

Abstract
Nip3 (nineteen kD interacting protein-3) is an E1B 19K and Bcl-2 binding protein of unknown function. Nip3 is detected as both a 60- and 30-kD protein in vivo and in vitro and exhibits strong homologous interaction in a yeast two-hybrid system indicating that it can homodimerize. Nip3 is expressed in mitochondria and a mutant (Nip3163) lacking the putative transmembrane domain and COOH terminus does not dimerize or localize to mitochondria. Transient transfection of epitope-tagged Nip3 in Rat-1 fibroblasts and MCF-7 breast carcinoma induces apoptosis within 12 h while cells transfected with the Nip3163 mutant have a normal phenotype, suggesting that mitochondrial localization is necessary for induction of cell death. Nip3 overexpression increases the sensitivity to apoptosis induced by granzyme B and topoisomerase I and II inhibitors. After transfection, both Nip3 and Nip3163 protein levels decrease steadily over 48 h indicating that the protein is rapidly degraded and this occurs in the absence of cell death. Bcl-2 overexpression initially delays the onset of apoptosis induced by Nip3 but the resistance is completely overcome in longer periods of incubation. Nip3 protein levels are much higher and persist longer in Bcl-2 expressing cells. In conclusion, Nip3 is an apoptosis-inducing dimeric mitochondrial protein that can overcome Bcl-2 suppression.
The genetic regulation of cell death is thought to be a central mechanism of cellular homeostasis and development (1–4). The Bcl-2 family of genes (1, 5), which are related to ced-9 of Caenorhabditis elegans (6), were originally identified as repressors of cell death. It is known that both pro- and anti- apoptotic Bcl-2 homologues exist, however their exact biochemical function has not been determined. Recent studies suggests that Ced-9 and Bcl-2/Bcl-XL may physically interact with proteins that are required for the execution of apoptosis, Ced-3 and Ced-4 (7–9). Ced-3 is a protease which in mammals is represented by a large family of cysteine proteases which cleave after aspartic acid, now called caspases (4, 10). In mammalian cells overexpression of bcl-2 prevents the processing and activation of caspase-3 (CPP32) (11, 12).
Bcl-2 family members bear COOH-terminal transmembrane domains that allow their association with the outer mitochondrial membrane (13) and this mitochondrial localization is important for the suppressive function of Bcl-2 (14, 15). There is growing evidence that mitochondrial function is disturbed early in the apoptotic response and may be important in mediating apoptosis (16–18). This is often seen as the loss of mitochondrial membrane potential (16, 17) and the release of cytochrome c (18), and cytochrome c has been implicated in the activation of caspases (18–20). Bcl-2 can suppress the release of cytochrome c from mitochondria and prevent caspase activation (19, 20).
The adenovirus E1B 19K protein is functionally similar to Bcl-2 as a survival factor (1). A two hybrid screen of proteins that interact with E1B 19K in the yeast Saccharomyces cerevisiae identified several unique cDNAs named Nip1, Nip2, and Nip3 (21). All three proteins interact with discrete conserved domains of E1B 19K protein and Bcl-2 that are involved in suppression of cell death, although a function was not identified. We report here that Nip3 is a pro-apoptotic mitochondrial protein that both induces apoptosis and increases the sensitivity of cells to apoptosis induced by drugs and granzyme B. Nip3 can overcome Bcl-2 suppression and may be related to pro-apoptotic members of the Bcl-2 family.
Materials and Methods
Cell Cultures and Reagents.
Rat-1, Rat-1/Bcl-2, and MCF-7 cells used in this study were cultured in α-minimal essential medium (α-MEM)1; (GIBCO BRL, Gaithersburg, MD) supplemented with 10% fetal bovine serum (GIBCO). Murine monoclonal anti-HA 12CA5 and anti-T7 antibody were purchased from Boehringer Mannheim (Laval, Quebec) and Novagen (Madison, WI), respectively. Rabbit anti-HSP60 antibody was donated by Dr. Radhey Gupta (McMaster University, Hamilton, Ontario). FITC-conjugated goat anti–rabbit IgG (FITC; Sigma Chem. Co., St. Louis, MO) and Cy3-conjugated goat anti–mouse IgG (BioCan Ltd., Mississauga, Ontario) were used for fluorescence studies.
Cloning and Construction of Expression Plasmids.
After isolation of a Nip3 cDNA, we utilized RACE to identify both 3′ and 5′ ends using a premade human brain marathon-ready cDNA according to the manufacturer's instructions (Clontech, Palo Alto, CA). The Nip3 3′ RACE was done using primer 5′CAGCGTTCCAGCCTCGGTTTCT3′ while the 5′ RACE was done using three different primers 5′CAAAGGTGCTGGTGGAGGT3′, 5′TGTGGTGTCTGCGAGCGAGGT3′, and 5′TCCAGCAGTATTTTTTCCA3′. The appropriate RACE products were TA cloned (Invitrogen, San Diego, CA) and several of the resulting clones were analysed to obtain the described Nip3 sequence.
To construct epitope tagged Nip3 in expression plasmids, KpnI and EcoRI sites were introduced to the 5′ and 3′ end of the Nip3 cDNA encoding only the open reading frame by PCR amplification and its sequence was confirmed by the T7 sequencing Kit (Pharmacia LKB Biotechnology, Inc., Piscataway, NJ), then inserted in frame into the KpnI-BamHI sites of plasmid pKM1310 containing the 5′ HA-tag. The recombinant cDNA 5′HA-Nip3 was then isolated by EcoRI digestion and blunt end ligated into the plasmid pKG86 creating the plasmid pKG5HA-Nip3 under the control of SP6 promoter which was used for in vitro transcription/translation of 5′HA-tagged Nip3 proteins. The plasmid pKG5HA-Nip3163 was generated by PCR and constructed as described above.
