

Abstract
HMG1 (high mobility group 1) is a ubiquitous and abundant chromatin component. However, HMG1 can be secreted by activated macrophages and monocytes, and can act as a mediator of inflammation and endotoxic lethality. Here we document a role of extracellular HMG1 in cell migration. HMG1 (and its individual DNA-binding domains) stimulated migration of rat smooth muscle cells in chemotaxis, chemokinesis, and wound healing assays. HMG1 induced rapid and transient changes of cell shape, and actin cytoskeleton reorganization leading to an elongated polarized morphology typical of motile cells. These effects were inhibited by antibodies directed against the receptor of advanced glycation endproducts, indicating that the receptor of advanced glycation endproducts is the receptor mediating the HMG1-dependent migratory responses. Pertussis toxin and the mitogen-activated protein kinase kinase inhibitor PD98059 also blocked HMG1-induced rat smooth muscle cell migration, suggesting that a Gi/o protein and mitogen-activated protein kinases are required for the HMG1 signaling pathway. We also show that HMG1 can be released by damage or necrosis of a variety of cell types, including endothelial cells. Thus, HMG1 has all the hallmarks of a molecule that can promote atherosclerosis and restenosis after vascular damage.
Keywords: atherosclerosis, chemotaxis, high mobility group 1, receptor of advanced glycation endproducts, smooth muscle cells
Introduction
High mobility group 1 (HMG1) protein is the archetypal protein of the HMG-box family, which is characterized by their DNA binding domains, called HMG boxes. HMG1 is a small 25-kD protein of 215 amino acids with a highly conserved sequence among mammals. The HMG1 molecule is organized into three domains: two DNA binding domains, HMG Box A and Box B, which are followed by an acidic COOH terminus composed of 30 glutamic and aspartic residues. The two HMG boxes, A and B, are similar 80 amino acid segments (29% identical, 65% similar) and form an L-shaped structure (Read et al. 1993; Weir et al. 1993; Hardman et al. 1995).
HMG1 has originally been identified as a ubiquitously expressed, abundant nuclear protein. It is present in >1 million copies per single nucleus and binds double stranded DNA without apparent sequence specificity. Instead, HMG1 binds with high affinity to specific DNA structures like kinked or bent DNA and four-way junctions. However, HMG1 can be recruited to double stranded DNA by interaction with several different DNA-binding proteins. When bound to double stranded DNA, it can bend it sharply, allowing the formation of nucleoprotein complexes where several DNA-binding proteins can contact each other while bound to their respective DNA cognate sites. Accordingly, HMG1 has been found to enhance the activity of several transcription factors including the glucocorticoid receptor (GR), as well as of the RAG recombinase (for review, see Bustin 1999; Bianchi and Beltrame 2000). The phenotype of Hmg1 −/− mice is in agreement with the role of HMG1 as a regulator of transcription. Mice die shortly after birth due to hypoglycemia and show a defect in the activation of GR-responsive genes (Calogero et al. 1999).
Recently, an additional role for HMG1 outside the cell nucleus has come into focus. HMG1 was shown to be a mediator of endotoxin lethality as well as acute lung inflammation in mice, and elevated serum levels of HMG1 in septic patients are a poor prognosis marker of survival. HMG1 can be secreted by macrophages and pituicytes in culture in response to cytokines and bacterial endotoxin (Wang et al. 1999a,Wang et al. 1999b; Abraham et al. 2000). Such secretion is atypical: HMG1 has no leader peptide and does not travel through the endoplasmic reticulum and the Golgi apparatus. Release of HMG1 from murine erythroleukemia cells correlates with cell differentiation and the protein can be found in a plasma membrane–associated form in these cells (Sparatore et al. 1996; Passalacqua et al. 1997). A protein called amphoterin, identical in sequence to HMG1, has been described in the brain, where it is found in the nucleus and cytoplasm of neuronal cells as well as in the extracellular space. Exogenously added HMG1 mediates outgrowth of neurites, and laminin-dependent migration of neuroblastoma and glioma cells is inhibited by antibodies against HMG1 (Rauvala et al. 1988; Merenmies et al. 1991; Parkkinen et al. 1993; Fages et al. 2000). Interactions between HMG1 and the plasminogen activation system, in particular tissue-type plasminogen activator results in enhanced plasmin formation (Parkkinen and Rauvala 1991; Parkkinen et al. 1993). Degradation of extracellular matrix proteins is an important step in the cell migration process, and HMG1-promoted increase of extracellular protease activity might enable the cells to migrate.
HMG1 has been identified as one of the ligands binding to the receptor for advanced glycation endproducts (RAGE) (Hori et al. 1995). RAGE is a multiligand receptor of the immunoglobulin superfamily and is expressed on many cell types, including endothelial cells, smooth muscle cells, mononuclear phagocytes, and neurons (Neeper et al. 1992; Brett et al. 1993). It has been implicated in several pathological processes, such as diabetes, amyloidoses, and atherosclerosis (reviewed in Schmidt et al. 1999). Interaction of HMG1 and RAGE induces neurite outgrowth, and the two proteins colocalize at the leading edge of advancing neurites during embryonic development (Hori et al. 1995; Huttunen et al. 1999). Tumour growth and metastasis can be blocked by preventing the interactions between HMG1 and RAGE. Inhibition of this interaction suppresses also activation of mitogen-activated protein (MAP) kinases and the expression of matrix metalloproteinases, molecules importantly linked to tumour proliferation and invasion (Taguchi et al. 2000).
In this work, we document that HMG1 has a potent biological effect on smooth muscle cells (SMC), one of the cell types where RAGE is expressed on the surface. Vascular SMC, the most predominant cells of the larger blood vessel, are located in the tunica media where they are normally embedded in the extracellular matrix. In intact tissues, SMC are in a contractile state or phenotype characterized by the absence of cell division and migration. They maintain vessel wall rigidity and elasticity and control blood pressure. When the endothelium is damaged, either after mechanical or inflammatory injuries, SMC switch to a synthetic phenotype and undergo cell division and cell migration. The migration of SMC from the tunica media to the tunica intima, resulting in intimal thickening, is believed to play an important role in the pathophysiology of many vascular disorders, such as atherosclerosis and restenosis after coronary angioplasty. In the synthetic state, SMC also produce higher amounts of extracellular proteinases, growth factors, and cytokines and secrete a fibrous extracellular matrix. After vessel wall injury, several growth factors and/or chemoattractants released either by circulating monocytes, macrophages and platelets, or by damaged endothelial cells can influence the switch from the contractile to the synthetic phenotypes and direct the migration of SMC towards the vessel intima. Among these factors, bFGF appears to be one of the most important. However, SMC can also migrate in response to angiogenic stimuli (for reviews, see van Leeuwen 1996; Schwartz 1997).
