

Abstract
In mice deficient in either lymphotoxin α (LT-α) or type I tumor necrosis factor receptor (TNFR-I), organized clusters of follicular dendritic cells (FDC) and germinal centers (GC) are absent from the spleen. We investigated the role of LT-α and TNFR-I in the establishment of spleen FDC and GC structure by using reciprocal bone marrow (BM) transfer. When LT-α–deficient mice were reconstituted with wild-type BM, FDC organization and the ability to form GC were restored, indicating that the LT-α–expressing cells required to establish organized FDC are derived from BM. The role of LT-α in establishing organized FDC structure was further investigated by the transfer of complement receptor 1 and 2 (CR1/2)–deficient BM cells into LT-α–deficient mice. Organized FDC were identified with both the FDC-M1 and anti-CR1 monoclonal antibodies in these BM-chimeric mice, indicating that these cells were derived from the LT-α–deficient recipient. Thus, expression of LT-α in the BM-derived cells, but not in the non–BM-derived cells, is required for the maturation of FDC from non-BM precursor cells. In contrast, when TNFR-I–deficient mice were reconstituted with wild-type BM, they showed no detectable FDC clusters or GC formation. This indicates that TNFR-I expression on non–BM-derived cellular components is necessary for the establishment of these lymphoid structures. TNFR-I–deficient BM was able to restore FDC organization and GC formation in LT-α–deficient mice, indicating that formation of these structures does not require TNFR-I expression on BM-derived cells. The data in this study demonstrate that FDC organization and GC formation are controlled by both LT-α–expressing BM-derived cells and by TNFR-I-expressing non–BM-derived cells.
Lymphotoxin (LT)1-α and TNF-α are structurally related cytokines that can modulate many immune and inflammatory reactions (1–3). In solution, LT-α exists as a homotrimer. TNF-α is also a homotrimer, synthesized first as a type II membrane protein that can be released from cells in a soluble form by the action of the TNF-α–converting enzyme (4–7). The LT-α and TNF-α homotrimers can each engage and activate the same two plasma membrane receptors: the 55-kD TNF receptor (TNFR type I) and the 75-kD TNF receptor (TNFR type II) (8, 9). LT can also exist in a membrane-associated form, which in its major form consists of an LT-α monomer and two identical 33-kD transmembrane LT-β subunits (10–12). Membrane LT binds and activates the recently identified LT-β receptor (LT-βR) and has no measurable affinity for TNFR-I or -II (13, 14).
Recent studies using mouse strains deficient in either LT-α, LT-β, or TNF-α have demonstrated that LT-α and -β have actions that are distinct from those of TNF-α. Mice rendered deficient in either LT-α or -β are born with a dramatic impairment of lymph node (LN) biogenesis (15–18). In contrast, mice deficient in TNF-α (19), like mice deficient in either TNFR-I or -II, have grossly normal LN structures (20–22). Thus, the actions of LT-α and -β in LN biogenesis are not dependent on signaling by either of the two TNF receptors, but rather most likely by a receptor for the membrane LT heteromer. The potential role of membrane LT is supported by additional recent studies by Rennert et al. (23) in which a soluble form of the LT-βR administered to mice during the latter portions of gestation blocked LN development in offspring. Further studies of LT-α–, LT-β–, and TNFR-I–targeted mice have revealed other important roles of LT or TNFR-I in the establishment of lymphoid organ structure. Firstly, LT-α–, LT-β–, and TNFR-I–deficient mice all manifest abnormal Peyer's patch (PP) development (15–18, 24). The absence of normal PP structure in the presence of LN in TNFR-I–deficient mice suggests that TNFR-I may act in lymphoid organogenesis in an organ-specific fashion. Secondly, LT-α–, LT-β–, TNF-α–, and TNFR-I–deficient mice all fail to form germinal centers (GC) in the spleen (17–19, 25–27).
GC are histologically well defined structures that develop in secondary lymphoid organs shortly after challenge with T cell–dependent antigens (28). It has been suggested that it is within GC that somatic hypermutation and affinity maturation of the antibody response occur (29–31). In a previous study, we investigated the mechanism of the defect of GC formation in LT-α–deficient mice through use of reciprocal bone marrow (BM) transfers (25). This study demonstrated that reconstitution of lethally irradiated LT-α– deficient mice with normal BM restored the ability to form GC. In contrast, when LT-α–deficient BM cells were used to reconstitute irradiated wild-type mice, GC formation was defective, as in the LT-α–deficient mice. These data indicate that BM-derived cells are the only essential source of LT-α required for the formation of GC.
Both primary and secondary lymphoid follicles characteristically contain clusters of follicular dendritic cells (FDC; reference 32). FDC trap and can retain antigen–antibody complexes for long periods (33), apparently by means of receptors for the third complement component (CR) as well as Fcγ receptors (32). We and others have shown that organized FDC structure, a major morphological characteristic of GC, is absent in LT-α–, LT-β–, TNF-α–, and TNFR-I–deficient mice (17–19, 26, 27, 34, 35).
