

Abstract
B cell precursors transiently express a pre–B cell receptor complex consisting of a rearranged mu heavy chain, a surrogate light chain composed of λ5/14.1 and VpreB, and the immunoglobulin (Ig)-associated signal transducing chains, Igα and Igβ. Mutations in the mu heavy chain are associated with a complete failure of B cell development in both humans and mice, whereas mutations in murine λ5 result in a leaky phenotype with detectable humoral responses. In evaluating patients with agammaglobulinemia and markedly reduced numbers of B cells, we identified a boy with mutations on both alleles of the gene for λ5/14.1. The maternal allele carried a premature stop codon in the first exon of λ5/14.1 and the paternal allele demonstrated three basepair substitutions in a 33-basepair sequence in exon 3. The three substitutions correspond to the sequence in the λ5/14.1 pseudogene 16.1 and result in an amino acid substitution at an invariant proline. When expressed in COS cells, the allele carrying the pseudogene sequence resulted in defective folding and secretion of mutant λ5/14.1. These findings indicate that expression of the functional λ5/14.1 is critical for B cell development in the human.
Early B cell development is dependent on the orchestrated control of sequential immunoglobulin gene rearrangements and selective expansion of cells that have successfully passed checkpoint controls that evaluate rearrangements (1, 2). The pre–B cell receptor, which consists of the membrane form of a rearranged mu heavy chain, a surrogate light chain composed of VpreB and λ5/14.1, and the immunoglobulin-associated signal transducing chains, Igα and Igβ, play a pivotal role in this process (3, 4). Failure to express the membrane form of mu heavy chain in both humans and mice results in a complete block in B cell differentiation (5–7). In the mouse, experimentally introduced defects in λ5 cause a block in B cell development at the transition between the pro–B cell and the pre–B cell stage (8). However, the block is not absolute, and by 4 mo of age, affected mice have ∼20% of the normal number of B cells and they are able to make antibodies to both T cell– dependent and –independent antigens (8). The effects of mutations in VpreB have not been evaluated in the mouse, perhaps because there are at least two genes for VpreB (9), both of which are transcribed and expressed in early B cell development (10).
The genes for the surrogate light chains are expressed exclusively in pro–B cells and pre–B cells (9, 11), and can act as markers for these stages of differentiation. The proteins encoded by these genes can escort the mu chain to the cell surface (12, 13), and they may assess the ability of the rearranged mu chain to bind effectively to light chains (13). Whether the surrogate light chain combines with the mu chain to form an extracellular ligand-binding motif is less clear (13). The NH2-terminal portion of VpreB has high homology to the variable region (9), and the COOH-terminal portion of λ5/14.1 has homology to J region and lambda constant regions (11, 14). VpreB and λ5/14.1 are noncovalently linked to one another (15), and, λ5/14.1 is covalently linked to the mu chain via a COOH-terminal cysteine (13, 15, 16).
The organization of the surrogate light chain genes is somewhat different in the human compared to the mouse. In the human, only a single VpreB gene has been reported; however, there are three λ5-like genes that have been named based on their size in EcoRI-digested genomic DNA (14, 17). The functional λ5-like gene is on a 14-kb EcoRI fragment. There are also two λ5-like pseudogenes, 16.1 and 16.2 (also called Fλ-1 [17,18]), that have over 95% homology to λ5/14.1 in exons 2 and 3, but lack exon 1 and associated regulatory elements (17). In addition, there are 7 to 10 lambda constant region genes, the most 5′ of which has high homology to λ5/14.1. It has been postulated that this gene, Gλ1, may be expressed in an unrearranged form like λ5/14.1 (19). Within the human lambda light chain locus on chromosome 22q11.22, the gene for VpreB is within the lambda variable region cluster of genes (20, 21), whereas the genes for λ5/14.1 and 16.2 are 800–1,000 kb distal to the lambda constant region genes. The pseudogene 16.1 is 1,500 kb distal or telomeric to λ5/14.1 (21).
In humans, mutations in Bruton's tyrosine kinase (Btk)1, a cytoplasmic tyrosine kinase that is defective in X-linked agammaglobulinemia (XLA; references 22, 23), as well as mutations in mu heavy chain (5), result in a block in B cell maturation that is first manifest at the transition from the pro–B cell to pre–B cell stage of differentiation (4, 24, 25). However, mutations in these two genes do not account for all of the patients with isolated defects in B cell development (5, 26–32); therefore, we examined the possibility that some patients might have mutations in other components of the pre–B cell receptor complex.
Materials and Methods
Patients.
DNA from eight unrelated patients with sporadic agammaglobulinemia was screened for mutations in λ5/14.1, VpreB, Igα, and Igβ. The patient with mutations in λ5/14.1 had the onset of recurrent otitis at 2 mo of age and was recognized to have hypogammaglobulinemia and absent B cells at 3 yr of age when he developed hemophilus meningitis complicated by arthritis. At that time he was seronegative for the T cell–dependent antigens tetanus toxoid, diphtheria toxoid, and conjugated hemophilus influenza, despite previous immunization. He also failed to make antibody to the T cell–independent antigens in blood group substances. On four evaluations since that time, the patient has never had detectable CD19+ B cells by routine clinical testing. At 5 yr of age, while receiving gammaglobulin replacement therapy, he had no measurable serum IgM and IgA (<8 mg/dl). He had normal numbers of T cells and normal proliferative responses to mitogens.