For cloning the Nip3 cDNA into the expression vector pcDNA3 (Invitrogen Corp.), the HindIII site was introduced at the 5′ end of HA-Nip3 cDNA by PCR amplification then cloning into the HindIII-EcoRI sites. The HindIII-KpnI fragment containing the HA-tag was inserted into the HindIII-KpnI sites of pcDNA3 vector making the pcD-5HA vector for cloning the 5′HA-tagged Nip3 cDNA and its mutants. For 5′HA-Nip3 and 5′HA-Nip3163, the cDNA fragments were isolated by KpnI-EcoRI digestion of pKG5HA-Nip3 and pKG5HA-Nip3163, respectively, then inserted into KpnI-EcoRI sites of pcD-5HA vector, making pcD5HA-Nip3 and pcD5HA-Nip3163. For the 3′ T7 tagged Nip3 and Nip3163, a cDNA fragment encoding the T7 epitope was inserted into the EcoRI/XhoI sites of pcDNA3 plasmid. The 3′ T7-tagged Nip3 and Nip3163 open reading frame were cloned into this vector as described above. These recombinant plasmids were used for in vitro translation and transient transfection into the mammalian cells.
For yeast transfection, plasmids encoding Nip3 and Nip3163 with suitable restriction sites were generated by PCR then ligated in-frame in the GAL4 binding domain of pAS1 or pGBT9 and GAL4 transcriptional activating domain of pACTII. The nucleotide sequence of all constructs was confirmed on an ABI 310 Genetic Analyzer (Applied Biosystems, Foster City, CA). The pAS1 or pGBT9 and pACTII constructs were cotransformed into the yeast strain KGY37 and colonies selected on SC medium lacking leucine and tryptophan. Protein–protein interactions were determined by growth on SC medium lacking leucine, tryptophan and histidine in the presence of 1 mM 3-amino-1,2,4-triazole (3AT). The relative β-galactosidase activity was measured using both the filter lift (X-Gal) and ONGP methods according to manufacturer's instructions.
In Vitro Translation.
35S-labeled Nip3 protein and mutants were prepared by in vitro transcription and translation of cDNA cloned in pcD-3HA vector using TnT coupled Reticulocyte Lysate System (Promega Corp., Madison, WI) according to manufacturer's instructions.
Two-dimensional Peptide Mapping.
[35S]methionine-labeled in vitro translated full-length Nip3 and Nip3163 proteins were immunoprecipitated using mouse monoclonal anti-Nip3 antibody. The proteins were then recovered from unfixed dried SDS–polyacrylamide gels by homogenizing the gel slices, then precipitated with trichloroacetic acid and oxidized with performic acid as described previously (22). The oxidized proteins were then resuspended in 25 ml of 50 mM NH4HCO3 and digested overnight at 37°C with 10 mg TPCK-treated trypsin. An additional 5 mg of trypsin was then added, and digestion continued for at least another 4 h. After digestion, samples were repeatedly lyophilized (four times) using a Speedvac concentrator. Tryptic digests of the methionine-labeled Nip3 proteins were subjected to electrophoresis using pH 1.9 buffer (15% acetic acid, 5% formic acid) followed by ascending chromatography with Scheidtmann buffer (isobutyric acid/pyridine/acetic acid/butanol/water (65:5:3:2:29) as described previously (23). [35S]methionine-labeled peptides were visualized by autoradiography.
Transient Transfection and Apoptosis Assays.
Rat-1 cells, Rat-1/Bcl-2 or MCF-7 cells were transfected with Nip3 and mutants in the expression plasmid pcDNA3 using LipofectAMINE Reagent (GIBCO BRL). Cells (3–8 × 105) were plated onto 100 × 20 mm tissue culture dishes (Nunc, Roskilde, Denmark) in 12 ml of complete medium 24 h before transfection. The DNA-LipofectAMINE mixture was allowed to incubate in a tissue culture incubator for 3 h for Rat-1 or 5 h for MCF-7 cells.
Aliquots of 5 × 105 cells were Western blotted as described previously using mouse monoclonal anti-HA antibody 12CA5 or anti-T7 antibody using an ECL detection kit according to manufacturers instructions (Amersham, Amersham, UK). For some experiments, cells were treated with granzyme B and perforin as previously described (24), etoposide or camptothecin, then stained with anti-T7 antibody with FITC-conjugated rabbit anti–mouse antibody and apoptotic cells were enumerated by altered DNA morphology with Hoechst dye staining, counting between 200 and 300 cells per sample (24).
For subcellular localization, cells were grown on cover slips, transfected with HA-Nip3 then fixed with 4% formaldehyde in PBS. Cells were double stained with mouse monoclonal anti-HA antibody 12CA5 and anti-HSP60 antibody and visualized with FITC-conjugated goat anti–mouse IgG secondary antibody and Cy3-conjugated goat anti–rabbit antibody. Fluorescence was visualized using a Zeiss Axiophot microscope equipped with a cooled CCD camera driven by IPLabs Spectrum H-SU2 (version 3.0; Signal Analytics Corp., Vienna, VA) and Multiprobe 1.1 E (Signal Analytics) software.
RNA Isolation and Northern Hybridization Analysis.
Total RNA from mice tissues was kindly provided by Dr. Robert Shiu (University of Manitoba). For Northern blot analysis 30 μg of total RNA was hybridized with nick-translated Nip3 probe (1–5 × 108 cpm μg DNA) at 42°C for 16–20 h. After hybridization and washing blots were exposed to Kodak XAR film at −70°C with an intensifying screen.
Results
Ubiquitous Expression of Nip3.