In this study, we have investigated the role of LT-α and TNFR-I in the establishment of follicle structure in the spleen through use of reciprocal BM transfers. This study defines distinct roles of LT-α and TNFR-I in the organization of these lymphoid tissue structures and demonstrates clearly that FDC are not transferred to adult animals by BM, but rather develop in transplant recipients from non-BM precursor cells.
Materials and Methods
Mice.
LT-α– and CR1/2-deficient mice were generated as previously described (15, 36). TNFR-I–deficient mice (37) were provided by J. Peschon (Immunex, Seattle, WA). The mice were maintained and bred in the Division of Comparative Medicine, Washington University School of Medicine, under pathogen-free conditions. All experiments were initiated with 8–12-wk-old mice.
Bone Marrow Transfer.
BM transfer was performed as previously described (38). In brief, BM cells were harvested by flushing the femurs of donor mice with RPMI 1640 medium (GIBCO/ BRL, Gaithersburg, MD) supplemented with 10% heat-inactivated fetal bovine serum (HyClone, Logan, UT), 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin, hereafter referred to as R10. Cells were washed once and suspended in R10 medium containing anti-Thy1.2 mAb (clone 5a-8; Accurate Chemical & Scientific Corporation, Westbury, NY) plus low-toxic rabbit complement (Accurate Chemical & Scientific Corporation). After incubation at 37°C for 45 min, cells were washed twice and adjusted to 3 × 107 viable cells/ml in R10. Each recipient mouse was lethally irradiated (10 Gy) and treated with 0.5 ml of donor BM cells intravenously on the same day. These recipient mice were used for analysis 6–10 wk after BM transfer.
Immunohistochemistry.
Mice were immunized intraperitoneally with 100 μl of PBS (pH 7.4) containing 10% sheep red blood cells. 10 d later, spleens were harvested, and frozen tissue sections were prepared. Immunohistochemistry using peanut agglutinin (PNA; Vector, Burlingame, CA), anti-B220 mAb (PharMingen, San Diego, CA) and anti-CR mAbs (8C12, 7G6, and 7E9; reference 39), and immune complex (IC) trapping in vitro were performed as previously described (25, 34, 40). For the combination of IC trapping in vitro and staining with anti-B220 mAb, frozen sections were first incubated with 1:10 diluted mouse horseradish peroxidase (HRP)–anti-peroxidase complex (code B650, lot 035A; DAKO A/S, Glostrup, Denmark) in the presence of 1:5 diluted fresh mouse serum as a source of complement. After washing in PBS, sections were further incubated with anti-B220-biotin and then with streptavidin conjugated with alkaline phosphatase (AP) (Zymed, South San Francisco, CA). Color development for bound AP and HRP was with an AP reaction kit (Vector) and with diaminobenzidine.
Results
Role of LT-α in the Organization of FDC.
The role of LT-α in the organization of FDC was studied using reciprocal BM transfer experiments. 6–10 wk after BM transfer, FDC organization in these BM-chimeric mice was visualized immunohistochemically by using 8C12, an mAb specific for murine CR1 (39). LT-α–deficient mice reconstituted with normal BM demonstrated prominent FDC reticula in the GC light zones (Fig. 1 A). Similar results were obtained using the anti-FDC mAbs FDC-M1 and FDC-M2 (41) and 7G6 or 7E9, mAbs specific for murine CR1 and CR2 (data not shown). FDC organization was further assessed by analysis of IC trapping using spleen sections from these animals. Consistent with the result obtained from anti-CR1 staining, IC trapping was demonstrated at sites corresponding to the FDC reticula in these mice (Fig. 1 E). Thus, restoration of GC formation in wild-type BM-reconstituted LT-α–deficient mice was associated with de novo development of organized FDC structure. In contrast, wild-type mice reconstituted with LT-α–deficient BM showed no clusters of anti-CR1 staining cells or PNA+ cells (Fig. 1 B), and showed absence of IC trapping (Fig. 1 F). Wild-type mice reconstituted with wild-type BM (Fig. 1, C and G) and LT-α–deficient mice reconstituted with LT-α–deficient BM (Fig. 1, D and H) showed similar anti-CR1 staining and in vitro IC trapping compared to wild-type mice and LT-α–deficient mice, respectively.
Figure 1.
