

Abstract
Human and non–human primate salivas retard the infectivity of HIV-1 in vitro and in vivo. Because thrombospondin 1 (TSP1), a high molecular weight trimeric glycoprotein, is concentrated in saliva and can inhibit the infectivity of diverse pathogens in vitro, we sought to determine the role of TSP1 in suppression of HIV infectivity. Sequence analysis revealed a TSP1 recognition motif, previously defined for the CD36 gene family of cell adhesion receptors, in conserved regions flanking the disulfide-linked cysteine residues of the V3 loop of HIV envelope glycoprotein gp120, important for HIV binding to its high affinity cellular receptor CD4. Using solid-phase in vitro binding assays, we demonstrate direct binding of radiolabeled TSP1 to immobilized recombinant gp120. Based on peptide blocking experiments, the TSP1–gp120 interaction involves CSVTCG sequences in the type 1 properdin-like repeats of TSP1, the known binding site for CD36. TSP1 and fusion proteins derived from CD36-related TSP1-binding domains were able to compete with radiolabeled soluble CD4 binding to immobilized gp120. In parallel, purified TSP1 inhibited HIV-1 infection of peripheral blood mononuclear cells and transformed T and promonocytic cell lines. Levels of TSP1 required for both viral aggregation and direct blockade of HIV-1 infection were physiologic, and affinity depletion of salivary TSP1 abrogated >70% of the inhibitory effect of whole saliva on HIV infectivity. Characterization of TSP1–gp120 binding specificity suggests a mechanism for direct blockade of HIV infectivity that might be exploited to retard HIV transmission that occurs via mucosal routes.
Human immunodeficiency virus (HIV) can be cultured from most tissues and body fluids of infected individuals. Saliva represents a significant exception. In an early report, HIV-1 was isolated from only 1 of 71 saliva samples of HIV+ donors (1). Recent work confirmed the paucity of infectious virus in salivas (2), with a mean viral load in 25 samples of 162 genome equivalents/ml, at the limits of detection by reverse transcription PCR (3). Clinical support for the limited transmissibility of HIV by salivas includes: lack of infection following contamination of open wounds with saliva from HIV+ individuals (4); low occupational risk for HIV infection among dentists in practices with large numbers of patients at risk for HIV infection (5); and the inability to infect adult chimpanzees by direct application of HIV to intact oral mucosa (6).
Such retarded transmission is not a general characteristic of viruses which can be shed orally. The annual attack rate for hepatitis B virus among unvaccinated dentists is 2.6% (7), human T cell lymphotrophic virus type I is found in saliva (8), and the type D retrovirus etiologic in a simian immune-deficiency syndrome can be readily isolated from macaque saliva and spread by this fluid (9). The ability of saliva to suppress HIV-1 also is relatively specific. It does not alter the infectivity of Herpes simplex virus (10), and both cytomegalovirus and Epstein-Barr virus are readily shed in oral secretions of HIV seronegative (11) and seropositive persons (12, 13). Other body fluids from HIV+ individuals do contain HIV in relatively high titers, including tears (14), genital secretions (15), feces (16), and breast milk (17), and the latter three have been implicated in HIV transmission.
Particulate and filterable oral secretions capable of inhibiting HIV infection represent potential explanations for the paucity of HIV in saliva. Reports from several different groups imply that two processes are involved (6, 10, 11, 18–25). Some studies found that whole saliva and submandibular secretions, but not parotid fluid, could sequester HIV virions (10, 11, 18, 19, 24, 25), whereas others identified soluble inhibitory factors capable of direct inhibition in secretions from all salivary glands, but only at very high concentrations (23, 25). Submandibular saliva contains sulfated polysaccharides of low (MG2) and high (MG1) molecular weights (26), with the latter forming an anionic charge barrier to binding of the high-affinity HIV receptor, CD4, to the HIV envelope glycoprotein gp120 (22). Secretory leukocyte protease inhibitor, (SLPI), a 12-kD protein found in whole saliva, has an effect independent of HIV binding to CD4 (20), although its significance in vivo has been questioned (27). Fibronectin, a matrix adhesion molecule, binds directly to gp120, but was shown to inhibit infectivity only at high concentrations (28).
We have focused on thrombospondin 1 (TSP1)1 as a mediator of HIV inhibition. TSP1 is a trimeric sulfated glycoprotein that belongs to a family of high molecular weight extracellular matrix molecules (for review see references 29 and 30). TSP1 is implicated in suppressing the infectivity of certain bacteria and protozoa, including Staphylococcus aureus, babesia, toxoplasma, leishmania, and the malaria sporozoite (31, 32). Unlike other multifunctional glycoproteins, TSP1 is found in very low quantities in plasma, but is stabilized during reversible binding to other matrix molecules, resulting in markedly elevated levels at certain cell surfaces (33). We recently established the presence of an evolutionarily conserved TSP1-binding domain, termed CLESH-1, functional in at least two members of the CD36 gene family, cell surface adhesion receptor CD36 (34) and lysosomal integral membrane protein II (LIMPII; reference 35). A homology search revealed CLESH-1–related sequences in HIV envelope glycoprotein gp120. Therefore, we were interested in whether a TSP-mediated direct interaction with these putative binding motifs could account for anti-HIV activity in saliva. In this study, we demonstrate the ability of purified TSP1 to block HIV-1 infection of primary and transformed target cell lines. These effects involve binding of TSP1 to C2 and C3 conserved regions adjoining the V3 loop of HIV-1 envelope glycoprotein gp120. These areas are key determinants for binding of gp120 to its high affinity cellular receptor, CD4 (36). The effect of TSP1 on CD4– gp120 complex formation suggests a potential role for TSPI in the prevention and therapeutics of HIV-1 infection.
Materials and Methods
Protein Sequence Analysis and Homology Search.
Deduced amino acid sequences encoded by exon 5 of CD36 or homolog LIMPII (Fig. 1) that contain previously characterized TSP1-binding motifs (35) were used as query for a BLAST-enhanced alignment utility search (BEAUTY; reference 37), which incorporates pattern-induced multiple alignment (PIMA; reference 38) sequence family clusters, conserved cluster domains, and PROSITE annotated libraries (39). HIV-1 consensus and subtype sequence alignments were retrieved from the Los Alamos Sequence Database.
Figure 1.

Amino acid sequence alignment of CD36/LIMPII TSP-binding motifs with homologous sequences in HIV-1 gp120. Results of a pattern-based blast enhanced alignment utility search (BEAUTY) that matched a split motif in gp120 domains C2 (top) and C3 (bottom). *, indicates disulfide-bonded cysteine residues of the V3 loop. The brackets above alignments show boundaries of CD36 exon 5 coding region (CD36 aa 95–143). Amino acids identical between HIV-1 gp120 and either CD36 or LIMPII are highlighted white on black. Bold residues represent conservative substitutions according to the following groups: basic (KRH), acidic (DE), charged (KRH, DE), aromatic (YFW), hydroxy (STY), aliphatic (AG), nonpolar/branched (IVL), hydrophobic (AGP, IVL, FM), polar/hydrophilic (ST, KRH, DNEQ, CWY), X = any aa, period = gap. These sequence data are available from GenBank/EMBL/ DDBJ under accession numbers: huCD36, M24795; huLIMPII, D12676; HIV-1, partial sequence of isolate U37041. HIV-1 clade B consensus, MN, and LAI isolate sequences were retrieved from the WHO HIV Sequence Database.
Preparation of TSP1.
Human platelet-rich plasma was obtained from the NY Blood Center (New York, NY). Human thrombin was from Boehringer Mannheim (Indianapolis, IN). Purified human calcium-replete TSP1 was prepared from releasate of thrombin-activated washed platelets as previously described (34, 40). Dot-blot analysis with mAbs to fibronectin (FN) and vitronectin (Calbiochem, La Jolla, CA) showed no reactivity. Endotoxin content was monitored by the Limulus amebocyte lysate test, and was <1 U/μg protein. Polyclonal rabbit anti-TSP antisera and murine anti-TSP1 and anti-TSP2 mAbs 11.4 and 45.2, respectively, were prepared and characterized as previously reported (41, 42).
Purified Fusion Proteins and Peptides.
