

Abstract
The endotoxic shock syndrome is characterized by systemic inflammation, multiple organ damage, circulatory collapse and death. Systemic release of tumor necrosis factor (TNF)-α and other cytokines purportedly mediates this process. However, the primary tissue target remains unidentified. The present studies provide evidence that endotoxic shock results from disseminated endothelial apoptosis. Injection of lipopolysaccharide (LPS), and its putative effector TNF-α, into C57BL/6 mice induced apoptosis in endothelium of intestine, lung, fat and thymus after 6 h, preceding nonendothelial tissue damage. LPS or TNF-α injection was followed within 1 h by tissue generation of the pro-apoptotic lipid ceramide. TNF-binding protein, which protects against LPS-induced death, blocked LPS-induced ceramide generation and endothelial apoptosis, suggesting systemic TNF is required for both responses. Acid sphingomyelinase knockout mice displayed a normal increase in serum TNF-α in response to LPS, yet were protected against endothelial apoptosis and animal death, defining a role for ceramide in mediating the endotoxic response. Furthermore, intravenous injection of basic fibroblast growth factor, which acts as an intravascular survival factor for endothelial cells, blocked LPS-induced ceramide elevation, endothelial apoptosis and animal death, but did not affect LPS-induced elevation of serum TNF-α. These investigations demonstrate that LPS induces a disseminated form of endothelial apoptosis, mediated sequentially by TNF and ceramide generation, and suggest that this cascade is mandatory for evolution of the endotoxic syndrome.
Endotoxic shock is a potentially lethal complication of systemic infection by gram-negative bacteria (1, 2). The toxin responsible for the induction of endotoxic shock is the glycolipid LPS, the only lipid present in the outer membrane of gram-negative bacteria. Release of LPS into the circulation activates a series of tissue responses that in their most severe forms lead to septic shock and death. Major events in the pathogenesis of the LPS syndrome include neutrophil, monocyte, and macrophage inflammatory responses, intravascular coagulopathy resulting from activation of plasma complement and clotting cascades, endothelial cell damage, and hypotension. Death of patients results from extensive tissue injury, multiple organ failure, and circulatory collapse.
Although a number of cytokines, including IL-1β, IL-6, and IL-8 are released by LPS-activated inflammatory cells during the onset of the endotoxic response (3), mounting evidence points to TNF-α as a primary mediator of this event (4–6). Not only are substantial quantities of TNF-α rapidly released into the circulation, but intravenous injection of TNF-α produces a systemic response very similar to LPS. Furthermore, approaches to interfere with TNF action, such as using neutralizing antibodies (4–6) or TNF binding proteins (TNF-bps), abrogate experimental endotoxic shock (7–11). Perhaps the most compelling evidence for a role for TNF-α is the attenuation of endotoxic shock observed in mice lacking the 55-kD TNF receptor (12, 13).
Although TNF-α was originally defined as a cytokine capable of inducing necrosis of tumors in vivo, recent studies suggest that in most instances TNF-α initiates an apoptotic form of cell death. In this regard, numerous studies have linked activation of the sphingomyelin pathway to the induction of apoptosis by TNF-α. The sphingomyelin pathway is an ubiquitous, evolutionarily conserved signaling system analogous to the cAMP and phosphoinositide pathways. Sphingomyelin (N-acylsphingosin-1-phosphocholine) is a phospholipid preferentially concentrated in the plasma membrane of mammalian cells (14). Sphingomyelin catabolism occurs via the action of sphingomyelin-specific forms of phospholipase C, termed sphingomyelinases, which hydrolyze the phosphodiester bond of sphingomyelin, yielding ceramide and phosphorylcholine. Several forms of sphingomyelinase exist, distinguished by their pH optima (15). Human and murine acid sphingomyelinase (ASMase; pH optimum 4.5–5.0) have been cloned and determined to be the products of a conserved gene, while Mg2+-dependent or -independent neutral SMases (pH optimum 7.4) have yet to be molecularly characterized. ASMase knockout mice retain NSMase activity, indicating that the neutral forms are products of a distinct gene or genes (16).
Signaling through the sphingomyelin pathway is mediated via generation of ceramide, which acts as a second messenger in stimulating a variety of cellular functions (for review see references 17–19). Receptors distinct as CD28, CD95, and the TNF-α, IL-1β, progesterone, γ-interferon, and glucocorticoid receptors signal via the sphingomyelin pathway after ligand binding. Thus ceramide signals pleiotropic cellular functions, including proliferation of fibroblasts, differentiation of promyelocytes, inhibition of the respiratory burst in human neutrophils, survival of T9 glioma cells, and apoptosis, to list a few.
Studies on the involvement of the sphingomyelin signaling system in apoptosis revealed that several cytokines and environmental stresses, including TNF-α (20–22), CD95/ Fas/APO-1 (23–25), ionizing radiation, ultraviolet-C, heat, and oxidative stress (26–28) induce rapid ceramide generation while effecting an apoptotic response. Furthermore, cell-permeable ceramide analogues, but not analogues of other lipid second messengers, mimicked the effect of cytokines and stress to induce apoptosis. Ceramide action was stereospecific, as analogues of the naturally occurring dihydroceramide, failed to initiate the apoptotic program. These studies suggested, but did not provide conclusive evidence, that ceramide mediates cytokine- and stress-induced apoptosis.
Definitive evidence for role of ASMase and ceramide in signaling one form of stress-induced apoptosis was derived from studies using genetic models of ASMase deficiency. Santana et al. (29) reported that lymphoblasts from patients with Niemann-Pick disease (NPD),1 an inherited deficiency of ASMase, manifested defects in ceramide generation and the apoptotic response to ionizing radiation. These abnormalities were reversible upon restoration of ASMase activity by retroviral transfer of human ASMase cDNA. Furthermore, ASMase knockout mice failed to generate ceramide and develop typical apoptotic lesions in the pulmonary endothelium after exposure to total body irradiation. The apoptotic response in the thymus, however, was preserved. The exact opposite occurred in irradiated p53 knockout mice. Whereas the thymus of the p53 knockout mouse was protected against radiation-induced apoptosis, the lung endothelium was not. Differences were observed in other tissues as well. While these studies demonstrated that radiation is capable of activating two apparently distinct and independent signaling mechanisms for induction of apoptosis, they also suggested a specific sensitivity of endothelial cells towards the ASMase-mediated signaling system for initiating apoptosis in response to stress.
Since both TNF-α and endothelial cell damage are critically involved in the pathogenesis of the endotoxic syndrome, we explored whether ceramide-mediated endothelial cell apoptosis plays a role in the LPS-induced response in vivo. Genetic and pharmacologic manipulations allowed for molecular ordering of the early and critical events in the progression of this syndrome. The data show that injection of LPS into C57BL/6 mice resulted in a disseminated form of microvascular endothelial apoptosis, mediated sequentially by TNF and ceramide generation, and suggested that this cascade plays a mandatory role in the evolution of LPS-induced death.
Materials and Methods
LPS Treatment.
Using a 26-gauge needle, C57BL/6 mice were injected intraperitoneally with LPS (Salmonella typhimurium; Westphal purified, Difco Laboratories, MI) resuspended in sterile water. For TNF-α and TNF-bp injections, mice were first anesthetized with pentobarbital (50 mg/kg) intraperitoneally. After obtaining adequate anesthesia, recombinant human TNF-α or TNF-bp (Amgen, Boulder, CO) was injected intravenously with a 28-gauge needle via a retro-orbital approach. Sham-injected animals received diluent. For studies measuring survival, animals were monitored for up to 2 wk. Survival as the end point in these experiments was calculated from the time of treatment using the product limit Kaplan-Meier method (30). Calculations of the dose leading to 50% lethality (LD50) at a given time after LPS treatment was performed using probit analysis. For studies evaluating histology or tissue ceramide content, mice were killed by hypercapnia asphyxiation.
