

Abstract
The SBM mouse is a unique transgenic model of polycystic kidney disease (PKD) induced by the dysregulated expression of c-myc in renal tissue. In situ hybridization analysis demonstrated intense signal for the c-myc transgene overlying tubular cystic epithelium in SBM mice. Renal proliferation index in SBM kidneys was 10-fold increased over nontransgenic controls correlating with the presence of epithelial hyperplasia. The specificity of c-myc for the proliferative potential of epithelial cells was demonstrated by substitution of c-myc with the proto-oncogene c-fos or the transforming growth factor (TGF)-α within the same construct. No renal abnormalities were detected in 13 transgenic lines established, indicating that the PKD phenotype is dependent on functions specific to c-myc. We also investigated another well characterized function of c-myc, the regulation of apoptosis through pathways involving p53 and members of the bcl-2 family, which induce and inhibit apoptosis, respectively. The SBM kidney tissues, which overexpress c-myc, displayed a markedly elevated (10–100-fold) apoptotic index. However, no significant difference in bcl-2, bax, or p53 expression was observed in SBM kidney compared with controls. Direct proof that the heightened renal cellular apoptosis in PKD is not occurring through p53 was obtained by successive matings between SBM and p53−/− mice. All SBM offspring, irrespective of their p53 genotype, developed PKD with increased renal epithelial apoptotic index. In addition, overexpression of both bcl-2 and c-myc in double transgenic mice (SBB+/SBM+) also produced a similar PKD phenotype with a high apoptotic rate, showing that c-myc can bypass bcl-2 in vivo. Thus, the in vivo c-myc apoptotic pathway in SBM mice occurs through a p53- and bcl-2–independent mechanism. We conclude that the pathogenesis of PKD is c-myc specific and involves a critical imbalance between the opposing processes of cell proliferation and apoptosis.
Polycystic kidney disease (PKD)1 is one of the most prevalent and clinically important inherited renal diseases. Studies of genetic animal models have provided a valuable in vivo system to investigate molecular and cellular mechanisms of cystogenesis. We have generated the SBM transgenic model which most closely resembles the human autosomal dominant PKD renal phenotype (35). The SBM transgene specifically targets c-myc to the renal tissue and was fully penetrant for the cystic phenotype in all 18 transgenic lines produced. The fact that all 18 transgenic lines developed PKD indicate that the site of transgene insertion is not critical to the development of the renal phenotype. The occurrence of spontaneous revertants in several different transgenic lines due to mutations in the transgene proves that the intact transgene is required for the PKD phenotype (34).
The course of PKD in the SBM mice evolves in a predictable manner with earliest detectable cyst formation in late fetal development (E16.5), progressive multicystic renal enlargement and inevitable development of renal failure in young adulthood. Cysts are first detectable in glomeruli and collecting tubules with later involvement of the more proximal nephron (7). Tubular epithelial hyperplasia is an integral feature of this model, often accompanied by microadenomas. Our studies into epithelial polarity have shown that the cyst lining cells frequently display reversed polarity similar to that of immature renal epithelium during nephrogenesis (2).
Although the physiological roles of the c-myc protein are poorly understood, c-myc is known to be involved in cell proliferation, apoptosis, differentiation and neoplasia. The critical role of the c-myc proto-oncogene in murine development is underscored by the severe intrauterine growth retardation of c-myc-null mice which fail to survive beyond E10.5 (8). Because these mice die (E9.5-E10.5) before renal metanephric development is initiated, these studies do not elucidate whether c-myc is essential for renal development. However, c-myc has been shown to be particularly highly expressed in tissues of mesodermal origin such as the kidney (29, 32). During renal organogenesis, c-myc is highly expressed in noninduced metanephric mesenchyme as well as in developing tubules of the mesonephros and metanephros (29). Upon metanephric induction and development, the high prevalence of cellular proliferation and apoptosis coincides with c-myc expression (5, 15). As fetal nephrogenesis progresses, there is continual decrease of c-myc expression to undetectable levels at birth (41). In contrast, in our SBM model, c-myc is not downregulated at birth but remains continually expressed in renal tissue. Moreover, the renal epithelium has an immature phenotype with respect to cellular hyperplasia and polarity, findings which correlate with previous observations on the reciprocal relationship between cell division and terminal differentiation.
Because of the compelling evidence that c-myc is a major mediator of cystogenesis, we set out to determine the specificity of c-myc for this phenotype. We approached this question by producing novel transgenic lines which target two different genes, c-fos and TGF-α, to the kidney. Moreover, we sought to investigate the roles of c-myc– driven proliferation and apoptosis and the relative contributions of potential regulators bcl-2, bax and p53 to decipher the underlying in vivo cellular mechanisms and molecular pathways that mediate PKD.
Materials and Methods
Production of Transgenic Mice
The SBM fusion construct used to produce the SBM transgenic mouse lines has been described previously (35). In brief, this construct consisted of the coding region of the c-myc proto-oncogene (exons 2, 3, and 3′ flanking sequences) linked to the β-globin promoter contained within a 687-bp fragment and the two 72-bp repeats of the SV40 enhancer.
The c-myc proto-oncogene in the SBM construct was substituted with the mouse c-fos gene in SBF mice. The β-globin promoter (containing its own transcription initiation site “cap”) and the SV40 enhancer of the SBM regulatory elements were retained. Genomic c-fos DNA from +108 bp (located between the cap and the translation initiation codon) (AccI) to +800 bp downstream of the poly(A) site (BamHI) was juxtaposed downstream of these regulatory elements (gift of Dr. J. Deschamps, Hubrecht Lab, Utrecht, The Netherlands). In addition, this transgenic construct contained a sea urchin marker of 130 bp (SalI) (17) between the translation termination codon (+98 bp from UAG) and the poly(A) site to differentiate the transgene from the endogenous gene and to disrupt the specific destabilizing sequence within c-fos mRNA (27). All novel junctions created in this construct were sequenced. Eight SBF transgenic lines were established for this construct as described for SBM (35).
The SBT construct was generated by substituting the human TGF-α cDNA for the c-myc proto-oncogene in the SBM construct, as described above for SBF. TGF-α sequences were contained within a 925-bp BglII cDNA fragment linked to a 900-bp fragment of the human growth hormone gene containing the 3′ untranslated region, the polyadenylation site and 3′ flanking sequence (gift of Dr. Glen Merlino, National Institutes of Health, Bethesda, MD) (13). All new junctions in this hybrid construct were also sequenced. Five SBT transgenic lines were established, as described for SBM (35).
The SBB was generated by substituting the human bcl-2 cDNA for the c-myc proto-oncogene in the SBM construct, as described above for SBF. The bcl-2 sequence was contained within a 758-bp (XbaI-Pst I) fragment located 58 bp upstream of the translation initiation codon to +98 bp downstream of the stop codon, linked to a 2,179-bp fragment of the human growth hormone gene containing the last five exons and introns, followed by the polyadenylation site and 3′ flanking sequence. Six SBB transgenic lines were established.
Transgene constructs were excised from plasmid sequences isolated by agarose gel electrophoresis and purified by centrifugation on CsCl gradients. The constructs were microinjected into the pronuclei of (C57BL/6J × CBA/J)F2 fertilized eggs. Positive heterozygote transgenic SBM, SBF, SBT, and SBB mice were identified by Southern Blot analysis of tail DNA using the entire hybrid gene construct as a probe. The 32P-labeled probe was generated by nick translation of the entire plasmid.