Characterization of glutathione-S-transferase (GST) fusion proteins derived from CD36 or CD36 homolog LIMPII are reported elsewhere (35), and are numbered to indicate encoded amino acids. CFP67–157 and LFP75–155 contain functionally homologous minimal TSP1-binding domains (CD36 aa93–120; reference 34), whereas CFP298– 439 and LFP156–243 represent downstream sequences. A truncation mutant LFP75–78 was used as an additional GST1 control. Large-scale production and purification of soluble fusion protein followed the method of Frangioni and Neel (43). For some experiments, LIMPII peptide (L75–155) was cleaved from the GST moiety with coagulation Factor Xa (Boehringer Mannheim), and purified by size exclusion chromatography (Centricon 10; Amicon, Beverly, MA). TSP1 synthetic peptides were from Chiron Mimotopes (Victoria, Australia). Recombinant baculovirus-expressed HIV envelope glycoprotein gp160 (gp120 noncovalently linked to transmembrane component gp41) was derived from HIV-1 isolate IIIB (IntraCel Corp., Cambridge, MA). The following purified recombinant proteins were provided by the National Institutes of Health (NIH) AIDS Research and Reference Reagent Program (Bethesda, MD): baculovirus-expressed HIV-1 gp120 derived from LAV and MN isolates, CHO cell–expressed soluble CD4, and HIV-1mn envelopesynthetic peptides.
Solid-phase Binding Assays.
In vitro binding experiments were performed as previously described for TSP1 binding to CD36 and LIMPII (34, 35). In brief, TSP1 (5–10 μg/ml) or fusion proteins and peptides (10–20 μg/ml) were immobilized on detachable 96-well microtiter plate strips (Immulon-4 Remov-a-well; Dynatech Laboratories, Inc., Chantilly, VA) by overnight incubation at 4°C in carbonate buffer (100 mM Na2CO3/1 mM MgCl2/0.02% NaN3, pH 9.8). Washed wells were blocked with 0.5% BSA, and then incubated in triplicate with soluble radiolabeled ligand for 2.5 h at 37°C. After extensive washing in 50 mM Tris, pH 7.5, 150 mM NaCl, 0.5% Tween 20 (TBS-Tween), bound radioactivity was quantified by gamma counter. Radiolabeling was performed with Na[ 125I] (Amersham Life Science Inc., Arlington Heights, IL) using immobilized chloramine T (Iodo-beadsTM; Pierce Chemical Co., Rockford, IL; references 44, 45). Specific activity determined for each experiment ranged from 0.06–1.0 μCi/μg. Specific binding was determined as quenchable in the presence of excess unlabeled ligand, above background binding to BSA-coated wells.
Immunohistochemistry.
Thin tissue sections of oral mucosa autopsy specimens were processed as previously described (46). Formalin-fixed paraffin-embedded sections were dewaxed, pronase-treated, and permeabilized in Triton X-100. Endogenous peroxidase activity was blocked by treatment with a 3% solution of H2O2 for 30 min. Slides were preincubated with normal human serum for 1 h at 22°C, and then incubated overnight at 4°C with 1:1000 dilutions of either polyclonal rabbit anti-TSP1 and anti-TSP2, or preimmune rabbit serum. After a brief blocking step with normal goat serum, successive rinses in PBS were performed between incubations with a 1:250 dilution of biotinylated goat anti–rabbit IgG (Dako, Carpinteria, CA), followed by avidin-biotin-peroxidase complex (Dako). Peroxidase deposition was visualized with 3,3′-diaminobenzidine tetrachloride. Samples were rinsed in distilled water, counterstained with hematoxylin, and viewed by light microscopy.
Quantitative Immunodetection of TSP1.
A sandwich ELISA was used to measure levels of TSP in saliva and cell culture supernatants. Polystyrene 96-well microtiter plates (Falcon, Oxnard, CA) were coated overnight at 4°C with 50 μl/well of 5 μg/ml anti-TSP1 mAb 45.2 in carbonate buffer. Plates were extensively washed with carbonate buffer and then blocked with 1% BSA in TBS-Tween. This was followed by incubating 50 μl of sample diluted 1:16 in TBS-Tween for 2 h at 37°C. Plates were washed with TBS-Tween between successive incubations of polyclonal rabbit anti-TSP1 for 1 h at 37°C, followed by alkaline-phosphatase–conjugated goat anti–rabbit IgG (Kirkegaard & Perry Labs, Gaithersburg, MD). Washed plates were developed with 100 μl/ well of 1 mg/ml p-nitrophenyl phosphate (Kirkegaard & Perry Labs) in 50 mM carbonate buffer, incubated for 30 min at 37°C, and then absorbance at 410 nm was measured using a Microtek plate reader. Quantitation was estimated relative to a standard curve constructed using purified TSP1 diluted in TBS-Tween and using comparison with saliva samples to which known quantities of TSP1 were added.
Saliva Collection.
Unstimulated whole saliva was collected by expectoration into chilled centrifuge tubes and placed on ice. All donors, whether HIV seropositive or negative, had no active periodontal disease or oral lesions. Whole saliva was clarified by microcentrifugation (12,000 g for 10 min at 4°C) and used immediately, and aliquots were stored at ≤20°C. Parotid saliva was collected from one gland using a modified Curby cup (47). Submandibular salivary fluid was collected using surgical sponges (Weck-Cel) cut into ∼0.25 × 0.5 cm rectangles and touched to blebs of fluid which formed at the duct orifi. Saliva was removed from sponges by centrifugation in polyethylene tubes, as previously described (47). Protein content was estimated using a micro-BCA reagent kit (Pierce Chemical Co.).
HIV-1 Infectivity Assays.
CD4+ T lymphoblastoid (SK23, Jurkat) and promonocytic (U937) cell lines, or PBMCs obtained from HIV seronegative donors and activated with PHA (2 μg/ml for 72 h), were cultured in RPMI 1640, 5% fetal bovine serum (FBS; complement-inactivated for 30 min at 56°C), 100 U/ml penicillin, and 100 μg/ml streptomycin (GIBCO BRL, Gaithersburg, MD). The PBMC medium also contained 32 U/ml IL-2 (Sigma Chemical Co., St. Louis, MO). FBS samples were prescreened to assure that levels of TSP were <10 μg/ml. Acute HIV infection was performed using HIV-1 isolate IIIB stock virus as previously described (48, 49). In brief, 2.5 × 105 target cells were exposed to stock virus at a multiplicity of infection (moi) of either 0.02 or 0.15 for 2 h at 37°C, washed once with PBS, and replated in tissue culture–treated microwells with 0.3 ml fresh culture medium. At 3–4 days after inoculation, one half of culture supernatant from each well was replaced with fresh medium. HIV activity was determined after 7 d using an ELISA antigen capture assay for HIV-1 p24 (Gag) core protein (Dupont Medical Products, Boston, MA) with Triton X-100 solubilized culture supernatants.
Saliva and TSP1 Inhibition Assays.
100 μl of HIV-1/IIIB inocula were mixed with 100 μl of various concentrations of whole saliva, salivary fluid fractions, or purified platelet TSP1 diluted in serum-free cell culture medium, and preincubated for 5 min to 2 h at 37°C. Next, 10-fold serial dilutions were added directly to target cell cultures for assay of infectivity. In some experiments, preincubated virus-saliva–TSP mixtures were filtered through 0.2 μm nitrocellulose syringe filters (Gelman Sciences, Inc., Ann Arbor, MI) before inoculation of target cells.
Salivary TSP1 Depletion Experiments.
An affinity column for adsorption of TSP1 was prepared using fusion protein LFP75– 155 coupled by N-hydroxysuccinimide to Sepharose (HiTRAPTM, Pharmacia Biotech, Piscataway, NJ). 2 ml of clarified whole saliva was diluted 1:1 in PBS and divided into two aliquots, one of which was applied to the TSP-binding GST/LIMPII column, the other to an identical column containing N-hydroxysuccinimide–linked GST alone. Columns were incubated for 45 min at 37°C, and then each was flushed with 1 ml PBS. The final products represented 1:4 dilutions of saliva. TSP concentrations were determined before and after column adsorption by sandwich ELISA, and total protein was assessed using the micro-BCA reagent kit.
Cell Surface Expression of CD4.
CD4 surface expression on HIV-1 infected or uninfected U937 cells was evaluated by indirect immunofluorescence flow cytometry using anti-CD4 mAb Leu3, as detailed elsewhere (48).
Results
Characterization of a Putative TSP1-binding Domain in HIV-1 env
Identification of Potential TSP1 Binding Motifs in gp120.