Mice were housed in a pathogen-free environment in our animal facility. This facility is approved by the American Association for Accreditation of Laboratory Animal Care and is maintained in accordance with the regulations and standards of the United States Department of Agriculture and the Department of Health and Human Services, National Institutes of Health.
Lipid Studies.
For studies measuring tissue ceramide levels, abdominal contents of killed animals were immediately exposed through a midline incision, and the gastric pylorus was identified. The duodenum was transected and the proximal 3–4 cm of small intestine were excised and placed on ice. Using a Nikon SMZ-2B dissecting microscope set at 10× magnification, the antimesenteric border of the bowel was incised, exposing the mucosal surface of the bowel. The bowel was irrigated with cold PBS and the mucosa bluntly dissected from the underlying muscularis propria with curved tissue forceps. Mucosa were homogenized in 8 vol (vol/vol) of ice-cold PBS. Homogenate (0.6 ml) was transferred to 16 × 100-mm glass tubes and lipids were extracted with 3 ml of chloroform/methanol (2:1 vol/vol). After mild alkaline hydrolysis to remove glycerophospholipids ceramide was quantified using E. coli diacylglycerol kinase (Calbiochem Novabiochem, La Jolla, CA) as described (27).
Apoptosis.
Apoptosis in vivo was assessed by the DNA terminal transferase nick-end translation method (also termed the TUNEL assay), as described (31). In brief, tissue specimens were fixed overnight in 4% buffered formaldehyde and embedded in paraffin blocks. 5-μm-thick tissue sections, adherent to polylysine-treated slides, were deparaffinized by heating at 90°C for 10 min and then at 60°C for 5 min. Tissue-mounted slides were first washed with 90% and then 80% ethanol (3 min each) and rehydrated. The slides were incubated in 10 mM Tris-HCl, pH 8, for 5 min, digested with 0.1% pepsin, rinsed in distilled water, and treated with 3% H2O2 in PBS for 5 min at 22°C to inactivate endogenous peroxidase. After three washes in PBS, the slides were incubated for 15 min at 22°C in buffer (140 mM Na-cacodylate, pH 7.2, 30 mM Trizma base, 1 mM CoCl2) and then for 30 min at 37°C in reaction mixture (0.2 U/μl terminal deoxynucleotidyl transferase, 2 nM biotin-11-dUTP, 100 mM Na-cacodylate, pH 7.0, 0.1 mM DTT, 0.05 mg/ml bovine serum albumin, and 2.5 mM CoCl2). The reaction was stopped by transferring the slices to a bath of 300 mM NaCl, 30 mM Na citrate for 15 min at 22°C. The slides were washed in PBS, blocked with 2% human serum albumin in PBS for 10 min, rewashed, and then incubated with avidin-biotin peroxidase complexes. After 30 min at 22°C, cells were stained with the chromogen 3,3′diamonobenzidine tetrachloride and counterstained with hematoxylin. Nuclei of apoptotic cells appear brown and granular, while normal nuclei stain blue.
Some studies employed double staining with TUNEL to assess apoptosis followed by immunostaining with a rat monoclonal anti-CD 31 antibody to identify endothelial cells. For these studies, TUNEL-stained sections were incubated with normal rabbit serum (10% in PBS-bovine serum albumin; Cappel Labs., Cochranville, PA) and subsequently with a primary rat anti-CD31 antibody at 4°C (1:500 dilution; PharMingen, San Diego, CA). A rat mAb of the same subclass as the primary antibody was used as a negative control at a similar working dilution. Biotinylated rabbit anti–rat antibodies were applied for 1 h (1:100 dilution; Vector Laboratories, Burlingame, CA), followed by avidin-biotin peroxidase complexes for 30 min (1:25 dilution; Vector Laboratories). True Blue Peroxidase substrate was used as the final chromogen (KPL Laboratories, Gaithersburg, MD) and nuclear fast red (Vector Labs. Inc., Burlingame, CA) was used as the nuclear counterstain. Cell surface immunoreactivity was identified as a dark blue staining. Double staining was considered positive when specific cells displayed a brown nuclear stain in the context of a surrounding or superimposed blue-to-black membrane immunoreactive pattern. The scoring of stained tissue was conducted independently by two investigators. The areas scored were always selected randomly and counts by each of the investigators were carried out in a blinded fashion, unveiling the code at the end of the study.
Serum TNF-α Levels.
Blood was obtained from anesthetized mice through an abdominal incision by aspiration from the inferior vena cava using a 28-gauge needle (Becton Dickinson, Rutherford, NJ). Serum TNF-α levels were measured by ELISA according to the manufacturer's instructions (Biosource International, Camarillo, CA).
Statistical Analysis.
Statistical analysis were performed by Student's t test and Chi Square test. Differences in product limit Kaplan Meier survival curves were evaluated by the Mantel log-rank test for censored data (32).
Results
Initial studies examined the time course and dose dependence of LPS-induced death of C57BL/6 mice. For these studies, S. typhimurium LPS or diluent were injected intraperitoneally. Death was detected as early as 16 h after a maximal dose of LPS (270 μg/25g mouse) and all of the mice were dead after 48 h. As little as 60 μg of LPS/25g mouse was effective and the LD50 was ∼90 μg of LPS/25g mouse.
To explore whether endothelial cell apoptosis is associated with the LPS response, C57BL/6 mice were injected with 90 μg of LPS/25 g of mouse body weight and multiple tissues were evaluated for an apoptotic response using the TUNEL method. Fig. 1 shows that LPS induced an apoptotic response in microvascular endothelial cells of intestinal crypts, the lung, pericolic fat, and thymus. Crypts of the intestinal mucosa are comprised of a layer of columnar epithelial cells on the intestinal luminal surface and a central network of capillaries in the lamina propria. Intestinal crypts from sham injected animals demonstrated minimal apoptosis (Fig. 1 A, left). Apoptotic cells display an intense brown nuclear stain, whereas the nuclei of unaffected cells are visualized blue due to the hematoxylin counterstaining. LPS-injected animals, however, demonstrated diffuse endothelial apoptosis with little if any changes in the epithelial cell layer (Fig. 1 A, middle). This effect was maximal at 6 h and preceded the onset of apoptosis in the epithelial cells of the crypt, which became apparent after 8–10 h (data not shown). Similarly, the lungs of sham-treated animals displayed little apoptosis in either capillary endothelial cells or in tissue pneumocytes (Fig. 1 A, left). Substantial and selective apoptotic damage was detected, however, in the pulmonary microvascular endothelium in response to LPS injection by 6 h (Fig. 1 A, middle). In both these tissues, hematoxylin- and eosin-stained sections from LPS-treated animals revealed large numbers of endothelial cells with shrunken pyncnotic nuclei, many of which were fragmented (data not shown). These apoptotic cells appeared to be phagocytized by neighboring cells in some sections. Apoptotic damage to the endothelium of pericolic fat tissue was similarly detected by 6–8 h after LPS injection, while adipocytes and fibroblasts, seen on the periphery of Fig. 1 A, middle, were spared. This effect was also observed in mediastinal and subcutaneous fat tissue (data not shown). In all of these organs, the extent of endothelial, and the subsequent nonendothelial, tissue damage was dose-dependent, increasing from 60 to 175 μg of LPS/25 g of mouse body weight (data not shown).