Expression Analysis by In Situ Hybridization of Adult Kidney
Renal tissues from SBM or SBF adults and nontransgenic littermate controls were fixed in paraformaldehyde and paraffin-embedded. Tissue sections were mounted on 3-aminopropyl triethoxy silane coated slides. SBM and control tissue sections were hybridized with a uniformly 35S-labeled antisense probe produced with the junction β-globin promoter/c-myc gene called “SBM probe” (35). Similarly, the SBF, SBM, and control renal sections were hybridized with a SBF antisense probe. This probe was produced with a BalI-HincII fragment containing the junction β-globin promoter/c-fos from the SBF construct and uniformly 35S-labeled. Sections were then washed and coated with Kodak NTBII emulsion for autoradiography. For controls, sense probe was utilized on mice positive and negative for the transgene; and antisense probe was used on renal tissue from transgene negative littermates. Photographs were taken using light and dark field optics.
Analysis of Gene Expression
RNA Extraction.
Total RNA from mouse kidney was extracted by the acid guanidium-phenol-chloroform method as previously described (35). Final pellets were resuspended in sterile DEPC treated water. Integrity of RNA was monitored on 0.8% agarose formaldehyde gels.
Reverse Transcription PCR.
All RNA samples (3 μg) were simultaneously reverse transcribed into cDNA in BRL RT buffer containing 0.5 mM each dNTP (Pharmacia LKB Nuclear, Gaithersburg, MD), 1.2 U/μl RNasin (Boehringer-Mannheim Corp., Indianapolis, IN), 200 U of M-MLV reverse transcriptase (GIBCO BRL, Gaithersburg, MD) and 5 μg of pd (N)6 random primers (Pharmacia). Total reaction volume was 20 μl. Reverse transcription PCR amplification was carried out at 37°C using 0.2-μl aliquots. All cDNAs to be quantified were amplified simultaneously in: PCR buffer (10 mM Tris, pH 8.3, 50 mM KCL, 1.5 mM MgCl2) 0.2 mM each dNTP, 20 pmol of specific primers (except for murine bcl-2, 40 pmol), 2 pmol of S16 primers (except for murine bcl-2, 1 pmol and bax, 5 pmol), and 0.5 units Taq polymerase (Perkin-Elmer Cetus Corp., Norwalk, CT) in a total volume of 20 μl. Parallel control reactions were carried out with water replacing DNA. Initial experiments were carried out to ensure that conditions for PCR were within the linear range of amplification.
For amplification, the primers used for the SBF and SBT gene product were 5′–CTTCTGACACAACTGTGTTC–3′ (forward; nt 42-61 including the cap site in the β-globin promoter) whereas the reverse 5′–CACTAGAGACGGACAGATCT–3′ (nt 1190-1209, exon 2 in fos) for SBF and 5′–AATGGGACACCACTGCTGCA–3′ (nt 167-186, exon 2 in TGF-α) for SBT; for SBB: 5′–TGTGGCCTTCTTTGAGTTCG–3′ (nt 1899-1918, exon 2) and 5′ TCACTTGTGGCTCAGATAGG-3′ (nt 2159-2178, exon 3); for p53: 5′–CAGCCAAGTCTGTTATGTGC–3′ (nt 456-475, exon 4) and 5′–ATGGTGGTATACTCAGAGCC–3′ (nt 778-797, exon 7); for murine bcl-2: 5′–TGTGGCCTTCTTTGAGTTCG–3′ (exon 1) and 5′–TCACTT- GTGGCCCAGGTATG–3′ (exon 2); for bax: 5′–GCGTCCACCAAGAAGCTGAG–3′ (exon 3) and 5′–ACCACCCTGGTCTTGGATCC–3′ (exon 5); for murine bad 5′–GAAGGGATGGAGGAGGAGCT–3′ (nt 831-850) and 5′–GGAGCCTCCTTTGCCCAAGT–3′ (nt 1051-1070). All reactions contained, as an internal control, a pair of primers to co-amplify the mouse S16 ribosomal protein gene product: 5′–AGGAGCGATTTGCTGGTGTGGA–3′ (nt 1451 -1472, exon 3) 5′–GCTACCAGGCCTTTGAGATGGA–3′ (nt 1620-1641, exon 4) as previously described (11). All RNA samples of SBT and SBB were treated with RNase-free DNaseI before the reverse transcriptase reaction. Additional controls consisted in amplification of RNA before and after DNaseI treatment to ensure that any contaminating DNA was eliminated.
Conditions for all amplification reactions were 94°C, 5 min, followed for SBF by 30 cycles of 94°C, 30 s, 55°C, 30 s, 72°C, 1 min, and a final elongation of 5 min at 72°C; for SBT by a ramp of 3 min, 64°C, 30 s, 72°C, 30 s, followed by 4 cycles of 94°C, 30 s, 63°C, 30 s, 72°C, 30 s, 26 cycles of 94°C, 30 s, 60°C, 30 s, 72°C, 30 s, and a final elongation of 10 min at 72°C; for SBB by 20 cycles of 94°C, 30 s, 58°C, 30 s, 72°C, 30 s and a final elongation of 7 min at 72°C; for p53 by 20 cycles of 94°C, 1 min, 60°C, 1 min, 72°C, 2 min, and a final elongation of 7 min at 72°C; for bcl-2, by 25 cycles of 94°C, 1 min, 61°C, 1 min, 72°C, 2 min, and a final elongation of 7 min at 72°C; for bax by 5 cycles of 94°C, 1 min, 55°C, 1 min, 72°C, 2 min, at which point S16 oligonucleotides were added, and PCR amplification resumed for another 20 cycles, and a final elongation of 7 min at 72°C; for bad by 25 cycles of 94°C, 1 min, 62°C, 1 min, 72°C, 1 min, and a final elongation of 7 min at 72°C.
Samples were analyzed on 10% polyacrylamide–TBE gels and visualized by ethidium bromide staining. For quantitative evaluation, gels were scanned by IS-1000 digital imaging densitometer system (Alpha InnoTech Corp., San Leandro, CA).
Tunel Assay for Detection of Apoptosis in Tissue Sections
3-μm-thick paraffin sections of formalin fixed renal tissue from transgenic mice and age-matched controls were deparaffinized and hydrated in graded alcohols. Sections were incubated for 30 min at 37°C with a reaction buffer (1 M potassium cacodylate, 125 mM Tris-HCl, bovine serum albumin 1.25 mg/ml) (20:100), CoCl2 solution (1:100), TdT (0.4:100), and biotin 16-2 dUTP labeling mix (1:100) (containing 1 mM each dATP, dGTP, dCTP, and 0.65 mM dTTP, 0.35 mM biotin-16-dUTP in Tris-HCl) (Boehringer Mannheim). Endogenous peroxidase was blocked with 3%H2O2 for 30 min. Sections were incubated for 30 min in Avidin-Biotin Complex (Vector Labs., Inc., Burlingame, CA) followed by diaminobenzidine. Sections were counterstained with Periodic acid-Schiff. Apoptotic cells were quantitated by counting the number of stained tubular nuclei per mm2 of tissue.