We isolated several Nip3 cDNAs from a human EBV transformed peripheral B lymphocyte library (Clonetech) by PCR and RACE of both 5′ and 3′ ends, and identified the longest as a 1,535-bp sequence (see Genebank accession number AF002697 for complete sequence). This is similar to the cDNA identified earlier (21) except in the 3′ UTR where we find no evidence of a homologous calbindin region in several independent experiments. The RNA has a long 3′ UTR and shorter 5′ UTR, and the encoded protein contains 194 amino acids and has a predicted molecular mass of ∼21.54 kD with a pI of 6.08. There is a putative transmembrane domain between amino acids 164 to 184 (Fig. 1 A). Nip3 mRNA is expressed in human breast carcinoma MCF-7 as a major transcript of ∼1.7 kb and two minor transcripts of 1.5 and 1.3 kb. The level of Nip3 mRNA in mouse tissues was also examined to determine how widely the gene is expressed. Two transcripts of 2.5 and 1.7-kb were identified with the 1.7-kb transcript found in all tissues examined and the larger transcript only in certain tissues such as brain, heart, kidney, liver, and submaxillary gland (Fig. 1 B). The identification of more than one Nip3 transcript in human and murine cells may indicate that the gene is alternately spliced or another closely related gene exists.
Figure 1.


(A) Nip3 mRNA of 1535 bp is shown with the open reading frame boxed with the predicted protein of 194 amino acids. The putative transmembrane domain between amino acids 164 and 184 of Nip3 protein is shaded. A mutant lacking the transmembrane sequence was constructed by truncation at amino acid 163 and is called Nip3163. (B) Nip3 Northern blot of RNA extracted from multiple murine tissues as indicated, and human MCF-7 mammary carcinoma cells. Transcript sizes are noted on the right.
Nip3 Protein Is Expressed as a Dimer and Monomer.
We prepared [35S]methionine-labeled Nip3 by in vitro transcription and translation and examined the protein on SDS-PAGE. Nip3 was seen as a major band at ∼60 kD and a smaller band of 30 kD (Fig. 2 A). Nip3 has a predicted molecular mass of ∼21.54 kD, much smaller than the size of the two bands estimated from SDS-PAGE, indicating that the protein runs anomalously. The two bands were seen under both reducing and nonreducing conditions. However, when the COOH terminus from amino acid 163 was removed (Nip3163) (Fig. 1 A), the large molecular mass form was absent and a major band of 28 kD and a minor band of 27 kD were detected. These data suggest that the 30-kD protein is a Nip3 monomer while the 60-kD protein is a dimer that was formed by interaction at the COOH terminus of the protein. To examine more directly if Nip3 is a homodimer, we compared tryptic peptide fragments of the 60-kD Nip3 to the 28-kD Nip3163. After in vitro translation, [35S]methionine-labeled Nip3 and Nip3163 were purified by immunoprecipitation with mouse mAb to Nip3 and SDS PAGE. Tryptic peptides were prepared and fragments separated by 2D electrophoresis and chromatography and detected by radioautography as described in Materials and Methods. Three peptide fragments of Nip3 and Nip3163 contain methionine, while the COOH-terminal fragments including the truncated region of Nip3163 do not. Fig. 2 B illustrates that the two proteins have three major [35S]methionine-labeled peptides and their positions are very similar after 2D mapping. No other major peptides are detected in the 60-kD Nip3 band. Minor proteins are also similar and likely represent partially digested Nip3.
Figure 2.
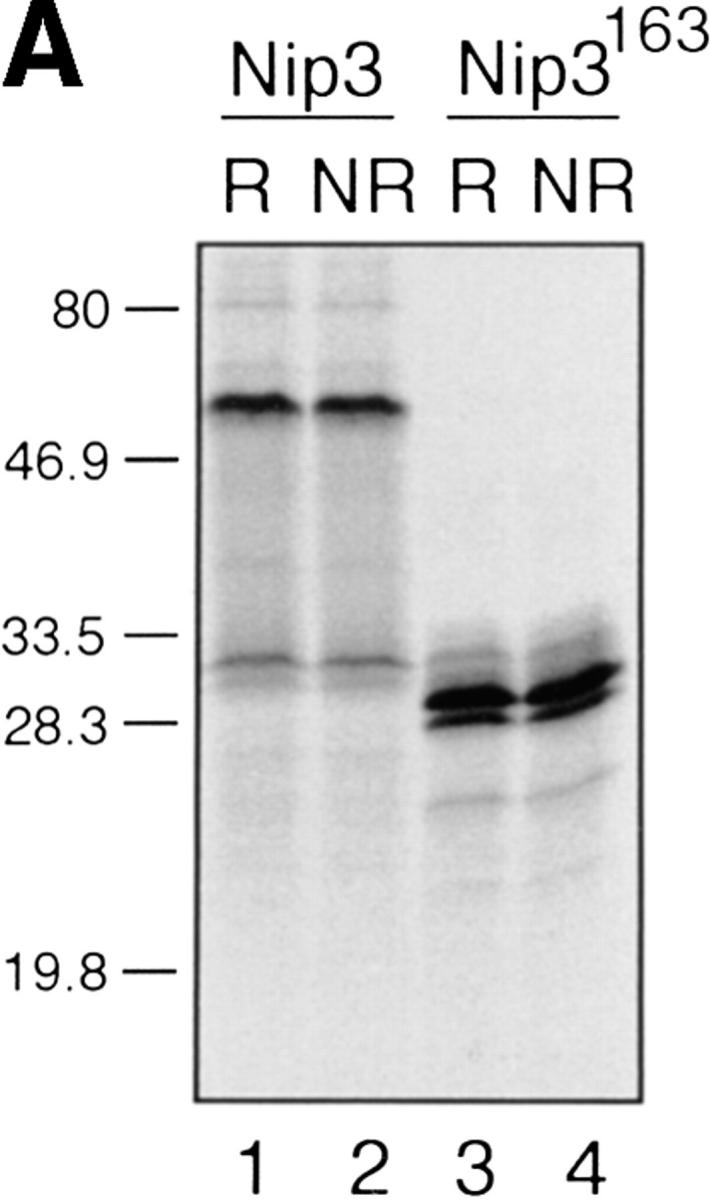


In vitro expression of Nip3 as a homodimer. (A) After in vitro transcription and translation of Nip3 the 35S-labeled protein was separated on SDS-PAGE (lanes 1 and 2) under reducing (R, lane 1) and nonreducing (NR, lane 2) conditions. Nip3 runs as two bands at 60 and 30 kD. Nip3163 is a truncated mutant in which the terminal 31 amino acids from 164 to 194 containing the putative transmembrane domain have been removed (lanes 3 and 4). The truncated Nip3163 is expressed as a major band at 29 kD and a minor band at 28 kD under both reducing (lane 3) and nonreducing (lane 4) conditions. (B) Comparative peptide mapping of in vitro