Figure 1.
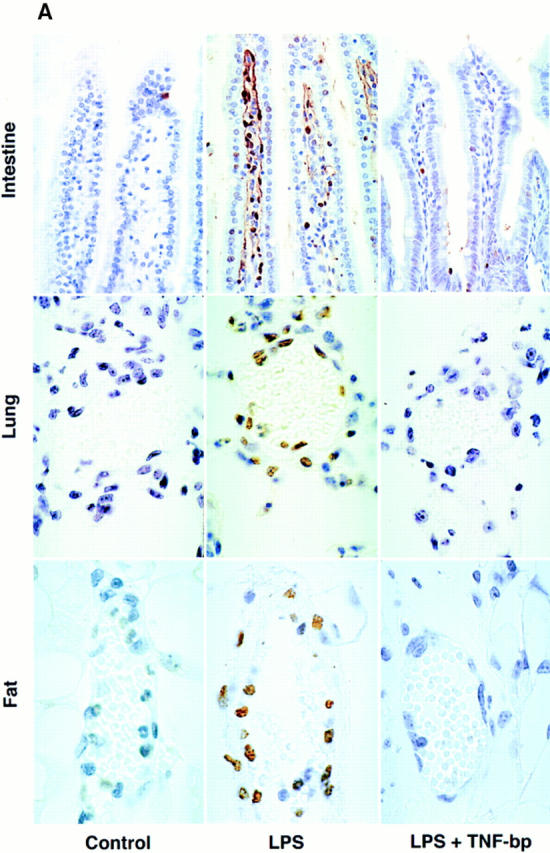

LPS induces, and TNF-bp blocks, apoptosis in the endothelium of (A) intestine, lung, pericolic fat, and (B) thymus. C57BL/6 mice were injected intraperitoneally with 90 μg of S. typhimurium LPS/25 g of mouse body weight or diluent (PBS), and after 6 h were killed by hypercapnia asphyxiation. For studies using TNF-bp, animals were injected with 75 μg of TNF-bp/25 g of mouse body weight or with diluent (PBS) 2 h before LPS. Tissue specimens were fixed overnight in 4% buffered formaldehyde and apoptosis assessed as in Materials and Methods by TUNEL assay (A) or a combination of TUNEL and immunohistochemical staining for the cell surface antigen CD31 (B). Nuclei of apoptotic cells appear brown and granular, and in B are surrounded by a blue-black perimeter. Normal nuclei in A stain blue and in B stain red due to hematoxylin and fast red counterstains, respectively. Original magnifications: intestine ×400; lung, pericolic fat and thymus ×1,000. This experiment represents one of three similar studies.
Apoptosis was also observed in thymic tissue by 6 h after LPS injection. Apoptotic cells, as assessed by the TUNEL assay, appeared in the thymus as discrete foci manifesting a configuration reminiscent of a vascular formation (data not shown). However, the dense packing of cells within this tissue precluded histologic and morphologic identification of the apoptotic cells as endothelium. To determine whether these apoptotic cells were of endothelial origin, we developed a double staining technique. Thymic tissue, stained by the TUNEL method to detect apoptotic nuclei, were costained immunohistochemically with an antibody to the endothelial cell surface antigen CD31, also known as platelet endothelial cell adhesion molecule (PECAM)-1 (33). Normal endothelium of thymic microvessels were identified by dark blue staining of the cell membrane, whereas thymocytes lacked this stain and manifested only a light red nuclear color resulting from the use of fast red as counterstain (Fig. 1 B, left). In specimens from LPS-treated mice, apoptotic endothelial cells displayed a central brown nuclear core surrounded by a blue-black perimeter (Fig. 1 B, middle). It should be noted that the microvessels identified in the thymus are comprised of only two to four endothelial cells and thus appear smaller than those in the lung and fat, which were frequently comprised of five to eight endothelial cells. Using this double staining technique, we were able to demonstrate that virtually all of the apoptotic cells present in the thymus at 6 h after LPS stimulation represented endothelial cells in microvessels, the lumens of which were partially or completely collapsed (Fig. 1 B, middle). It should also be noted that endothelial apoptosis occurred in all of these tissues in the absence of an inflammatory response, which was subsequently detected at 10–12 h. Taken together, these studies indicate that intraperitoneal injection of LPS induces a disseminated form of endothelial apoptosis, which precedes nonendothelial parenchymal tissue damage.
The extent of microvascular involvement was quantified. Table 1 shows that 71% of the intestinal villae and 64–79% of the microvessels of the lung, fat and thymus displayed apoptotic damage at 6 hours after a dose of 90 μg of LPS/ 25g mouse. Similar effects were observed at 8 h after injection of 90 μg of LPS/25g mouse and with 175 μg of LPS/ 25g mouse (data not shown). It should be noted that apoptosis was detected in less than 5% of microvessels in tissues from control sham-treated animals (data not shown).
Table 1.
Quantitation of Microvascular Apoptosis after LPS and Inhibition by TNF-bp
| Apoptotic Blood Vessels | ||||
|---|---|---|---|---|
| Tissue | LPS | LPS + TNF bp | ||
| Intestine | 107 (71%) | 13 (8%)* | ||
| Lung | 104 (70%) | 12 (8%)* | ||
| Fat | 118 (79%) | 32 (21%)* | ||
| Thymus | 95 (64%) | 20 (13%)* | ||
Tissues, obtained from mice treated as in Fig. 1, were analyzed for the extent of apoptosis. 150 intestinal villae or capillaries from lung, fat or thymus, were scored for apoptosis. Data are presented as the number of positive villae or vessels, and the percentage positively is shown in parentheses.
P <0.001 vs. LPS alone.
To determine whether ceramide generation plays a role in LPS-induced apoptosis, C57BL/6 mice were treated with 175 μg of LPS/25g mouse and at various periods of time thereafter, the intestinal mucosa was dissected away from the muscularis layer. Ceramide content of the intestinal mucosa significantly increased from a basal level of 1,200 pmol/mg tissue by 1 h after LPS injection and peaked at twofold by 2 h (P <0.001 vs. control; Fig. 2 A). As little as 60 μg/25g mouse was effective and a maximal effect occurred with 175 μg/25g mouse (Fig. 2 B). Similar ceramide elevation was detected in the lung of C57BL/6 mice within the first hour after LPS injection (n = 3; data not shown). In contrast, the level of the lipid second messenger 1,2-diacylglycerol was not elevated (data not shown). These studies demonstrate that ceramide generation precedes the apoptotic response.
Figure 2.

LPS induces rapid ceramide generation in the mucosa of the intestine. These studies were performed as in Fig. 1 except mice were killed at the indicated times. The intestinal mucosa was dissected as described in Materials and Methods. Mucosal specimens were homogenized in 8 vol (vol/vol) of ice-cold PBS and lipids were extracted with 2 ml of chloroform:methanol (2:1, vol/vol)/400 μl of homogenate. After mild alkaline hydrolysis to remove glycerophospholipids, ceramide was quantified using E. coli diacylglycerol kinase (Calbiochem Novabiochem) as described (27), and results normalized for protein content. The data (mean ± SEM) represent triplicate determinations from two mice per point from two experiments for time course (A), and from one representative of two experiments for dose dependence (B).