Proliferation Index in Kidney Sections
Kidney from seven SBM transgenic adult mice and four control littermates were fixed in formalin and embedded in paraffin. 3-μm sections were deparaffinized and hydrated in graded alcohols. After blocking for endogenous peroxidase activity slides were microwaved for 25 min at 750 W. After PBS rinse and blocking with 10% goat serum, tissue sections were incubated overnight at 4°C with rabbit polyclonal antibody MIB-1 to Ki-67 (clone MM1) (Novacastra Labs, Newcastle Upon Tyne, UK) at 1:100 followed by biotinylated secondary antibody (Vector Labs., Inc.), followed by avidin/peroxidase complex (Vector Labs., Inc.) and DAB. Sections were counterstained with Mayer's hematoxylin (Sigma Chem. Co., St. Louis, MO). Proliferation index was calculated as number of MIB-1 positive cells per mm2 of tissue.
Results
Cell Proliferation as One Mediator of SBM Renal Phenotype
Morphological studies in the SBM model demonstrated renal cysts are first detectable at E16.5, where they are manifested predominantly as glomerular cysts, followed by development of tubular cysts (35a). Both cyst number and size increased progressively with age, leading to end stage renal failure by age 3–6 mo (35). Cyst formation was associated with renal epithelial hyperplasia, occasionally leading to adenoma formation.
To determine the site and cellular localization of c-myc expression within the adult kidneys, we examined normal and cystic SBM kidneys by in situ hybridization. A strong expression of the c-myc transgene was specifically localized to the SBM renal cystic epithelium as well as focal noncystic tubules (Fig. 1 a), with undetectable signal in adult controls. This overexpression of c-myc in the renal epithelium correlated with the development of hyperplasia and abnormal polarity in many of the cystic tubules.
Figure 1.


c-myc expression induces increased proliferation in transgenic SBM renal epithelial cells. (a) In situ hybridization for the c-myc transgene shows high expression in the cystic renal epithelium of adult SBM mice (dark field optics, X112.5). (b) Proliferation analysis of SBM adult kidneys with antibody MIB-1 reveals many positive cells lining the renal cysts. There is a tendency for positive nuclear staining to be clustered. MIB-1 immunostain, periodic acid-Schiff counterstain, ×300.
Because hyperplasia is a consistent feature of the SBM phenotype, the rate of epithelial proliferation was determined using antibody to murine MIB-1 (Ki-67), expressed in cycling cells. As shown in Table 1, the proliferation index was over 10-fold higher in SBM adult kidneys compared with controls. Proliferating cells were almost exclusively localized to the tubular epithelium with greater involvement of cystic tubules than non-cystic tubules (Fig. 1 b). MIB-1–positive cells were also frequently clustered in the tubular epithelial lining, suggesting an inductive or paracrine/juxtacrine signal.
Table 1.
Proliferation Index of Adult Mouse Kidney
| Mouse | No. of animals | Mean MIB-1 positive cells/mm2 | ||
|---|---|---|---|---|
| (C57 × CBA) F1 Controls | 4 | 8.7 ± 3.8 | ||
| SBM | 8 | 99.4 ± 47.3 |
Specificity of c-myc in the Induction of Renal Cystogenesis
To verify the specificity of the c-myc proto-oncogene in cystogenesis, we investigated the potential of other proto-oncogenes/growth factors to induce a PKD phenotype. In the first study, we substituted c-myc with c-fos sequences, while retaining the SV40 and β-globin regulatory elements of the SBM construct, thereby generating a new construct, SBF. c-fos was chosen because it is a well-characterized proto-oncogene with properties closely resembling those of c-myc, including early response gene, cellular proliferation, and the ability to promote epithelial proliferation in H2c-fos transgenic mice (27).
SBF transgenic mice carrying the SBF construct (Fig. 2) were identified by Southern blot analysis of tail DNA. Pathological examination of brain, lung, liver, spleen, and kidney showed no gross or histological abnormalities. Specifically, no PKD phenotype or epithelial hyperplasia could be identified in any of the 8 SBF transgenic lines. Unlike the SBM mice which die of renal failure by 3–4 mo of age, mice of the different SBF lines analyzed have a normal life span of 2–3 yr.
Figure 2.

Schematic representation of SBF, SBT, SBB, and SBM constructs. The SBM construct consists of the murine c-myc gene exon 2, 3, and 3′ flanking sequences (35). All share common regulatory elements, SV40 enhancer (dark oval) and β-globin promoter (stippled) cojoined as in SBM. For SBF, these regulatory elements are linked to the mouse genomic c-fos sequences (hatched box, exons 1-4; solid bar, introns 2-3 and flanking sequences). The CIIIa sea urchin marker (dark box) was inserted into the 3′ end, where unstable sequences have been identified. For SBT, the same regulatory elements are linked to the human TGF-α cDNA (hatched box) bound to the human growth hormone (hGH) 3′ untranslated region of the last exon (open box), the polyadenylation signal and the 3′ flanking region (hGH) (solid bar). For the SBB, the same regulatory elements are linked to the human bcl-2 cDNA (hatched box) bound to the hGH last 5 exons (open boxes, exons; solid bar, introns), polyadenylation signal and 3′ flanking sequences (hGH) (solid bar).
Fig. 3 a shows RT-PCR analysis of the SBF transgene in the various organs (including kidney, liver, spleen, heart, lung, and brain). The highest levels of SBF transgene expression were obtained in kidney, where levels were demonstrated to be within the linear range (Fig. 3 b). Some expression was also occasionally detectable in spleen and lung, with little or undetectable expression in other organs (Fig. 3 a). This particular organ distribution of transgene expression closely resembles that described previously for SBM mice (35), and it is likely that the transgene is preferentially targeted to the kidney due to common regulatory elements with SBM. Furthermore, to determine more precisely the localization of the c-fos transgene in SBF kidneys, we have carried out in situ hybridization. Adult transgenic SBF kidneys demonstrated high expression of the transgene in tubular epithelial cells throughout the kidney, with particularly intense signal over the medullary collecting tubules (Fig. 4 a). This cellular localization is very similar to that observed for the c-myc transgene. No expression was detected in renal tissue of non transgenic controls (C57BL/6 × CBA)F1 (Fig. 4 b). Furthermore, c-fos expression was also investigated in the SBM mouse kidneys (Fig. 4 c); the absence of signal indicates that c-fos is not induced in the SBM c-myc–dependent cystogenic pathway.
Figure 3.


Expression analysis of the SBF transgenic mice. (a) RT-PCR expression analysis of SBF (234-bp fragment) in 4 different transgenic lines (SBF 38, 19, 29, and 20) shows strong expression of the SBF construct in kidney (K) and spleen (S), but not in liver (Li), heart (H), lung (Lu), or brain (B), shown for SBF 38 only). SBF38 shows high expression of the transgene in kidney and spleen and weak expression in lung and brain. M, molecular weight marker; C, negative control; F1, nontransgenic kidney control; S16, internal control (103 nt). (b) Demonstration of linearity of PCR amplification. Total kidney cDNA (0.1 to 4 μl) aliquots were amplified with primers specific for c-fos and S16 as internal control. For semiquantitative evaluation, acrylamide gels (bottom) were scanned by computerized densitometer and graphed (top). SBF (filled squares); S16 (open squares).