Structural mapping studies have elucidated a TSP1-binding domain within the region encoded by CD36 exon 5 (34). We recently showed that the corresponding sequence of the CD36 homolog LIMPII (50, 51) bound TSP1 with high affinity, confirming a functionally identical domain (35). Computer-assisted motif analysis of 14 CD36 gene family homologs revealed highly conserved blocks implicating an evolutionarily maintained functional module that we have designated CLESH-1 (35). Using this sequence as query in a pattern-based homology search to identify putative TSP-binding sequences in proteins outside the CD36 gene family, we identified CLESH-1 related sequences in HIV-1 and -2 and simian immunodeficiency virus (SIV). Fig. 1 shows the amino acid alignment of CD36/LIMPII CLESH-1 motifs with HIV-1–matched sequences. Strongest homology appeared as a split motif localized to domains C2 and C3 on either side of the V3 loop region of gp120. Homology was highest to LIMPII, with 62.5% identity, 87.5% similarity for the first half-site (C2 domain, 16 aa), and 55.6% identity, 88.9% similarity for the second half-site (C3 domain, 18 aa). Comparison with the clade B subtype consensus and two laboratory isolates used in this study (mn and lav) shows that homology extends throughout both full-length motifs. The overall range of 30–36% identity, 79–84% similarity between LIMPII and HIV-1 isolates is comparable to 37% identity, 75% similarity observed between gene family members CD36 and LIMPII. This discovery prompted a biochemical approach to determine whether TSP1 could interact directly with gp120.
125I-TSP1 Binds to Recombinant HIV-1 env.
We used solid-phase binding assays to assess direct binding of TSP1 to the HIV-1 envelope complex. Fig. 2 A shows concentration-dependent, saturable binding of radiolabeled soluble TSP1 to immobilized recombinant gp160. An apparent affinity of ∼250 nM was comparable to that demonstrated for binding of TSP1 to purified platelet CD36. Binding was effectively quenched in the presence of 10-fold molar excess unlabeled soluble TSP1 (Fig. 2 B, 94 ± 6% inhibition), demonstrating specificity. In addition, a LIMPII fusion protein LFP75–155 containing the TSP-binding domain partially blocked binding (47 ± 14% inhibition), whereas control fusion protein LFP156–243 representing downstream LIMPII sequences did not, supporting the existence of a functionally similar domain in HIV-1 env.
Figure 2.


(A) Concentration-dependent binding of 125I-TSP1 to gp160. Increasing concentrations of soluble 125I-labeled TSP1 (1 nM–1 μM) were added to immobilized recombinant HIV-1 gp160 for 3 h at 22°C, and bound TSP1 was measured after extensive washing. Nonlinear curve fit was generated with ExcelTM version 5.0. Apparent affinity was estimated from Scatchard analysis. (n = 2; error calculated as SD). (B) Competitive inhibition demonstrates specificity of the TSP1–HIV interaction. A fixed concentration of 125I-labeled TSP1 (50 nM) was added to immobilized rgp160 in the absence or presence of 10-fold molar excess (0.5 μM) of unlabeled TSP1, fusion protein LFP75–155 containing the LIMPII TSP-binding domain (aa 75–155), or downstream fusion protein LFP156–243. Samples were incubated and bound TSP1 was measured as in A. Plots represent single data sets of triplicate samples. (n = 3, error as SD).
125I-gp120 Interacts with the CD36-binding Peptide of TSP1.
CSVTCG peptides found in the type 1 repeats of TSP1 are binding sites for CD36 and LIMPII (35, 52–54). Therefore, we tested whether gp120 shared this same specificity. Saturation isotherms showed significant binding of radiolabeled gp120 (lav isolate) to immobilized CSVTCG peptide (Fig. 3), with an apparent affinity of 300 nM. The activity was sequence-specific, as demonstrated by inefficient binding to scrambled control peptide VGSCCT, or to an RGD-containing peptide similar to the GRGDA cell adhesion sequence of the last TSP1 type 3 calcium-binding repeat. This is further evidence that the TSP1-gp120 interaction is mediated through a CD36/LIMPII-related structural domain.
Figure 3.

125I-gp120 interacts with the CD36 binding peptide of TSP1. Increasing concentrations of soluble 125I-labeled recombinant gp120lai (1 nM–1 μM) were incubated with immobilized ligand for 2 h at 37°C, and bound radioactivity was measured as in Fig. 2. Shown are 125I-gp120 binding to CSVTCG peptide derived from TSP1 properdin-like type 1 repeat, scrambled control peptide VGSCCT, and GRGDS derived from TSP1 calcium binding type 3 repeat. This plot represents a single data set of triplicate samples (n = 2, errors as SD).
Peptides Define the TSP1-binding Site in gp120.
Fig. 4 A shows the location of a series of 20 aa synthetic peptides with respect to gp120 domain structure (mn isolate). Given the constraints of the solid phase assay, peptides were immobilized either singly or in pairs, and tested for an ability to bind radiolabeled TSP1. As shown in Fig. 4 B, active peptides corresponded to regions of gp120 containing homologous CLESH-1 motifs. Peptide pairs that extended represented motif sequences showed augmented binding compared to either peptide alone. In addition, single peptides representing sequences outside strongly homologous split motif half-sites also showed significant TSP1 binding. Interestingly, a V3-containing peptide (No. 4, aa311–330) with TSP1-binding activity also displayed augmented binding in combination with an inactive C2 peptide within the first CLESH-1 motif (No. 3, aa291–310). These data suggest the presence of two competent TSP1-binding elements in the predicted regions of gp120, including V3 sequences in the first motif, with the potential for interchangeable combinations of site usage, and possible TSP-mediated structural alterations that might disrupt conformation-dependent binding of gp120 to CD4 receptor.
Figure 4.


(A) Map showing the position of snythetic gp120 peptides with respect to gp120 C2-V3-C3 domains and TSP-binding motifs. A set of gp120mn peptides (∼20 aa) were immobilized either singly (numbered 1–7) or in pairs as indicated. Corresponding amino acid positions are: 1, aa 271–290; 2, aa 281–300; 3, aa 291–310; 4, aa 311–330; 5, aa 331–351; 6, aa 351–370; 7, aa 3361–380; 1+2 and 6+7 overlap by 10 aa; gp120, full length rgp120mn; V3 loop, aa 305–332; motif 1, aa 272–321; motif 2, aa 331–384. (B) Binding of 125I-TSP1 to gp120 peptides. Increasing concentrations of soluble 125I-labeled TSP1 (25 nM–1 μM) were added to immobilized gp120mn peptides. Samples were incubated and bound TSP1 was measured as in Fig. 2. Shown is a single data set for 25 nM TSP1 binding to ∼75–130 μmol peptide or ∼2 μmol gp120.
TSP1 Inhibits Binding of 125I-CD4 to gp120.
An obvious question is whether TSP1 could compromise the ability of gp120 to bind to CD4. Fig. 5 shows competitive inhibition of radiolabeled CD4-binding to gp120 derived from two different viral isolates (mn and lav). In the presence of 10-fold molar excess TSP1, complete inhibition of 125I-CD4–binding to immobilized gp120 was seen. CSVTCG peptide showed a partial but significant inhibition of CD4–gp120 complex formation (53 ± 9%), confirming a TSP-specific effect, while the RGD-containing peptide had little effect (4 ± 4%), and the scrambled control actually enhanced binding. Consistent with structural homology data, TSP1-binding CD36- and LIMPII-derived fusion proteins proved strong competitors (both ∼89% inhibition), whereas downstream LIMPII control protein had a minimal effect (32 ± 5%). For comparison, a 1:2 dilution of whole saliva was a potent inhibitor in this assay system. These observations support a role for salivary TSP1 as a direct inhibitor of HIV infectivity.
Figure 5.

Competitive inhibition of 125I-CD4 binding to gp120. A fixed concentration of 125I-labeled soluble recombinant CD4 (50 nM) was added to immobilized rgp160 (solid bars, MN; hatched bars, LAV isolate-derived) in the absence or presence of 10-fold molar excess (0.5 μM) of unlabeled TSP1, TSP1-derived peptides CSVTCG or GRGDS, scrambled control peptide VGSCCT, fusion proteins containing the TSP1-binding domain of CD36 (CFP, aa 67–157) or LIMPII (aa 75–155), downstream fusion protein LFP156–243, or an equal volume of saliva (final twofold dilution). Samples were incubated and bound TSP1 was measured as in Fig. 2. Shown is a single data set of triplicate samples. (n = 3, error as SD).