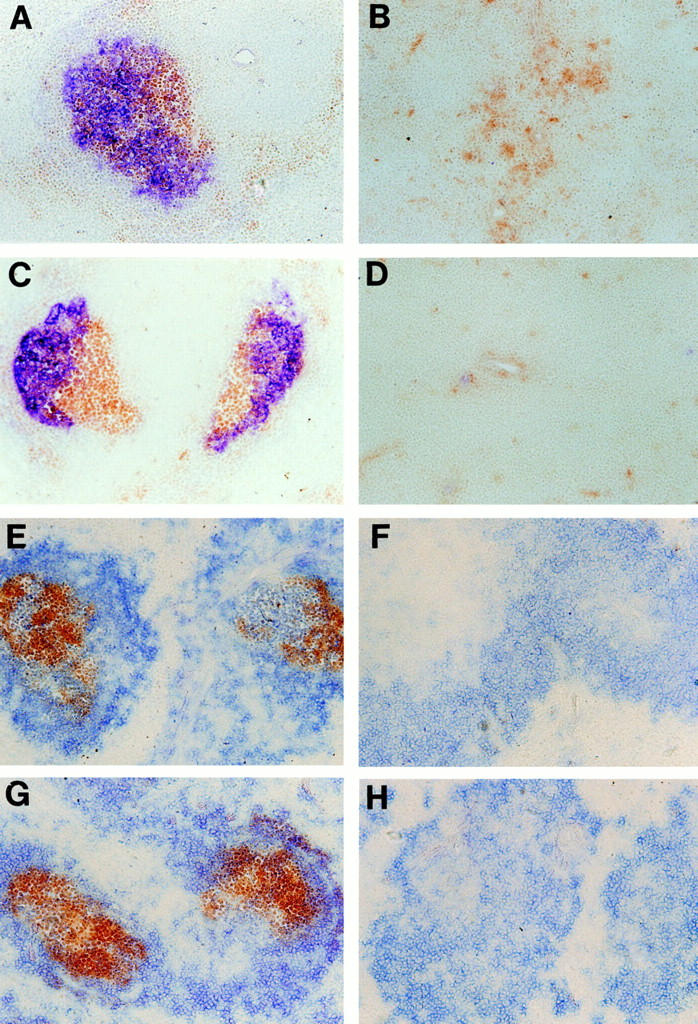
Restoration of organized FDC clusters and GC in LT-α–deficient mice after transplantation with normal BM. After BM transfer, mice were immunized intraperitoneally with 100 μl of PBS containing 10% sheep red blood cells. 10 d later, spleens were harvested and frozen sections were stained with (A–D) anti-CR1 antibody 8C12 (blue) and PNA (brown), or (E–H) with horseradish peroxidase–anti-peroxidase complex (brown) and anti-B220 (blue). (A and E) LT-α–deficient mice reconstituted with wild-type BM showed restored FDC clusters and an ability to form GC. (B and F) Conversely, wild-type mice reconstituted with LT-α–deficient BM showed no detectable FDC clusters or formation of GC. Wild-type mice reconstituted with wild-type BM (C and G) and LT-α–deficient mice reconstituted with LT-α–deficient BM (D and H) showed similar anti-CR1 staining and in vitro IC trapping compared to wild-type mice and LT-α–deficient mice, respectively. Original magnification, ×100.
Although the precise lineage from which FDC differentiate is unknown, several pieces of data suggest that FDC are not BM-derived cells (32, 42, 43). Because transferred LT-α–expressing BM-derived cells can induce both the formation of FDC clusters and the formation of GC in LT-α–deficient recipient mice, we speculate that functional FDC precursors are present in the spleen or other tissues of LT-α–deficient mice. These cells can be induced to form mature FDC clusters once an LT-α–dependent signal is provided by the transferred BM cells.
To further demonstrate the role of LT-α as a signal required to establish organized FDC structure and to investigate the cell lineage of FDC, we used BM cells from CR1/ 2-deficient mice to reconstitute irradiated LT-α–deficient mice. In this context, donor CR1/2-deficient BM-derived cells are LT-α wild type, but can be distinguished from the LT-α–deficient recipient cells by their failure to stain with anti-CR1 mAb (36). After reconstitution, spleen sections were stained with anti-CR1 mAb and PNA to assay for the presence of FDC clusters and GC. Similar to the results obtained after transfer of wild-type BM cells, clustered FDC were identified with anti-CR1 mAb after transfer of CR1/ 2-deficient BM (Fig. 2 A). These FDC were able to support the formation of typical CR1/2-expressing GC (Fig. 2 B). These results clearly indicate that the clustered FDC induced in these BM-chimeric mice are derived from the LT-α–deficient recipient, and that LT-α provides a signal that supports the development of FDC clusters. These results also indicate that the lack of organized FDC structure in LT-α–deficient mice is a plastic characteristic determined by the LT-α expression status of the BM-derived cells populating the animal.
Figure 2.