Figure 4.



Detection of SBF mRNA by in situ hybridization in transgenic kidney tissues. (a) In situ hybridization analysis of c-fos mRNA in transgenic SBF adult kidneys detected intense signal over the epithelial cells lining the normal-appearing tubules. (Hematoxylin-eosin counterstain, ×200). (b) In situ hybridization for c-fos mRNA in non-transgenic controls of the same genetic background (C57BL/6 × CBA)F1 is negative, with low background levels. (c) In SBM kidneys, in situ hybridization for c-fos is negative. The renal cysts are visible even with dark-field optics.
In a further attempt to test the specificity of c-myc within the SBM transgene, TGF-α was selected as a substitute for c-myc because of its known mitogenic effect in fetal tissues where it is localized to the developing tubular epithelium during early nephrogenesis (36). This growth factor has also been implicated in epithelial cell proliferation in the mammary glands of TGF-α transgenic mice (13, 19). Similarly to the SBF construct, the SBT construct was produced as illustrated in Fig. 2. Five SBT transgenic lines were generated and Fig. 5 show renal transgene expression in a SBT line by RT-PCR analysis. However, the SBT transgenic lines, like the SBF lines, had no gross or microscopic abnormalities in any of the organs (lung, brain, liver, kidney, spleen) analyzed.
Figure 5.

Expression analysis of the SBT transgenic mice. RT-PCR expression analysis of SBT (241-bp fragment) in a transgenic line (SBT 68) shows expression of the SBT construct in kidney (K), but not in liver (Li). M, molecular weight marker; C, negative control with water replacing cDNA; S16, internal control (103 nt).
Apoptosis as a Second Mediator in SBM Renal Phenotype
c-myc has been implicated in both cellular growth and proliferation as well as apoptosis (10). To determine the biological importance of these opposing cell processes for cystogenesis in SBM mice, we have complemented the epithelial proliferation studies with investigations into apoptosis and its molecular mechanisms.
Apoptosis in SBM Mice.
In SBM adult mice apoptotic index determined by Tunel assay was approximately 100-fold higher than the negligeable apoptotic rate in nontransgenic littermate controls (Table 2). Apoptotic nuclei were often clustered in cystic epithelium suggesting a zonal effect that may be mediated by paracrine effects or local cell-cell/ cell-matrix interactions (Fig. 6).
Table 2.
Apoptotic Index in Adult Kidneys from SBB × SBM Mating
| Mouse genotype | ||||||||
|---|---|---|---|---|---|---|---|---|
| SBB transgene | SBM transgene | No. of animals | Range of apoptotic | Mean apoptotic | ||||
| cells/mm2 | cells/mm2 | |||||||
| − | − | 4 | 0.0–0.1 | 0.02 ± 0.03 | ||||
| + | − | 6 | 0.0–0.1 | 0.03 ± 0.03 | ||||
| − | + | 7 | 1.0–5.9 | 3.40 ± 1.72 | ||||
| + | + | 8 | 1.2–7.0 | 3.42 ± 1.94 | ||||
Figure 6.

c-myc–induced apoptosis in SBM transgenic mice. Apoptotic nuclei are clustered on one edge of a renal tubular cyst from an adult SBM mouse (DNA end-labeling, ×500).
Expression of bcl-2 and bax in SBM Renal Tissue.
It is well documented that Bcl-2, a major suppressor of apoptosis, is capable of abrogating c-myc induced apoptosis (3). Bcl-2 plays an important role in protecting different cell types from apoptotic death (26), and homozygous null bcl-2 mice develop renal cysts (23). Thus, we have compared bcl-2 expression levels in renal tissue of SBM transgenic mice with those of control littermates by RT-PCR. Comparable levels of bcl-2 expression were detected in SBM lines (SBM9 and 75) and controls (Fig. 7 a, top). The bcl-2 expression in SBM mice did not change significantly over a wide age range from birth to end stage renal disease (data not shown).
Figure 7.


bcl-2–independent c-myc–dependent apoptosis in transgenic mice. (a) RT-PCR analysis of renal bcl-2 (280 bp fragment) and bax (311-bp fragment) expression in SBM 9 and 75, control littermates and progenies of SBM/p53−/− matings. Comparable levels of bcl-2 (top) and bax (bottom) are seen in all the lines studied. Similar levels are also seen for the S16 internal control. c, control reaction with water replacing cDNA, M, molecular weight marker. (b) RT-PCR expression analysis for the SBB transgene (280-bp fragment) for four different SBB lines (10, 11, 17, and 20) is shown in kidney (K) and other organs (Lu, lung; S, spleen; B, brain; Li, liver; C, control). A (C57BL/6J × CBA/J)F1 non-transgenic kidney control is also included. S16, internal control (103 nt).
Because the bax/bcl-2 ratio was shown to be critical for cell survival and cell death, and because the promoter of the bax gene contains several binding sites for the myc family of transcription factors, we have investigated the levels of bax in SBM mice. RT-PCR analysis indicated that bax expression levels were similar in SBM mice and in control littermates (Fig. 7 a, bottom) and over a wide range of developmental stages (data not shown). Moreover, measurements of bad expression, another important proapoptotic member of bcl-2 family (40), have also shown no significant difference in expression levels between SBM kidney and controls from birth to adulthood (data not shown). Altogether these data indicate that some key members of the bcl-2 gene family do not play a significant role in the apoptotic pathway induced by c-myc in SBM mice.
Targeted Expression of Bcl-2 in SBM Kidney
To directly address whether overexpression of bcl-2 in vivo can prevent c-myc–induced cystogenesis, we have generated transgenic mice called SBB. This SBB construct carries the complete human bcl-2 cDNA and is illustrated in Fig. 2. Transgene expression was found in kidney and spleen with low to undetectable levels in lung, brain and liver (Fig. 7 b). These SBB transgenic lines have a normal renal phenotype. The physiological importance of Bcl-2 within the c-myc–induced cystogenic pathway was then tested by crossmating the 3 SBB lines showing highest transgene expression (SBB 10, 11, and 20) with the SBM 75 line. Of 165 progenies produced, the renal phenotype was analyzed for each of the four genotypes. Morphologic studies on SBB−/SBM− and SBB+/SBM− mice revealed normal renal histology. The SBB+/SBM+ and SBB−/SBM+ mice had comparable morphologic features with typical cystic phenotype indistinguishable from that of the pure SBM line. Moreover, the clinical evolution of the renal failure was similar in the double transgenic (SBB+/SBM+) and SBM lines, with mean death due to renal failure at ∼3 mo. Renal proliferation index in the double transgenic SBB+/SBM+ mice was 89.2 ± 66.0, comparable to the proliferation index of 99.4 ± 47.3 described for SBM mice, (Table 1). It is of interest that the proliferation index in the SBB+/SBM− mice is equivalent to that of negative control littermates (10.4 ± 5.0 versus 8.7 ± 3.8). We then investigated the implication of bcl-2 in the c-myc–dependent apoptotic pathway. Table 2 summarizes the renal apoptotic indices in the four genotypes generated from the SBB × SBM crosses. Renal tissues from mice carrying the SBM transgene had virtually identical apoptotic rates irrespective of the SBB genotype. Conversely, kidney tissues from mice without the SBM transgene had identical apoptotic rates at least 30-fold lower than the SBM lines, irrespective of their SBB genotype. Thus it appears that overexpression of bcl-2 does not modify the SBM-dependent apoptotic rate in vivo.
