. 1987;24:667–673. doi: 10.1016/0161-5890(87)90049-6. [DOI] [PubMed] [Google Scholar]
- 33.van der Kemp PA, Thomas D, Barbey R, De Oliveira R, Boiteux S. Cloning and expression in Escherichia coli of the OGG1 gene of Saccharomyces cerevisiae, which codes for a DNA glycosylase that excises 7,8-dihydro-8-oxoguanine and 2,6-diamino-4-hydroxy-5-N-methylformamidopyrimidine. Proc Natl Acad Sci USA. 1996;93:5197–5202. doi: 10.1073/pnas.93.11.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nash HM, Bruner SD, Scharer OD, Kawate T, Addona TA, Spooner E, Lane WS, Verdine GL. Cloning of a yeast 8-oxoguanine DNA glycosylase reveals the existence of a base-excision DNA-repair protein superfamily. Curr Biol. 1996;6:968–980. doi: 10.1016/s0960-9822(02)00641-3. [DOI] [PubMed] [Google Scholar]
- 35.Dryja TP. Human genetics. Deficiencies in sight with the candidate gene approach. Nature (Lond) 1990;347:614. doi: 10.1038/347614a0. [DOI] [PubMed] [Google Scholar]
- 36.Liang P, Pardee AB. Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science (Wash DC) 1992;257:967–971. doi: 10.1126/science.1354393. [DOI] [PubMed] [Google Scholar]
- 37.Meeker TC, Grimaldi JC, O'Rourke R, Loeb J, Juliusson G, Einhorn S. Lack of detectable somatic hypermutation in the V region of the Ig H chain gene of a human chronic B lymphocytic leukemia. J Immunol. 1988;141:3994–3998. [PubMed] [Google Scholar]
- 38.Pratt LF, Rassenti L, Larrick J, Robbins B, Banks PM, Kipps TJ. Ig V region gene expression in small lymphocytic lymphoma with little or no somatic hypermutation. J Immunol. 1989;143:699–705. [PubMed] [Google Scholar]
- 39.Ohno T, Oka K, Yamaguchi M, Kita K, Shirakawa S. Frequent expression of shared idiotypes in mantle cell lymphoma and extranodal small lymphocytic/non-mantle cell diffuse small cleaved lymphoma. Leukemia. 1995;9:1935–1939. [PubMed] [Google Scholar]
- 40.Michaels ML, Miller JH. The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine) J Bacteriol. 1992;174:6321–6325. doi: 10.1128/jb.174.20.6321-6325.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cai JP, Kakuma T, Tsuzuki T, Sekiguchi M. cDNA and genomic sequences for rat 8-oxo-dGTPase that prevents occurrence of spontaneous mutations due to oxidation of guanine nucleotides. Carcinogenesis. 1995;16:2343–2350. doi: 10.1093/carcin/16.10.2343. [DOI] [PubMed] [Google Scholar]
- 42.Sakumi K, Furuichi M, Tsuzuki T, Kakuma T, Kawabata S, Maki H, Sekiguchi M. Cloning and expression of cDNA for a human enzyme that hydrolyzes 8-oxo-dGTP, a mutagenic substrate for DNA synthesis. J Biol Chem. 1993;268:23524–23530. [PubMed] [Google Scholar]
- 43.McGoldrick JP, Yeh YC, Solomon M, Essigmann JM, Lu AL. Characterization of a mammalian homolog of the Escherichia coliMutY mismatch repair protein. Mol Cell Biol. 1995;15:989–996. doi: 10.1128/mcb.15.2.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Slupska MM, Baikalov C, Luther WM, Chiang JH, Wei YF, Miller JH. Cloning and sequencing a human homolog (hMYH) of the Escherichia coliMutY gene whose function is required for the repair of oxidative DNA damage. J Bacteriol. 1996;178:3885–3892. doi: 10.1128/jb.178.13.3885-3892.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bessho T, Tano K, Kasai H, Ohtsuka E, Nishimura S. Evidence for two DNA repair enzymes for 8-hydroxyguanine (7,8-dihydro-8-oxoguanine) in human cells. J Biol Chem. 1993;268:19416–19421. [PubMed] [Google Scholar]
- 46.Nagashima M, Sasaki A, Morishita K, Takenoshita S, Nagamachi Y, Kasai H, Yokota J. Presence of human cellular protein(s) that specifically binds and cleaves 8-hydroxyguanine containing DNA. Mut Res. 1997;383:49–59. doi: 10.1016/s0921-8777(96)00045-6. [DOI] [PubMed] [Google Scholar]
- 47.Le Page F, Margot A, Grollman AP, Sarasin A, Gentil A. Mutagenicity of a unique 8-oxoguanine in a human Ha-ras sequence in mammalian cells. Carcinogenesis. 1995;16:2779–2784. doi: 10.1093/carcin/16.11.2779. [DOI] [PubMed] [Google Scholar]
- 48.Kamiya H, Murata-Kamiya N, Koizume S, Inoue H, Nishimura S, Ohtsuka E. 8-hydroxyguanine (7,8-dihydro-8-oxoguanine) in hot spots of the c-Ha-ras gene: effects of sequence contexts on mutation spectra. Carcinogenesis. 1995;16:883–889. doi: 10.1093/carcin/16.4.883. [DOI] [PubMed] [Google Scholar]
- 49.Emmert S, Epe B, Saha-Moller CR, Adam W, Runger TM. Assessment of genotoxicity and mutagenicity of 1,2-dioxetanes in human cells using a plasmid shuttle vector. Photochem Photobiol. 1995;61:136–141. doi: 10.1111/j.1751-1097.1995.tb03951.x. [DOI] [PubMed] [Google Scholar]
- 50.Bachl J, Wabl M. An immunoglobulin mutator that targets G.C. base pairs. Proc Natl Acad Sci USA. 1996;93:851–855. doi: 10.1073/pnas.93.2.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fairhurst RM, Vallesayoub Y, Neshat M, Braun J. A DNA repair abnormality specific for rearranged immunoglobulin variable genes in germinal center B cells. Mol Immunol. 1996;33:231–244. doi: 10.1016/0161-5890(95)00145-x. [DOI] [PubMed] [Google Scholar]
- 52.Betz AG, Rada C, Pannell R, Milstein C, Neuberger MS. Passenger transgenes reveal intrinsic specificity of the antibody hypermutation mechanism: clustering, polarity, and specific hot spots. Proc Natl Acad Sci USA. 1993;90:2385–2388. doi: 10.1073/pnas.90.6.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yelamos J, Klix N, Goyenechea B, Lozano F, Chui YL, Gonzalez A, Fernandez, Pannell R, Neuberger MS, Milstein C. Targeting of non-Ig sequences in place of the V segment by somatic hypermutation. Nature (Lond) 1995;376:225–229. doi: 10.1038/376225a0. [DOI] [PubMed] [Google Scholar]