Since TNF-α is a primary mediator of the septic shock response to LPS (4–6), and since ceramide has been described as a mediator of TNF-induced apoptosis in numerous cellular systems (17–19), we investigated the effect of TNF-α on tissue ceramide generation, endothelial apoptosis, and survival of C57BL/6 mice. Recombinant human TNF-α, when injected intravenously, induced time- and dose-dependent lethality in this strain of mice. As little as 5 μg of TNF-α/25g mouse was effective and the LD50, although somewhat variable between experiments, ranged from 25–50 μg of TNF-α/25g mouse. At a dose of 25 μg of TNF-α/25g mouse, death occurred as early as 10 h after injection and the mean time until death in multiple experiments was 24 h (data not shown). Fig. 3 A shows that TNF-α induced time- and dose-dependent ceramide generation in the intestinal mucosa. 25 μg of TNF-α/25g mouse stimulated an increase in ceramide content with a slightly more rapid time course than induced by LPS. TNF-α–induced ceramide generation was detected by 0.5 h and peaked at 1.5 h (P <0.001 vs. control). As little as 2.5 μg of TNF-α/25g mouse was effective and a maximal effect occurred with 25 μg of TNF-α/25g mouse (Fig. 3 B). TNF-α, like LPS, induced endothelial apoptosis in intestinal mucosa, lung, and fat tissues, beginning 6 h after injection (data not shown). These studies demonstrate that TNF-α, like LPS, induces tissue ceramide generation followed by microvascular endothelial apoptosis and demise of the animal.
Figure 3.

TNF-α induces ceramide generation in intestinal mucosa. (A) Time course of the effect of 25 μg of recombinant human TNF-α/ 25g mouse; (B) dose response at 2 h. These studies were performed as in Fig. 2 except C57BL/6 mice were injected with TNF-α retro-orbitally. The data (mean ± SEM) represent triplicate determinations from two mice per point from one representative of three experiments for A and one representative of four experiments for B.
Agents that inhibit TNF-α action have been shown to prevent the endotoxic shock response in a variety of different experimental models. These include neutralizing antibodies to TNF-α (4–6), chimeric inhibitors comprised of the extracellular domain of the TNF receptor fused with an immunoglobulin heavy chain fragment (10) or as a polyethylene glycol-linked dimer (TNF-bp) (9, 11, 34), and a TNF convertase metalloproteinase inhibitor (35), to list a few. To evaluate whether the effect of TNF-α to induce tissue ceramide generation and endothelial apoptosis is essential for the LPS effect, we injected TNF-bp with LPS. Fig. 4 shows that intravenous injection of TNF-bp (serum t 1/2 ∼30 h) abolished the effect of a maximal dose of 175 μg of LPS/25g mouse on ceramide generation in the intestinal mucosa. Furthermore, TNF-bp markedly attenuated LPS-induced apoptosis in the endothelium of the intestine, lung, pericolic fat, and thymic tissue at 6 h (Fig. 1, right panels) and at 8–10 h (data not shown) after stimulation. Quantitation of apoptotic microvessels in tissues treated with TNF-bp and LPS, demonstrated near complete protection from apoptosis in all tissues (Table 1; P <0.001 vs. LPS-treated for each tissue). These studies provide evidence that LPS-induced ceramide generation, endothelial apoptosis, and endotoxic death require TNF-α action.
Figure 4.

TNF-bp blocks LPS-induced ceramide generation. For these studies, animals were injected with TNF-bp and LPS as described in Fig. 1, and ceramide levels determined as in Fig. 2. These data (mean ± SEM) represent triplicate determinations from two mice per point from two experiments.
To determine whether ceramide generation is necessary for progression of the endotoxic syndrome, we treated wild-type and ASMase knockout mice with 175 μg of LPS/25g mouse. LPS-induced elevation of serum TNF-α was unaffected in the ASMase knockout mice, increasing to a maximum of 12 ± 3 ng/ml at 1.5 h after LPS injection. These data indicate that monocyte/macrophage activation is normal in the ASMase mouse. However, ASMase knockout mice were defective in LPS-induced ceramide generation and endothelial apoptosis. In contrast to the twofold maximal ceramide elevation observed in the intestines of wild-type animals 2 h after 175 μg of LPS/25 g mouse, in the ASMase knockout mouse the ceramide level did not increase significantly and after 2 h was only 1.27 ± 0.18-fold of control (mean ± SD; 4 mice/group). Furthermore, upon evaluation of 150 intestinal villae for apoptotic microvessels at 6 h after injection of 175 μg of LPS/25g mouse, 118 (79%) were positive in the wild-type mice, while only 17 (11%) were positive in the ASMase knockout mice (P <0.001 vs. LPS-treated wild type; 6 mice/ group). Upon evaluation of 150 capillaries in lungs from the same animals, 109 (72%) demonstrated apoptotic damage in the wild type animal, whereas only 15 (10%) were apoptotic in the ASMase knockout mice (P <0.001 vs. LPS-treated wild type). Apoptosis progressed with time in LPS-treated ASMase knockout mice, reaching a maximum at 8 h when 33 of 150 (22%) intestinal villae, and 29 of 150 (19%) lung microvessels, were positive (P <0.001 vs. LPS-treated wild-type mice tissues in which 70–80% of microvessels display apoptosis at 8 h). Furthermore, ASMase knockout mice were protected against LPS-induced death (P = 0.05 vs. LPS-treated wild type; Fig. 5). These studies suggest that LPS-induced apoptosis, like radiation-induced apoptosis, requires a functional sphingomyelin pathway.
Figure 5.

Acid SMase knockout mice are defective in LPS-induced death. Actuarial (Kaplan-Meier) survival curves of wild-type and ASMase knockout mice injected intraperitoneally with 175 μg of LPS/25g mouse. The number in parenthesis indicates the number of animals in each group.
To provide additional support for the notion that endothelial damage is essential for evolution of the endotoxic response, C57BL/6 mice were treated concomitantly with LPS and bFGF. Prior studies from our laboratory showed that bFGF protected endothelium in vitro and in vivo from radiation-induced apoptosis (31). In vivo, intravenously injected bFGF has been shown to be retained within blood vessels, apparently bound to the heparan sulfate proteoglycan coating the vascular surface of the endothelium and its basement membrane (31). Consequently, intravenously injected bFGF served as a selective endothelial survival factor, preventing radiation-induced apoptosis and lethal radiation pneumonitis (36). Fig. 6 A shows that bFGF abrogated LPS-induced apoptosis in the endothelium of the intestine and lung of C57BL/6 mice. Fig. 6 B demonstrates that intravenous bFGF, when injected concomitantly with a dose of 175 μg of LPS/25g mouse, provided protection from the lethal effects of LPS (P <0.001 vs. untreated). bFGF also rescued C57BL/6 mice from maximal doses of 270 and 350 μg of LPS/25g mouse, although the protection was not as complete (data not shown). Additional studies delineated the site of bFGF action. Table 2 shows that while TNF-α was not detected in the serum of sham- or bFGF-injected animals, 175 μg of LPS/25g mouse induced an elevation of serum TNF-α to a maximum of 4.2 ± 0.9 ng/ ml. The LPS-induced elevation of serum TNF-α was not blocked by bFGF. In contrast, bFGF prevented the elevation of tissue ceramide in response to LPS. These studies indicate that intravenous bFGF does not affect cell types that generate TNF-α in response to LPS (i.e., monocytes and macrophages), but specifically targets endothelial cells and the ceramide response to TNF stimulation. These data also substantiate endothelial damage as mandatory for LPS-induced death, and define inhibition of TNF signaling as the mechanism of the protective effect of bFGF on endothelium.
Figure 6.