Expression of p53 in SBM Renal Tissue
p53 is a tumor suppressor gene that mediates c-myc-induced apoptosis (12, 38). Since c-myc is overexpressed in SBM kidney and renal tissues displayed a high apoptotic index, the level of p53 expression was investigated by RT-PCR analysis. Two transgenic mouse lines (SBM 9, SBM 75) at different stages of development (0.5 d, 10 d, 4 mo, end-stage) expressed approximately two- to threefold lower levels of p53 compared with negative age-matched littermates (Fig. 8 a), suggesting that p53 is not a major inducer of apoptosis in SBM. Proof that the expression analysis falls within the linear range is provided in Fig. 8 b.
Figure 8.

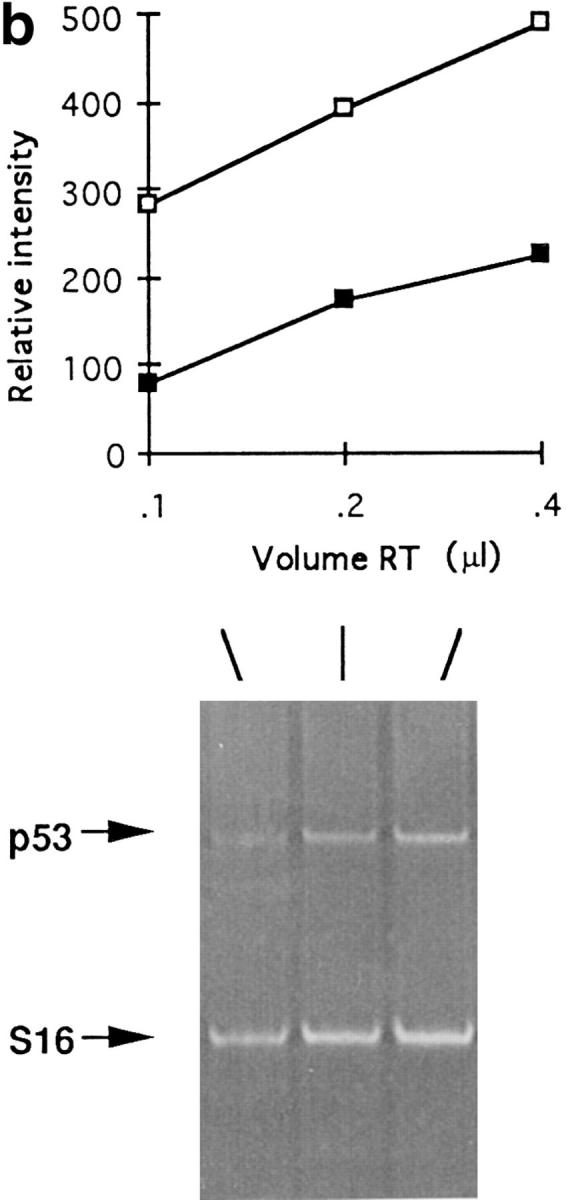
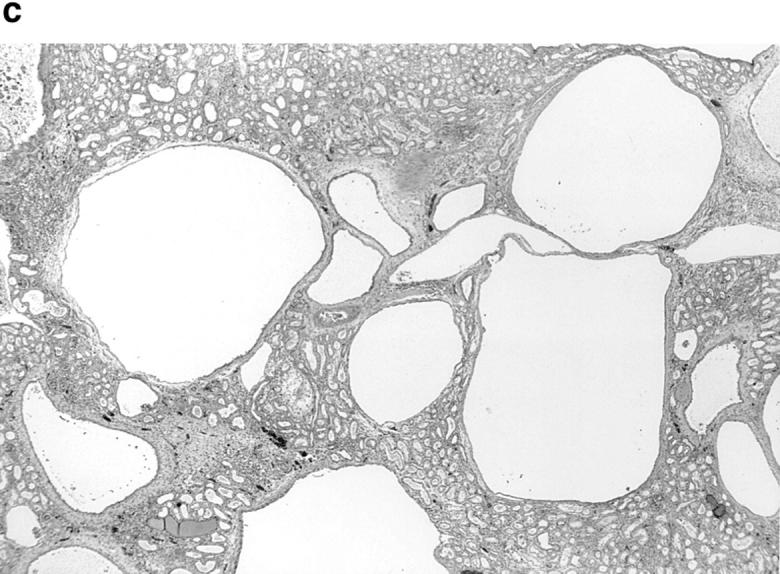


p53-independent apoptosis in c-myc–induced PKD. (a) RT-PCR analysis of p53 expression (340-bp fragment) at different stages of development (0.5 d, 10 d, 4 mo, and end-stage renal disease, indicated above each lane) of a transgenic SBM line (SBM9) compared with control (negative age-matched littermates). M, molecular weight marker; ESRD, end stage renal disease; d, day; m, month. (b) Demonstration of linearity of PCR amplification. Total kidney cDNA (0.1–4 μl) aliquots were amplified with primers specific for p53 and S16 as internal control. For semiquantitative evaluation, an acrylamide gel (bottom) was scanned by computerized densitometer and graphed (top). (Filled squares) p53, (open squares) S16. (c) Renal tissue of p53−/−SBM+ mice that shows numerous tubular cysts (hematoxylin-eosin, X30). (d) Apoptotic tubular epithelial cells are highlighted along the wall of a cyst and in the cyst lumen of a SBM+p53−/− mouse (DAB chromogen, ×800). (e) p53−/−SBM+ renal tissue showing an apoptotic epithelial cell with nuclear fragmentation (arrow) being shed into the cyst lumen (hematoxylin-eosin, ×1,250).
To demonstrate that the apoptosis observed in SBM mice is p53-independent, successive matings of SBM mice were performed with p53-null mice. From several matings, SBM/p53−/− mice as well as SBM/p53+/−, SBM/p53+/+ and p53−/− littermates were obtained. The mean survival of the SBM/p53−/− mice (n = 4) was 2.6 mo, virtually identical to that of SBM/p53+/− mice (n = 16) and of SBM/p53+/+ mice (n = 3). In adult SBM/p53−/− mice, renal tubular cysts were numerous and large, resembling those of SBM mice (Fig. 8 c). The range of apoptotic indices in the SBM/p53−/− mice was within the range observed in SBM/p53+/+ and SBM/p53+/− (Table 3). Renal tissues of all three of these genotypes had greatly increased apoptotic rates compared with the negligeable apoptosis seen in SBM negative littermates. By Tunel assay, apoptosis was detected in renal cystic epithelial cells (Fig. 8 d), some of which had morphologically detectable nuclear fragmentation (Fig. 8 e). These results indicate that apoptosis in this model is not abrogated by the absence of p53, but occurs through a c-myc–dependent, p53-independent pathway. These findings provide definitive evidence that p53 is not required for cystogenesis in SBM/p53−/− mice and support the inference from the quantitative RT-PCR results that p53 does not play a major role in the induction of apoptosis in SBM mice.
Table 3.