Mutation Detection.
Genomic DNA was analyzed by single-strand conformation polymorphism (SSCP; reference 26) using the following primers to amplify λ5/14.1: exon 1, 5′-ACCAGGGCACCACTCTCTA-3′ and 5′-GTCATCCTTTCCCGCCTCT-3′; exon 2, 5′-TGGGTCACAGCCTACACACT-3′ and 5′-CAGAAGAGGGTGGGACAGC-3′; exon 3, 5′-TCTCACCCCCTCCTCTGTCC-3′ and 5′-TGCAGAGAGAGAGACCCTTCC-3′. PCR conditions were 5 min at 95°C followed by 30 cycles of 95°C for 45 s, 62°C for 30 s, and 72°C for 30 s with a final extension of 5 min at 72°C. Before electrophoresis, PCR products from exons 1 and 3 were digested with PvuII and BstUI, respectively. BstUI would be expected to cleave the functional λ5/14.1 exon 3 but not exon 3 from the pseudogenes. Sequences demonstrating altered mobility in SSCP were cloned into TA vector (Invitrogen, Carlsbad, CA) and sequenced using M13 primers. The mutations were confirmed using a second independent PCR reaction.
Analysis of λ5/14.1 cDNA.
Reverse transcriptase PCR (RT-PCR) was used to amplify λ5/14.1 cDNA from the patient's mononuclear bone marrow cells using the primers 5′-ACGCATGTGTTTGGCAGC-3′ and 5′-GGCGTCAGGCTCAGGTA-3′. The PCR products were cloned and sequenced as above. PCR products were digested with MspI (New England Biolabs, Beverly, MA) for 2 h at 37°C and visualized by ethidium bromide staining in a 1.8% agarose gel.
Immunofluorescence Staining.
Peripheral blood lymphocytes and bone marrow cells were stained using previously described techniques and reagents (5). In addition, fixed cells were stained with an IgG1 murine monoclonal anti-VpreB antibody produced in mice immunized with a recombinant human VpreB protein (Wang, Y.-H., J. Nomura, O.M. Faye-Petersen, and M.D. Cooper, manuscript in preparation).
Vector Construction.
The pre–B cell line Nalm6 was used as the source of cDNA to amplify the genes for λ5/14.1 and VpreB by RT-PCR. PCR primers for λ5/14.1 were 5′-AGGGCACCACTCTCTAGGGA-3′ and 5′-TGCAGAGAGAGAGACCCTTCC-3′, and for VpreB were 5′-GGCCACAGGAGTCAGAGCT-3′ and 5′-GATGCGTGCCTCTGCTGTCTT-3′. PCR products were cloned into the TA vector and the Pro 142Leu mutant was generated by replacing the endogenous SacI– BamHI fragment of exon 3 with that of the patient. After the sequence had been verified, the three cDNA products were cloned into the pcDNA3 expression vector (Invitrogen) that contains a CMV promoter and SV40 origin of replication.
Transfection and Immunoprecipitation.
COS7 cells were transfected with expression vectors for the normal or mutant λ5/14.1, with or without VpreB using lipofectamine (GIBCO BRL, Gaithersburg, MD) according to the manufacturer's protocol. The cells were metabolically labeled with 0.3 mCi of 35S-Translabel (ICN, Irvine, CA) for 2 h, 44 h after transfection. Labeled cells were lysed in 1% Triton X-100, 10 mM Tris (pH 8.0), 140 mM NaCl, 1% bovine hemoglobin, 0.2 U/ml aprotinin, and 1 mM PMSF. After preclearing, lysates were incubated with murine anti-VpreB or goat antilambda chain antibodies (Sigma Chemical Co., St. Louis, MO). Specific proteins were immunoprecipitated with protein G Sepharose (Pharmacia, Uppsala, Sweden) and eluted with a sample buffer containing 50 mM Tris (pH 6.8), 10% glycerol, 1% SDS, 0.001% bromophenol blue, with or without 5% 2-mercaptoethanol. Proteins were separated by 12.5% SDS-PAGE and detected by fluorography.
Results
Identification of Mutations in λ5/14.1.
Genomic DNA samples from eight patients with defects in B cell development but without mutations in Btk or mu heavy chain were screened for mutations in genes that encode components of the pre–B cell receptor complex. PCR was used to amplify individual exons by flanking splice sites and the products were analyzed with SSCP. A 5-yr-old boy with <1% of the normal number of B cells and severe hypogammaglobulinemia was found to have alterations in exons 1 and 3 of the surrogate light chain gene, λ5/14.1. Although both the patient's mother and father had normal percentages of circulating CD19+ B cells, 6 and 10% respectively, SSCP analysis of genomic DNA from the patient's parents indicated that the abnormality in exon 1 was derived from the maternal allele and the abnormality in exon 3 was derived from the paternal allele. Both exons were cloned and sequenced.