Here we show that HMG1 can be released by damage or necrosis of a variety of cell types, including endothelial cells, and we demonstrate that HMG1 is a strong chemoattractant for rat SMC (RSMC) in vitro, and that it induces cell migration, cell shape changes, and cytoskeleton reorganization. These events can be inhibited by addition of an anti–RAGE antibody and by pertussis toxin, suggesting that both RAGE and a Gi/o protein might be involved. Furthermore, HMG1 promotes the translocation of phosphorylated ERK 1 and 2 into the nucleus, indicating the involvement of the MAP kinase pathway. Therefore, HMG1 has all the hallmarks of a molecule that can promote atherosclerosis and restenosis after vascular damage.
Materials and Methods
Materials
RSMC from thoracic aorta were a gift of Dr. Marco Bertulli (Bayer Research Laboratory, Milan, Italy) (Degryse et al. 1999). Human umbilical vein endothelial cells (HUVEC) were provided by Dr. Fabio Pellegatta (San Raffaele Scientific Institute, Milan, Italy). Rat pro-urokinase was provided by Dr. S.A. Rabbani (McGill University and Royal Victoria Hospital, Montreal, Québec, Canada) (Degryse et al. 1999). Bordetella pertussis toxin and its mutant were provided by Dr. M.G. Pizza (I.R.I.S., Siena, Italy) (Fazioli et al. 1997). Anti–RAGE antibody was a gift of Dr. A.M. Schmidt (Columbia University, New York, NY) (Taguchi et al. 2000). HMG1 purified from calf thymus was a gift of Jordi Bernués (C.S.I.C., Barcelona, Spain). Collagen I and fibronectin were purchased from Roche. The polyclonal rabbit anti–HMG1 was purchased from BD PharMingen. The monoclonal mouse antiphosphorylated MAP kinases (ERK 1 and 2) was from New England Biolabs, Inc. Fluorescein-conjugated F(ab)′2 fragments of anti–rabbit immunoglobulins and fluorescein-conjugated F(ab)′2 fragments of anti–mouse immunoglobulins were from Chemicon. Nonspecific rabbit polyclonal immunoglobulins, nonspecific monoclonal mouse IgG1κ (MOPC-21), TRITC-conjugated phalloidin, and fMLP (formyl-methionine-leucine-proline) were from Sigma-Aldrich.
Expression and Purification of HMG1 and Derivatives
HMG1/M1-176 is a truncated form of HMG1 that lacks the COOH-terminal acidic domain of the intact HMG1 molecule; for the sake of simplicity, it will be called Box A+B. The plasmids pRNHMG1/M1-V176, pT7HMG1bA, and pT7HMG1bB coding for Box A+B, Box A, and Box B, respectively, have been previously described, as well as the protocols for expression and purification of the single and double boxes (Bianchi et al. 1992).
The plasmid pT7-7-rHMG1cm used for the expression of full-length HMG1 in bacteria was a kind gift of Prof. J.O. Thomas (Cambridge University, Cambridge, UK). Expression and purification of full-length HMG1 was performed in BL21(−) Escherichia coli strain following the protocol of Studier and Moffatt 1986, with slight modifications: transformed bacteria was grown in M9 medium supplemented with Cas-aminoacids 20 g/liter, glycerol 0.5%, yeast extract 5 g/liter, glucose 0.4%. Chloramphenicol 100 μg/ml was used as selective agent. Temperature was shifted from 37° to 23°C during the 16-h induction period.
The procedure for the expression and purification of full-length HMG1 in yeast (Pichia pastoris) has been described elsewhere (Mistry et al. 1997).
Chemotaxis Assay
Chemotaxis assays were performed as previously described (Degryse et al. 1999). In brief, modified Boyden chambers were used with filters (5-μm pore size; Corning) treated with collagen I (100 μg/ml in 0.5 M acetic acid) and fibronectin (10 μg/ml). 20,000–40,000 cells in serum-free DMEM were added to the upper well of Boyden chambers. The molecules to be tested were diluted in serum-free medium and added to the lower well. Antibodies, pertussis toxin (PT), or inhibitor were added in both wells of Boyden chambers. Overnight migration was allowed at 37°C. Then, cells remaining on the upper surface of filters were scraped off and filters were fixed in methanol and stained in a solution of 10% (wt/vol) crystal violet in 20% (vol/vol) methanol. All experiments were performed at least twice in triplicate. Results are the mean ± SD of the number of cells counted in 10 high power fields per filter and expressed as fold over control. Random cell migration (i.e., migration in the absence of chemoattractant) was given the arbitrary value of 100%.
Immunofluorescence Microscopy
As previously described (Degryse et al. 1999) 15,000–25,000 cells (20–40% confluence) were seeded on glass coverslips in 2-cm2 wells and cultured for 24 h in DMEM plus 10% FCS, washed with PBS, and cultured for another 24 h in DMEM without FCS. In some experiments, cells were pretreated with either antibodies (overnight), PT, or inhibitor [6 h, except with PD 98059 (1 h); New England Biolabs, Inc.]. After stimulation at 37°C, RSMC were fixed for 20 min at room temperature with a solution of 3% paraformaldehyde, 2% sucrose in PBS, pH 7.5, followed by three washes with PBS-BSA 0.2%. Cells were permeabilized with (mM): 20 Hepes, pH 7.4, 300 saccharose, 50 NaCl, 3 MgCl2, 0.5% (vol/vol) Triton X-100 for 3 min at 4°C, and washed again three times with PBS-BSA 0.2%. Then, RSMC were incubated with PBS-BSA 2% for 15 min at 37°C, with primary antibodies for 30 min at 37°C, washed three times with PBS-BSA 0.2%, and further incubated with PBS-BSA 2% for 15 min. Cells were stained with secondary antibodies and/or phalloidin for visualization of filamentous actin for 30 min at 37°C. In some cases, DAPI (Roche) was used to label the nucleus. After incubation, coverslips were washed three times with PBS-BSA 0.2%, once with distilled water, mounted with 20% (wt/vol) Mowiol in PBS, and analyzed on a Zeiss Axiophot microscope (Carl Zeiss, Inc.). Fluorescence photographs were taken either on T-Max 400 or EPH P1600X film (Eastman Kodak Co.) using Zeiss 40 and 100 neofluar lenses.
Wounding Assay
As previously described (Degryse et al. 1999), confluent cultures of RSMC, grown on glass coverslips in 2-cm2 wells, were washed once with PBS and FCS-starved for 24 h in serum-free DMEM. Then, single wounds were made. The injured monolayers, washed once with PBS, were allowed to recover for further 48 h in serum-free medium supplemented or not with the molecule to be tested. RSMC were fixed and stained with TRITC-phalloidin as described above. Quantification was made by taking photographs at lower magnification and by counting the number of cells that had migrated into the cell-free space.