Apoptotic Index in Adult Kidneys from SBM × P53 Mating
| SBM transgene | P53 genotype | No. of animals | Range of apoptotic | Mean apoptotic | ||||
|---|---|---|---|---|---|---|---|---|
| cells/mm2 | cells/mm2 | |||||||
| − | +/− | 6 | 0.0–0.1 | 0.04 ± 0.06 | ||||
| − | −/− | 5 | 0.0–0.1 | 0.06 ± 0.05 | ||||
| + | +/+ | 2 | 0.5–2.2 | 1.39 ± 1.18 | ||||
| + | +/− | 10 | 0.3–3.6 | 1.39 ± 0.99 | ||||
| + | −/− | 3 | 0.3–0.7 | 0.54 ± 0.23 |
p53 has been shown to induce alterations in bcl-2 and bax expression in some tissues (e.g., prostate epithelium, thymus) but not in others (e.g., neurons) (20, 21). Because this effect has not been documented in the kidney and because a common cell death pathway has been suggested for p53 and bcl-2, we next analyzed the expression of bcl-2 and bax in the various SBM/p53 progenies. As shown in Fig. 7 a, levels of bcl-2 and bax expression did not vary appreciably in SBM/p53−/− mice compared with those in SBM or even to control mice. These data indicate that in the kidney bcl-2 and bax are not modulated by p53 or by c-myc and imply the existence of other regulators of bcl-2 family members in kidney as reported for p53 in neurons (20).
Discussion
To address the in vivo molecular and cellular processes induced by the c-myc protein, transgenic mice overexpressing this proto-oncogene in renal epithelial cells were generated. These SBM transgenic mice consistently produced a renal phenotype which closely mimics human PKD. Our results demonstrate that the properties of c-myc which induce PKD are specific since other proto-oncogenes with growth-related properties could not reproduce this phenotype. We have also shown that concomitant proliferation and apoptosis are specifically induced by c-myc and that both these processes are crucial determinants to the development of the renal phenotype. Using our c-myc transgenic model, we have determined that the in vivo role of c-myc in these cellular processes appears to be independent of the molecular pathways regulated by key members of the bcl-2 family. Moreover, we have established that in vivo c-myc–induced apoptosis is p53-independent.
To address the specificity of c-myc in the epithelial phenotype that underlies PKD, we investigated whether c-myc could be substituted with another proto-oncogene (c-fos) and a growth factor (TGF-α) known to be operant in renal development. Similar to the SBM transgene, these two novel transgenes, SBF and SBT, were expressed in renal tissue and specifically in tubular epithelium. In contrast to the SBM, neither SBF nor SBT produced renal abnormalities. Hence, it appears that the high mitogenic potential of TGF-α in epithelial cells is insufficient to cause a renal PKD phenotype. Further, despite the implication of c-fos in epithelial proliferation during renal organogenesis (37), in acute nephrotoxic injury and in cystogenesis of cpk mice (6), our results indicate that c-fos also does not play a major role in cyst formation. Indeed, c-fos is not detected in adult SBM kidneys and importantly, its overexpression in SBF mice does not induce cystogenesis. This inability of the SBF transgene to induce PKD cannot be explained by the absence of c-jun, a heterodimeric partner required for c-fos transcriptional activity, since c-jun is ubiquitously expressed (4). The renal SBM phenotype thus appears to be independent of the c-fos signaling pathway. On the basis of these results, we conclude that c-myc has specific renal cystogenic properties.
Targeting of c-myc to the kidney, the mammary gland and the hematopoietic cells has resulted in cellular hyperplasia in each of these tissues (1, 30, 35). In the SBM model, the consistent renal hyperplasia suggested a role for increased epithelial proliferation. Indeed, the SBM renal epithelium was highly proliferative (>10-fold), similar to our previous findings and those of others in human PKD (16, 22). In vitro cell culture studies have demonstrated that the levels of c-myc correlated with susceptibility to apoptosis, thereby suggesting a similar relationship in vivo in SBM renal epithelium. The rates of apoptosis in kidneys from our SBM mice were in fact, also highly elevated (10– 100-fold). This represents the first demonstration of concurrent c-myc–induced dysregulation of both proliferation and apoptosis in vivo. Interestingly, both these cellular processes frequently occur in a focal distribution suggestive of a paracrine signal or local tissue phenomena such as cell– cell or cell–matrix interactions. Furthermore, the occurrence of both these cellular mechanisms in the same tissue may explain the low frequency of adenomas in adult SBM mice. Indeed, the c-myc–driven hyperproliferation could theoretically promote a high frequency of cellular neoplasia and transformation, but this propensity is likely checked by the elevated apoptotic rate. In addition, our studies suggest that a similar temporal and spatial balance between c-myc– induced proliferation and apoptosis is critical to the development and progression of cyst formation and renal tissue remodeling. While in the human autosomal dominant polycystic kidney disease (ADPKD), we have demonstrated a consistent association between the upregulation of c-myc and the cellular dysregulation of proliferation and apoptosis (16), our studies in the SBM model provide strong evidence of causality.
Based on the specificity of c-myc for the cystic phenotype, we next sought to determine which molecular pathways underlie the proliferative and apoptotic cellular processes. It is likely that critical factors govern the cell's differential susceptibility to these opposing c-myc–regulated functions. Two of these factors, p53 and Bcl-2, previously shown to be involved in these complex cellular pathways have been investigated. p53 is a well-recognized factor which mediates c-myc–induced apoptosis. c-myc can induce apoptosis by causing an approximately three-fold increase in p53 mRNA levels followed by an increase in protein levels (12). Since our data show an approximately twofold decrease in p53 expression, this suggests that p53 does not play a major role in the SBM apoptotic pathway. The apoptosis observed in SBM mice was confirmed to be p53-independent by studies in SBM/p53−/− and SBM/p53+/− mice, which displayed similar apoptotic index and renal pathology to the SBM mice. These results implicate the existence of an in vivo c-myc induced apoptotic pathway that is independent of p53. In agreement with our results, in vivo studies by others have reported the existence of a p53-independent pathway in different tissues which express two other oncoproteins (SV40 T-antigen and HPV-16E7) (18, 25) but the pathway for c-myc in vivo had not been investigated. Furthermore, the observation in our model that bax expression levels do not appear modulated in this c-myc–dependent p53-independent pathway is consistent with the proposed model that c-myc can cause bax induction only through p53 activation in immortalized epithelial cells (BRK) (28). A p53-independent apoptotic pathway(s) had also been inferred from our studies on human ADPKD, since no induction of p53 or bax was associated with the increased c-myc expression and the dysregulation of apoptosis and proliferation in both fetal and end-stage polycystic kidneys (16). Thus the demonstration of a p53-independent apoptotic pathway in the c-myc– induced PKD mouse model not only corroborates our human data but also validates the relevance of the SBM model for molecular studies of human ADPKD.
Indirectly, the existence in vivo of a p53-independent apoptotic mechanism in renal epithelium is also supported by the observation that p53-null mice display normal kidney development and homeostasis (9), even though renal organogenesis requires high levels of apoptosis (15). In fact, renal organogenesis is dependent on concomitant cellular apoptosis and proliferation associated with high levels of c-myc expression; on completion of kidney ontogeny, these cellular processes are virtually abrogated and c-myc expression is turned off. In our PKD model, it is likely that the c-myc induced persistence of apoptosis independent of p53, together with the epithelial proliferation and incomplete epithelial differentiation (2) results from a failure to switch out of a developmental program. Indeed, cystogenesis becomes morphologically evident in utero when renal organogenesis is almost completed.