Analysis of TSP1-mediated Inhibition of HIV-1 Infectivity
Quantitation and Localization of Salivary TSP1.
As shown in Table 1, levels of TSP1 in whole saliva from either HIV+ or HIV− donors were at least 10-fold greater (1–12 μg/ml) compared to plasma. Amounts of TSP1 in parotid saliva fractions were equivalent to that of plasma, with the bulk of TSP1 found in the submandibular secretions. These values are consistent with previous reports of anti-HIV activity predominantly in submandibular and not parotid gland fluids (11, 19) although only a single saliva fractionation was performed. To document that elevated levels of TSP1 in saliva may be secondary to local production, rather than leakage and concentration from plasma, we performed immunohistochemical staining of fixed tissue from oral mucosa. Moderate to intense levels of TSP1-directed immunoreactivity were evident on gingival epithelium (Fig. 6), confirming that cell-associated TSP1 may correspond to significantly high local concentrations in the oral cavity.
Table 1.
Quantitation of TSP1 in Human Plasma and Saliva
| Sample Source | HIV Status | TSP1.2 | n | |||
|---|---|---|---|---|---|---|
| μg/ml (range) | ||||||
| Plasma | negative | 0.25 (0.1–0.34) | 8 | |||
| Whole saliva | negative | 4.1 (1.1–12.8) | 6 | |||
| positive | 3.3 | 2 | ||||
| Parotid | negative | 0.1 | 1 | |||
| Submandibular | negative | 2.5 | 1 |
Figure 6.
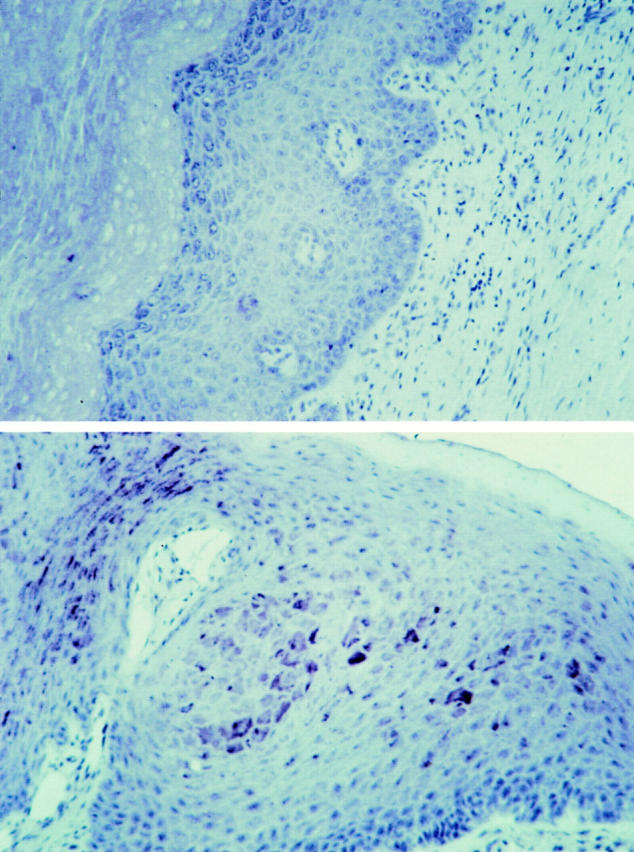
Immunohistochemical detection of cell-associated TSP1 in human gingival mucosa. Fixed oral epithelial tissue thin section was incubated with polyclonal antiserum reactive against both TSP1 and 2 (lower panel), or with preimmune serum (upper panel), followed by biotinylated second antibody, and developed using avidin-conjugated peroxidase. Brown deposits indicate sites of thrombospondin reactivity. Original magnification, ×200.
Inhibition of HIV-1 Infectivity by Purified TSP1.
To investigate whether purified TSP1 could inhibit HIV-1 infection similarly to saliva, viral isolate IIIB was added to target cells after preincubation with TSP1. Many studies have shown that filtration of virus–saliva mixtures is required for maximum inhibition (10, 11, 18, 24). The physiologic equivalent of such filtration is thought to be the constant cleansing of the oral cavity by salivary flow. Therefore, in some experiments preincubated virus-TSP1 mixtures were passed through 0.2-μm filters. Different multiplicities of infectious virus per target cell were tested in both systems. At concentrations found in saliva (2–10 μg/ml), TSP1 reduced HIV-1 infection of PHA-activated donor PBMCs by >83% when prefiltered, as measured by ELISA detection of p24 viral antigen (Fig. 7). This was comparable to a 1:2 dilution of whole saliva. In contrast, fibrinogen, another high molecular weight adhesive glycoprotein in saliva, had no effect in this system. Specificity was documented by abrogation of the inhibitory effect in the presence of a specific anti-TSP1 polyclonal antibody (107% of control p24), but not control IgG (data not shown). Prolonged incubation of the TSP–virion mixture was unnecessary, as exposures as brief as 5 min appeared sufficient to reduce infectivity by ⩾50% at 1 μg/ml TSP1 (data not shown). The TSP1 effect also was apparent for HIV-1 IIIB infection of CD4+ T-lymphoblastoid and monocytoid cell lines (SK23 ⩾ 90.7% and U937 ⩾83.0% inhibition, respectively). In addition, two monocytotropic strains of HIV-1, p13 and HA593, representing patient isolates obtained from the NIH AIDS Retroviral Repository (Bethesda, MD), were susceptible to inhibition by purified TSP1. At viral mois of 0.8, 100 μg/ml TSP1 inhibited HIV infectivity by ⩾98 ± 1% (data not shown). In contrast to prefiltration experiments, direct addition of TSP1 was able to inhibit HIV-1 by 50–75% only when a high concentration of TSP1 and a low moi of inoculum was used (Fig. 7). In parallel assays, saliva could inhibit HIV-1 as a direct addition only when added in dilutions of <1:4. However, preincubation of virus with saliva permitted dilutions >1:10. The results again are consistent with levels of TSP1 found in these dilutions of saliva.
Figure 7.

Inhibitory effect of TSP1 on HIV-1 infectivity. HIV-1 isolate IIIB was preincubated in the absence of TSP1, or with various concentrations of purified TSP1 for 1 h at 37°C. Preincubated virus–TSP1 mixtures were added to target cells either directly (−), or first passed through 0.2 μm filters (+). PMA-activated primary PBMC, SK23 (T cell line) or U937 (promonocytic line) were inoculated with the mois indicated. After an additional 1 h at 37°C, infected cells were washed, cultured for 7 d, and HIV-1 p24 antigen was measured by ELISA. Inhibition is expressed for a single data set as percentage of maximum p24 detected in the absence of TSP1.
Depletion of Salivary TSP1 Abrogates the Anti-HIV Effect.
To determine the extent of TSP1 contribution to saliva inhibition of HIV infectivity, clarified saliva samples were passed over affinity columns of immobilized TSP-binding LIMPII fusion protein LFP75–155 before virus preincubation. Adsorption removed ∼95% of TSP (Table 2), as assessed by sandwich ELISA, whereas the decrease in total protein was substantially less (∼15%). TSP depletion correlated with ⩾70% reduction in anti-viral activity, in contrast to saliva adsorbed using a control fusion protein affinity column (GST-1). The data suggest that TSP may account for a major proportion of HIV-specific inhibitory activity in saliva.
Table 2.
Effect of TSP1 Depletion on Salivary Inhibition of HIV-1
| Saliva Adsorption | Protein | TSP1.2 | % Inhibition | |||
|---|---|---|---|---|---|---|
| None (preadsorbed) | 8.16 | 1.20 | 99.8 | |||
| TSP-affinity | 6.20 (76.0%) | 0.08 (6.3%) | 31.1 | |||
| (LFP75–155) | ||||||
| GST control | 7.52 (92.2%) | 1.18 (98.3%) | 99.7 |
HIV-1iiib (0.15 moi) was admixed with clairfied whole saliva that was untreated or first adsorbed with immobilized fusion proteins for 1 h at 37°C (final 1:4 dilute). Preincubated and filtered virus–saliva mixtures then were used to infect PHA-activated PBMCs, and HIV-1 activity was determined on day 7 after inoculation as in Fig. 7. Percentages in parenthesis are relative to preadsorbed concentrations. Anti–HIV-1 inhibitory effect is expressed as percentage of decrease of maximum HIV-1 p24 antigen in the absence of saliva.
TSP-binding Peptide Suppresses TSP1 Anti-HIV Activity.