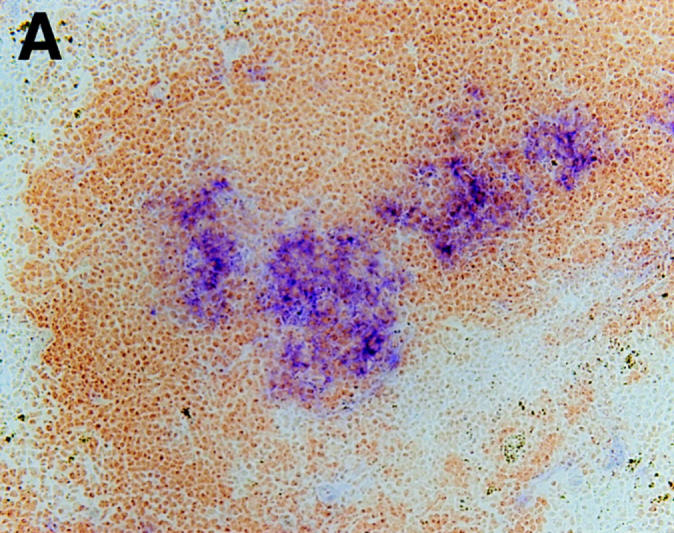
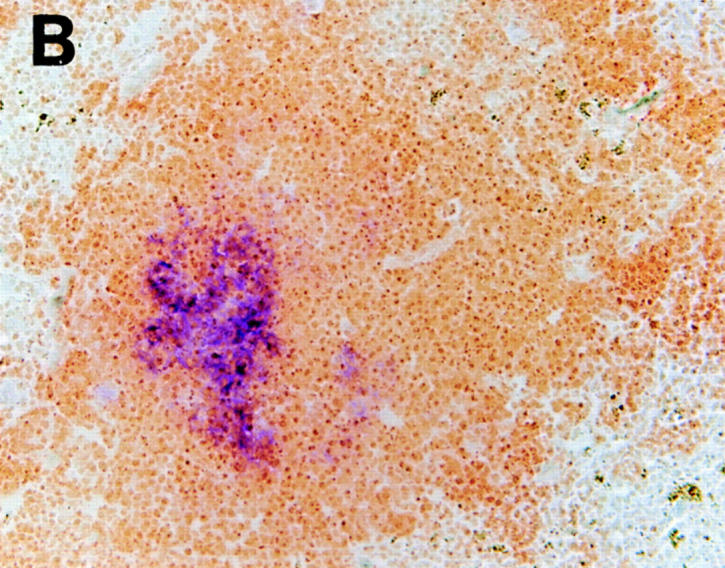
FDC clusters induced after BM transfers are non–BM-derived. When BM from CR1/2-deficient mice was used to reconstitute LT-α–deficient mice, FDC clusters, which stained with anti-CR1 mAb, formed. These FDC clusters supported the development of PNA+ GC. After BM transfer, mice were immunized and spleens were harvested as described in Fig. 1. (A) Staining was with anti-CR1 (blue) and anti-B220 (brown). (B) Staining was with PNA (blue) and anti-B220 (brown). Original magnification, ×100.
Role of TNFR-I in the Development of FDC Clusters and GC.
TNFR-I–deficient mice also lack FDC clusters and spleen GC formation (25, 26). To investigate the mechanism of this structural defect in the TNFR-I–deficient mice, we performed similar reciprocal BM transfer experiments. In contrast to the LT-α–deficient mice, reconstitution of TNFR-I–deficient mice with wild-type BM did not restore the organization of FDC clusters or the formation of GC (Fig. 3, A and C). This suggests that development of normal clusters of FDC in the spleen requires TNFR-I expression on some radioresistant non–BM-derived component(s) in the recipient mouse, most likely a cellular element within the spleen itself. Alternatively, the failure to restore FDC clusters in TNFR-I–deficient mice after transfer of normal BM might indicate that essential TNFR-I–dependent interactions may be required within a developmental window before 8–12 wk of age, when these BM transfers were performed.
Figure 3.
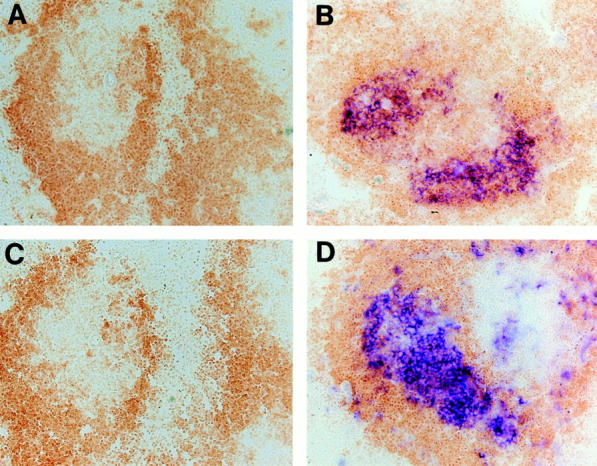
Failure to restore organized FDC clusters and development of GC in TNFR-I–deficient mice by transplantation with normal BM. (A and C) TNFR-I–deficient mice reconstituted with wild-type BM showed no detectable FDC clusters or GC. (B and D) Wild-type mice reconstituted with TNFR-I–deficient BM showed FDC clusters and GC formation. After BM transfer, mice were immunized and spleens were harvested as described in Fig. 1. (A and B) Staining was with anti-CR1 antibody 8C12 (blue) and anti-B220 (brown). (C and D) Staining was with PNA (blue) and anti-B220 (brown). Original magnification, ×100.
Conversely, when irradiated wild-type mice were reconstituted with BM cells from TNFR-I–deficient mice, both organized FDC clusters and morphologically normal GC formed (Fig. 3, B and D). Additionally, these chimeric mice demonstrated robust trapping of IC at sites corresponding to the FDC reticula (data not shown). This suggests that neither the development of FDC clusters nor the formation of GC requires expression of TNFR-I on BM-derived cells.
Distinct Roles of LT-α and TNFR-I in Development of FDC Clusters.