Cell Culture and Induction of Cell Damage
All cells were cultured in DMEM supplemented with 10% FCS. HeLa cells and HUVEC were induced to undergo necrosis by treatment with 5 μM ionomycin (Sigma-Aldrich) and 20 μM CCCP (Sigma-Aldrich), or 6 mM deoxyglucose (Sigma-Aldrich) and 10 mM sodium azide. After 16 h at 37°C, ∼50% of the cells were necrotic. For Western blot analysis, the medium from treated and untreated cells was collected and concentrated 50-fold using Amicon Ultrafree-MC filters; the cells were dissolved on the plate in SDS-PAGE sample buffer. For immunofluorescence analysis, the cells were fixed with 4% PFA, incubated with an anti–HMG1 antibody, and stained with secondary antibody and DAPI. The permeabilization of cells with 0.1% NP-40 was performed as described in Falciola et al. 1997.
Binding of Extracellular HMG1 to RSMC
1 million RSMC were trypsinized and incubated for 20 min at 4°C in 50 μl PBS containing 5 μg BSA and 800 ng Box A+B (∼2.5 × 1013 molecules). Then, cells were pelleted and the supernatant was saved. After two washes in 500 μl cold PBS, cells were resuspended in SDS-PAGE sample buffer, heated for 5 min at 100°C, and loaded on a 12% tricine-SDS gel, along with 20 μl of supernatant. The gel was blotted to an Immobilon P filter, which was stained with India ink. The amount of Box A+B recovered in each lane was determined densitometrically, and was found to be very similar. About 7 × 1012 molecules were associated to the cell sample, and ∼18 × 1012 molecules remained unbound. We confirmed this conclusion by performing a binding titration of fluorescently labeled HMG1 to RSMC using a FMAT (fluorometric microvolume assay technology) instrument (PE Biosystems): the binding was nonsaturable at 4°C.
Western Blots
Cells were trypsinized, lysed in SDS-PAGE sample buffer (50 mM Tris, pH 6.8, 2% 2-mercaptoethanol, 4% SDS, 12% glycerol, 0.05% bromophenol blue), and heated for 5 min at 100°C. Proteins were separated on a tricine gel (Schägger and von Jagow 1987) and blotted on a Immobilon P membrane (Millipore) using a tankblot system (Hoefer) in 25 mM Tris, pH 7.5, 0.192 M glycine, 20% methanol. The blot was blocked for 1 h at room temperature in 5% skim milk/TBST (20 mM Tris, pH 7.5, 137 mM NaCl, 0.1% Tween 20), washed in TBST, and incubated for 1 h at room temperature with the anti–HMG1 antibody in TBST/0.01% BSA. After another wash in TBST, the blot was further incubated for 1 h with an anti–rabbit Ig antibody (Amersham Pharmacia Biotech) in TBST/0.01% BSA. The blot was developed after additional washes in TBST using the ECL system (Amersham Pharmacia Biotech).
Statistical Analysis
Statistical analysis was performed with the Prism software using Student's t test for pairwise comparisons of treatments, or an ANOVA model for the evaluation of treatments with increasing doses of a reagent.
Results
HMG1 Induces RMSC Migration
The chemotactic effect of HMG1 was determined with a chemotaxis assay using modified Boyden chambers. We tested several preparations of HMG1: HMG1 purified from calf thymus, recombinant HMG1 expressed from E. coli, and a slightly modified HMG1 produced by the yeast P. pastoris (Mistry et al. 1997).
HMG1 from calf thymus stimulated migration of RSMC in a concentration-dependent manner, starting at doses as low as 0.1 ng/ml and with a 2.5-fold maximal response at 100 ng/ml (Fig. 1 A). The effect of HMG1 was comparable in amplitude to the effects of the well-characterized attractants fMLP and bFGF (Baggiolini et al. 1994; van Leeuwen 1996; Degryse et al. 1999) (Fig. 1 B, and results not shown). Polyclonal antibodies against HMG1, but not nonspecific control antibodies, totally blocked the migratory response (Fig. 1 C), showing that this was specifically due to HMG1. These antibodies failed to alter the effect of the chemoattractant peptide fMLP used as positive control, and they affected cell migration only marginally. Similar results were obtained with recombinant HMG1 produced in yeast and E. coli (Fig. 1 D, and results not shown), and anti–HMG1 antibodies abolished the effect of recombinant HMG1 as well (not shown).
Figure 1.

HMG1 has chemotactic activity on RSMC. Chemotaxis assays were performed using modified Boyden chambers. The value of 100% corresponds to the number of cells migrating in the absence of any stimulator (random cell migration). The data represent the mean ± SD (n = 3). (A) Concentration-dependent migratory response of RSMC to HMG1 purified from calf thymus. The statistical significance of the result is P < 0.0001 in an ANOVA model. (B) Comparison of the chemotactic effect of HMG1 proteins, either purified from calf thymus or expressed in yeast, with those of the well-characterized chemoattractants fMLP and bFGF. All treatments increase the migratory response relative to the control (P < 0.0001 in Student's t test). (C) Effect of anti–HMG1 antibodies on fMLP- and HMG1-induced migration. *Treatments where the migratory response was statistically different from the control beyond the P = 0.0001 limit in Student's t test. Treatment of RSMC with the anti–HMG1 antibody alone, or an unspecific antibody, also gave statistically significant results (0.05 < P < 0.01). Treatment with HMG1 plus anti–HMG1 gave results that did not differ statistically from the untreated control. (D) Concentration-dependent migratory response of RSMC to HMG1 expressed in yeast (P. pastoris). The statistical significance of the result is P < 0.0001 in an ANOVA model.
HMG1 Induces Time-dependent Cytoskeleton Reorganization and Cell Shape Changes
Chemoattractant-induced cell motility requires cytoskeleton reorganization and cell shape changes (Ridley 1994). Using RSMC, we have previously functionally connected induction of cell migration and cytoskeleton reorganization (Degryse et al. 1999). Subconfluent cultures of serum-starved RSMC were stimulated with 100 ng/ml HMG1 from calf thymus for increasing times from 5 to 120 min, and actin filaments were labeled with rhodamine-conjugated phalloidin. Low magnification pictures showed that stress fibers content, cell shape and size, and cytoskeleton organization changed within 30 min, but reversed after 120 min (Fig. 2 A). Higher magnification pictures showed that in control conditions most RSMC exhibited numerous stress fibers and a nonpolarized cell shape (Fig. 2 B). After 5 min, HMG1 induced a clear decrease in stress fiber content and stimulated membrane ruffling (data not shown). Within 15–30 min, a complete change of morphology and cytoskeleton organization had occurred: RSMC had an elongated, polarized morphology that reflected the spatial rearrangement of the actin cytoskeleton. Semi-ring structures of actin and membrane ruffling were observed at the leading part of the cell. Filaments of actin were also present flanking the nucleus and in the dragging trail. These effects of HMG1 slowly decreased. After 1–2 h, the stress fiber content increased back to a level similar to that of unstimulated cells, and cell morphology returned similar to that of unstimulated control cells.