The patient's exon 1 sequence revealed a C to T transition at codon 22, resulting in the substitution of a premature stop codon for the wild-type glutamine (Fig. 1). In exon 3, there were three basepair substitutions: T to C at nucleotide 393 (codon 131), T to C at nucleotide 420 (codon 140), and C to T at nucleotide 425 (codon 142). The first two substitutions do not change the coding sequence; however, the third results in the replacement of the wild-type proline with leucine. The proline at this site, which occurs in the loop linking the second and third strands of one of the two beta pleated sheets that compose the immunoglobulin domain (33), is conserved not only in lambda constant region domains in all species evaluated, but also in most immunoglobulin domains (11). SSCP analysis of exon 3 of λ5/14.1 in DNA from 100 individuals did not demonstrate the pattern seen in the patient's paternal allele in any other samples.
Figure 1.

Comparison of the normal and mutant λ5/14.1 genes. (A) Partial DNA sequence of the first (left) and third (right) exon of λ5/14.1 in the patient and in a normal control. The basepair substitutions are indicated by an asterisk. (B) Sequence alignment of the third exon of the functional λ5/14.1 gene, the patient's λ5/14.1 gene, and the 16.1 pseudogene. Only the basepairs at which λ5/14.1 and the pseudogene 16.1 differ are indicated. The basepairs at which the patient's λ5/14.1 gene correspond to the 16.1 gene are shown in bold type.
The three basepair substitutions in the paternal allele of λ5/14.1 are the same as those found at the corresponding site in exon 3 of the λ5/14.1 pseudogene, 16.1 (17; Fig. 1). To determine whether the altered sequence could be attributed to gross rearrangements within the lambda locus, DNA from the patient was digested with EcoRI and HindIII and examined by Southern blot using an exon 3 λ5/ 14.1 probe. The results demonstrated the expected fragments representing the λ5-like genes and the lambda constant region genes, and did not reveal any altered fragments (Fig. 2).
Figure 2.

Southern blot analysis of the lambda and lambda-like genes. Genomic DNA from the patient (lane 1) and four controls (lanes 2–5) was digested with EcoRI (A) or HindIII (B) and analyzed with a probe from the third exon of λ5/14.1. The molecular size standards are shown at the right side of the figure. In the EcoRI digest, the genes for λ5/14.1 and the pseudogene 16.1 are indicated at the left side of the figure. The bands at 8, 18, and 23 kb in the EcoRI digest represent previously reported polymorphic variants in the number of lambda constant region genes (49).
The mutation in the paternal allele was confirmed by analyzing cDNA from the patient's bone marrow. RT-PCR, using primers from within exons 2 and 3, allowed the amplification of the 3′ portion of the patient's λ5/14.1 gene. The C to T basepair substitution at nucleotide 425 would be expected to destroy an MspI site at this locus. To determine the proportion of the transcripts derived from the maternal and paternal alleles, the RT-PCR products from a control and the patient were digested with MspI and examined in an agarose gel. Although all of the product from the normal control was digested, none of the product from the patient was cleaved, indicating that all of the transcripts were derived from the paternal allele (Fig. 3). The patient's PCR product was sequenced and revealed the three basepair substitutions specific to the 16.1 pseudogene sequence flanked by sequence characteristic of the functional λ5/14.1 gene (Fig. 1). These results provide strong support for the occurrence of a gene conversion event in the lambda locus.
Figure 3.

Analysis of λ5/14.1 cDNA from the patient's bone marrow. RT-PCR was used to amplify the 3′ portion of the λ5/14.1 transcripts from an age-matched control (lane 1), the patient (lane 2), or a negative control (lane 3). Molecular size standards (lane M), the undigested PCR products (A), and the MspI-digested products (B) were separated by electrophoresis. All of the 266-basepair product from the control, but none from the patient, was digested to yield 143- and 123-basepair fragments.
In Vivo Consequences of Mutations in λ5/14.1.
The effects of the λ5/14.1 gene mutations on B cell development were examined in the patient's peripheral blood and bone marrow. In normal individuals, between 5 and 18% of the peripheral blood lymphocytes are B cells as defined by expression of CD19. Our past studies have shown that in patients with X-linked agammaglobulinemia, who have mutations in Btk, B cells represent between 0.01 and 0.10% of lymphocytes (34, 35), whereas in patients with mutations in mu heavy chain, B cells cannot be detected (5). In the patient with mutations in λ5/14.1, 0.06% of the peripheral blood lymphocytes expressed CD19. Like normal circulating B cells, these cells had low intensity expression of surface IgM and dim expression of CD38. By contrast, B cells seen in patients with mutations in Btk expressed an immature phenotype as defined by high intensity expression of both surface IgM and CD38 (34, 35).
Bone marrow cells from the patient and a normal age-matched control were stained in suspension with antibodies to surface immunoglobulin and CD19. Although a small number of CD19+ cells could be detected, there were almost no mature B cells as defined by coexpression of CD19 and surface immunoglobulin (Fig. 4 A). Over 85% of the CD19+ cells were positive for CD34 (Fig. 4 B), indicating a block at the transition between the pro–B cell and pre–B cell stage of differentiation. This block was also seen in permeabilized cells stained for terminal deoxynucleotidyl transferase (TdT) and surface or cytoplasmic IgM (Fig. 4 C). Although the number of TdT+ cells in the patient was comparable to the control, there were very few cells at the next stage of differentiation, cells which express cytoplasmic mu heavy chain. Bone marrow from λ5 knockout mice has markedly decreased expression of VpreB (36), suggesting that in the absence of λ5, VpreB is unstable. In bone marrow of controls, a mean of 82% of TdT+ cells was dimly positive for cytoplasmic VpreB, whereas, in the patient, 14% of the TdT+ cells were VpreB+.