translated 35S-labeled 60-kD Nip3 and 28-kD Nip3163 protein. After in vitro translation, Nip3 and Nip3163 were immunoprecipitated, the 60- and 28-kD bands recovered after SDS-PAGE then trypsin digested. The resulting peptides were separated on the same plate by electrophoresis at pH 1.9 (horizontal dimension with anode to the left), followed by ascending chromatography. The positions of the origin of Nip3 and Nip3163 are marked by arrows. Three [35S]methionine-labeled peptides predicted from the trypsin digest are circled and labeled A, B, and C (Nip3) and A′, B′, and C′ (Nip3163). Peptide A and A′, amino acids 1–45; B and B′, amino acids 140–146; C and C′, 147–152. All three peptides are represented in both Nip3 and Nip3163. Minor spots are similar in the two proteins and likely are partially digested fragments. One of two experiments with similar results is shown. (C) Yeast two-hybrid system identifies Nip3 homodimerization. (left) Plasmids encoding full-length Nip3 and the COOH-terminal truncated mutant Nip3163 fused to the GAL4 DNA-binding domain were cotransformed with plasmids encoding Nip3, Nip3163 or empty vector sequences fused to the GAL4 transcriptional activation domain. Protein–protein interactions were determined by growth of yeast in the absence of leucine, tryptophan and histidine. (Right) Growth in the absence of leucine and tryptophan is shown as a control. The results are representative of 3 independent experiments.
The ability of Nip3 to homodimerize was next examined in the yeast two hybrid system by inserting the Nip3 cDNA in the GAL4 binding and activating domains which were then simultaneously expressed in yeast cells (KGY37). This was compared to Nip3 interaction with non specific protein controls. A strongly positive interaction was found with Nip3 in both vectors indicating that the protein was capable of binding to itself (Table 1). In contrast, the Nip3163 was unable to interact with Nip3 or with itself (Fig. 2 C) indicating that the COOH terminus was required for homodimerization.
Table 1.
Interaction of Nip3 in Yeast Two Hybrid System
| Protein ‡ | Relative level of X-Gal | lacZ expression * ONPG | ||
|---|---|---|---|---|
| pAS1 | W | 0.7 | ||
| Nip3 | W | 1.0 | ||
| Nip3/Nip3 | B | 103.3 | ||
| MK/Nip3 | W | 0.3 | ||
| PTP2/Nip3 | W | 0.5 |
Relative level of interaction is based on β-galactosidase activity. W, white; B, blue.
Binding domain/Activating domain; pAS1, empty vector; MK, Myotonin Kinase; PTP2, protein tyrosine phosphatase 2.
Subcellular Localization of Nip3 to Mitochondria.
Boyd et al. (21) reported that HA-Nip3 localizes in discrete cytoplasmic patches which are typical of a mitochondrial distribution. To confirm the mitochondrial localization of Nip3 and to test the hypothesis that it depends on the putative transmembrane domain, we transfected Rat-1 cells with HA-Nip3 or HA-Nip3163 then stained with FITC-conjugated anti-HA antibody. Cells were simultaneously treated with anti-HSP60 antibody which primarily stains mitochondria (15) and Cy3-conjugated second antibody. Both the green Nip3 and red HSP60 fluorescence appeared in a similar punctate pattern, and when the images were combined, a uniform yellow staining pattern indicated virtually complete coincidence of the two stains (Fig. 3, A–C). In contrast, HA-Nip3163 stained diffusely throughout the cytoplasm rather than the punctate distribution of the intact Nip3, however, a small amount of HA staining appeared to colocalize with HSP60 by double fluorescence (Fig. 3, D–F).
Figure 3.






Subcellular localization of Nip3 and Nip3163. Rat-1 cells were transfected with HA-Nip3 and stained with anti-HA antibody using FITC (A) and the mitochondrial protein marker anti-HSP60 antibody using Cy3 (B). The stained images were combined to compare the staining pattern of both proteins (C) and their coincidence is indicated by the conversion of green FITC and red Cy3 stain to yellow thoughout the cell cytoplasm. HA-Nip3163 was expressed in Rat-1 cells then stained with anti-HA antibody (D), or anti-HSP60 antibody (E) and shown as a combined image using antibodies (F), as described above.
Overexpression of Nip3 but not Nip3163 Induces Apoptosis.