Various studies have shown that the apoptotic pathway induced by c-myc can be abrogated in hematopoietic and fibroblast cells (3, 38) by overexpression of bcl-2 and that a decrease in the Bcl-2/Bax ratio is believed to determine cell death (24). In SBM mice, although the apoptotic index was elevated in renal tissue, bcl-2 expression levels were not significantly decreased compared with controls. Moreover bax levels were not appreciably increased, suggesting that the Bcl-2/Bax ratio is not an important determinant of the c-myc–dependent cystic phenotype. Confirmation that Bcl-2 does not play a major role in SBM cystogenesis was obtained by its overexpression in the many double bcl-2/ c-myc transgenic mouse lines. Strikingly the overexpression of bcl-2 in SBM renal epithelium could not block the cell death pathway and rescue the cystic phenotype, indicating that the cellular effects of c-myc are Bcl-2 independent in this model. Furthermore, the double transgenic mice did not have a higher incidence of tumor formation, in contrast to the previous transgenic mice expressing c-myc and bcl-2 in lymphoid cells (33). These findings suggest first that the c-myc dependent apoptotic pathway can bypass Bcl-2 in vivo and likely involves other regulatory factors that are not inhibited by Bcl-2. Second, susceptibility to tumor formation mediated by c-myc and bcl-2 expression is likely to be a function of specific cell types. The inability of the double bcl-2/c-myc transgenic mice to rescue the SBM phenotype is in apparent contradiction to the cystic phenotype of the bcl-2 knockout mouse (23). However, the bcl-2−/− mouse model more closely resembles cystic hypoplasia than PKD, since tubular cysts are produced without renal enlargement (31). Our results are congruent with the bax knock-out mouse, which has no renal abnormalities (14). Altogether these data suggest that apoptosis can be modulated either by bcl-2 overexpression, as described for several cell lines (3, 28) or can proceed via a bcl-2–independent pathway as demonstrated in vivo by our mouse model of PKD.
The molecular and cellular mechanisms of cystogenesis induced by c-myc in SBM mice closely resemble those previously reported for human ADPKD. Indeed, we have shown that c-myc is consistently highly overexpressed in human ADPKD (16). In both murine and human PKD, there is markedly elevated renal epithelial proliferation (16, 22) and apoptosis (16, 39) of approximately similar magnitude (10–100-fold) compared with normal kidneys. These similarities also extend to the renal expression levels of pro- and anti-apoptotic genes. In ADPKD, bax expression remains virtually unaltered and p53 varies from normal to slightly decreased by no more than two- to threefold, remarkably similar to our results in SBM mice. While bcl-2 expression levels appear unchanged in SBM mice, these are increased in human ADPKD in the face of elevated apoptotic rates, indicating that apoptosis in human ADPKD may also bypass Bcl-2. Together these findings in human ADPKD and the SBM model strongly support the existence of a common c-myc induced Bcl-2- and p53-independent apoptotic pathway in the mediation of renal cystogenesis.
In conclusion our data indicate that the PKD phenotype observed in the SBM mouse model is dependent on specific properties of the c-myc transgene. Our results on the c-myc induced maintenance of concomitant proliferation and apoptosis in the SBM mouse, also suggested in human ADPKD (16), are highly reminiscent of the normal processes operant in the renal developmental program. Importantly, these concurrent and opposing cellular mechanisms are likely essential for progressive growth and dynamic remodeling of renal cysts. The cellular apoptotic processes mediated by c-myc appear independent of bcl-2–regulated pathways. Moreover we have demonstrated for the first time the existence in vivo of a renal c-myc–dependent apoptotic pathway that is p53-independent.
Footnotes
1 Abbreviations used in this paper: ADPKD, autosomal dominant polycystic kidney disease; PKD, polycystic kidney disease.
We are very grateful to Dr. M. Aubry for helpful discussions and Dr. J. Deschamps for the gift of the c-fos gene. We thank C. Lagacé, J.C. Dunn, K. Butterfield, V. Ouellet, and N. Chrétien for technical assistance. This work was supported by grants from The Kidney Foundation of Canada (M. Trudel), the Medical Research Council of Canada (M. Trudel), the National Institutes of Health (no. DK44864, M. Trudel), Coopération Québec/\x83 tats-Unis (M. Trudel), M.Trudel is a chercheur-boursier of Fonds de la Recherche en Santé du Québec.
References
- 1.Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- 2.Barisoni L, Trudel M, Chretien N, Ward L, Adelsberg J V, D'Agati V. Analysis of the role of membrane polarity in polycystic kidney disease of transgenic SBM mice. Am J Pathol. 1995;147:1728–1735. [PMC free article] [PubMed] [Google Scholar]
- 3.Bissonnette RP, Echeverri F, Mahboubi A, Green DR. Apoptotic cell death induced by c-myc is inhibited by bcl-2. Nature. 1992;359:552–553. doi: 10.1038/359552a0. [DOI] [PubMed] [Google Scholar]
- 4.Carrasco D, Bravo R. Tissue-specific expression of the fos-related transcription factor fra-2 during mouse development. Oncogene. 1995;10:1069–1079. [PubMed] [Google Scholar]
- 5.Coles HSR, Burne JF, Raff MC. Large-scale normal cell death in the developing rat kidney and its reduction by epidermal growth factor. Development. 1993;118:777–784. doi: 10.1242/dev.118.3.777. [DOI] [PubMed] [Google Scholar]
- 6.Cowley BD, Chadwick LJ, Grantham JJ, Calvet JP. Elevated proto-oncogene expression in polycystic kidneys of the C57BL/6J (cpk) mouse. J Am Soc Nephrol. 1991;1:1048–1053. doi: 10.1681/ASN.V181048. [DOI] [PubMed] [Google Scholar]
- 7.D'Agati V, Trudel M. Lectin characterization of cystogenesis in the SBM transgenic model of polycystic kidney disease. J Am Soc Nephrol. 1992;3:975–983. doi: 10.1681/ASN.V34975. [DOI] [PubMed] [Google Scholar]
- 8.Davis AC, Wims M, Spotts GD, Hann SR, Bradley A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 1993;7:671–682. doi: 10.1101/gad.7.4.671. [DOI] [PubMed] [Google Scholar]
- 9.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 10.Evan G, Harrington E, Fanidi A, Land H, Amati B, Bennett M. Integrated control of cell proliferation and cell death by the c-myc oncogene. Biol Sci. 1994;345:269–275. doi: 10.1098/rstb.1994.0105. [DOI] [PubMed] [Google Scholar]