Figure 2.

Effect of HMG1 on RSMC morphology and actin cytoskeleton organization. (A) Subconfluent (50–70%) cultures of RSMC were challenged for the indicated times with HMG1 (100 ng/ml), either purified from calf thymus or expressed in yeast or E. coli as indicated. Actin filaments were visualized using TRITC-phalloidin. (B) Anti–HMG1 rabbit antibodies, but not unspecific rabbit antibodies, inhibit HMG1-stimulated cytoskeleton reorganization. RSMC were pretreated overnight with either anti–HMG1 (2 μg/ml) or unspecific control antibodies (2 μg/ml), and then 100 ng/ml HMG1 (from calf thymus) was added. (C) RSMC were stimulated with 100 ng/ml HMG1. Quantification of the actin cytoskeleton reorganization was performed by taking low-magnification photographs and counting the cells in each state of cytoskeleton organization. Resting cells (state 1) exhibit numerous stress fibers. Nonresting cells (state 2) show a reorganization of actin cytoskeleton: a decrease of stress fibers content, membrane ruffling, actin semi-ring with an elongated polarized morphology characteristic of motile RSMC.
Antibodies against HMG1 totally inhibited the cytoskeletal reorganization and the morphological change of RSMC induced by HMG1 (Fig. 2 B), but did not influence the state of organization of actin cytoskeleton of unstimulated RSMC. Control antibodies were not able to inhibit HMG1-induced reorganization of the actin filaments.
To determine whether the observed effects of HMG1 on RSMC actually reflects a dynamic transition from resting to motile states, we quantified the proportion of each different state during the course of the same experiment. We took low-magnification pictures and classified the cells in two states: state 1 is characterized by a high number of stress fibers and a nonpolarized cell shape, typical of unstimulated cells; RSMC exhibiting a low stress fiber content, membrane ruffling, actin semi-rings, or an elongated shape were classified in state 2. Fig. 2 C shows that in unstimulated cultures 60% of the cells were in state 1 and 40% in state 2. Within 5 min after stimulation, the proportion of cells in state 2 increased to ∼60%, and rose to ∼80% after 15–30 min. 1 h after the addition of HMG1, these proportions reversed back to the values of unstimulated cultures, with 60% of RSMC in state 1 and 40% in state 2. Afterwards, these proportions did not change. These data show that exposure to HMG1 leads to the characteristic morphological changes and cytoskeletal rearrangements connected to cell migration. These effects are transient and represent a change from a resting to a migrating state.
Taken together, qualitative and quantitative data from the chemotaxis and chemokinesis assays fully confirm that HMG1 is a chemoattractant for RSMC.
Both HMG Boxes of HMG1 Have Chemoattractant Activity
To identify the chemotactically active region of HMG1, we used truncated mutant proteins. Full-length and truncated forms of HMG1 were expressed in E. coli and purified. Box A+B lacks the acidic COOH-terminal tail of the intact HMG1 molecule, but bears the two DNA-binding domains. Box A, the first DNA-binding domain of HMG1, corresponds to amino acids 1–89; Box B, the second DNA-binding domain, covers residues 89–176.
Full-length HMG1 from E. coli induced a dose-dependent chemotactic effect on RSMC with a maximum, about twofold above control, at 100 ng/ml. Box A+B (not shown), Box A, and Box B also stimulated RSMC migration in a concentration-dependent manner. However, these maxima were not obtained within the same range of concentrations: the effect of Box A was maximal at 1–10 ng/ml, whereas the chemotactic effect of Box B peaked at 10–100 ng/ml (Fig. 3 A).
Figure 3.

Chemotactic response of RSMC to the HMG box domains of HMG1. (A) Concentration-dependent response to Box A and Box B, both expressed in E. coli. Random cell migration is referred to as 100% migration. The data represent the mean ± SD (n = 3). The statistical significance of the result is P < 0.0001 in an ANOVA model, for both Box A and Box B. (B) Effects of full-length HMG1 expressed in E. coli (100 ng/ml), Box A+B (100 ng/ml), Box A (10 ng/ml), or Box B (10 ng/ml) on actin cytoskeleton organization. Cells were incubated with the indicated molecule for 30 min. Actin filaments were visualized using TRITC-phalloidin.
Box A+B, Box A, and Box B fully reproduced the effects of HMG1 on stress fiber content, membrane ruffling, and transition to the motile state (Fig. 3 B).
Taken together, these results indicate that both DNA-binding domains of HMG1, Box A and Box B, are chemotactically active and are responsible for the migration-promoting activity of full-length HMG1.
Wounding Experiments
To independently confirm the results obtained with the chemotaxis and chemokinesis assays, we used an in vitro wound-healing assay. Confluent monolayers of serum-starved RSMC were wounded and then stimulated for 48 h with HMG1, either from calf thymus or E. coli. Both HMG1 preparations increased the number of migrating cells by ∼1.5–2-fold (Fig. 4). We also tested Box A and Box B (at 10 ng/ml each): both stimulated cell migration ∼1.8-fold. HMG1 and its derivatives were more effective than bFGF (50 ng/ml), which increased cell migration ∼1.5-fold.
Figure 4.

Effects of HMG1 and its HMG boxes on RSMC migration into a wound. Monolayers were wounded and allowed to recover for 48 h in the presence of the indicated molecules. Then, RSMC that had migrated into the wound were counted as described in Materials and Methods. The value of 100% corresponds to the number of cells migrating in the absence of any stimulator (random cell migration). The data represent the mean ± SD (n = 5). Statistical significance was 0.05 < P < 0.01 for treatment with bFGF and full-length E. coli–made HMG1, 0.01 < P < 0.001 for treatment with Box A and Box B, and 0.001 < P < 0.0001 for treatment with calf thymus HMG1.
HMG1 Binds to the Surface of RMSC and Activates Motility through the Receptor for Advanced Glycation Endproducts
To act as a migratory signal, HMG1 must arrive to the membrane of responsive cells and bind to a receptor. To test whether HMG1 binds to the surface of RSMC, we incubated these cells at 4°C with the Box A+B polypeptide, which is slightly smaller than the endogenous, full-length HMG1 and can thus be distinguished easily on SDS-PAGE gels. From the amount of Box A+B recovered in the cell pellet and in the supernatant (Fig. 5 A), it can be estimated that as much as 7 million Box A+B molecules can bind to a single RSMC (see Materials and Methods). This result demonstrates that extracellular HMG1 (or truncated forms thereof) can bind to RSMC, but most likely does not reflect the actual receptor number. Indeed, HMG1 has already been shown to bind to heparin and proteoglycans (Bianchi 1988; Salmivirta et al. 1992; Nair and Jungalwala 1997); thus, HMG1 might also be associated with the extracellular matrix produced by RSMC. In accordance with this hypothesis, only small amounts of HMG1 bind to cells, like HeLa, that produce little extracellular matrix (not shown).