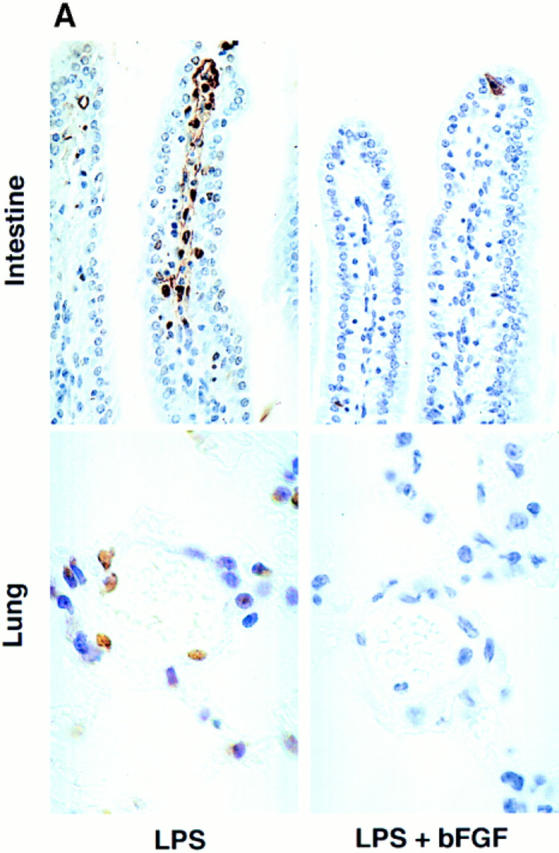

Basic FGF blocks LPS-induced endothelial apoptosis and animal death. (A) C57BL/ 6 mice injected intravenously with 800 ng bFGF 30 min before, and 5 min, 1 and 2 h after, an intraperitoneal injection of 175 μg of LPS/25g mouse. Endothelial apoptosis was assessed as in Fig. 1 by TUNEL assay. (B) Actuarial (Kaplan-Meier) survival curves of C57BL/6 treated as in A. The number in parenthesis indicates the number of animals in each group.
Table 2.
Basic FGF Blocks LPS-induced Tissue Ceramide Generation but Not Serum TNF-α Elevation
| Control | bFGF | LPS | LPS + bFGF | |||||
|---|---|---|---|---|---|---|---|---|
| TNFα (ng/ml) | n.d. | n.d. | 4.2 ± 0.9 | 7.9 ± 2.0 | ||||
| Ceramide (pmol/mg prot) | 1942 ± 97 | 1352 ± 113 | 3175 ± 274* | 1810 ± 108 |
Blood and intestinal mucosa were obtained from anesthetized mice (3 per group) 1.5 h after intraperitoneal injection of 175 μg of LPS/25g mouse with or without a single intravenous injection of 800 ng bFGF, as in Fig. 6 a. Serum TNF-α levels were measured in triplicate by ELISA as described in Materials and Methods. Ceramide content was measured in triplicate as described in Fig. 2. n.d., not detected.
P <0.01 vs. LPS + bFGF and control.
Discussion
The present studies define a set of early biochemical and biological responses to LPS using a standard model of endotoxic shock. Fig. 7 orders these events. Within 1 h of intraperitoneal injection of LPS, elevation of tissue ceramide content was detected in the intestinal mucosa and lung. Although our evidence supports endothelium as the primary source of the increase in ceramide, it remains formally possible that cells other than endothelium contribute to the ceramide elevation. Ceramide elevation appeared dependent on TNF action since TNF mimicked the LPS effect, and TNF-bp blocked the LPS-induced increase in tissue ceramide. Elevation of ceramide preceded the appearance of a generalized form of apoptosis, expressed initially in the microvascular endothelium of a variety of organs, beginning at 6 h after LPS injection. Both ceramide elevation and endothelial apoptosis preceded damage to nonendothelial parenchymal tissue and the death of the animal, which became apparent at 16 h after a dose of 175 μg LPS/25g mouse. Endothelial apoptosis appeared mandatory for the progression of the endotoxic response, since intravenous injection of bFGF, which specifically protects the endothelium against stress-induced apoptosis, prevented death. Furthermore, ceramide appeared to be a key intracellular mediator of this response, as the ASMase knockout mouse, which is defective in ceramide generation but not in TNF-α production, exhibited decreased endothelial apoptosis and death. Nevertheless, additional models of endotoxic shock must be studied before it can be concluded that these observations are representative of this process in general, as significant species differences have been observed (3).
Figure 7.

Proposed schema for progression of the endotoxic response. LPS, released by gram negative bacteria, interacts with inflammatory cells leading to generation of TNF-α and other cytokines. TNF-α, acting upon endothelium, stimulates sphingomyelin hydrolysis to ceramide via an ASMase. Apoptosis of the endothelium ensues, which can be blocked by bFGF via inhibition of ceramide generation. We further propose that endothelial apoptosis results in generalized microvascular dysfunction sufficient to compromise the circulation to major organs, leading to nonendothelial tissue damage, circulatory collapse, and death.
Previous studies reported that LPS induces endothelial damage in vivo and under some conditions in vitro. Microvascular injury occurs in numerous tissues during sepsis, including the lung, gut and liver, and this event has been generally considered an important element in the pathogenesis of the septic shock syndrome (1). The mechanism of microvascular injury and its relevance to the evolution of the septic shock syndrome have been a subject of substantial debate. Disseminated intravascular thrombosis, extensive endothelial necrosis, and humoral microvascular dysfunction have all been ascribed a role as mediating vascular collapse (1). Generalized endothelial apoptosis has not hitherto been reported, although apoptosis of liver endothelium ex vivo was recognized subsequent to induction of TNF-α on Kupfer cells by LPS (37). The large majority of studies reported that LPS did not induce apoptosis in primary cultures of endothelial cells (37–40) unless a second stress such as heat shock or cycloheximide was applied subsequently (39, 40). However, one group has argued that LPS can induce direct DNA damage leading to apoptosis in primary cultures of sheep pulmonary endothelial cells (41, 42).
In the present studies, endothelial apoptosis appeared to be preferentially increased in tissues known to play prominent roles in the pathogenesis of endotoxic shock. In this regard, the microvascular endothelium of the bowel and lung were markedly affected. However, even the endothelium of tissues which play no overt role in the endotoxic response, such as the pericolic fat and the thymus, seemed to be affected. Endothelial damage preceded nonendothelial damage suggesting that loss of vascular integrity may play a role in the parenchymal tissue damage and the multi-organ failure that characterizes the endotoxic syndrome. It is reasonable to suggest that the generalized nature of the apoptotic response in the microvascular endothelium may account, in part, for the circulatory failure that is a major factor in the progression of the endotoxic response. Whether endothelial apoptosis in the lung is the critical lesion leading to the asphyxiation that results in the ultimate demise of affected mice (5) cannot be ascertained from the present information.
The critical role of endothelial cell apoptosis in the pathogenesis of endotoxic shock is similar to its role in the evolution of the inflammatory phase of radiation-induced pneumonitis. As in the case of the LPS response, microvascular endothelial apoptosis preceded the expression of other histopathological manifestations of lethal radiation-induced pneumonitis, and intravenous injections of bFGF abrogated the evolution of pneumonitis and death after whole lung irradiation (31, 36). Furthermore, both LPS- and radiation-induced endothelial apoptosis in vivo appeared initiated by activation of ASMase. Thus, the sphingomyelin pathway may integrate diverse responses to signal death in stressed endothelial cells. Consistent with this hypothesis, the prevention of ceramide generation by bFGF suggests that the anti-apoptotic survival function of bFGF may be mediated, in part, via this mechanism.