T7 epitope-tagged Nip3 was transiently expressed in Rat-1 or MCF-7 cells. Cells were stained with both anti-T7 antibody to identify cells expressing Nip3, and Hoechst dye to quantitate apoptosis. Over time, progressively more Nip3 expressing cells became apoptotic starting around 12 h and reaching completion by 48 to 60 h (Fig. 4 A). Nip3 protein measured by anti-T7 Western blot were highest between 12 and 24 h after transfection then progressively decreased with time paralleling the induction of apoptosis (Fig. 4 B). To determine if the transmembrane domain of Nip3 which is necessary for mitochondrial localization was also necessary to induce apoptosis, the experiment was repeated using the Nip3163. The mutant was unable to induce apoptosis (Fig. 4, A and B). Of additional interest, Nip3163 protein levels decreased very rapidly with time despite the inability of the mutant to induce apoptosis, thus indicating that the loss of protein in the cell lysates was not related to the death of cells.
Figure 4.


Overexpression of Nip3 but not Nip3163 induces apoptosis. (A) Rat-1 or MCF-7 (left) cells were transfected with T7-tagged Nip3 then at different times cells harvested and stained with anti-T7 antibody and FITC anti–mouse IgG antibody to identify cells expressing Nip3. The frequency of apoptotic cells was quantitated by Hoechst dye staining. Rat-1 cells (right) were then transfected and apoptotic cells expressing (•) and not expressing (○) Nip3 were quantitated. Rat-1 cells expressing (▾) or not expressing (▿) Nip3163 were analysed in the same manner. At least 200 cells were counted in each sample. All assays were repeated three to seven times with identical results. (B) Western blot of Rat-1 cells transfected with T7-Nip3 (left) or T7-Nip3163 (right). Cells were harvested at the times indicated and Western blots developed with anti-T7 antibody.
Nip3 Sensitizes Rat-1 Cells to Apoptosis.
To determine whether Nip3 can influence apoptosis induced by other cell death signals, Rat-1 cells were transiently transfected with T7-Nip3 for 12 h then treated with increasing doses of the cytotoxic lymphocyte protease granzyme B, or the topoisomerase I and II inhibitors etoposide or camptothecin. The 12-h transfection was chosen because of minimal effects of Nip3 on cell survival at this time. Apoptotic cells were then quantitated by Hoechst dye in cells either expressing or not expressing T7-Nip3 protein within the same samples, using immunofluorescence staining by anti-T7 antibody to detect Nip3. The frequency of apoptotic cells was clearly higher in Nip3-expressing cells (Fig. 5). A 5–10-fold increase in sensitivity to the drugs and a 2–3-fold increase in granzyme B apoptosis was observed in three independent experiments.
Figure 5.

Nip3 sensitizes Rat-1 cells to drug-induced apoptosis. Rat-1 cells were transfected with T7-tagged Nip3 then 12 h later treated with increasing amounts of etoposide, camptothecin or granzyme B and perforin. Cells expressing Nip3 were identified by anti-T7 antibody and the nucleus stained with Hoechst dye. Apoptotic cells expressing Nip3 (Nip3 +) or cells not expressing Nip3 (Nip3 −) were enumerated. This is representative of three experiments showing similar results.
Nip3 Overcomes Bcl-2 Suppression of Apoptosis.
Since it had been demonstrated that Bcl-2 binds Nip3 (21), we next determined if Bcl-2 was capable of suppressing Nip3-induced apoptosis. Rat-1 and Rat-1/Bcl-2 cells (Fig. 6 C) were transfected with T7-Nip3 and apoptotic cells appearing in both populations was determined over time. Apoptotic Rat-1 cells expressing Nip3 were detected within 12 h and reached maximum levels by 48 h, while Nip3 did not induce cell death in Rat-1/Bcl-2 cells until 24 h after transfection. However, by 48 h virtually all cells expressing Nip3 were apoptotic despite Bcl-2 overexpression (Fig. 6 A). The Rat-1 cells expressing Bcl-2 were highly resistant to granzyme B and perforin (Fig. 6 A). We noticed that Nip3 protein level was considerably higher in Rat-1/Bcl-2 cells than in Rat-1 cells by 12 h after transfection and remained higher throughout the time course. However, a significant decrease in Nip3 level was detected by 48–60 h in Rat-1/ Bcl-2 cells at the same time that recovery of apoptosis was detected (Fig. 6 B).
Figure 6.