Figure 4.
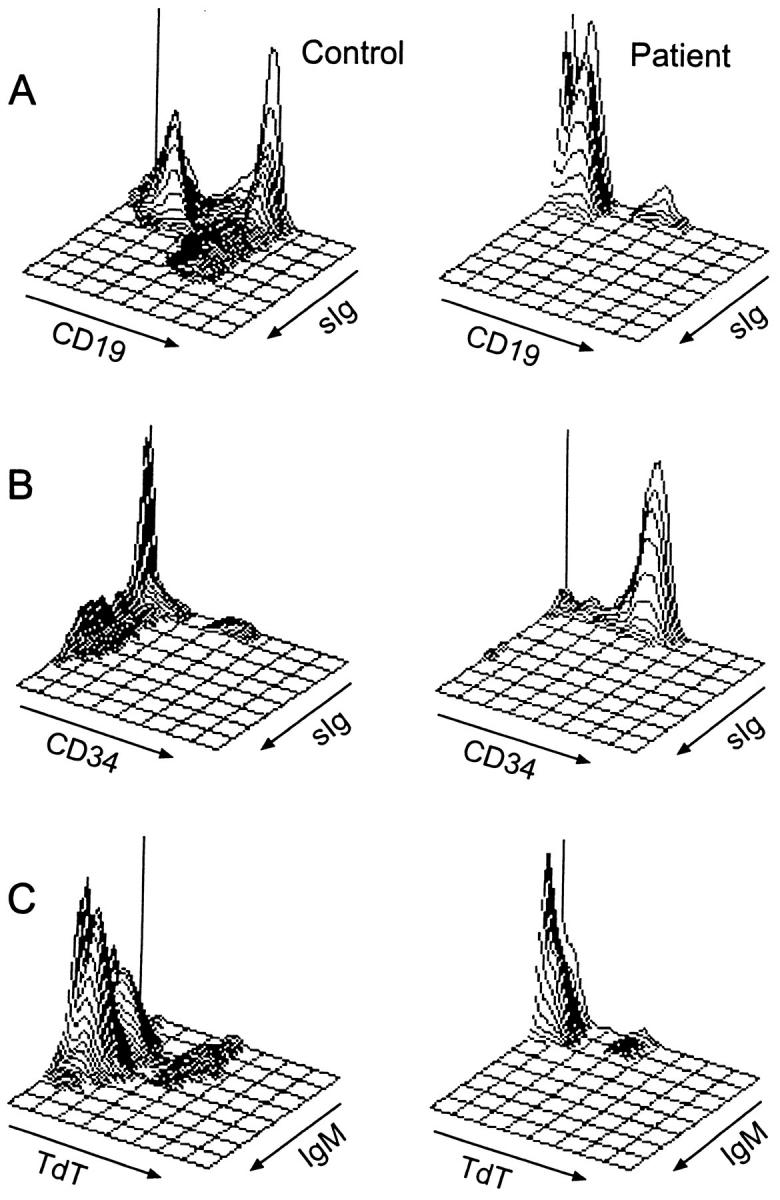
Flow cytometric analysis of bone marrow B cell precursors from a normal age-matched control (left) and the patient with mutations in λ5/14.1 (right). (A) Isometric contour plots of lymphoid-gated cells stained with antibodies to CD19 and surface immunoglobulin (kappa + lambda light chains). In the control, 46% of cells were CD19+, of which 29% were surface Ig (sIg)+. In the patient, 6% of cells were CD19+, with only 0.7% of which were sIg+. (B) Three-color immunofluorescence was used to stain cells for CD19, CD34, and sIg and CD19+ cells were analyzed. In the control sample, 11% of CD19+ cells were CD34+ and 60% were CD34−, sIg−. In the patient, 87% of CD19+ cells were CD34+. (C) Permeabilized cells were stained for TdT and surface or cytoplasmic IgM. In the control, 5% of cells were TdT+, IgM−, 1% were TdT+, IgM+, and 40% were TdT−, IgM+. In the patient, 5% of cells were TdT+, IgM−, 0.2% were TdT+, IgM+, and 0.4% were TdT−, IgM+.
Functional Consequences of Mutations in λ5/14.1.
To determine the functional consequences of the amino acid substitution in exon 3 of λ5/14.1, expression vectors for VpreB and the normal and mutant λ5/14.1 genes were introduced into COS7 cells by lipofection. In metabolically labeled cells transfected with VpreB and the normal or mutant λ5/14.1, both VpreB and λ5/14.1 could be immunoprecipitated from cell lysates using a monoclonal anti-VpreB antibody, demonstrating that the mutant as well as the normal λ5/14.1 could bind to VpreB. However, analysis of supernatants from cells transfected with the same vectors indicated that normal λ5/14.1 was secreted into the supernatant with VpreB, as previously described (13); whereas the mutant λ5/14.1 was not secreted (Fig. 5 A).