The present investigations establish a role for TNF-α in LPS-induced generation of ceramide and apoptosis in vivo. Recent studies have clarified the mechanism by which the 55-kD TNF receptor signals the apoptotic response (43– 52). This receptor contains a carboxy-terminal death domain which appears to be required for transmission of the apoptotic signal. Binding of TNF-α to the receptor triggers formation of a multiprotein complex in which cytoplasmic proteins and the receptor interact through their respective death domain motifs. Upon TNF stimulation, the receptor death domain binds to the death domain of a cytoplasmic protein known as TRADD (TNF receptor 1–associated death domain), which in turn binds the death domain of another cytoplasmic protein, termed FADD/MORT-1. The latter protein also contains a death effector domain (DED) motif, which binds the DED of the ICE/Ced-3 protease FLICE/MACH-1 (Caspase 8). It has been suggested that activation of FLICE/MACH-1 initiates activation of a cascade of caspases, which serves as the effector system for the apoptotic destruction of the cell.
This model suggests that ligand binding to the TNF receptor is capable of activating the final death effector pathway without apparent involvement of lipid second messengers. However, several recent studies demonstrated a role for ceramide in TNF-induced cell death, in some systems. In this regard, activation of the death domain system of the 55-kD TNF and CD95 receptors has been shown to couple to ASMase (24, 53). This notion was based on the observation that mutations in these receptor death domains that abolished apoptosis also abolished ceramide generation. Furthermore, dominant negative FADD/MORT-1 blocked ceramide generation and apoptosis in BJAB B lymphoma cells, but not apoptosis induced by ceramide analogues. Whether ASMase activation might couple to FLICE/MACH-1 activation is presently uncertain. However, Pronk et al. (54), using peptide inhibitors of ICE/Ced-3 proteases, molecularly ordered ceramide generation downstream of an undefined CPP32-like protease during REAPER-induced apoptosis in Drosophila Schneider L2 cells. In the present studies, TNF appeared essential for ceramide generation during the evolution of the endotoxic syndrome. Whether the TNF receptor death domain adaptor protein system is involved in LPS-induced ceramide generation via TNF-α in vivo, remains uncertain.
Although the present studies define ceramide as critical for the induction of endothelial apoptosis by LPS, its precise role in signaling apoptosis is as yet unknown. Kroemer and coworkers (55) have provided evidence that ceramide acts upstream of mitochondria to initiate apoptosis. Their investigations suggested that ceramide, once generated, signals mitochondrial membrane permeability transition (MPT), a committed step in the apoptotic process. MPT may signal apoptosis via release of an apoptosis-initiating factor, a Z-VAD- but not DEVD-inhibitable ICE-like protease (55). Consistent with this paradigm, Pastroini et al. (56) showed that TNF-α– and ceramide-stimulated MPT was not inhibited by the protein synthesis inhibitor cycloheximide. Alternatively, ceramide-initiated MPT may involve the release of cytochrome C from mitochondria and activation of a CPP32-like protease (Caspase 3) (57–59). Either scenario is consistent with the inhibition of ceramide-mediated apoptosis by Bcl-2 that has been reported in numerous systems (55, 60–62). Whether ceramide-mediated mitochondrial damage is linked to the SAPK/JNK signaling system, also reported to be critically involved in TNF-mediated apoptosis in endothelial cells (26), remains unknown.
The present investigations enhance the understanding of the mechanism of the endotoxic syndrome, defining upstream elements of the LPS signaling system and their molecular ordering, as well as the early tissue responses that trigger its pathogenesis. The identification of biochemical pathways that signal pro- and anti-apoptosis during the LPS response, and the characterization of the primary tissue target for the endotoxic syndrome, provide a molecular and cellular context for testable experimental hypotheses, and a basis for developing strategies for pharmacologic intervention, with potential for clinical application. In particular, the ability of bFGF to inhibit ceramide generation suggests that treatment with bFGF may affect the progression of the LPS syndrome in gram-negative septicemia with evidence of rising serum TNF, or in patients already manifesting symptoms of septic shock.
Acknowledgments
This work was supported by grant CA52462 to Z. Fuks from the National Institutes of Health.
Footnotes
1 Abbreviations used in this paper: MPT, mitochondrial membrane permeability transition; NPD, Niemann-Pick disease; PECAM, platelet endothelial cell adhesion molecule-1.
Correspondence should be addressed to Richard Kolesnick, Laboratory of Signal Transduction, Memorial Sloan-Kettering Cancer Center, 1275 York Ave., New York, NY 10021. Tel.: (212) 639-8573; Fax: (212) 639-2767.
References
- 1.Morrison DC, Ryan JL. Endotoxins and disease mechanisms. Ann Rev Med. 1987;38:417–432. doi: 10.1146/annurev.me.38.020187.002221. [DOI] [PubMed] [Google Scholar]
- 2.Bone RC. Sepsis, the sepsis syndrome, multi-organ failure: a plea for comparable definitions. Ann Int Med. 1991;114:332–333. doi: 10.7326/0003-4819-114-4-332. [DOI] [PubMed] [Google Scholar]
- 3.Tracey KJ, Lowry SF. The role of cytokine mediators in septic shock. Adv Surg. 1990;23:21–56. [PubMed] [Google Scholar]
- 4.Beutler B, Milsark IW, Cerami AC. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science (Wash DC) 1985;229:869–871. doi: 10.1126/science.3895437. [DOI] [PubMed] [Google Scholar]
- 5.Tracey KJ, Beutler B, Lowry SF, Merryweather J, Wolpe S, Milsark IW, Hariri RJ, Fahey TJ, III, Zentella A, Albert JD, et al. Shock and tissue injury induced by recombinant human Cachectin. Science (Wash DC) 1986;234:470–474. doi: 10.1126/science.3764421. [DOI] [PubMed] [Google Scholar]
- 6.Mathison JC, Wolfson E, Ulevitch RJ. Participation of tumor necrosis factor in the mediation of gram negative bacterial lipopolysaccharide-induced injury in rabbits. J Clin Invest. 1988;81:1925–1937. doi: 10.1172/JCI113540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lesslauer W, Tabuchi H, Gentz R, Brockhaus M, Schlaeger EJ, Grau G, Piguet PF, Pointaire P, Vassalli P, Loetscher H. Recombinant soluble tumor necrosis factor receptor proteins protect mice from lipopolysaccharide-induced lethality. Eur J Immunol. 1991;21:2883–2886. doi: 10.1002/eji.1830211134. [DOI] [PubMed] [Google Scholar]
- 8.Hale KK, Smith CG, Baker SL, Vanderslice RW, Squires CH, Gleason TM, Tucker KK, Kohno T, Russell DA. Multifunctional regulation of the biological effects of TNF-alpha by the soluble type I and type II TNF receptors. Cytokine. 1995;7:26–38. doi: 10.1006/cyto.1995.1004. [DOI] [PubMed] [Google Scholar]