Figure 5.

HMG1 binds to the surface of RSMC and stimulates cell motility through RAGE. (A) Large amounts of HMG1 bind to the surface of RSMC. 1 million cells were incubated at 4°C with 800 ng Box A+B and 5 μg BSA. P shows the proteins associated with the cells, after washing, and S shows 20 μl of the medium containing unbound protein. (B) RSMC express RAGE. 1 million cells were lysed on the plate in SDS-PAGE sample buffer, heated for 5 min at 100°C, and then loaded on a 12% tricine gel. RAGE was detected by Western blot with an anti–RAGE antibody. (C) Anti–RAGE antibody inhibits HMG1-induced RSMC migration. The value of 100% corresponds to the number of cells migrating in the absence of any stimulator (random cell migration). The data represent the mean ± SD (n = 3). Statistical significance was 0.001 < P < 0.0001 for treatment with HMG1 and HMG1 + unspecific antibody. Treatments with HMG1 + anti–RAGE antibody, and HMG1 + anti–HMG1 antibody, had no statistically significant difference from the control. Treatment with anti–HMG1 and anti–RAGE antibodies alone also did not differ from the control.
HMG1 has been reported to bind to RAGE (Hori et al. 1995), a multi-ligand membrane receptor that is expressed by a vast range of cell types. RAGE is present on RSMC (Fig. 5 B), and is thus a good candidate for the receptor that initiates the migratory program induced by extracellular HMG1. This hypothesis was supported by the finding that HMG1 chemotaxis was inhibited not only by anti–HMG1 antibodies, but also by anti–RAGE antibodies (Fig. 5 C). Furthermore, when preincubated with anti–RAGE antibody, RSMC did not exhibit the cytoskeletal reorganization and morphological change in response to HMG1 migratory signal (not shown). The anti–RAGE antibodies had no effect on RMSC in the absence of HMG1 and an irrelevant antibody did not block HMG1-induced cytoskeleton reorganization.
These data indicate that the RAGE receptor is required for the HMG1-induced responses of RSMC.
HMG1 Induces Cell Migration through a Pertussis Toxin-sensitive Mechanism
Since HMG1 acts as a chemoattractant for RSMC, and many chemoattractants act via membrane receptors associated to heterotrimeric GTP binding proteins (G proteins), we tested whether G proteins could be implicated in HMG1 signaling. A specific subclass of G proteins, the Gi/o proteins, are inhibited by pertussis toxin and, conversely, PT inhibition of a certain signaling pathway reveals the involvement of Gi/o proteins (Baggiolini et al. 1994; Neer 1995; Haribabu et al. 1999). We then tested the effect of PT on HMG1-induced RSMC migration; as a control, we used mPT, an inactive mutant of PT that cannot ADP-ribosylate G proteins. PT (50 ng/ml) inhibited the chemotactic effect of HMG1 (Fig. 6), and of Box A, Box B, and Box A+B (not shown). As expected, a similar inhibitory effect was observed on fMLP-induced migration, and no inhibition was exerted on bFGF-dependent migration (Baggiolini et al. 1994; Bokoch 1995; Degryse et al. 1999) (data not shown). mPT at the same dose (50 ng/ml) had no inhibitory effect (Fig. 6), and random cell migration was altered neither by PT nor by mPT.
Figure 6.

Pertussis toxin (PT) inhibits HMG1-induced RSMC migration and actin cytoskeleton reorganization. (A) Chemotaxis assays. The data represent the mean ± SD. The value of 100% corresponds to the number of cells migrating in the absence of any stimulator (random cell migration). (B) Cytoskeleton reorganization. RSMC were pretreated for 6 h either with PT or mPT (both at 50 ng/ml), and were then stimulated (at time 0) with either 100 ng/ml HMG1, 10 ng/ml Box A, or 10 ng/ml Box B. The stimulation with HMG1 was carried on for 30 min. Actin filaments were visualized using TRITC-phalloidin.
These data suggest the involvement of Gi/o proteins in the signaling pathway controlled by HMG1.
HMG1 Induces the Translocation of Phosphorylated ERK1 and -2 into the Nucleus of RSMC
We next investigated whether HMG1 signaling involves the MAP kinase pathway, which is known to respond to RAGE and to activate directly the intracellular motility machinery. Within 30 min, HMG1 activated ERK1/2 proteins in RSMC and induced their nuclear translocation (Fig. 7). In contrast, phosphorylated ERK proteins were hardly detectable and located in the cytoplasm, in unstimulated RSMC. PD98059, the selective inhibitor of MEK, the upstream regulator of ERK, inhibited HMG1-induced ERK phosphorylation and nuclear translocation. HMG1-induced RSMC migration and cytoskeleton reorganization was also inhibited by PD98059 (Fig. 7, and results not shown). Thus, the MAP kinase pathway appears to play an essential role in HMG1-induced cell migration.
Figure 7.

The MAP kinase pathway is involved in HMG1 signaling. RSMC, pretreated or not for 1 h with 50 μM PD98059, were stimulated for 30 min with 100 ng/ml calf thymus HMG1. Cells were then stained with antibodies against phosphorylated ERK1/2 and DAPI. A separate sample of cells were stained with TRITC-phalloidin to visualize the reorganization of the cytoskeleton.
Extracellular HMG1 Can Originate from Damaged or Necrotic Cells
HMG1 is located in the nucleus of most cell types, but a group of recent papers has shown that HMG1 can be secreted by macrophages stimulated with IL-1 and TNFα, and acts extracellularly as a potent proinflammatory signal (Wang et al. 1999a,Wang et al. 1999b; Abraham et al. 2000).
We had previously shown that HMG1 is loosely bound to chromatin (Falciola et al. 1997). Therefore, we tested whether damaged cells, or cells undergoing necrosis, could release HMG1 in the medium. HeLa cells or HUVEC, which contain a significant amount of HMG1, were permeabilized by the addition of the nonionic detergent NP-40 (Falciola et al. 1997). Alternatively, cells were forced into necrosis by addition of ionomycin and the membrane uncoupler CCCP, or by treatment with deoxyglucose and azide. The number of cells undergoing necrosis was scored morphologically, and when it approached 50% the supernatant was collected. HMG1 was recovered in the supernatant of both necrotic cells and damaged cells (Fig. 8 A). The amount of HMG1 found in the supernatant of HUVEC, even when permeabilized with detergent, was always a fraction of the total HMG1 originally present in the cells, because HMG1 binds to the extracellular matrix secreted by HUVEC (not shown).
Figure 8.