These data demonstrate that LT-α produced by BM-derived cells is required for the organization of FDC clusters, but that expression of LT-α is not required by the FDC themselves or their precursors. In contrast, the development of FDC clusters requires expression of TNFR-I on non–BM-derived cells, but not on BM-derived cells. Thus, clustering of FDC within spleen lymphoid follicles is controlled by at least two distinct cell populations, one defined by the expression of LT-α and the other defined by the expression of TNFR-I. To confirm that the requirement for LT-α and TNFR-I was a manifestation of the need for at least two distinct cell populations, we transferred BM cells from TNFR-I deficient donor mice (LT-α wild-type) into irradiated LT-α–deficient recipients (TNFR-I wild-type). Organized FDC clusters and the ability to form GC were demonstrated in these mice (Fig. 4, A and B). These results confirmed that neither expression of TNFR-I on the BM-derived cells nor of LT-α by radioresistant cells in the recipient is necessary for the development of FDC clusters or GC.
Figure 4.

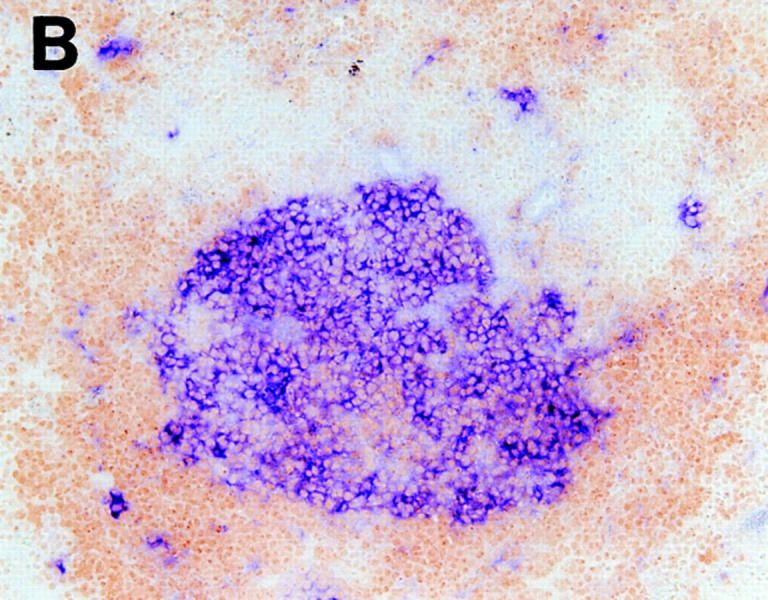
Restoration of organized FDC clusters and GC formation in LT-α–deficient mice reconstituted with TNFR-I–deficient BM. After BM transfer, mice were immunized and spleens were harvested as described in Fig. 1. Serial sections are shown. (A) Staining was with anti-CR1 (blue) and anti-B220 (brown). (B) Staining is with PNA (blue) and anti-B220 (brown). Original magnification, ×100.
Discussion
Our results demonstrate that both LT-α produced by BM-derived cells and TNFR-I expressed by some non– BM-derived cells are required for the development of FDC clusters in the spleen. Because TNFR-I is strongly expressed on the FDC themselves (44) and because FDC are non–BM-derived, as demonstrated in this study, we speculate that the FDC themselves or their lineage precursors are the recipient cells that require signaling directly through TNFR-I for establishment of organized clusters. The inability of wild-type BM to restore clustered FDC organization after transfer into TNFR-I–deficient mice is consistent with this hypothesis.
The presence or absence of FDC clusters is not an intrinsically fixed morphological feature. Plasticity of FDC clusters is seen in LT-α–deficient mice, in which clusters of FDC can be restored by reconstitution with LT-α–expressing BM. This implies that the precursors of clustered FDC cells that respond to an LT-α–dependent signal by formation of mature FDC clusters remain present in LT-α–deficient animals even in the absence of an LT-α signal. Thus, LT-α is not absolutely required for the production of the FDC lineage, but rather it is necessary for its maturation or for organization of the mature cells. In this context, it is of interest that mice transgenic for expression of LT-α driven by the rat insulin promoter develop chronic inflammatory lesions and that these lesions manifest clustered FDC within the cellular infiltrate (45). Currently, whether the LT-α acts as the soluble homotrimer or as a component of the membrane LT heteromer, and which LT-sensitive receptor determines this phenotype remain unknown in this transgenic model.
Although the identification of similarly disturbed FDC clustering in both LT-α– and TNFR-I–deficient mice is consistent with the possibility that LT-α (presumably in its secreted homotrimeric form) acts to regulate FDC organization by binding directly to TNFR-I, the recent observations that both TNF-α–deficient and LT-β–deficient mice also manifest absence of FDC clusters (17–19) demonstrate that both the LT axis and the TNF-α axis are required for the development of this structure, and underscores the likelihood that LT-α signals as a component of the membrane LT heteromer via a receptor distinct from TNFR-I, such as the LT-βR. This latter possibility is supported by the observation that LT-α appears to contribute to the development of other aspects of spleen follicle structure by interaction through a receptor other than TNFR-I or -II. We have observed that LT-α contributes to the formation of a morphologically normal marginal zone in the spleen; staining with MOMA-1, an mAb specific for the metallophilic macrophages that constitute a major component of the marginal zone (46), was essentially absent in LT-α–deficient mice, whereas the pattern of MOMA-1 staining in TNFR-I– or TNFR-II–deficient mice was indistinguishable from that in wild-type mice (25). Analysis of mice carrying targeted mutation in the LT-βR gene should permit clear definition of the role of these TNF/TNFR family members in the establishment of the lymphoid organ structure.