Figure 5.

Transfection of COS7 cells with expression vectors for normal or mutant λ5/ 14.1 and normal VpreB. (A) Cells were transfected with VpreB and normal λ5/14.1 (lane 1), VpreB and mutant λ5/14.1 (lane 2), VpreB alone (lane 3), or an empty vector (lane 4). After metabolic labeling, both lysates and supernatants were immunoprecipitated with antibody to VpreB. (B) COS7 cells were transfected with normal λ5/14.1 (lane 1), mutant λ5/14.1 (lane 2), or an empty vector (lane 3), immunoprecipitated using a polyclonal antilambda antibody, and electrophoresed under reducing (R) or nonreducing (NR) conditions.
Because appropriate protein folding is required for protein secretion, and the amino acid substitution in the mutant λ5/14.1 was at a site that would be expected to influence tertiary structure, the capacity of the normal and mutant λ5/14.1 proteins to fold was compared in reducing and nonreducing conditions. As shown in Fig. 5 B, under nonreducing conditions, ∼50% of the normal λ5/14.1 moved through the gel more rapidly because of protein folding, but none of the mutant λ5/14.1 demonstrated altered migration. When coupled with the decreased expression of VpreB in the patient's bone marrow, these studies suggest that the mutant λ5/14.1 was improperly folded and degraded within the endoplasmic reticulum (37).
Discussion
The results of these studies demonstrate that mutations in λ5/14.1 can result in profound B cell deficiency in humans. Transcription of the λ5/14.1 pseudogenes, 16.2 and Gλ1, has been described (18, 19), but it is not clear whether these transcripts are needed for normal development. The severe block in B cell differentiation in our patient indicates that the surrogate light chain pseudogenes in the patient could not compensate for the mutations in the λ5/14.1 gene. The fact that the patient had two different mutations in λ5/14.1 and that he was not a member of an isolated or inbred population, suggests that there may be other similar patients.
The mutation derived from the maternal allele in the patient was a C to T transition in the first exon resulting in the replacement of a wild-type glutamine with a premature stop codon. Premature stop codons in many genes, including Btk, β globin, and dihydrofolate reductase, are associated with faulty processing of the pre–messenger RNA and poor accumulation of the transcript in the cytoplasm (26, 38, 39), as was seen in this case. Thus, this mutation is functionally a null mutation.
The mutation in the paternal allele most likely occurred by gene conversion rather than by two unequal crossover events or three separate basepair substitutions. None of the three basepair substitutions seen in the patient's paternal allele were found individually in 100 normal people. Gene conversion is an uncommon mechanism of mutation; however, it has been suggested that gene conversion between the two highly homologous human fetal globin genes, Aγ and Gγ, explains the genetic variation within these tandemly duplicated genes (40). Immediate proximity of the donor and recipient DNA does not appear to be necessary for gene conversion. Although the functional von Willebrand factor gene is on chromosome 12, a portion of the gene is duplicated as a pseudogene on chromosome 22. In two families with von Willebrand disease, two different segments of the functional gene were replaced by portions of the pseudogene resulting in three basepair substitutions (41), as in our patient. The fact that gene conversion is used to generate antibody diversity in some species (42, 43) suggests that immunoglobulin domains may be unusually susceptible to this type of mutational event.
It is intriguing that the defects in λ5/14.1 seen in the patient described in this paper resulted in a disease more severe than that seen in mice lacking λ5 due to homologous recombination. One could postulate that the mutant λ5/ 14.1 protein produced in the patient's bone marrow might have a more deleterious effect than the absence of λ5 in the knockout mice. However, based on the observation that the patient's father, who was heterozygous for this mutation, had normal B cell numbers, the possibility of a dominant negative mutation is unlikely. The immune function in the λ5 knockout mice improved with time (8); it is possible that older patients with defects in λ5/14.1 may repair their B cell defects and antibody-forming capacity. Other explanations can be considered. The leaky phenotype in λ5-deficient mice has been attributed to B cell precursors in which light chain gene rearrangement preceded heavy chain gene rearrangement, thus negating the requirement for λ5 expression (8, 44). Although studies using human EBV-transformed B cell lines first suggested that rearrangement of light chain genes could occur before that of the heavy chain genes (45), the frequency or efficiency of this event may differ in the two species. There may be other factors that compensate for the lack of expression of λ5, and these factors may differ in the human compared to the mouse. Finally, the recovery of λ5-deficient mice is dependent on the expansion of early B cell precursors that have successfully rearranged both heavy and light chain genes. There may be differences in the regulation of this expansion in the human compared to the mouse.
Early B cell development in the mouse, but not the human, is highly dependent on signaling through the IL-7R (46, 47). By contrast, defects in Btk disrupt the pro–B cell to pre–B cell transition in the human, but not in the mouse (4, 25, 48). Our studies demonstrating the greater consequences of mutations in λ5/14.1 in the human compared to the mouse further emphasize the use of studying multiple models of B cell development. Comparison of the effects of mutations in the same gene in different species highlights the aspects of B cell signaling that are invariant and those that may be influenced by other genetic or environmental factors.