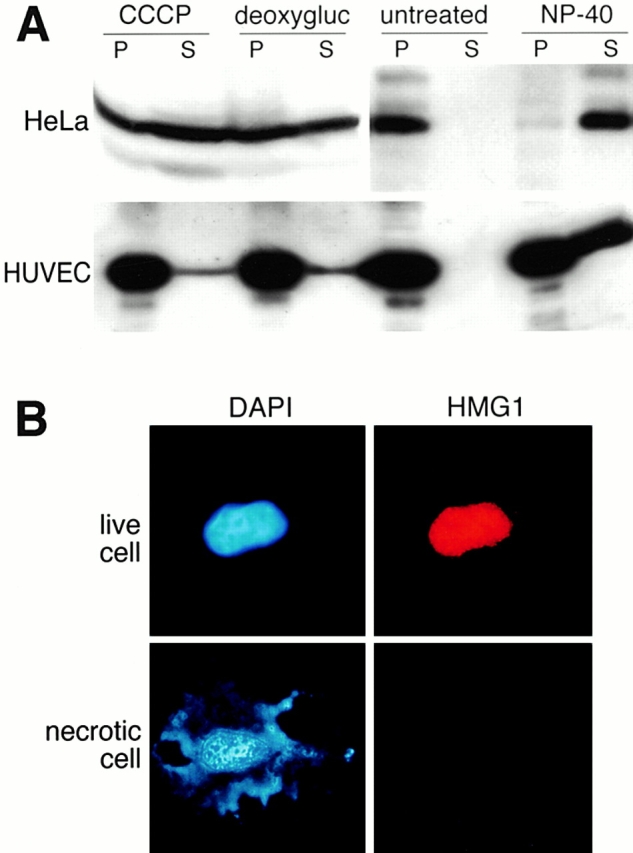
HMG1 is released by necrotic and damaged cells. (A) Western-blot analysis showing the release of HMG1 by necrotic or permeabilized HeLa cells and HUVEC. Cells were induced into necrosis by adding to the medium 5 μM ionomycin plus 20 μM CCCP (CCCP), or 6 mM deoxyglucose plus 10 mM sodium azide (deoxygluc), for 16 h. Alternatively, 0.1% NP-40 was added for 10 min. The presence of the protein in supernatants (S) and cell pellets (P) was detected by Western blot using an anti–HMG1 antibody. The presence of HMG1 in the left two P lanes is due to the fact that only ∼50% of the treated cells underwent necrosis. (B) Immunofluorescence performed on living and necrotic HeLa cells. The cells (untreated or treated with ionomycin + CCCP) were fixed and stained for HMG1 and DNA.
At the single cell level, HMG1 was not associated to the remnants of necrotic cells (Fig. 8 B). The proteins released by damaged or necrotic cells were effective in inducing the morphological changes associated with motility in RSMC, and these effects were blocked by anti–HMG1 antibodies (data not shown).
Immunohistochemistry confirmed that HMG1 is contained in the nuclei of endothelial cells that line human arteries (Fig. 9). Surprisingly, we found that most nuclei of smooth muscle cells in the same arteries (Fig. 9 B, arrows) contain an undetectable amount of HMG1. RMSC in culture in vitro also contain a very small amount of HMG1 compared with HeLa cells (Fig. 9 C).
Figure 9.

HMG1 is present in the nuclei of endothelial cells, but not in those of vascular SMC. (A and B). HMG1 is present in the nuclei of endothelial cells, but is not detectable in the nuclei of vascular SMC. Shown is a section of a human pancreatic artery, stained with anti–HMG1 antibody and counterstained with hematoxylin, at low (A) and high (B) magnification. The red frame indicates the location of the area shown in B, and the arrows point to the nuclei of SMC. (C) Western blot analysis showing the expression level of HMG1 in RSMC in comparison to HeLa cells. The indicated number of cells were lysed directly in SDS-PAGE sample buffer and loaded onto the gel. After the detection of HMG1 (which is identical in rat and human), the blot was stripped and subsequently stained with antihistone antibodies to check for loading.
Altogether, these data suggest that the HMG1 molecules that signal to vascular smooth muscle cells may originate simply by necrosis or mechanical damage of nearby cells.
Discussion
In this study, we show that the nuclear protein HMG1, when present in the medium of rat smooth muscle cells, can induce chemotaxis, chemokinesis, and wound healing in vitro. We also show that HMG1 can be passively released by damaged or necrotic cells. These elements, when taken together, point to HMG1 as a potent mediator of vascular remodeling after mechanical injury and/or inflammation.
HMG1 as a Chemoattractant
HMG1 is a nuclear protein that facilitates the formation and promotes the stability of multiprotein complexes on DNA (reviewed by Bustin 1999; Bianchi and Beltrame 2000). HMG1 also has a second, unrelated activity: it can be secreted by monocytes and macrophages upon activation with IL-1 and TNFα, and acts as a potent mediator of inflammation (Wang et al. 1999a,Wang et al. 1999b; Abraham et al. 2000). HMG1 is also present extracellularly in the brain, from where it was purified under the name of amphoterin (Rauvala et al. 1988). HMG1 has been described as a chemoattractant for neuronal cells, and chemoattraction and extravasation of blood cells is an essential component of inflammatory processes. For these reasons, we tested whether HMG1 could act as a chemoattractant for RSMC, a cell type exhibiting well-known patterns of cell migration upon activation by a diverse set of stimulants.
To demonstrate that HMG1 is a chemoattractant, three independent cell migration assays were used: chemotaxis, chemokinesis, and wound healing in vitro. We also investigated whether HMG1-induced cell migration is functionally connected with the characteristic morphological changes of motile cells, which consist in the reorganization of actin filaments and formation of an elongated polarized shape (Degryse et al. 1999). HMG1 appears to be as potent a chemoattractant as bFGF or fMLP in chemotaxis and wound healing assays, and promotes changes of cell shape and of cytoskeleton organization similar to those observed with pro-urokinase (Degryse et al. 1999).
These effects are specifically due to HMG1, and not to potential contaminants: we tested three preparations (purified from calf thymus, or produced recombinantly by yeast and E. coli) with similar results. In addition, antibodies directed against HMG1 inhibit its effects on cell migration, whereas nonspecific control antibodies are unable to do so.
We also mapped the chemoattractant activity of HMG1 to each of its two DNA-binding domains, HMG Boxes A and B. HMG1 is composed of the two DNA-binding domains, of ∼80 amino acids each, and an acidic tail of 30 aspartic and glutamic acids, connected by short linkers. The two boxes have a low similarity at the level of primary sequence (29% identity), but they have a very similar tertiary structure (Read et al. 1993; Weir et al. 1993; Hardman et al. 1995). Most likely, the molecular recognition of the HMG boxes by the receptor that initiates the signaling pathway for RSMC migration involves the spatially conserved atoms of the polypeptide backbone, or the side residues conserved in both boxes.