In the BM transfer experiments reported here, the restoration of GC structure in LT-α–deficient mice reconstituted with wild-type BM was associated with restoration of organized FDC clusters; however, when total splenocytes from wild-type donors were transferred into irradiated LT-α–deficient mice, the LT-α–deficient recipients manifested in their spleen follicles the presence of clusters of PNA+ B cells typical of GC but without detectable clustered FDC (Fu, Y.-X., G. Huang, and D.D. Chaplin, unpublished data). This suggests that LT-α acts additionally to provide a signal for the development or maintenance of proliferating clusters of PNA+ B cells. In this short term reconstitution model, this proliferation is not dependent on the presence of FDC clusters. These data suggest that the signals leading to the formation of a fully functional GC are complex, and that GC formation may occur in discrete stages, several of which are controlled by members of the TNF/TNFR family.
Acknowledgments
We thank V.M. Holers and T. Kinoshita for anti-CR mAbs and M. Kosco-Vilbois for anti-FDC antibodies FDC-M1 and FDC-M2. We also thank J. Peschon for providing TNFR-I–deficient mice.
Footnotes
D.D. Chaplin is an investigator of the Howard Hughes Medical Institute. Portions of this work were supported by grant AI34580 from the National Institutes of Health (D.D. Chaplin).
Abbreviations used in this paper: BM, bone marrow; FDC, follicular dendritic cell(s); GC, germinal center(s); IC, immune complex; LN, lymph node; LT, lymphotoxin; PNA, peanut agglutinin; TNFR, TNF receptor.
References
- 1.Beutler, B. 1992. Tumor Necrosis Factors: the Molecules and their Emerging Role in Medicine. Raven Press, Ltd., New York. 590 pp.
- 2.Paul NL, Ruddle NH. Lymphotoxin. Annu Rev Immunol. 1988;6:407–438. doi: 10.1146/annurev.iy.06.040188.002203. [DOI] [PubMed] [Google Scholar]
- 3.Ruddle NH. Tumor necrosis factor (TNF-α) and lymphotoxin (TNF-β) Curr Opin Immunol. 1992;4:327–332. doi: 10.1016/0952-7915(92)90084-r. [DOI] [PubMed] [Google Scholar]
- 4.Kriegler M, Perez C, DeFay K, Albert I, Lu SD. A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: ramifications for the complex physiology of TNF. Cell. 1988;53:45–53. doi: 10.1016/0092-8674(88)90486-2. [DOI] [PubMed] [Google Scholar]
- 5.McGeehan GM, Becherer JD, Bast RC, Jr, Boyer CM, Champion B, Connolly KM, Conway JG, Furdon P, Karp S, Kidao S, et al. Regulation of tumour necrosis factor–α processing by a metalloproteinase inhibitor. Nature. 1994;370:558–561. doi: 10.1038/370558a0. [DOI] [PubMed] [Google Scholar]
- 6.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, et al. A metalloproteinase disintegrin that releases tumour necrosis factor–α from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 7.Moss ML, Jin S-LC, Milla ME, Burkhart W, Carter HL, Chen W-J, Clay WC, Didsbury JR, Hassler D, Hoffman CR, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor–α. Nature. 1997;385:733–736. doi: 10.1038/385733a0. [DOI] [PubMed] [Google Scholar]
- 8.Tartaglia LA, Goeddel DV. Two TNF receptors. Immunol Today. 1992;13:151–153. doi: 10.1016/0167-5699(92)90116-O. [DOI] [PubMed] [Google Scholar]
- 9.Banner DW, D'Arcy A, Janes W, Gentz R, Schoenfeld HJ, Broger C, Loetscher H, Lesslauer W. Crystal structure of the soluble human 55 kd TNF receptor–human TNF β complex: implications for TNF receptor activation. Cell. 1993;73:431–445. doi: 10.1016/0092-8674(93)90132-a. [DOI] [PubMed] [Google Scholar]
- 10.Androlewicz MJ, Browning JL, Ware CF. Lymphotoxin is expressed as a heteromeric complex with a distinct 33-kDa glycoprotein on the surface of an activated human T cell hybridoma. J Biol Chem. 1992;267:2542–2547. [PubMed] [Google Scholar]
- 11.Browning JL, Ngam-ek A, Lawton P, DeMarinis J, Tizard R, Chow EP, Hession C, O'Brine-Greco B, Foley SF, Ware CF. Lymphotoxin β, a novel member of the TNF family that forms a heteromeric complex with lymphotoxin on the cell surface. Cell. 1993;72:847–856. doi: 10.1016/0092-8674(93)90574-a. [DOI] [PubMed] [Google Scholar]
- 12.Browning JL, Dougas I, Ngam-ek A, Bourdon PR, Ehrenfels BN, Miatkowski K, Zafari M, Yampaglia AM, Lawton P, Meier W, et al. Characterization of surface lymphotoxin forms. Use of specific monoclonal antibodies and soluble receptors. J Immunol. 1995;154:33–46. [PubMed] [Google Scholar]