- 11.Foley KP, Leonard MW, Engel JD. Quantitation of RNA using the polymerase chain reaction. Trends Genet. 1993;9:380–385. doi: 10.1016/0168-9525(93)90137-7. [DOI] [PubMed] [Google Scholar]
- 12.Hermeking H, Eick D. Mediation of c-myc-induced apoptosis by p53. Science. 1994;265:2091–2093. doi: 10.1126/science.8091232. [DOI] [PubMed] [Google Scholar]
- 13.Jhappan C, Stahle C, Harkins RN, Fausto N, Smith GH, Merlino GT. TGFα overexpression in transgenic mice induces liver neoplasia and abnormal development of the mammary gland and pancreas. Cell. 1990;61:1137–1146. doi: 10.1016/0092-8674(90)90076-q. [DOI] [PubMed] [Google Scholar]
- 14.Knudson CM, Tung KSK, Tourtellotte WG, Brown GAJ, Korsmeyer SJ. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science. 1995;270:96–99. doi: 10.1126/science.270.5233.96. [DOI] [PubMed] [Google Scholar]
- 15.Koseki C, Herzlinger D, Al-Awqati Q. Apoptosis in metanephric development. J Cell Biol. 1992;119:1327–1333. doi: 10.1083/jcb.119.5.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lanoix J, D'Agati V, Szabolcs M, Trudel M. Dysregulation of cellular proliferation and apoptosis mediates human autosomal dominant polycystic kidney disease (ADPKD) Oncogene. 1996;13:1153–1160. [PubMed] [Google Scholar]
- 17.Lee JJ, Calzone FJ, Britten RJ, Angerer RC, Davidson EH. Activation of sea urchin actin genes in strongylocentrotus purpuratus. J Mol Biol. 1986;188:173–183. doi: 10.1016/0022-2836(86)90302-5. [DOI] [PubMed] [Google Scholar]
- 18.Li M, Hu J, Heermeier K, Hennighausen L, Furth PA. Apoptosis and remodeling of mammary gland tissue during involution proceeds through p53-independent pathways. Cell Growth & Differ. 1996;7:13–20. [PubMed] [Google Scholar]
- 19.Matsui Y, Halter SA, Holt JT, Hogan BLM, Coffrey RJ. Development of mammary hyperplasia and neoplasia in MMTV-TGFα transgenic mice. Cell. 1990;61:1147–1156. doi: 10.1016/0092-8674(90)90077-r. [DOI] [PubMed] [Google Scholar]
- 20.Miyashita T, Krajewski S, Krajewska M, Wang HG, Lin HK, Liebermann DA, Hoffman B, Reed JC. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene. 1994;9:1799–1805. [PubMed] [Google Scholar]
- 21.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 22.Nadasdy T, Laszik Z, Lajoie G, Blick KE, Wheeler DE, Silva FG. Proliferative activity of cyst epithelium in human renal cystic diseases. J Am Soc Nephrol. 1995;5:1462–1468. doi: 10.1681/ASN.V571462. [DOI] [PubMed] [Google Scholar]
- 23.Nakayama K, Nakayama KI, Negishi I, Kuida K, Sawa H, Loh DY. Targeted disruption of bcl-2ab in mice: occurrence of gray hair, polycystic kidney disease, and lymphocytopenia. Proc Natl Acad Sci USA. 1994;91:3700–3704. doi: 10.1073/pnas.91.9.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oltvai ZN, Milliman CL, Korsmeyer CL. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 25.Pan H, Griep AE. Temporally distinct patterns of p53-dependent and p53-independent apoptosis during mouse lens development. Genes & Dev. 1995;9:2157–2169. doi: 10.1101/gad.9.17.2157. [DOI] [PubMed] [Google Scholar]
- 26.Reed JC. Bcl-2 and the regulation of programmed cell death. J Cell Biol. 1994;124:1–6. doi: 10.1083/jcb.124.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruther U, Wagner EF, Muller R. Analysis of the differentiation-promoting potential of inducible c-fos genes introduced into embryonal carcinoma cells. EMBO (Eur Mol Biol Organ) J. 1985;4:1775–1781. doi: 10.1002/j.1460-2075.1985.tb03850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakamuro D, Eviner V, Elliott KJ, Showe L, White E, Prendergast GC. c-Myc induces apoptosis in epithelial cells by both p53-dependent and p53-independent mechanisms. Oncogene. 1995;11:2411–2418. [PubMed] [Google Scholar]
- 29.Schmid P, Schulz WA, Hameister H. Dynamic expression pattern of the myc protooncogene in midgestation mouse embryos. Science. 1989;24:226–229. doi: 10.1126/science.2911736. [DOI] [PubMed] [Google Scholar]
- 30.Sinn E, Muller W, Pattengale P, Tepler I, Wallace R, Leder P. Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell. 1987;49:465–475. doi: 10.1016/0092-8674(87)90449-1. [DOI] [PubMed] [Google Scholar]
- 31.Sorenson CM, Rogers SA, Korsmeyer SJ, Hammerman MR. Fulminant metanephric apoptosis and abnormal kidney development in bcl-2-deficient mice. Am J Physiol. 1995;37:F73–F81. doi: 10.1152/ajprenal.1995.268.1.F73. [DOI] [PubMed] [Google Scholar]
- 32.Stanton BR, Perkins AS, Tessarollo L, Sassoon DA, Parada LF. Loss of N-myc function results in embryonic lethality and failure of the epithelial component of the embryo to develop. Genes & Dev. 1992;6:2235–2247. doi: 10.1101/gad.6.12a.2235. [DOI] [PubMed] [Google Scholar]
- 33.Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331–333. doi: 10.1038/348331a0. [DOI] [PubMed] [Google Scholar]
- 34.Trudel M, Chretien N, D'Agati V. Disappearance of polycystic kidney disease in revertant c-myc transgenic mice. Mamm Genome. 1994;5:149–152. doi: 10.1007/BF00352345. [DOI] [PubMed] [Google Scholar]
- 35.Trudel M, D'Agati V, Costantini F. C-myc as an inducer of polycystic kidney disease in transgenic mice. Kidney Int. 1991;39:665–671. doi: 10.1038/ki.1991.80. [DOI] [PubMed] [Google Scholar]
- 35a.Trudel, M., L. Barisoni, J. Lanoix, and V. D'Agati. 1997. Polycystic kidney disease in SBM transgenic mice: role of c-myc in disease induction and progression. AM. J. Pathol. In press. [PMC free article] [PubMed]
- 36.Twardzik DR. Differential expression of transforming growth factor-α during prenatal development of the mouse. Cancer Res. 1985;45:5413–5416. [PubMed] [Google Scholar]
- 37.Vamvakas S, Bittner D, Koster U. Enhanced expression of the proto-oncogenes c-myc and c-fos in normal and malignant renal growth. Toxicol Lett (Amst) 1993;67:161–172. doi: 10.1016/0378-4274(93)90053-z. [DOI] [PubMed] [Google Scholar]
- 38.Wagner AJ, Kokontis JM, Hay N. Myc-mediated apoptosis requires wild-type p53 in a manner independent of cell cycle arrest and the ability of p53 to induce p21waf1/cip1 . Genes Dev. 1994;8:2817–2830. doi: 10.1101/gad.8.23.2817. [DOI] [PubMed] [Google Scholar]
- 39.Woo D. Apoptosis and loss of renal tissue in polycystic kidney diseases. N Eng J Med. 1995;333:18–25. doi: 10.1056/NEJM199507063330104. [DOI] [PubMed] [Google Scholar]
- 40.Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-XL . Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 41.Zimmerman KA, Yancopoulos GD, Collum RG, Smith RK, Kohl NE, Denis KA, Nau MM, Witte ON, Toran-Allerand D, Gee CE, et al. Differential expression of myc family genes during murine development. Nature. 1986;319:780–783. doi: 10.1038/319780a0. [DOI] [PubMed] [Google Scholar]