- 9.Russell DA, Tucker KK, Chinookoswong N, Thompson RC, Kohno T. Combined inhibition of interleukin-1 and tumor necrosis factor in rodent endotoxemia: improved survival and organ function. J Infect Dis. 1995;171:1528–1538. doi: 10.1093/infdis/171.6.1528. [DOI] [PubMed] [Google Scholar]
- 10.Van Zee K, Moldawer L, Oldenburg H, Thompson W, Stackpole S, Montegut W, Rogy M, Meschter C, Gallati H, Schiller C, et al. Protection against lethal Escherichia coli bacteremia in baboons (Papio anubis) by pretreatment with a 55-kD TNF receptor (CD120a)-Ig fusion protein, Ro 45-2081. J Immunol. 1996;156:2221–2230. [PubMed] [Google Scholar]
- 11.Espat NJ, Cendan JC, Beierle EA, Auffenberg TA, Rosenberg J, Russell D, Kenney JS, Fischer E, Montegut W, Lowry SF, et al. PEG-BP-30 monotherapy attenuates the cytokine-mediated inflammatory cascade in baboon Escherichia coliseptic shock. J Surg Res. 1995;59:153–158. doi: 10.1006/jsre.1995.1147. [DOI] [PubMed] [Google Scholar]
- 12.Rothe J, Lesslauer W, Lotscher H, Lang Y, Koebel P, Kontgen F, Althage A, Zinkernagel R, Steinmetz M, Bluethmann H. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature (Lond) 1993;364:798–802. doi: 10.1038/364798a0. [DOI] [PubMed] [Google Scholar]
- 13.Pfeffer K, Matsuyama T, Kundig TM, Wakeham A, Kishihara K, Shahinian A, Wiegmann K, Ohashi PS, Kronke M, Mak TW. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 1993;73:457–467. doi: 10.1016/0092-8674(93)90134-c. [DOI] [PubMed] [Google Scholar]
- 14.Merrill J-AH, Jones DD. An update of the enzymology and regulation of sphingomyelin metabolism. Biochim Biophys Acta. 1990;1044:1–12. doi: 10.1016/0005-2760(90)90211-f. [DOI] [PubMed] [Google Scholar]
- 15.Kolesnick RN. Sphingomyelin and derivatives as cellular signals. Prog Lipid Res. 1991;30:1–38. doi: 10.1016/0163-7827(91)90005-p. [DOI] [PubMed] [Google Scholar]
- 16.Horinouchi K, Erlich S, Perl DP, Ferlinz K, Bisgaier CL, Sandhoff K, Desnick RJ, Stewart CL, Schuchman EH. Acid sphingomyelinase deficient mice: a model of types A and B Niemann-Pick disease. Nat Genet. 1995;10:288–293. doi: 10.1038/ng0795-288. [DOI] [PubMed] [Google Scholar]
- 17.Spiegel S, Foster DA, Kolesnick RN. Signal tranduction through lipid second messengers. Curr Opin Cell Biol. 1996;8:159–167. doi: 10.1016/s0955-0674(96)80061-5. [DOI] [PubMed] [Google Scholar]
- 18.Ballou LR, Laulederkind SJF, Rosloniec EF, Raghow R. Ceramide signalling and the immune response. Biochim Biophys Acta. 1996;1301:273–287. doi: 10.1016/0005-2760(96)00004-5. [DOI] [PubMed] [Google Scholar]
- 19.Hannun YA. Function of ceramide in coordinating cellular reponses to stress. Science (Wash DC) 1996;274:1855–1859. doi: 10.1126/science.274.5294.1855. [DOI] [PubMed] [Google Scholar]
- 20.Jarvis WD, Kolesnick RN, Fornari FA, Traylor RS, Gewirtz DA, Grant S. Induction of apoptotic DNA damage and cell death by activation of the sphingomyelin pathway. Proc Natl Acad Sci USA. 1994;91:73–77. doi: 10.1073/pnas.91.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Obeid LM, Linardic CM, Karolak LA, Hannun YA. Programmed cell death induced by ceramide. Science (Wash DC) 1993;259:1769–1771. doi: 10.1126/science.8456305. [DOI] [PubMed] [Google Scholar]
- 22.Cai Z, Korner M, Tarantino N, Chouaib S. IkappaB alpha overexpression in human breast carcinoma MCF7 cells inhibits nuclear factor-kappaB activation but not tumor necrosis factor-alpha-induced apoptosis. J Biol Chem. 1997;272:96–101. doi: 10.1074/jbc.272.1.96. [DOI] [PubMed] [Google Scholar]
- 23.Cifone MG, DeMaria R, Roncaioli P, Rippo MR, Azuma M, Lanier LL, Santoni A, Testi R. Apoptotic signaling through CD95 (Fas/Apo-1) activates an acidic sphingomyelinase. J Exp Med. 1994;177:1547–1552. doi: 10.1084/jem.180.4.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cifone MG, Roncaioli P, De Maria R, Camarda G, Santoni A, Ruberti G, Testi R. Multiple pathways originate at the Fas/APO-1 (CD95) receptor: sequential involvement of phosphatidylcholine-specific phospholipase C and acidic sphingomyelinase in the propagation of the apoptotic signal. EMBO (Eur Mol Biol Organ) J. 1995;14:5859–5868. doi: 10.1002/j.1460-2075.1995.tb00274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gulbins E, Bissonnette R, Mahboubi A, Martin S, Nishioka W, Brunner T, Baier G, Bitterlich-Baier G, Byrd C, Lang F, et al. Fas-induced apoptosis is mediated by a ceramide-initiated Ras signaling pathway. Immunity. 1995;2:341–351. doi: 10.1016/1074-7613(95)90142-6. [DOI] [PubMed] [Google Scholar]
- 26.Verheij M, Bose R, Lin XH, Yao B, Jarvis WD, Grant S, Birrer MJ, Szabo E, Zon LI, Kyriakis JM, et al. Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature (Lond) 1996;380:75–79. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- 27.Haimovitz-Friedman A, Kan CC, Ehleiter D, Persaud RS, McLoughlin M, Fuks Z, Kolesnick RN. Ionizing radiation acts on cellular membranes to generate ceramide and initiate apoptosis. J Exp Med. 1994;180:525–535. doi: 10.1084/jem.180.2.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chmura SJ, Nodzenski E, Beckett MA, Kufe DW, Quintans J, Weichselbaum RR. Loss of ceramide production confers resistance to radiation-induced apoptosis. Cancer Res. 1997;57:1270–1275. [PubMed] [Google Scholar]
- 29.Santana P, Pena LA, Haimovitz-Friedman A, Martin S, Green D, McLoughlin M, Cordon-Cardo C, Schuchman EH, Fuks Z, Kolesnick R. Acid sphingomyelinase-deficient human lymphoblasts and mice are defective in radiation-induced apoptosis. Cell. 1996;86:189–199. doi: 10.1016/s0092-8674(00)80091-4. [DOI] [PubMed] [Google Scholar]
- 30.Kaplan E, Meier P. Nonparametric estimation from incomplete observations. J Am Statist Assoc. 1958;53:457–816. [Google Scholar]
- 31.Fuks Z, Persaud RS, Alfieri A, McLoughlin M, Ehleiter D, Schwartz JL, Seddon AP, Cordon-Cardo C, Haimovitz-Friedman A. Basic fibroblast growth factor protects endothelial cells against radiation-induced programmed cell death in vitro and in vivo. . Cancer Res. 1994;54:2582–2590. [PubMed] [Google Scholar]
- 32.Mantel N. Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother Rep. 1966;50:163–170. [PubMed] [Google Scholar]
- 33.Vecchi A, Garlanda C, Lampugnani M, Resnati M, Matteucci C, Stoppacciaro A, Schnurch H, Risau W, Ruco L, Mantovani A. Monoclonal antibodies specific for endothelial cells of mouse bloodvessels. Their application in the identification of adult and embryonic endothelium. Eur J Cell Biol. 1994;63:247–254. [PubMed] [Google Scholar]
- 34.Colagiovanni, D.B., R.J. Evans, J.M. McCabe, A.M. Bendele, and C.K. Edwards III. 1997. Protection of fas mutant MRL-lpr/lpr mice to LPS or Staphylococcal enterotoxin B-induced septic shock by tumor necrosis factor binding protein (TNF-bp). In press.