- 13.Crowe PD, Van Arsdale TL, Walter BN, Ware CF, Hession C, Ehrenfels B, Browning JL, Din WS, Goodwin RG, Smith CA. A lymphotoxin-β-specific receptor. Science. 1994;264:707–710. [PubMed] [Google Scholar]
- 14.Ware CF, VanArsdale TL, Crowe PD, Browning JL. The ligands and receptors of the lymphotoxin system. Curr Top Microbiol Immunol. 1995;198:175–218. doi: 10.1007/978-3-642-79414-8_11. [DOI] [PubMed] [Google Scholar]
- 15.De Togni P, Goellner J, Ruddle NH, Streeter PR, Fick A, Mariathasan S, Smith SC, Carlson R, Shornick LP, Strauss-Schoenberger J, et al. Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science. 1994;264:703–707. doi: 10.1126/science.8171322. [DOI] [PubMed] [Google Scholar]
- 16.Banks TA, Rouse BT, Kerley MK, Blair PJ, Godfrey VL, Kuklin NA, Bouley DM, Thomas J, Kanangat S, Mucenski ML. Lymphotoxin-α-deficient mice. Effects on secondary lymphoid organ development and humoral immune responsiveness. J Immunol. 1995;155:1685–1693. [PubMed] [Google Scholar]
- 17.Koni PA, Sacca R, Lawton P, Browning JL, Ruddle NH, Flavell RA. Distinct roles in lymphoid organogenesis for lymphotoxins α and β revealed in lymphotoxin β–deficient mice. Immunity. 1997;6:491–500. doi: 10.1016/s1074-7613(00)80292-7. [DOI] [PubMed] [Google Scholar]
- 18.Alimzhanov MB, Kuprash DV, Kosco-Vilbois MH, Luz A, Turetskaya RL, Tarakhovsky A, Rajewsky K, Nedospasov SA, Pfeffer K. Abnormal development of secondary lymphoid tissues in lymphotoxin β–deficient mice. Proc Natl Acad Sci USA. 1997;94:9302–9307. doi: 10.1073/pnas.94.17.9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pasparakis M, Alexopoulou L, Episkopou V, Kollias G. Immune and inflammatory responses in TNF-α–deficient mice: a critical requirement for TNF-α in the formation of primary B cell follicles, follicular dendritic cell networks, and germinal centers, and in the maturation of the humoral immune response. J Exp Med. 1996;184:1397–1411. doi: 10.1084/jem.184.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pfeffer K, Matsuyama T, Kundig TM, Wakeham A, Kishihara K, Shahinian A, Wiegmann K, Ohashi PS, Kronke M, Mak TW. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenesinfection. Cell. 1993;73:457–467. doi: 10.1016/0092-8674(93)90134-c. [DOI] [PubMed] [Google Scholar]
- 21.Rothe J, Lesslauer W, Lotscher H, Lang Y, Koebel P, Kontgen F, Althage A, Zinkernagel R, Steinmetz M, Bluethmann H. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. . Nature. 1993;364:798–802. doi: 10.1038/364798a0. [DOI] [PubMed] [Google Scholar]
- 22.Erickson SL, de Sauvage FJ, Kikly K, Carver-Moore K, Pitts-Meek S, Gillett N, Sheehan KCF, Schreiber RD, Goeddel DV, Moore MW. Decreased sensitivity to tumour necrosis factor but normal T-cell development in TNF receptor-2–deficient mice. Nature. 1994;372:560–563. doi: 10.1038/372560a0. [DOI] [PubMed] [Google Scholar]
- 23.Rennert PD, Browning JL, Mebius R, Mackay F, Hochman PS. Surface lymphotoxin α/β complex is required for the development of peripheral lymphoid organs. J Exp Med. 1996;184:1999–2006. doi: 10.1084/jem.184.5.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neumann B, Luz A, Pfeffer K, Holzmann B. Defective Peyer's patch organogenesis in mice lacking the 55-kD receptor for tumor necrosis factor. J Exp Med. 1996;184:259–264. doi: 10.1084/jem.184.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsumoto M, Mariathasan S, Nahm MH, Baranyay F, Peschon JJ, Chaplin DD. Role of lymphotoxin and the type I TNF receptor in the formation of germinal centers. Science. 1996;271:1289–1291. doi: 10.1126/science.271.5253.1289. [DOI] [PubMed] [Google Scholar]
- 26.Le Hir M, Bluethmann H, Kosco-Vilbois MH, Muller M, di Padova F, Moore M, Ryffel B, Eugster H-P. Differentiation of follicular dendritic cells and full antibody responses require tumor necrosis factor receptor–1 signaling. J Exp Med. 1996;183:2367–2372. doi: 10.1084/jem.183.5.2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marino MW, Dunn A, Grail D, Inglese M, Noguchi Y, Richards E, Jungbluth A, Wada H, Moore M, Williamson B, et al. Characterization of tumor necrosis factor– deficient mice. Proc Natl Acad Sci USA. 1997;94:8093–8098. doi: 10.1073/pnas.94.15.8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.MacLennan ICM. Germinal centers. Annu Rev Immunol. 1994;12:117–139. doi: 10.1146/annurev.iy.12.040194.001001. [DOI] [PubMed] [Google Scholar]