Binding to RAGE Initiates the HMG1 Signaling Pathway in RSMC
RAGE had already been shown to be a receptor for amphoterin, that is molecularly identical to HMG1, in neurons (Hori et al. 1995). Moreover, RAGE is involved in inflammation (Hofmann et al. 1999), cell migration (Taguchi et al. 2000), and diabetic atherosclerosis (Hori et al. 1995), whereas it does not promote angiogenesis (Taguchi et al. 2000). The experiments reported here show that RAGE is expressed in RSMC, and anti–RAGE antibodies inhibit the effects of HMG1 on RSMC.
When RAGE binds its ligands (advanced glycation endproducts, calgranulin/S100, β-amyloid fibrils, and amphoterin/HMG1), it triggers the activation of a number of key signaling pathways, such as p21ras, NF-κB, cdc42/rac, and MAP kinases. We confirmed that MAP kinases are recruited for HMG1-induced cell migration in RSMC, as ERK1/2 were phosphorylated and translocated to the cell nucleus upon HMG1 stimulation, and the MEK inhibitor PD98059 blocked cell migration.
Our data also indicate that a Gi/o protein is probably a component of the HMG1 signaling pathway, since HMG1-induced cell migration can be blocked by B. pertussis toxin. Heterotrimeric G proteins are well known to be activated in response to chemoattractants (reviewed in Dekker and Segal 2000; Murdoch and Finn 2000). RAGE has been found together with heterotrimeric G proteins in caveolae (Lisanti et al. 1994), but so far no direct association between RAGE and G proteins has been described in the literature. Only one member of the immunoglobulin superfamily, the thrombospondin receptor (also called integrin-associated protein, IAP, or CD47), has been shown to interact directly with a G protein (Frazier et al. 1999). On the other hand, inhibition by pertussis toxin demonstrates a functional association, and not necessarily a direct physical one: for example, the urokinase receptor, which also mediates chemotaxis and chemokinesis, can be inhibited by pertussis toxin without being directly associated to a G protein (Degryse et al. 1999, Degryse et al. 2001). The relation between RAGE and G proteins will be the focus of additional research.
The Source of Extracellular HMG1
HMG1/amphoterin is present on the surface of neural cells, and can be released by monocytes and macrophages upon stimulation with cytokines or lipopolysaccharide, through a nonconventional route that does not include the endoplasmic reticulum nor the Golgi compartment (Wang et al. 1999a). We ourselves have reproduced these findings (unpublished data). Most likely, HMG1 is translocated directly from the cytoplasm by a specific transporter in a manner analogous to interleukin-1β and a small number of other secreted proteins that lack a leader peptide (Andrei et al. 1999).
However, we also showed that HMG1 can be released in a second, passive manner. Almost every mammalian cell contains a large amount of HMG1 (on average, 1 million molecules per nucleus). HMG1 is a chromatin protein, and binds tightly to nucleosomes reconstituted from purified DNA and histones. However, it is not tightly bound to the chromatin of interphase cells and can be released in the extracellular medium upon detergent action (Falciola et al. 1997), or simply when cells are mechanically damaged or undergo necrosis (Fig. 8). Thus, HMG1 can signal the damage or destruction of an individual cell to the neighboring cells in a paracrine manner. Significantly, HMG1 binds to heparin and proteoglycans, and thus it is not likely to travel very far from the site of cell damage. This does not apply in case of toxic shock, where HMG1 was found in the serum of affected individuals: in this condition, the release of HMG1 is massive in quantitative terms and probably systemic; in fact, the appearance of HMG1 in the serum is correlated with a negative prognosis (Wang et al. 1999a).
The cells that respond to extracellular HMG1 appear to contain very little HMG1 themselves, and almost none in the nucleus. RSMC contain very little HMG1 compared with HeLa cells or endothelial cells, and what little HMG1 they contain is mainly located in the cytoplasm. Migrating RSMC tend to concentrate HMG1 on their surface at the leading edge of the cell (results not shown). This observation agrees with previous data showing that neuroblastoma and glioma cells, which are responsive to HMG1, contain little nuclear HMG1; moreover, HMG1 was localized in growth cones during neurite outgrowth and at the leading edge of laminin-stimulated C6 cells (Rauvala et al. 1988; Merenmies et al. 1991; Parkkinen et al. 1993; Fages et al. 2000). The significance of these findings is so far unclear. However, one can speculate that HMG1-responsive cells might contain little HMG1 to reduce the chance of inappropriate responses to their own HMG1. Concentration of HMG1 at the leading edge of migrating cells might evoke HMG1-induced responses in neighboring cells: relocation of molecules involved in cell migration, such as integrins, the urokinase receptor, or c-Src, is a feature of motile RSMC (Degryse et al. 1999). Migration also involves the activation of extracellular proteases, and the interaction between HMG1 and the plasminogen activation system (Parkkinen and Rauvala 1991; Parkkinen et al. 1993) might facilitate cell migration within the extracellular matrix.
A Potential Role for HMG1 in Vasculopathies
The responsiveness of smooth muscle cells to HMG1, the observation that endothelial cells contain high amounts of HMG1 while vascular SMC contain little, and the release of HMG1 from cells undergoing mechanical damage (as well as by activated macrophages) all point to a possible role of HMG1 during the tissue remodeling occurring in atherosclerosis and restenosis. Endothelial damage results in a burst of SMC migration and proliferation (Gimbrone 1999). Macrophages infiltrate the intima in early steps of atherogenesis, and necrotic cells accumulate in later stages; both macrophages and necrotic cells are expected to release HMG1 and promote SMC invasion of the intima.
RAGE has been shown to be involved in accelerated atherosclerosis in diabetic mice deficient in apolipoprotein E (Park et al. 1998), and the binding of advanced glycation endproducts (AGEs) to RAGE has been considered as the trigger for this process. However, HMG1 might be an additional player in diabetic atherosclerosis since it might be secreted by macrophages/foam cells and it may be released by necrotic cells. Most interestingly, HMG1 binding to RAGE might be the trigger of tissue remodeling in nondiabetic atherosclerosis, where AGEs are not present in pathological amounts. Our observations point to HMG1 as a prime target in the control of vascular disease.
Acknowledgments
The authors thank Drs. J. Bernués, M. Bertulli, F. Pellegatta, M.G. Pizza, S.A. Rabbani, A.M. Schmidt, and J.O. Thomas for providing cells and reagents. We are very grateful to Drs. F. Blasi, A. Mondino, and G. Romeo for technical support and helpful advice. We are especially grateful to Dr. M. de Virgilio for critical review of this manuscript. We thank Mr. M. Azzini for excellent assistance in preparing the photographic illustrations.