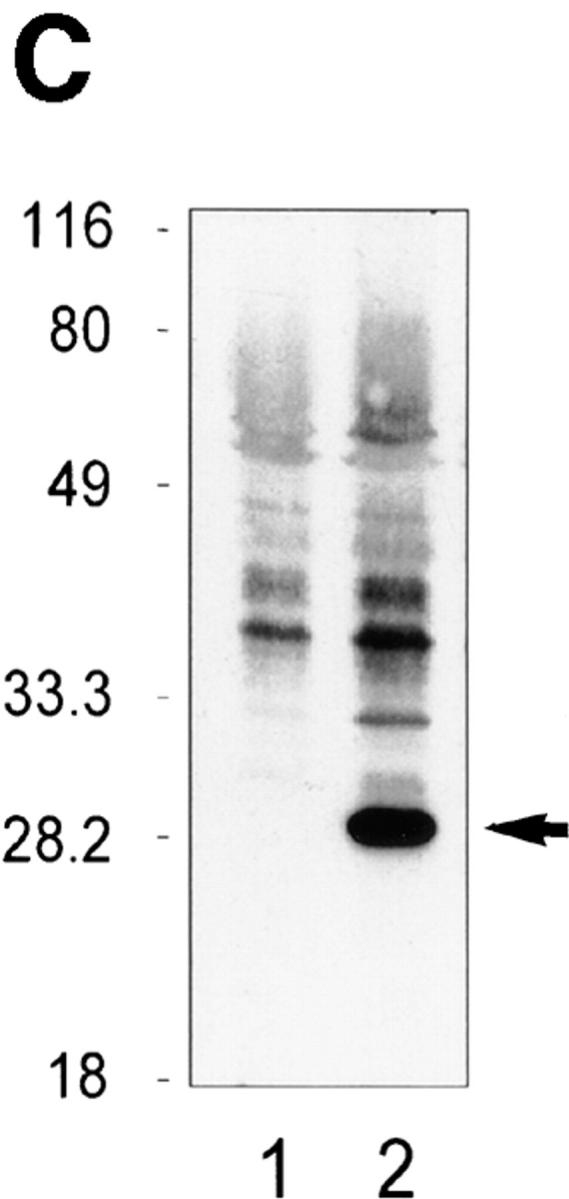
Nip3 overcomes Bcl-2 suppression of apoptosis. (A) (Left) Rat-1 and Rat-1/Bcl-2 cells were transfected with T7-Nip3 and at increasing time the cells were harvested and Nip3-expressing apoptotic cells quantitated as described in Fig. 4. (Right) The Rat-1 and Rat-1/ Bcl-2 cells were treated with granzyme B as shown at a constant concentration of perforin (125 ng/ml) for 3 h and apoptotic cells quantitated by Hoechst dye as described previously (22). Three other experiments showed similar results. (B) After transfection of Rat-1 and Rat-1/ Bcl-2 cells with T7-Nip3, lysates were harvested at increasing time intervals then Western blotted with anti-T7 antibody. Molecular mass markers are shown on the left. (C) Western blot of Bcl-2 (arrow) in Rat-1 (lane 1) and Rat-1/Bcl-2 (lane 2) cell lines. Blots were developed with rabbit anti–human Bcl-2 (PharMingen). Relative molecular mass (kD) is shown on the left.
Discussion
In the present study we have identified the E1B 19-kD/ Bcl-2–binding protein Nip3 as a pro-apoptotic protein that localizes to mitochondria. Nip3 protein is unusual in several respects. It migrates on SDS-PAGE at a molecular mass that is much higher than the 21.54-kD predicted by the primary amino acid sequence and appears in vivo after transfection and in vitro after transcription and translation as 60- and 30-kD proteins. The 60-kD band does not dissociate after reduction or reduction and alkylation, nor after treatment with 6M urea (Chen, G., and A.H. Greenberg, unpublished data). Thus Nip3 appears to interact with a second protein very strongly and the evidence favors homodimerization. Three experiments support this conclusion. First, truncation of the COOH-terminal 31 amino acids results in the loss of the 60-kD Nip3 band and only a lower molecular weight protein is observed. Second, [35S]methionine-labeled tryptic peptide fragments of the 60-kD Nip3 were identical to the 28-kD Nip3163, and finally, Nip3 exhibits strong interaction with itself in the yeast two hybrid system. The inability to bind Nip3163 to interact with Nip3 or itself indicates that it cannot dimerize and is consistent with the requirement of the COOH-terminal portion of Nip3 for homodimerization.
The COOH-terminal domain of Nip3 not only influences dimer formation but also directs its expression to mitochondria. Nip3 colocalized with HSP60 which is primarily expressed in mitochondria (25) while the mutant Nip3163 lacking the transmembrane domain was mostly expressed as a free cytosolic protein. A minor component of the Nip3163 protein was colocalized with the mitochondrial markers suggesting that it may interact by a mechanism other than membrane insertion, perhaps by binding to other mitochondrial proteins. Nip3163 does not interact with Nip3 or form a homodimer in vitro so it likely does not homodimerize with endogenous Nip3 in vivo.
Nip3 protein expression progressively decreases over time, which may reflect the increasing death of Nip3 expressing cells. Partial Bcl-2 suppression of Nip-3 induced apoptosis and a significant increase in Nip3 protein levels suggests that at least part of the Nip3 is lost by cell death. However, a progressive decrease in protein levels was also observed with the Nip3163 mutant which does not induce apoptosis, therefore another mechanism is affecting protein turnover. Nip3 contains PEST sequences suggesting that the protein may be susceptible to rapid degradation by proteases. PEST sequences commonly contain high local concentrations of amino acids P, E, S, T, and D flanked by charged amino acids (26) and these are abundantly present in Nip3. Thus the posttranslational control of Nip3 expression through rapid protein degradation may constitute a mechanism for regulating the intracellular levels of a potentially lethal protein.
Nip3 was originally identified as an E1B 19K interacting protein, but the structure of Nip3 does not closely resemble other death agonists of the Bcl-2 family. Bcl-2 family agonists such as Bax act by heterodimerizing with anti-apoptotic family members and mutations in BH1 and BH2 domains in Bcl-2 or Bcl-XL result in loss of binding and anti-apoptotic activity (27, 28). However, other studies suggest that pro- and anti-apoptotic Bcl-2 family members can act independently (28) or through other family members such as BAD (29, 30). Nip3 does not contain either a BH1 or BH2 domain, although it may have a BH3 domain in which leucine110, aspartic acid115, and isoleucine117 are conserved based on the critical amino acids of the BH3 domain of BAK that determines BAK-Bcl-XL heterodimerization (31). Other BH3 domain-only Bcl-2 family members that are death agonists have been described such as Bik/Nbk (32, 33), Bid (34) and, more recently, Hrk (35). Bik and Hrk have putative transmembrane domains while Bid is a cytoplasmic factor that is hypothesized to be recruited to membrane-associated Bcl-2 or Bax. Thus it is possible that Nip3 is a death factor of the Bcl-2 family related to Bik/Nbk, Bid, or Hrk.