Acknowledgments
These studies were supported by grants from the National Institutes of Health AI25129, National Cancer Institute grant P30 CA21765, the Assisi Foundation, March of Dimes FY97-0384, American Lebanese Syrian Associated Charities, and by funds from the Federal Express Chair of Excellence. M.D. Cooper is a Howard Hughes Medical Institute Investigator.
Footnotes
We appreciate the willingness of the patients and their families to participate in research studies. We also thank J.C. Treadaway and D.K. Mathias for technical assistance, and Drs. J. Rohrer, T. Inukai, and A. Kitanaka for helpful discussions.
Abbreviations used in this paper: Btk, Bruton's tyrosine kinase; RT-PCR, reverse transcriptase PCR; SSCP, single strand conformation polymorphism; TdT, terminal deoxyhucleotidyl transferase.
References
- 1.Spanopoulou E, Roman CA, Corcoran LM, Schlissel MS, Silver DP, Nemazee D, Nussenzweig MC, Shinton SA, Hardy RR, Baltimore D. Functional immunoglobulin transgenes guide ordered B-cell differentiation in Rag-1–deficient mice. Genes Dev. 1994;8:1030–1042. doi: 10.1101/gad.8.9.1030. [DOI] [PubMed] [Google Scholar]
- 2.Young F, Ardman B, Shinkai Y, Lansford R, Blackwell TK, Mendelsohn M, Rolink A, Melchers F, Alt FW. Influence of immunoglobulin heavy- and light-chain expression on B-cell differentiation. Genes Dev. 1994;8:1043–1057. doi: 10.1101/gad.8.9.1043. [DOI] [PubMed] [Google Scholar]
- 3.Rajewsky K. Clonal selection and learning in the antibody system. Nature. 1996;381:751–758. doi: 10.1038/381751a0. [DOI] [PubMed] [Google Scholar]
- 4.Burrows PD, Cooper MD. B cell development and differentiation. Curr Opin Immunol. 1997;9:239–244. doi: 10.1016/s0952-7915(97)80142-2. [DOI] [PubMed] [Google Scholar]
- 5.Yel L, Minegishi Y, Coustan-Smith E, Buckley RH, Trubel H, Pachman LM, Kitchingman GR, Campana D, Rohrer J, Conley MD. Mutations in the mu heavy chain gene in patients with agammaglobulinemia. N Engl J Med. 1996;335:1486–1493. doi: 10.1056/NEJM199611143352003. [DOI] [PubMed] [Google Scholar]
- 6.Kitamura D, Roes J, Kuhn R, Rajewsky K. A B cell–deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. 1991;350:423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- 7.Chen J, Trounstine M, Alt FW, Young F, Kurahara C, Loring JF, Huszar D. Immunoglobulin gene rearrangement in B cell deficient mice generated by targeted deletion of the JH locus. Int Immunol. 1993;5:647–656. doi: 10.1093/intimm/5.6.647. [DOI] [PubMed] [Google Scholar]
- 8.Kitamura D, Kudo A, Schaal S, Muller W, Melchers F, Rajewsky K. A critical role of λ5 protein in B cell development. Cell. 1992;69:823–831. doi: 10.1016/0092-8674(92)90293-l. [DOI] [PubMed] [Google Scholar]
- 9.Kudo A, Melchers F. A second gene, VpreB in the λ5locus of the mouse, which appears to be selectively expressed in pre–B lymphocytes. EMBO (Eur Mol Biol Organ) J. 1987;6:2267–2272. doi: 10.1002/j.1460-2075.1987.tb02500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okabe T, Bauer SR, Kudo A. Pre–B lymphocyte–specific transcriptional control of the mouse VpreB gene. Eur J Immunol. 1992;22:31–36. doi: 10.1002/eji.1830220106. [DOI] [PubMed] [Google Scholar]
- 11.Sakaguchi N, Melchers F. λ5, a new light-chain– related locus selectively expressed in pre–B lymphocytes. Nature. 1986;324:579–582. doi: 10.1038/324579a0. [DOI] [PubMed] [Google Scholar]
- 12.Lassoued K, Illges H, Benlagha K, Cooper MD. Fate of surrogate light chains in B lineage cells. J Exp Med. 1996;183:421–429. doi: 10.1084/jem.183.2.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Melchers F, Karasuyama H, Haasner D, Bauer S, Kudo A, Sakaguchi N, Jameson B, Rolink A. The surrogate light chain in B-cell development. Immunol Today. 1993;14:60–68. doi: 10.1016/0167-5699(93)90060-X. [DOI] [PubMed] [Google Scholar]
- 14.Chang H, Dmitrovsky E, Hieter PA, Mitchell K, Leder P, Turoczi L, Kirsch IR, Hollis GF. Identification of three new Ig λ-like genes in man. J Exp Med. 1986;163:425–435. doi: 10.1084/jem.163.2.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kerr WG, Cooper MD, Feng L, Burrows PD, Hendershot LM. Mu heavy chains can associate with a pseudo-light chain complex (ψ L) in human pre–B cell lines. Int Immunol. 1989;1:355–361. doi: 10.1093/intimm/1.4.355. [DOI] [PubMed] [Google Scholar]