- 29.Kuppers R, Zhao M, Hansmann M-L, Rajewsky K. Tracing B cell development in human germinal centres by molecular analysis of single cells picked from histological sections. EMBO (Eur Mol Biol Organ) J. 1993;12:4955–4967. doi: 10.1002/j.1460-2075.1993.tb06189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacob J, Kelsoe G, Rajewsky K, Weiss U. Intraclonal generation of antibody mutants in germinal centers. Nature. 1991;354:389–392. doi: 10.1038/354389a0. [DOI] [PubMed] [Google Scholar]
- 31.Berek C, Berger A, Apel M. Maturation of the immune response in germinal centers. Cell. 1991;67:1121–1129. doi: 10.1016/0092-8674(91)90289-b. [DOI] [PubMed] [Google Scholar]
- 32.Schriever F, Nadler LM. The central role of follicular dendritic cells in lymphoid tissues. Adv Immunol. 1992;51:243–284. doi: 10.1016/s0065-2776(08)60489-7. [DOI] [PubMed] [Google Scholar]
- 33.Nossal GJ, Abbot A, Mitchell JJ. Antigens in immunity. XIV. Electron microscopic radioautographic studies of antigen capture in the lymph node medulla. J Exp Med. 1968;127:263–276. doi: 10.1084/jem.127.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsumoto M, Lo SF, Carruthers CJL, Min J, Mariathasan S, Huang G, Plas DR, Martin SM, Geha RS, Nahm MH, Chaplin DD. Affinity maturation without germinal centres in lymphotoxin-α–deficient mice. Nature. 1996;382:462–466. doi: 10.1038/382462a0. [DOI] [PubMed] [Google Scholar]
- 35.Ettinger R, Browning JL, Michie SA, van Ewijk W, McDevitt HO. Disrupted splenic architecture, but normal lymph node development in mice expressing a soluble lymphotoxin-β receptor–IgG1 fusion protein. Proc Natl Acad Sci USA. 1996;93:13102–13107. doi: 10.1073/pnas.93.23.13102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Molina H, Holers VM, Li B, Fang Y-F, Mariathasan S, Goellner J, Strauss-Schoenberger J, Karr RW, Chaplin DD. Markedly impaired humoral immune response in mice deficient in complement receptors 1 and 2. Proc Natl Acad Sci USA. 1996;93:3357–3361. doi: 10.1073/pnas.93.8.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, Lenardo MJ. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature. 1995;377:348–351. doi: 10.1038/377348a0. [DOI] [PubMed] [Google Scholar]
- 38.Mariathasan S, Matsumoto M, Baranyay F, Nahm MH, Kanagawa O, Chaplin DD. Absence of lymph nodes in lymphotoxin-α (LTα)–deficient mice is due to abnormal organ development, not defective lymphocyte migration. J Inflamm. 1995;45:72–78. [PubMed] [Google Scholar]
- 39.Kinoshita T, Takeda J, Hong K, Kozono H, Sakai H, Inoue K. Monoclonal antibodies to mouse complement receptor type 1 (CR1). Their use in a distribution study showing that mouse erythrocytes and platelets are CR1-negative. J Immunol. 1988;140:3066–3072. [PubMed] [Google Scholar]
- 40.Fu Y-X, Molina H, Matsumoto M, Huang G, Min J, Chaplin DD. Lymphotoxin-α (LTα) supports development of splenic follicular structure that is required for IgG responses. J Exp Med. 1997;185:2111–2120. doi: 10.1084/jem.185.12.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kosco MH, Pflugfelder E, Gray D. Follicular dendritic cell-dependent adhesion and proliferation of B cells in vitro. J Immunol. 1992;148:2331–2339. [PubMed] [Google Scholar]
- 42.Yoshida K, van den Berg TK, Dijkstra CD. The functional state of follicular dendritic cells in severe combined immunodeficient (SCID) mice: role of the lymphocytes. Eur J Immunol. 1994;24:464–468. doi: 10.1002/eji.1830240230. [DOI] [PubMed] [Google Scholar]
- 43.Ahearn JM, Fischer MB, Croix D, Goerg S, Ma M, Xia J, Zhou X, Howard RG, Rothstein TL, Carroll MC. Disruption of the Cr2 locus results in a reduction in B-1a cells and in an impaired B cell response to T-dependent antigen. Immunity. 1996;4:251–262. doi: 10.1016/s1074-7613(00)80433-1. [DOI] [PubMed] [Google Scholar]
- 44.Ryffel B, Mihatsh MJ. TNF receptor distribution in human tissues. Int Rev Exp Pathol. 1993;34B:149–156. doi: 10.1016/b978-0-12-364935-5.50015-8. [DOI] [PubMed] [Google Scholar]
- 45.Kratz A, Campos-Neto A, Hanson MS, Ruddle NH. Chronic inflammation caused by lymphotoxin is lymphoid neogenesis. J Exp Med. 1996;183:1461–1472. doi: 10.1084/jem.183.4.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kraal G, Janse M. Marginal metallophilic cells of the mouse spleen identified by a monoclonal antibody. Immunology. 1986;58:665–669. [PMC free article] [PubMed] [Google Scholar]