Many Bcl-2 family members are integral membrane proteins that bear COOH-terminal regions that allow localization to the mitochondrial outer membrane, nuclear envelope and endoplasmic reticulum (15, 36, 37). Localization of Bcl-2 to mitochondria also appears to be important in its suppression of cell death as mutants lacking the transmembrane domain are ineffective when overexpressed (14, 38). Nip3 shares the ability to localize to mitochondria and recent work suggests that mitochondria may play an important role in the regulation of apoptosis. They are necessary for the apoptotic activity of cytosolic extracts of Xenopus laevis oocytes (39), and can initiate nuclear destruction when taken from cells undergoing apoptosis in in vitro reconstituted cell systems (16). Nip3 localization in mitochondria places it in a position to influence mitochondrial function early in the apoptotic response. Nip3 can overcome Bcl-2 suppression of apoptosis so it might act on Bcl-2 or other anti-apoptotic family members that regulate mitochondrial permeability transition pores (16, 17) or cytochrome c release (18–20). Nip3 preferentially forms homodimers even under reducing conditions and could provide a stable link between Bcl-2 homo- and/or heterodimers and other proteins perhaps forming larger complexes or altering their interaction with regulators such as BAD (30).
In conclusion, we have identified the E1B 19K/Bcl-2 binding protein Nip3 as a homodimer localized to mitochondrial membrane. Nip3 expression in mitochondria induces apoptosis and can overcome Bcl-2 suppression of cell death, indicating that Nip3 is a novel pro-apoptotic protein.
Acknowledgments
We thank Dr. Radhey Gupta for his gift of anti-HSP60 antibody.
This work was supported by grants from the National Cancer Institute and MRC of Canada. A.H. Greenberg is a Terry Fox Cancer Research Scientist of the National Cancer Institute of Canada.
Footnotes
Abbreviations used in this paper: α-MEM, α-minimal essential medium; 3AT, 3-amino-1,2,4-triazole.
References
- 1.White E. Life, death, and the pursuit of apoptosis. Genes Dev. 1996;10:1–15. doi: 10.1101/gad.10.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Chinnaiyan AM, Dixit VM. The cell-death machine. Curr Biol. 1996;6:555–562. doi: 10.1016/s0960-9822(02)00541-9. [DOI] [PubMed] [Google Scholar]
- 3.Jacobson MD, Weil M, Raff MC. Programmed cell death in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- 4.Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 5.Oltvai ZN, Korsmeyer SJ. Checkpoints of dueling dimers foil death wishes. Cell. 1994;79:189–192. doi: 10.1016/0092-8674(94)90188-0. [DOI] [PubMed] [Google Scholar]
- 6.Hengartner MO, Horvitz HR. C. eleganscell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell. 1994;76:665–676. doi: 10.1016/0092-8674(94)90506-1. [DOI] [PubMed] [Google Scholar]
- 7.Chinnaiyan AM, O'Rourke K, Lane BR, Dixit VM. Interaction of CED-4 with CED-3 and CED-9: a molecular framework for cell death. Science. 1997;275:1122–1126. doi: 10.1126/science.275.5303.1122. [DOI] [PubMed] [Google Scholar]
- 8.Wu D, Wallen HD, Nunez G. Interaction and regulation of subcellular localization of CED4 by CED9. Science. 1997;275:1126–1129. doi: 10.1126/science.275.5303.1126. [DOI] [PubMed] [Google Scholar]
- 9.Spector MS, Desnoyers S, Hoeppner DJ, Hengartner MO. Interaction between the C. eleganscell death regulators CED9 and CED4. Nature. 1997;385:653–656. doi: 10.1038/385653a0. [DOI] [PubMed] [Google Scholar]
- 10.Martin SJ, Green DR. Protease activation during apoptosis: death by a thousand cuts? . Cell. 1995;82:349–352. doi: 10.1016/0092-8674(95)90422-0. [DOI] [PubMed] [Google Scholar]
- 11.Chinnaiyan AM, Orth K, O'Rourke K, Duan HJ, Poirier GG, Dixit VM. Molecular ordering of the cell death pathway, Bcl-2 and Bcl-xL function upstream of the CED-3-like apoptotic proteases. J Biol Chem. 1996;271:4573–4576. doi: 10.1074/jbc.271.9.4573. [DOI] [PubMed] [Google Scholar]
- 12.Armstrong R, Aja T, Xiang J, Gaur S, Krebs J, Hoang K, Bai S, Korsmeyer S, Kranewsky D, Fitz L, Tomaselli K. Fas-induced activation of the cell death-related protease CPP32 is inhibited by Bcl-2 and by ICE family protease inhibitors. J Biol Chem. 1996;271:16850–16855. doi: 10.1074/jbc.271.28.16850. [DOI] [PubMed] [Google Scholar]
- 13.Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W, Reed JC. Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 1993;53:4701–4714. [PubMed] [Google Scholar]
- 14.Tanaka S, Saito K, Reed JC. Structure-function analysis of the Bcl-2 oncoprotein. Addition of a heterologous transmembrane domain to portions of the Bcl-2 beta protein restores function as a regulator of cell survival. J Biol Chem. 1993;268:10920–10926. [PubMed] [Google Scholar]
- 15.Zhu W, Cowie A, Wasfy GW, Penn LZ, Leber B, Andrews DA. Bcl-2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types. EMBO (Eur Mol Bio Organ) J. 1996;15:4130–4141. [PMC free article] [PubMed] [Google Scholar]