To further delineate a TSP-specific effect, LIMPII TSP-binding peptide L75–155 (10-kD product purified after removal of GST moiety by proteolytic cleavage) was included in HIV–TSP1 preincubation mixtures as a competitor. Fig. 8 shows that 1 μM LIMPII peptide abrogated the inhibitory effect of even high concentrations of TSP1 (50– 100 μg/ml) by 83–90%. Incubation of virus in the presence of LIMPII peptide alone resulted in minimal decrease of HIV-1 infectivity (∼9%), suggesting that the amounts of peptide able to block the TSP1 antiviral effect were not sufficient to compete for HIV-1 envelope binding sites on PBMC target cells. The ability of the LIMPII peptide to restore infectivity supports a direct role for a CD36/ LIMPII-related TSP1-binding domain on HIV-1 gp120, and provides further evidence of a common binding site on TSP1.
Figure 8.

Competitive inhibition of TSP1 anti-HIV effect by LIMPII TSP1-binding domain. PMA-stimulated primary PBMCs were infected with 0.02 moi HIV/IIIB that had been preincubated with TSP1 alone, or with LIMPII peptide LFP75–155 alone, or in the presence of TSP1, and filtered before incubation as in Fig. 7. TSP-mediated inhibition is expressed as percentage of decrease in p24 relative to maximum p24 detected in the absence of TSP1. Data shown represent the average of two independent experiments (error as SD).
Effect of TSP1 on CD4 Expression.
Another mechanism to explain TSP-mediated blockade to HIV infection would be a direct effect on target cells, whereby alterations in cell function would decrease the capacity to support productive infection. To address whether exogenous purified TSP1 induced downmodulation of the high affinity HIV receptor, we monitored CD4+ Jurkat and SK23 T cell lines, as well as PHA-activated PBMCs, for differences in CD4 surface expression after culture for 3 d in the absence or presence of 100 μg/ml TSP1. By flow cytometric analysis of cells stained with fluorescein-conjugated anti-CD4 IgG, no change in relative fluorescence intensity or percentage of CD4+ cells was detected (data not shown). Thus, TSP1 likely does not reduce cell susceptibility to HIV-1 infection.
Effect of Acute and Chronic HIV-1 infection on Production of TSP1.
To examine whether HIV-1 infection modulates TSP1 expression, TSP1 secretion was monitored in three groups of cells: an uninfected line of U937 promonocytic cells; U1.1 cells representing chronically infected U937 containing two stably integrated copies of HIV-1/LAI; and U937 acutely infected with HIV-1 to high copy number (>1000 proviral copies/cell; reference 49). Cells were preincubated for 1 h in the presence or absence of PMA, a mitogenic inducer of HIV from chronically infected cells (49), which has also been shown to stimulate TSP1 expression in cell lines (55). After 24 h in culture medium, cells were switched to serum-free medium for an additional 18 h and then supernatants were collected for TSP1 quantitation by ELISA. As shown in Table 3, HIV-1 infection did not diminish TSP1 production, although acute infection blunted the response to PMA induction of TSP1.
Table 3.
Production of TSP by Monocytoid Cells in the Presence or Absence of HIV-1 Infection
| Cell line | Infection status | PMA | TSP1,2 | |||
|---|---|---|---|---|---|---|
| 5 ng/ml | ng/ml | |||||
| U937 | Parental, uninfected | − | <5 | |||
| promonocytic line | + | 51 | ||||
| U1.1 | U937, chronic | − | 50 | |||
| 2 stable copies LAI | + | 125 | ||||
| U937/HIV | acute, high copy | − | 180 | |||
| IIIB (0.02 moi) | + | 135 |
Cells were exposed to buffer or PMA for 1 h at 37°C, cultured for 24 h in RPMI 1640 with 5% FCS, and then cultured for 18 h in serum-free RPMI 1640 with 0.25% BSA. TSP levels were measured by sandwich ELISA. LAI and IIIB indicate HIV-1 isolate subtypes.
Discussion
We report the identification of TSP1-binding sites in the C2 and C3 regions of gp120, conserved areas of the HIV envelope that are important in binding to CD4, and demonstrate direct interaction with a specific cell adhesion sequence found in the type I repeat of TSP1. Characterization by in vitro binding and competition studies substantiates that these CD36/LIMPII-related CLESH-1 motifs in gp120 represent authentic TSP1-binding domains. The physiological significance of TSP1–gp120 complex formation is supported by observations that salivary inhibition of HIV-1 infectivity was markedly reduced by affinity depletion of TSP1; saliva samples that block infection after filtration contained levels of TSP1 correlating with inhibitory concentrations of purified TSP1; and higher amounts of TSP1 required to block HIV-1 infectivity in vitro are comparable to the greater quantities of saliva required to obtain an antiviral effect (19, 23, 25). Our findings establish a distinct mechanism to explain HIV-specific blockade of transmission via saliva.
The likelihood that HIV inhibitors in saliva identified in vitro are active in vivo is bolstered by two lines of evidence. First, in a study of 48 HIV+ patients, 88% of PBMC samples, but no saliva samples, were positive for replication-competent HIV− (56). Second, recovery rates for HIV in salivas do not differ before and after dental procedures accompanied by bleeding into the oral cavity (57), indicating that free virus from blood was removed or inactivated. The fact that HIV is not found within salivary acinar and ductal elements (58) implies that although virions and infected cells may traffic into the salivary glands, they cannot establish a productive infection.
TSP1 is synthesized in low amounts by monocytes and macrophages, epithelial cells, fibroblasts, smooth muscle cells, pneumocytes, and endothelial cells, and in larger quantities by platelets (30). However, HIV may be exposed to levels of TSP1 over two log higher on surfaces in the oral cavity. The fluid distribution of TSP1, with very low concentrations in plasma, sweat, tears, and urine, reflects the relative frequency with which HIV can be isolated from these secretions, but not from saliva. Breast secretions present another issue. HIV can be cultured from some samples of breast milk, which has been implicated in HIV transmission. Colostrum often contains high concentrations of TSP1 (>145 μg/ml), whereas lower, more variable amounts (to <1 μg/ml) have been measured in other breast secretions (59). However, HIV has not been recovered from breast milk devoid of cells (60), and breast milk contains factors that inhibit HIV infection (17), one of which may be TSP.
The concept that an extracellular matrix molecule may serve as an inhibitor of microbial pathogens is not new. For example, FN binds free gp120 (28), and might thereby sequester HIV virions. The compartmentalization of FN in gingival crevicular fluid and whole and submandibular saliva, but not parotid fluid (61), parallels our findings with TSP. However, concentrations required to inhibit HIV infectivity in vitro exceed those found in saliva by 10-fold. Although high levels of FN are present in plasma (∼300 μg/ml; reference 62), recoverable infectious HIV is present as well. FN actually may facilitate HIV-mediated syncycium formation (63), and promote the growth of AIDS-Kaposi sarcoma–related cells constitutively expressing high levels of FN receptor (64), bringing further into question the physiological significance of FN–gp120 binding. In contrast, TSP1 proteolytic fragments and peptides show opposite effects, inhibiting Kaposi sarcoma and endothelial cell proliferation (33, 65).
Defining the relationship between salivary inhibitors of HIV in vivo versus in vitro model systems is important. Concentrations of TSP1 required to inhibit HIV infectivity by >50% after direct addition were equivalent to those found in dilutions of saliva used in HIV inhibition experiments by other investigators (10, 11, 18, 24). Much lower doses of TSP1 were required to abrogate HIV infectivity when HIV–TSP mixtures were prefiltered, suggesting aggregation of virion–TSP1 complexes as a potential mechanism of inhibition. The requirement for prefilteration may have its in vivo counterpart in the continued cleansing of oral surfaces by salivary flow, with elimination of enmeshed viral particles from potential attachment sites. Experiments testing direct inhibition by whole saliva are complicated by the fact that additional salivary components may contribute to the anti-HIV effect. Indeed, TSP1 affinity depletion removed only ∼70% of the HIV-inhibitory activity. In addition, nonspecific antiviral phenomena may occur with saliva dilutions of 1:1 to 1:4 (3). For example, highly charged sulfated polysaccharides such as dextran sulfate and salivary mucins present nonspecific anionic charge barriers to CD4– gp120 interactions at high concentrations (22, 66).