- 16.Pillai S, Baltimore D. Formation of disulphide-linked μ2 ω2tetramers in pre–B cells by the 18K ω-immunoglobulin light chain. Nature. 1987;329:172–174. doi: 10.1038/329172a0. [DOI] [PubMed] [Google Scholar]
- 17.Bossy D, Milili M, Zucman J, Thomas G, Fougereau M, Schiff C. Organization and expression of the λ-like genes that contribute to the μ-ψ light chain complex in human pre–B cells. Int Immunol. 1991;3:1081–1090. doi: 10.1093/intimm/3.11.1081. [DOI] [PubMed] [Google Scholar]
- 18.Schiff C, Milili M, Zucman-Rossi J, Djabali M, Fougereau M. Composite exon structure of an unusual Ig λ-like gene located at human 22q11 position. Mamm Genome. 1996;7:598–602. doi: 10.1007/s003359900177. [DOI] [PubMed] [Google Scholar]
- 19.Evans RJ, Hollis GF. Genomic structure of the human Igλ1 gene suggests that it may be expressed as an Igλ14.1-like protein or as a canonical B cell Igλ light chain: implications for Igλ gene evolution. J Exp Med. 1991;173:305–311. doi: 10.1084/jem.173.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frippiat JP, Williams SC, Tomlinson IM, Cook GP, Cherif D, Le Paslier D, Collins JE, Dunham I, Winter G, Lefranc MP. Organization of the human immunoglobulin lambda light-chain locus on chromosome 22q11.2. Hum Mol Genet. 1995;4:983–991. doi: 10.1093/hmg/4.6.983. [DOI] [PubMed] [Google Scholar]
- 21.Bauer TR, Jr, McDermid HE, Budarf ML, Van Keuren ML, Blomberg BB. Physical location of the human immunoglobulin lambda-like genes, 14.1, 16.1, and 16.2. Immunogenetics. 1993;38:387–399. doi: 10.1007/BF00184519. [DOI] [PubMed] [Google Scholar]
- 22.Tsukada S, Saffran DC, Rawlings DJ, Parolini O, Allen RC, Klisak I, Sparkes RS, Kubagawa H, Mohandas T, Quan S, et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell. 1993;72:279–290. doi: 10.1016/0092-8674(93)90667-f. [DOI] [PubMed] [Google Scholar]
- 23.Vetrie D, Vorechovsky I, Sideras P, Holland J, Davies A, Flinter F, Hammarstrom L, Kinnon C, Levinsky R, Bobrow M, et al. The gene involved in X-linked agammaglobulinemia is a member of the srcfamily of protein-tyrosine kinases. Nature. 1993;361:226–233. doi: 10.1038/361226a0. [DOI] [PubMed] [Google Scholar]
- 24.Pearl ER, Vogler LB, Okos AJ, Crist WM, Lawton AR, III, Cooper MD. B lymphocyte precursors in human bone marrow: an analysis of normal individuals and patients with antibody-deficiency states. J Immunol. 1978;120:1169–1175. [PubMed] [Google Scholar]
- 25.Campana D, Farrant J, Inamdar N, Webster ADB, Janossy G. Phenotypic features and proliferative activity of B cell progenitors in X-linked agammaglobulinemia. J Immunol. 1990;145:1675–1680. [PubMed] [Google Scholar]
- 26.Conley ME, Fitch-Hilgenberg ME, Cleveland JL, Parolini O, Rohrer J. Screening of genomic DNA to identify mutations in the gene for Bruton's tyrosine kinase. Hum Mol Genet. 1994;3:1751–1756. doi: 10.1093/hmg/3.10.1751. [DOI] [PubMed] [Google Scholar]
- 27.Bradley LAD, Sweatman AK, Lovering RC, Jones AM, Morgan G, Levinsky RJ, Kinnon C. Mutation detection in the X-linked agammaglobulinemia gene, BTK, using single strand conformation polymorphism analysis. Hum Mol Genet. 1994;3:79–83. doi: 10.1093/hmg/3.1.79. [DOI] [PubMed] [Google Scholar]
- 28.Hagemann TL, Chen Y, Rosen FS, Kwan S. Genomic organization of the Btk gene and exon scanning for mutations in patients with X-linked agammaglobulinemia. Hum Mol Genet. 1994;3:1743–1749. doi: 10.1093/hmg/3.10.1743. [DOI] [PubMed] [Google Scholar]
- 29.Conley ME, Sweinberg SK. Females with a disorder phenotypically identical to X-linked agammaglobulinemia. J Clin Immunol. 1992;12:139–143. doi: 10.1007/BF00918144. [DOI] [PubMed] [Google Scholar]
- 30.Hoffman T, Winchester R, Schulkind M, Frias JL, Ayoub EM, Good RA. Hypoimmunoglobulinemia with normal T cell function in female siblings. Clin Immunol Immunopathol. 1977;7:364–371. doi: 10.1016/0090-1229(77)90070-8. [DOI] [PubMed] [Google Scholar]
- 31.de la Morena M, Haire RN, Ohta Y, Nelson RP, Litman RT, Day NK, Good RA, Litman GW. Predominance of sterile immunoglobulin transcripts in a female phenotypically resembling Bruton's agammaglobulinemia. Eur J Immunol. 1995;25:809–815. doi: 10.1002/eji.1830250327. [DOI] [PubMed] [Google Scholar]