- 16.Zamzami N, Susin SA, Marchetti P, Hirsch T, Gomez-Monterrey I, Castedo M, Kroemer G. Mitochondrial control of nuclear apoptosis. J Exp Med. 1996;183:1533–1544. doi: 10.1084/jem.183.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marchetti P, Castedo M, Susin SA, Zamzami N, Hirsch T, Macho A, Haeffner A, Hirsch F, Geuskens M, Kroemer G. Mitochondrial permeability transition is a central coordinating event of apoptosis. J Exp Med. 1996;184:1155–1160. doi: 10.1084/jem.184.3.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu X, Kim C, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 19.Yang J, Liu XS, Bhalla K, Kim CN, Ibrado AM, Cai JY, Peng TI, Jones DP, Wang XD. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 20.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 21.Boyd JM, Malstrom S, Subramanian T, Venkatesh LK, Schaeper U, Elangovan B, D'Sa-Eipper C, Chinnadurai G. Adenovirus E1B 19 kDa and Bcl-2 proteins interact with a common set of cellular proteins. Cell. 1994;79:341–351. doi: 10.1016/0092-8674(94)90202-x. [DOI] [PubMed] [Google Scholar]
- 22.Boyle WJ, Van Der Geer P, Hunter T. Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates. Methods Enzymol. 1991;201:110–149. doi: 10.1016/0076-6879(91)01013-r. [DOI] [PubMed] [Google Scholar]
- 23.Scheidtmann KH, Echle B, Walter G. Simian virus 40 large T antigen is phosphorylated at multiple sites clustered in two separate regions. J Virol. 1982;1:116–133. doi: 10.1128/jvi.44.1.116-133.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi LF, Mai S, Israels S, Browne K, Trapani JA, Greenberg AH. Granzyme B (GraB) autonomously crosses the cell membrane and perforin initiates apoptosis and GraB nuclear localization. J Exp Med. 1997;185:855–866. doi: 10.1084/jem.185.5.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soltys BJ, Gupta RS. Immunoelectronmicroscopic localization of the 60 kDa heat shock chaperonin protein (Hsp60) in mammalian cells. Exp Cell Res. 1996;222:16–27. doi: 10.1006/excr.1996.0003. [DOI] [PubMed] [Google Scholar]
- 26.Rogers S, Wells R, Rechsteiner M. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science. 1986;234:364–368. doi: 10.1126/science.2876518. [DOI] [PubMed] [Google Scholar]
- 27.Yin XM, Oltvai ZN, Korsmeyer SJ. Heterodimerization with Bax is required for Bcl-2 to repress cell death. Curr Top Microbiol Immunol. 1995;194:331–338. doi: 10.1007/978-3-642-79275-5_38. [DOI] [PubMed] [Google Scholar]
- 28.Cheng EHY, Levine B, Boise LH, Thompson CB, Hardwick JM. Bax-independent inhibition of apoptosis by Bcl-xL. Nature. 1996;379:554–556. doi: 10.1038/379554a0. [DOI] [PubMed] [Google Scholar]
- 29.Wang HG, Rapp UR, Reed JC. Bcl-2 targets the protein kinase Raf-1 to mitochondria. Cell. 1996;87:629–638. doi: 10.1016/s0092-8674(00)81383-5. [DOI] [PubMed] [Google Scholar]
- 30.Zha JP, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-XL . Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 31.Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, et al. Structure of Bcl-xL-Bak peptide complex: Recognition between regulators of apoptosis. Science. 1997;275:983–986. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- 32.Boyd JM, Gallo GJ, Elangovan B, Houghton AB, Malstrom S, Avery BJ, Ebb RG, Subramanian T, Chittenden T, Lutz RJ, Chinnadurai G. Oncogene. 1995;11:1921–1928. [PubMed] [Google Scholar]
- 33.Han J, Sabbatini P, White E. Induction of apoptosis by human Nbk/Bik, a BH3-containing protein that interacts with E1B 19K. Mol Cell Biol. 1996;16:5857–5864. doi: 10.1128/mcb.16.10.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang K, Yin XM, Chao DT, Milliman CL, Korsmeyer SJ. BID: a novel BH3 domain-only death agonist. Genes Dev. 1996;10:2859–2869. doi: 10.1101/gad.10.22.2859. [DOI] [PubMed] [Google Scholar]
- 35.Inohara, N., Ding, L., Chen, S. and Nunez, G. 1997. harakiri, a novel regulator of cell death encodes a protein that activates apoptosis and interacts selectively with survival-promoting proteins Bcl-2 and Bcl-XL EMBO (Eur. Mol. Biol. Organ.) J. 16:1686–1694. [DOI] [PMC free article] [PubMed]
- 36.Hockenbery DM, Nunez G, Minniman RD, Schreiber RB, Korsmeyer SJ. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990;348:334–336. doi: 10.1038/348334a0. [DOI] [PubMed] [Google Scholar]
- 37.Akao Y, Otsuki YS, Kataoka S, Ito Y, Tsujimoto Y. Multiple subcellular localization of bcl-2: detection in nuclear outer membrane, endoplasmic reticulum and mitochondrial membrane. Can Res. 1994;54:2468–2471. [PubMed] [Google Scholar]
- 38.Nguyen M PE, Branton, Walton PA, Oltvai ZN, Korsmeyer SJ, Shore GC. Role of membrane anchor domain of Bcl-2 in suppression of apoptosis caused by E1B-defective adenovirus. J Biol Chem. 1994;269:16521–16524. [PubMed] [Google Scholar]
- 39.Newmeyer DD, Farschon DM, Reed JC. Cell-free apoptosis in Xenopusegg extracts: inhibition by Bcl-2 and requirement for an organelle fraction enriched in mitochondria. Cell. 1994;79:353–364. doi: 10.1016/0092-8674(94)90203-8. [DOI] [PubMed] [Google Scholar]