The presence of properdin-related sequences and properdin-binding activity described for gp120 and gp41 (67) support the functional significance of our sequence search results. The CSVTCG properdin motif is found in two of the three type 1 properdin/malaria-like repeats of TSP1. Consistent with homologies detected between CD36/LIMPII TSP1-binding domains and conserved sequences surrounding the V3 region of gp120, in vitro binding and peptide inhibition data indicate a highly specific gp120 interaction mediated through this CLESH-1 site. We initially envisioned a high affinity TSP1-binding site on gp120 traversing the cysteine bridge of the V3 loop. However, envelope peptide mapping data suggested the presence of two full-length functional sites, with potential for direct involvement of the V3 loop, which presents a more complex model in which multiple or sequential site use is possible. Discontinuity created by intervening residues between the first and second motif half-sites could induce conformational strain to distort or physically disrupt V3 loop integrity, with profound negative effects on gp120–CD4 association. Binding of TSP to the first highly homologous half-site in the C2 domain might lead to subsequent binding to weakly homologous downstream residues extending into the V3 loop, freeing the second full-length TSP-binding site in the C3 domain. The second motif encompasses a serine pair (KQSS) shown to be critical for CD4 binding (68). Mechanistically, TSP1 could compete directly with CD4, occupying an identical or overlapping site. Alternatively, the bulky TSP1 trimer may sterically block access to the CD4-binding site, as well as V3 loop determinants. This model could explain why only partial inhibition is seen using CSVTCG peptides. Elucidation of the molecular basis for TSP1 interference of CD4–gp120 association awaits additional structural studies.
Anti-HIV activities vary quantitatively among individual saliva donors. There also are differences in whether filtration is required to detect an inhibitory effect (69). However, in general titers of salivary inhibitory factors decline with disease progression, in parallel with decreased total protein concentrations (70). There is a greater chance of HIV recovery from salivas with advancing clinical stage, although the rate is still low in comparison with other body fluids (57). This raises the possibility that HIV may directly suppress production of saliva inhibitory factors, or elicit blocking molecules. TSP1 production is downregulated by DNA viruses (71), and production of FN is depressed by retroviral infection (72). However, production of TSP1 by PMA-activated monocytes was not affected by HIV infection in our system, and acute or chronic HIV infection actually upregulated TSP1 production by these cells, although to levels lower than those that have been shown to affect HIV infectivity (<2 ng/ml). Regardless of normal levels of TSP1 production in the oral cavity, mechanical alterations also may contribute to decreased saliva inhibitory activity in vivo, as decreased salivary flow rates and buffering capacity correlate with advanced HIV disease (73).
The location of TSP1-binding motifs in highly conserved HIV domains makes these sites attractive targets for blocking agents that would be broadly reactive to HIV-1 substrains. The ability of TSP1 to block CD4–gp120 complex formation suggests the potential use of this matrix molecule in the development of nontoxic natural inhibitors of local transmission of HIV-1, perhaps as a candidate topical adjuvant that could serve as a preventive physical barrier for recto-genital and gastrointestinal tract mucosa.
Acknowledgments
We thank Dr. David Berman for provision of saliva samples. Gingival tissue was kindly supplied by Dr. Daniel Malamud.
This work was supported by the following grants from the National Institutes of Health: DE-11348, HL-55646, AI-41327 to J. Laurence; HL-422540 to R.L. Silverstein; T32HL-07029 to R. Crombie.
Footnotes
Abbreviations used in this paper: FN, fibronectin; GST, glutathione-S-transferase; LIMP, lysosomal integral membrane protein; moi, multiplicity of infection; TBS-Tween, 50 mM Tris pH 7.5, 150mM NaCl, 0.5% Tween-20; TSP1, thrombospondin 1.
References
- 1.Ho DD, Byington BS, Schooley RT, Rota TR, Hirsch MS. Infrequencey of isolation of HTLV-III virus from saliva in AIDS. New Engl J Med. 1985;313:1606. doi: 10.1056/NEJM198512193132512. [DOI] [PubMed] [Google Scholar]
- 2.Groopman JR, Salahuddin SZ, Sarngadharan MG, Markham PD, Gonda M, Sliski M, Gallo RC. HTLV-III in saliva of people with AIDS-related complex and healthy homosexual men at risk for AIDS. Science. 1994;226:447–449. doi: 10.1126/science.6093247. [DOI] [PubMed] [Google Scholar]
- 3.Liuzzi GA, Chirianni A, Clementi M, Bagnareni P, Valenza A, Catello PT, Piazza M. Analysis of HIV-1 load in blood, semen and saliva: evidence for different viral compartments in a cross-sectional and longitudinal study. AIDS. 1996;10:F10–F56. doi: 10.1097/00002030-199612000-00001. [DOI] [PubMed] [Google Scholar]
- 4.CDC Update: universal precautions for prevention of transmission of human immunodeficiency virus, hepatitis B virus, and other blood-borne pathogens in healthcare settings. Morb Mortal Wkly Rep. 1988;37:377–388. [PubMed] [Google Scholar]
- 5.Klein RS, Phelan JA, Freeman K, Schable C, Friedland GH, Trieger N, Steigbigel NH. Low occupational risk of human immunodeficiencey virus infection among dental professionals. New Engl J Med. 1988;318:86–90. doi: 10.1056/NEJM198801143180205. [DOI] [PubMed] [Google Scholar]
- 6.Fultz PN. Components of saliva inactivate human immunodeficiency virus. Lancet. 1986;2:1215. doi: 10.1016/s0140-6736(86)92218-x. [DOI] [PubMed] [Google Scholar]
- 7.Remis RS, Rossignol MS, Kane MA. Hepatitis B infection in a day school for mentally retarded students: transmission from students to staff. Am J Public Health. 1987;77:1183–1186. doi: 10.2105/ajph.77.9.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Achiron A, Pinhas-Hamiel O, Barak Y, Doll L, Offen D, Djaldetti R, Frankel G, Shohat B. Detection of proviral-human T-cell lymphotropic virus Type I DNA in mouthwash samples of HAM/TSP patients and HTLV-I carriers. Arch Virol. 1996;141:147–153. doi: 10.1007/BF01718595. [DOI] [PubMed] [Google Scholar]
- 9.Lercke NW, Osborn KG, Marx PA, Prahalaha S, Maul DH, Lowenskine LJ, Munn RJ, Henrickson RV, Arthur CO, Gilden RV, et al. Inapparent carrier of simian acquired immune deficiency type D retrovirus and disease transmission with saliva. J Natl Cancer Inst. 1986;77:489–495. [PubMed] [Google Scholar]
- 10.Malamud D, Davis C, Berthold P, Roth E, Friedman H. Human submandibular saliva aggregates HIV. AIDS Res Hum Retroviruses. 1993;9:633–637. doi: 10.1089/aid.1993.9.633. [DOI] [PubMed] [Google Scholar]
- 11.Fox PC, Wolff A, Yeh C-F, Atkinson JC, Baum BJ. Saliva inhibits HIV-1 infectivity. J Am Dent Assoc. 1988;116:635–637. doi: 10.14219/jada.archive.1988.0002. [DOI] [PubMed] [Google Scholar]
- 12.Alsip GR, Enchy Y, Sumaya CV, Boswell RN. Increased Epstein-Barr virus DNA in oropharygeal secretions from patients with AIDS, AIDS-related complex, or asymptomatic human immunodeficiency virus infections. J Infect Dis. 1988;157:1072–1076. doi: 10.1093/infdis/157.5.1072. [DOI] [PubMed] [Google Scholar]
- 13.Scholes, A., N. Beeching, P. Carey, M. Martin, and C. Hart. 1993. Oral shedding of CMV and HSV in relation to HIV disease. IXth International Conference AIDS. Berlin, June 6–11. PO-B18–1801 (Abstr.)