- 32.Meffre E, Fougereau M, Argenson JN, Aubaniac JM, Schiff C. Cell surface expression of surrogate light chain (ψL) in the absence of mu on human pro–B cell lines and normal pro–B cells. Eur J Immunol. 1996;26:2172–2180. doi: 10.1002/eji.1830260932. [DOI] [PubMed] [Google Scholar]
- 33.Amzel LM, Poljak RJ. Three-dimensional structure of immunoglobulins. Annu Rev Biochem. 1979;48:961–997. doi: 10.1146/annurev.bi.48.070179.004525. [DOI] [PubMed] [Google Scholar]
- 34.Conley ME. B cells in patients with X-linked agammaglobulinemia. J Immunol. 1985;134:3070–3074. [PubMed] [Google Scholar]
- 35.Tedder TF, Crain MJ, Kubagawa H, Clement LT, Cooper MD. Evaluation of lymphocyte differentiation in primary and secondary immunodeficiency diseases. J Immunol. 1985;135:1786–1791. [PubMed] [Google Scholar]
- 36.Rolink A, Karasuyama H, Grawunder U, Haasner D, Kudo A, Melchers F. B cell development in mice with a defective λ5gene. Eur J Immunol. 1993;23:1284–1288. doi: 10.1002/eji.1830230614. [DOI] [PubMed] [Google Scholar]
- 37.Knittler MR, Dirks S, Haas IG. Molecular chaperones involved in protein degradation in the endoplasmic reticulum: quantitative interaction of the heat shock cognate protein BiP with partially folded immunoglobulin light chains that are degraded in the endoplasmic reticulum. Proc Natl Acad Sci USA. 1995;92:1764–1768. doi: 10.1073/pnas.92.5.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baserga SJ, Benz EJ., Jr β-globin nonsense mutation: deficient accumulation of mRNA occurs despite normal cytoplasmic stability. Proc Natl Acad Sci USA. 1992;89:2935–2939. doi: 10.1073/pnas.89.7.2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Urlaub G, Mitchell PJ, Ciudad CJ, Chasin LA. Nonsense mutations in the dihydrofolate reductase gene affect RNA processing. Mol Cell Biol. 1989;9:2868–2880. doi: 10.1128/mcb.9.7.2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Powers PA, Smithies O. Short gene conversions in the human fetal globin gene region: a by-product of chromosome pairing during meiosis? . Genetics. 1986;112:343–358. doi: 10.1093/genetics/112.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eikenboom JC, Vink T, Briet E, Sixma JJ, Reitsma PH. Multiple substitutions in the von Willebrand factor gene that mimic the pseudogene sequence. Proc Natl Acad Sci USA. 1994;91:2221–2224. doi: 10.1073/pnas.91.6.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reynaud CA, Anquez V, Grimal H, Weill JC. A hyperconversion mechanism generates the chicken light chain preimmune repertoire. Cell. 1987;48:379–388. doi: 10.1016/0092-8674(87)90189-9. [DOI] [PubMed] [Google Scholar]
- 43.Thompson CB. Creation of immunoglobulin diversity by intrachromosomal gene conversion. Trends Genet. 1992;8:416–422. doi: 10.1016/0168-9525(92)90324-w. [DOI] [PubMed] [Google Scholar]
- 44.Papavasiliou F, Jankovic M, Nussenzweig MC. Surrogate or conventional light chains are required for membrane immunoglobulin mu to activate the precursor B cell transition. J Exp Med. 1996;184:2025–2030. doi: 10.1084/jem.184.5.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kubagawa H, Cooper MD, Carroll AJ, Burrows PD. Light-chain gene expression before heavy-chain gene rearrangement in pre–B cells transformed by Epstein-Barr virus. Proc Natl Acad Sci USA. 1989;86:2356–2360. doi: 10.1073/pnas.86.7.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.von Freeden-Jeffry U, Vieira P, Lucian LA, McNeil T, Burdach SE, Murray R. Lymphopenia in interleukin (IL)-7 gene-deleted mice identifies IL-7 as a nonredundant cytokine. J Exp Med. 1995;181:1519–1526. doi: 10.1084/jem.181.4.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pribyl JA, LeBien TW. Interleukin 7 independent development of human B cells. Proc Natl Acad Sci USA. 1996;93:10348–10353. doi: 10.1073/pnas.93.19.10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hendriks RW, de Bruijn MF, Maas A, Dingjan GM, Karis A, Grosveld F. Inactivation of Btk by insertion of lacZ reveals defects in B cell development only past the pre–B cell stage. EMBO (Eur Mol Biol Organ) J. 1996;15:4862–4872. [PMC free article] [PubMed] [Google Scholar]
- 49.Taub RA, Hollis GF, Hieter PA, Korsmeyer S, Waldmann TA, Leder P. Variable amplification of immunoglobulin λ light-chain genes in human populations. Nature. 1983;304:172–174. doi: 10.1038/304172a0. [DOI] [PubMed] [Google Scholar]