- 35.Solorzano CC, Ksontini R, Pruitt JH, Hess PJ, Edwards PD, Kaibara A, Abouhamze A, Auffenberg T, Galardy RE, Vauthey JN, et al. Involvement of 26-kD cell-associated TNF-alpha in experimental hepatitis and exacerbation of liver injury with a matrix metalloproteinase inhibitor. J Immunol. 1997;158:414–419. [PubMed] [Google Scholar]
- 36.Fuks Z, Alfieri A, Haimovitz-Friedman A, Seddon A, Cordon-Cardo C. Intravenous basic fibroblast growth factor protects the lung but not mediastinal organs against radiation-induced apoptosis in vivo. Cancer J. 1995;1:62–72. [PubMed] [Google Scholar]
- 37.Takei Y, Kawano S, Nishimura Y, Goto M, Nagai H, Chen SS, Omae A, Fusamoto H, Kamada T, Ikeda K, et al. Apoptosis: a new mechanism of endothelial and Kupffer cell killing. J Gastroenterol Hepatol. 1995;10:S65–S67. doi: 10.1111/j.1440-1746.1995.tb01802.x. [DOI] [PubMed] [Google Scholar]
- 38.Eissner G, Kohlhuber F, Grell M, Ueffing M, Scheurich P, Hieke A, Multhoff G, Bornkamm GW, Holler E. Critical involvement of transmembrane tumor necrosis factor-alpha in endothelial programmed cell death mediated by ionizing radiation and bacterial endotoxin. Blood. 1995;86:4184–4193. [PubMed] [Google Scholar]
- 39.Xu DZ, Lu Q, Swank GM, Deitch EA. Effect of heat shock and endotoxin stress on enterocyte viability apoptosis and function varies based on whether the cells are exposed to heat shock or endotoxin first. Arch Surg. 1996;131:1222–1228. doi: 10.1001/archsurg.1996.01430230104018. [DOI] [PubMed] [Google Scholar]
- 40.Buchman TG, Abello PA, Smith EH, Bulkley GB. Induction of heat shock response leads to apoptosis in endothelial cells previously exposed to endotoxin. Am J Physiol. 1993;265:165–170. doi: 10.1152/ajpheart.1993.265.1.H165. [DOI] [PubMed] [Google Scholar]
- 41.Hoyt DG, Mannix RJ, Rusnak JM, Pitt BR, Lazo JS. Collagen is a survival factor against LPS-induced apoptosis in cultured sheep pulmonary artery endothelial cells. Am J Physiol. 1995;269:171–177. doi: 10.1152/ajplung.1995.269.2.L171. [DOI] [PubMed] [Google Scholar]
- 42.Hoyt DG, Mannix RJ, Gerritsen ME, Watkins SC, Lazo JS, Pitt BR. Integrins inhibit LPS-induced DNA strand breakage in cultured lung endothelial cells. Am J Physiol. 1996;270:689–694. doi: 10.1152/ajplung.1996.270.4.L689. [DOI] [PubMed] [Google Scholar]
- 43.Tartaglia L, Ayres T, Wong G, Goeddel D. A novel domain within the 55 kd TNF receptor signals cell death. Cell. 1993;74:845–853. doi: 10.1016/0092-8674(93)90464-2. [DOI] [PubMed] [Google Scholar]
- 44.Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 45.Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 46.Chinnaiyan AM, O'Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 47.Chinnaiyan AM, Tepper CG, Seldin MF, O'Rourke K, Kischkel FC, Hellbardt S, Krammer PH, Peter ME, Dixit VM. FADD/MORT1 is a common mediator of CD95 (Fas/APO-1) and tumor necrosis factor receptor-induced apoptosis. J Biol Chem. 1996;271:4961–4965. doi: 10.1074/jbc.271.9.4961. [DOI] [PubMed] [Google Scholar]
- 48.Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORT1/ FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell. 1996;85:803–15. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- 49.Boldin MP, Mett IL, Varfolomeev EE, Chumakov I, Shemer-Avni Y, Camonis JH, Wallach D. Self-association of the “death domains” of the p55 tumor necrosis factor (TNF) receptor and Fas/APO1 prompts signaling for TNF and Fas/APO1 effects. J Biol Chem. 1995;270:387–391. doi: 10.1074/jbc.270.1.387. [DOI] [PubMed] [Google Scholar]
- 50.Chinnaiyan AM, Orth K, O'Rourke K, Duan H, Poirier GG, Dixit VM. Molecular ordering of the cell death pathway. Bcl-2 and Bcl-xL function upstream of the CED-3-like apoptotic proteases. J Biol Chem. 1996;271:4573–4576. doi: 10.1074/jbc.271.9.4573. [DOI] [PubMed] [Google Scholar]
- 51.Muzio M, Chinnaiyan AM, Kischkel FC, Orourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, et al. FLICE, a novel FADD-homologous ICE/ CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- 52.Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, Peter ME. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. E. MBO (Eur Mol Biol Organ) J. 1995;14:5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wiegmann K, Schutze S, Machleidt T, Witte D, Kronke M. Functional dichotomy of neutral and acidic sphingomyelinases in tumor necrosis factor signaling. Cell. 1994;78:1005–1015. doi: 10.1016/0092-8674(94)90275-5. [DOI] [PubMed] [Google Scholar]
- 54.Pronk GJ, Ramer K, Amiri P, Williams LT. Requirement of an ICE-like protease for induction of apoptosis and ceramide generation by REAPER. Science (Wash DC) 1996;271:808–810. doi: 10.1126/science.271.5250.808. [DOI] [PubMed] [Google Scholar]
- 55.Susin SA, Zamzami N, Castedo M, Hirsch T, Marchetti P, Macho A, Daugas E, Geuskens M, Kroemer G. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J Exp Med. 1996;184:1331–1341. doi: 10.1084/jem.184.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pastorino J, Simbula G, Yamamoto K, Glascott PJ, Rothman R, Farber J. The cytotoxicity of tumor necrosis factor depends on induction of the mitochondrial permeability transition. J Biol Chem. 1996;271:29792–29798. doi: 10.1074/jbc.271.47.29792. [DOI] [PubMed] [Google Scholar]
- 57.Liu XS, Kim CN, Yang J, Jemmerson R, Wang XD. Induction of apoptotic program in cell-free extracts - requirement for datp and cytochrome C. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 58.Kluck RM, Bossywetzel E, Green DR, Newmeyer DD. The release of cytochrome C from mitochondria - a primary site for Bcl-2 regulation of apoptosis. Science (Wash DC) 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 59.Yang J, Liu XS, Bhalla K, Kim CN, Ibrado AM, Cai JY, Peng TI, Jones DP, Wang XD. Prevention of apoptosis by Bcl-2 - release of cytochrome C from mitochondria blocked. Science (Wash DC) 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 60.Castedo M, Hirsch T, Susin S, Zamzami N, Marchetti P, Macho A, Kroemer G. Sequential acquisition of mitochondrial and plasma membrane alterations during early lymphocyte apoptosis. J Immunol. 1996;157:512–521. [PubMed] [Google Scholar]
- 61.Zhang KZ, Westberg JA, Holtta E, Andersson LC. Bcl-2 regulates neural differentiation. Proc Natl Acad Sci USA. 1996;93:4504–4508. doi: 10.1073/pnas.93.9.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martin SJ, Takayama S, McGahon AJ, Miyashita T, Corbeil J, Kolesnick RN, Reed JC, Green DR. Inhibition of ceramide-induced apoptosis by Bcl-2. Cell Death Differ. 1995;2:253–257. [PubMed] [Google Scholar]