- 14.CDC Recommendations for preventing possible transmission of human T-lymphotropic virus type III/ lymphadenopathy-associated virus from tears. Morb Mortal Wkly Rep. 1986;34:533–534. [PubMed] [Google Scholar]
- 15.Mostad SB, Kreiss JK. Shedding of HIV in the genital tract. AIDS. 1996;10:1305–1315. doi: 10.1097/00002030-199610000-00001. [DOI] [PubMed] [Google Scholar]
- 16.Yolken RH, Li S, Perman J, Viscidi R. Persistent diarrhea and fecal shedding of retroviral nucleic acids in children infected with human immunodeficiency virus. J Infect Dis. 1991;164:61–66. doi: 10.1093/infdis/164.1.64. [DOI] [PubMed] [Google Scholar]
- 17.VandePerre P, Simonon A, Hitimana D-G, Dabis F, Msellati P, Mukamababo B, Butera J-P, Goethem CV, Karita E, Lepage P. Infective and anti-infective properties of breast milk from HIV-1 infected women. Lancet. 1993;341:914–918. doi: 10.1016/0140-6736(93)91210-d. [DOI] [PubMed] [Google Scholar]
- 18.Fox PC, Wolff A, Yeh C-F, Atkinson JC, Baum BJ. Salivary inhibition of HIV-1 infectivity: functional properties and distribution in men, women and children. J Am Dent Assoc. 1989;118:709–711. doi: 10.14219/jada.archive.1989.0165. [DOI] [PubMed] [Google Scholar]
- 19.Archibald DW, Cole DA. In vitro inhibition of HIV-1 infectivity by human salivas. AIDS Res Hum Retroviruses. 1990;6:1425–1432. doi: 10.1089/aid.1990.6.1425. [DOI] [PubMed] [Google Scholar]
- 20.McNeely TB, Dealy M, Dripps DJ, Orenstein JM, Eisenberg SP, Wahl SM. Secretory leukocyte protease inhibitor: a human saliva protein exhibiting anti-human immunodedficiency virus 1 activity in vitro. J Clin Invest. 1995;96:456–464. doi: 10.1172/JCI118056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malamud D, Freidman HM. HIV in the oral cavity: virus, viral inhibitory activity, and antibodies: a review. Crit Rev Oral Biol Med. 1993;4:461–466. doi: 10.1177/10454411930040032901. [DOI] [PubMed] [Google Scholar]
- 22.Amory J, Martin N, Levy J, Wara D. The large molecular weight glycoprotein MG1, a component of human saliva, inhibits HIV-1 infectivity. Clin Res. 1992;40:51A. [Google Scholar]
- 23.Yeh C-K, Hardelman B, Fox PC, Baum BJ. Further studies of salivary inhibition of HIV-1 infectivity. J Acquired Immune Defic Syndr. 1992;5:898–903. [PubMed] [Google Scholar]
- 24.Bergey EJ, Cho M-I, Blumberg BM, Hammarskjold M-L, Rekosh D, Epstein LJ, Levine MJ. Interaction of HIV-1 and human salivary mucins. J Acquired Immune Defic Syndr. 1994;7:995–1002. [PubMed] [Google Scholar]
- 25.Phillips JN, Qureshi C, Barr C, Henrard DR. Low level of cell-free virus detected at high frequency in saliva from HIV-1 infected indidviduals. AIDS. 1994;8:1011–1012. doi: 10.1097/00002030-199407000-00021. [DOI] [PubMed] [Google Scholar]
- 26.Levine JM, Reddy MS, Tabak LA, Loomis RE, Bergey EJ, Jones PC, Cohen RE, Stinson MW, Al-Hashimi I. Structural aspects of salivary glycoproteins. J Dent Res. 1987;66:436–441. doi: 10.1177/00220345870660020901. [DOI] [PubMed] [Google Scholar]
- 27.Bu, M., J.A. Turpin, C.A. Schaeffer, L. Graham, and R.W. Buckheit, Jr., and W.G. Rice. 1995. Secretory leukocyte protease inhibitor (SLPI) does not effectively inhibit HIV-1 replication. 35th International Conference on Antimicrobial Agents and Chemotherapy. San Francisco, CA, September 17–20. I142 (Abstr.)
- 28.Su HL, Boackle RJ. Interaction of the envelope glycoprotein of human immunodeficiency virus with C1q and fibronectin under conditions present in saliva. Mol Immunol. 1991;28:811–817. doi: 10.1016/0161-5890(91)90044-k. [DOI] [PubMed] [Google Scholar]
- 29.Bornstein P. Thrombospondins: structure and regulation of expression. FASEB (Fed Am Soc Exp Biol) J. 1992;6:3222–3229. doi: 10.1096/fasebj.6.14.1426766. [DOI] [PubMed] [Google Scholar]
- 30.Lahav, J., editor. 1993. Thrombospondin. CRC Press. Boca Raton, FL. pp. 1–298.
- 31.Lahav J. The functions of thrombospondin and its involvement in physiology and pathophysiology. Biochim Biophys Acta. 1993;1182:1–14. doi: 10.1016/0925-4439(93)90146-r. [DOI] [PubMed] [Google Scholar]
- 32.Sinnis P, Willnow TE, Briones MRS, Herz J, Nussenzweig V. Remnant lipoproteins inhibit malaria sporozoite invasion of hepatocytes. J Exp Med. 1996;184:945–954. doi: 10.1084/jem.184.3.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taraboletti G, Roberts D, Liotta LL, Gavazzi R. Platelet thrombospondin modulates endothelial cell adhesion, motility, and growth: a potential angiogenesis regulatory factor. J Cell Biol. 1990;111:765–772. doi: 10.1083/jcb.111.2.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pearce SFA, Wu J, Silverstein RL. Recombinant GST/CD36 fusion proteins define a thrombospondin binding domain: evidence for a single calcium-dependent binding site on CD36. J Biol Chem. 1995;270:2981–2986. doi: 10.1074/jbc.270.7.2981. [DOI] [PubMed] [Google Scholar]
- 35.Crombie, R., and R.L. Silverstein. 1997. Lysosomal integral membrane protein LIMP II binds thrombospondin-1: structure–function homology with the cell adhesion molecule CD36 defines a conserved recognition motif. J. Biol. Chem. In press. [DOI] [PubMed]
- 36.Ivanhoff LA, Dubay JW, Morris JF, Roberts SJ, Gutshall L, Sternberg EJ, Hunter E, Matthews TJ, Petteway SR. V3 loop region of the HIV-1 envelope protein is essential for virus infectivity. Virology. 1992;187:423–432. doi: 10.1016/0042-6822(92)90444-t. [DOI] [PubMed] [Google Scholar]
- 37.Worley KC, Wiese BA, Smith RF. BEAUTY: an enhanced BLAST-based search tool that integrates multiple biological information resources into sequence similarity search results. Genome Res. 1995;5:173–184. doi: 10.1101/gr.5.2.173. [DOI] [PubMed] [Google Scholar]
- 38.Smith RF, Smith TF. Pattern-induced multi-sequence alignment (PIMA) algorithm employing secondary structure-dependent gap penalties for comparative protein modelling. Protein Eng. 1992;5:35–41. doi: 10.1093/protein/5.1.35. [DOI] [PubMed] [Google Scholar]
- 39.Bairoch A. The PROSITE dictionary of sites and patterns, its current status. Nucleic Acids Res. 1993;21:3097–3103. doi: 10.1093/nar/21.13.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Silverstein RL, Leung LLK, Harpel PC, Nachman RL. Platelet thrombospondin forms a trimolecular complex with plasminogen and histidine-rich glycoprotein. J Clin Invest. 1985;75:2065–2073. doi: 10.1172/JCI111926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silverstein RL, Nachman RL. Thrombospondin binds to monocytes–macrophages and mediates platelet-monocyte adhesion. J Clin Invest. 1987;79:867–874. doi: 10.1172/JCI112896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pearce SFA, Wu J, Silverstein RL. A carboxy terminal truncation mutant of CD36 is secreted and binds thrombospondin: evidence for a single transmembrane domain. Blood. 1994;84:384–389. [PubMed] [Google Scholar]
- 43.Frangioni JV, Neel BG. Solubilization and purification of enzymatically active glutathione-S-transferase (pGEX) fusion proteins. Anal Biochem. 1993;210:179–187. doi: 10.1006/abio.1993.1170. [DOI] [PubMed] [Google Scholar]
- 44.Silverstein RL, Leung LLK, Nachman RL. Thrombospondin: a versatile multifunctional glycoprotein. Arteriosclerosis. 1986;6:245–253. doi: 10.1161/01.atv.6.3.245. [DOI] [PubMed] [Google Scholar]
- 45.MacGregor JL, Catimel B, Parmentier S, Clezardin P, Dechavanne M, Leung LL. Rapid purification and partial characterization of human platelet glycoprotein IIIb. J Biol Chem. 1989;264:501–506. [PubMed] [Google Scholar]
- 46.Hajjar KA, Gavish D, Breslow JL, Nachman RL. Lipoprotein(a) modulation of endothelial cell surface fibrinolysis and its potential role in atherosclerosis. Nature. 1989;339:303–305. doi: 10.1038/339303a0. [DOI] [PubMed] [Google Scholar]
- 47.Smith DJ, Taubman MA, King WF. Immunological features of minor salivary gland saliva. J Clin Immunol. 1987;7:449–454. doi: 10.1007/BF00915054. [DOI] [PubMed] [Google Scholar]
- 48.Laurence J, Friedman SM, Chartash EK, Crow MK, Posnett DN. Human immunodeficiency virus infection of helper T-cell clones: early proliferative defects despite intact antigen-specific recognition and interleukin 4 secretion. J Clin Invest. 1989;83:1843–1848. doi: 10.1172/JCI114090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laurence J, Sanders A, Early E, Salmon JE. Human immunodeficiency virus infection of monocytes: relationship to Fc-gamma receptors and antibody-dependent viral enhancement. Immunology. 1990;70:338–343. [PMC free article] [PubMed] [Google Scholar]
- 50.Vega MA, Seguí-Real B, García JA, Calés C, Rodríguez F, Vanderkerckhove J, Sandoval IV. Cloning, sequencing, and expression of a cDNA encoding the rat LIMP II, a novel 74-kDa lysosomal membrane protein related to the surface adhesion protein CD36. J Biol Chem. 1991;266:16818–16824. [PubMed] [Google Scholar]
- 51.Calvo D, Dopazo J, Vega MA. The CD36, CLA-1 (CD36L1), and LIMP II (CD36L2) gene family: cellular distribution, chromosomal location, and genetic evolution. Genomics. 1995;25:100–106. doi: 10.1016/0888-7543(95)80114-2. [DOI] [PubMed] [Google Scholar]
- 52.Asch AS, Tepler J, Silbiger S, Nachman RL. Cellular attachment to thrombospondin: cooperative interactions between receptor systems. J Biol Chem. 1991;266:1740–1745. [PubMed] [Google Scholar]
- 53.Catimel, B., L. Leung, H. elGhissasi, N. Mercier, and J. McGregor. 1992. Human platelet glycoprotein IIIb binds to thrombospondin fragments bearing the C-terminal region, and/or the type I repeats (CSVTCG motif), but not to the N-terminal heparin-binding region. Biochem. J. 284:231–236. [DOI] [PMC free article] [PubMed]
- 54.Li WX, Leung LL. Identification of SVTCG in thrombospondin as the conformation-dependent, high affinity binding site for its receptor, CD36. J Biol Chem. 1993;268:16179–16184. [PubMed] [Google Scholar]
- 55.Varani J, Stoolman L, Wang T, Schuger L, Flippen C, Dame M, Johnson KJ, Todd RF, III, Ryan US, Ward PA. Thrombospondin production and thrombospondin-mediated adhesion in U937 cells. Exp Cell Res. 1991;195:177–182. doi: 10.1016/0014-4827(91)90514-u. [DOI] [PubMed] [Google Scholar]
- 56.Qureshi MN, Joshi B, Hewlett I, Henrard D, Qiu Z, Barr CE. Prevalence of HIV-1 proviral DNA an virion-associated RNA in saliva. J Dent Res. 1994;73:2564A. . (Abstr.) [Google Scholar]
- 57.Moore BE, Flaitz CM, Copenhaver DH, Nichols CM, Kalmaz GD, Bresman JD, Cloyd MW, Lynch DP, Prabhakar BS, Baron S. HIV recovery from saliva before and after dental treatment: inhibitors may have a critical role in viral inactivation. J Am Dent Assoc. 1993;124:67–74. doi: 10.14219/jada.archive.1993.0197. [DOI] [PubMed] [Google Scholar]
- 58.Fox PC. Saliva and salivary gland alterations in HIV infection. J Am Dent Assoc. 1991;122:46–48. doi: 10.14219/jada.archive.1991.0331. [DOI] [PubMed] [Google Scholar]
- 59.Dawes JP, Clezardin P, Pratt DA. Thrombospondin in milk, other breast secretions and breast tissue. Sem Thromb Hemostasis. 1987;13:378–384. doi: 10.1055/s-2007-1003514. [DOI] [PubMed] [Google Scholar]
- 60.Guay LA, Hom DL, Mmiro F, Piwowar EM, Kabengera S, Parsons J, Ndugwa C, Marum L, Olness K, Kataaha P, Jackson JB. Detection of human immunodeficiency virus type 1 (HIV-1) DNA and p24 antigen in breast milk of HIV-1–infected Ugandan women and vertical transmission. Pediatrics. 1996;98:438–444. [PubMed] [Google Scholar]
- 61.Tynelius-Bratthall G, Ericson D, Araujo MM. Fibronectin in saliva and gingival crevices. J Periodontal Res. 1986;21:563–568. doi: 10.1111/j.1600-0765.1986.tb01492.x. [DOI] [PubMed] [Google Scholar]
- 62.Torre D, Issi M, Sampietro C, Fior GP, Chelazzi G, Ferraro G. Plasma fibronectin concentrations in patients with human immunodeficiency virus infection. J Clin Pathol. 1990;43:560–562. doi: 10.1136/jcp.43.7.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ushijima H, Unten S, Hormen H. Effect of serum components on syncytium formation and virus production by cells infected with human immunodeficiency viruses in vitro. AIDS Res Hum Retroviruses. 1992;8:513–520. doi: 10.1089/aid.1992.8.513. [DOI] [PubMed] [Google Scholar]
- 64.Barillari G, Holmes A, Gendelman R, Buonaguro L, Gallo RC, Ensoli B. The RGD motif and the integrin receptors are involved in the vascular cell growth and adhesive properties of extracellular HIV-1 Tat protein. J AIDS. 1993;6:688A. (Abstr.) [Google Scholar]
- 65.Roberts, D.D., N. Guo, P.J. Browning. V.S. Zabrenetzky, J.K. Inman, and H.C. Krutzsch. 1995. Modulation of tumor growth in vitro and in vivo by stable analogs of thrombospondin peptides. AIDS Res. Hum. Retroviruses. 11(51):40 (Abstr.).
- 66.Baba N, DeClercq E, Schols D, Pauwels R, Snoeck R, VanBoeckel C, VanDedem G, Kraaijeveld N, Hobbelen P, Ottenheijm H, Hollander FD. Novel sulfated polysaccharides: dissociation of anti-human immunodeficiency virus activity from anti-thrombin activity. J Infect Dis. 1990;161:208–213. doi: 10.1093/infdis/161.2.208. [DOI] [PubMed] [Google Scholar]
- 67.Stoiber HR, Schneider R, Janatova J, Dierich MP. Human complement proteins C3b, C4b, factor H and properdin react with specific sites in gp120 and gp41, the envelope proteins of HIV-1. Immunobiology. 1995;193:98–113. doi: 10.1016/s0171-2985(11)80158-0. [DOI] [PubMed] [Google Scholar]
- 68.Kowalski M, Potz J, Basiripour L, Dorfman T, Goh WC, Terwilliger E, Dayton A, Rosen C, Haseltine W, Sodroski J. Functional regions of the envelope glycoprotein of human immunodeficiency virus type 1. Science. 1987;237:1351–1355. doi: 10.1126/science.3629244. [DOI] [PubMed] [Google Scholar]
- 69.Nagashunmugam T, Friedman HM, Davis C, Kennedy S, Goldstein LT, Malamud D. Human submandibular saliva specifically inhibits HIV type 1. AIDS Res Hum Retroviruses. 1997;13:371–376. doi: 10.1089/aid.1997.13.371. [DOI] [PubMed] [Google Scholar]
- 70.Lal K, Pollock JJ, Santarpia RP, III, Heller HM, Kaufman HW, Fuhrer J, Steigbigel RT. Pilot study comparing the salivary cationic protein concentrations in healthy adults and AIDS patients: correlation with antifungal activity. J AIDS. 1992;5:904–914. [PubMed] [Google Scholar]
- 71.Mosher DF. Physiology of thrombospondin. Ann Rev Med. 1990;41:85–97. doi: 10.1146/annurev.me.41.020190.000505. [DOI] [PubMed] [Google Scholar]
- 72.Adams SL, Pacifici M, Boettiger D, Pallante KM. Modulation of fibronectin gene expression in chondrocytes by viral transformation and substrate attachment. J Cell Biol. 1987;105:483–488. doi: 10.1083/jcb.105.1.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Madigan A, Murray P. Caries and criogenic flora in HIV-positive children versus uninfected children. J Dent Res. 1994;73:1898A. . (Abstr) [Google Scholar]