This work was supported by grants from Associazione Italiana per la Ricera sul Cancro and the European Union Training and Mobility of Researchers (EU TMR) program to M.E. Bianchi. B. Degryse was a recipient of a fellowship from Università degli Studi di Milano, T. Bonaldi of a fellowship from Fondazione San Raffaele del Monte Tabor, and S. Müller is supported by the EU TMR program.
Footnotes
Dr. Degryse's present address is Department of Vascular Biology, The Scripps Research Institute, La Jolla, CA 92037.
Abbreviations used in this paper: HMG1, high mobility group 1; HUVEC, human umbilical vein endothelial cells; MAP, mitogen-activated protein; PT, Bordetella pertussis toxin; RAGE, receptor for advanced glycation endproducts; RSMC, rat smooth muscle cells; SMC, smooth muscle cells.
References
- Abraham E., Arcaroli J., Carmody A., Wang H., Tracey K.J. HMG-1 as a mediator of acute lung inflammation. J. Immunol. 2000;165:2950–2954. doi: 10.4049/jimmunol.165.6.2950. [DOI] [PubMed] [Google Scholar]
- Andrei C., Dazzi C., Lotti L., Torrisi M.R., Chimini G., Rubartelli A. The secretory route of the leaderless protein interleukin 1beta involves exocytosis of endolysosome-related vesicles. Mol. Biol. Cell. 1999;10:1463–1475. doi: 10.1091/mbc.10.5.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggiolini M., Dewald B., Moser B. Interleukin-8 and related chemotactic cytokines-CXC and CC chemokines. Adv. Immunol. 1994;55:97–179. [PubMed] [Google Scholar]
- Bianchi M.E. Interaction of a protein from rat liver nuclei with cruciform DNA. EMBO (Eur. Mol. Biol. Organ.) J. 1988;7:843–849. doi: 10.1002/j.1460-2075.1988.tb02883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi M.E., Beltrame M. Upwardly mobile proteins. The role of HMG proteins in chromatin structure, gene expression and neoplasia. EMBO Rep. 2000;1:109–114. doi: 10.1093/embo-reports/kvd030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi M.E., Falciola L., Ferrari S., Lilley D.M.J. The DNA binding site of HMG1 protein is composed of two similar segments (HMG boxes), both of which have counterparts in other eukaryotic regulatory proteins. EMBO (Eur. Mol. Biol. Organ.) J. 1992;11:1055–1063. doi: 10.1002/j.1460-2075.1992.tb05144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokoch G.M. Chemoattractant signaling and leukocyte activation. Blood. 1995;86:1649–1660. [PubMed] [Google Scholar]
- Brett J., Schmidt A.M., Yan S.D., Zou Y.S., Weidman E., Pinsky D., Nowygrod R., Neeper M., Przysiecki C., Shaw A. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am. J. Pathol. 1993;143:1699–1712. [PMC free article] [PubMed] [Google Scholar]
- Bustin M. Regulation of DNA-dependent activities by the functional motifs of the high-mobility-group chromosomal proteins. Mol. Cell. Biol. 1999;19:5237–5246. doi: 10.1128/mcb.19.8.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calogero S., Grassi F., Aguzzi A., Voigtländer T., Ferrier P., Bianchi M.E. The lack of chromosomal protein Hmg1 does not disrupt cell growth, but causes lethal hypoglycaemia in newborn mice. Nat. Genet. 1999;22:276–280. doi: 10.1038/10338. [DOI] [PubMed] [Google Scholar]
- Degryse B., Orlando S., Resnati M., Rabbani S.A., Blasi F. Urokinase/urokinase receptor and vitronectin/αvβ3 integrin induce chemotaxis and cytoskeleton reorganization through different signalling pathways. Oncogene. 2001;In press doi: 10.1038/sj.onc.1204261. [DOI] [PubMed] [Google Scholar]
- Degryse B., Resnati M., Rabbani S.A., Villa A., Fazioli F., Blasi F. Src-dependence and pertussis-toxin sensitivity of urokinase receptor-dependent chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. Blood. 1999;94:649–662. [PubMed] [Google Scholar]
- Dekker L.V., Segal A.W. Signals to move cells. Science. 2000;287:982–985. doi: 10.1126/science.287.5455.982. [DOI] [PubMed] [Google Scholar]
- Fages C., Nolo R., Huttunen H.J., Eskelinen E., Rauvala H. Regulation of cell migration by amphoterin. J. Cell Sci. 2000;113:611–620. doi: 10.1242/jcs.113.4.611. [DOI] [PubMed] [Google Scholar]
- Falciola L., Spada F., Calogero S., Längst G., Voit R., Grummt I., Bianchi M.E. High mobility group 1 (HMG1) protein is not stably associated with the chromosomes of somatic cells. J. Cell Biol. 1997;137:19–26. doi: 10.1083/jcb.137.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazioli F., Resnati M., Sidenius N., Higashimoto Y., Appella E., Blasi F. A urokinase-sensitive region of the human urokinase receptor is responsible for its chemotactic activity. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:7279–7286. doi: 10.1093/emboj/16.24.7279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier W.A., Gao A.G., Dimitry J., Chung J., Brown E.J., Lindberg F.P., Linder M.E. The thrombospondin receptor integrin associated protein (CD47) functionally couples to heterotrimeric Gi . J. Biol. Chem. 1999;274:8554–8560. doi: 10.1074/jbc.274.13.8554. [DOI] [PubMed] [Google Scholar]
- Gimbrone M.A., Jr. Vascular endothelium, hemodynamic forces, and atherogenesis. Am. J. Pathol. 1999;155:1–5. doi: 10.1016/S0002-9440(10)65090-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardman C.H., Broadhurst W.R., Raine A.R.C., Grasser K.D., Thomas J.O., Laue E.D. Structure of the A-domain of HMG1 and its interactions with DNA as studied by heteronuclear three- and four-dimensional NMR spectroscopy. Biochemistry. 1995;34:16596–16607. doi: 10.1021/bi00051a007. [DOI] [PubMed] [Google Scholar]
- Haribabu B., Zhelev D.V., Pridgen B.C., Richardson R.M., Ali H., Snyderman R. Chemoattractant receptors activate distinct pathways for chemotaxis and secretion. Role of G-protein usage. J. Biol. Chem. 1999;274:37087–37092. doi: 10.1074/jbc.274.52.37087. [DOI] [PubMed] [Google Scholar]
- Hofmann M.A., Drury S., Fu C., Qu W., Taguchi A., Lu Y., Avila C., Kambham N., Bierhaus A., Nawroth P. RAGE mediates a novel proinflammatory axisa central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- Hori O., Brett J., Slattery T., Cao R., Zhang J., Chen J.X., Nagashima M., Lundh E.R., Vijay S., Nitecki D. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. J. Biol. Chem. 1995;270:25752–25761. doi: 10.1074/jbc.270.43.25752. [DOI] [PubMed] [Google Scholar]
- Huttunen H.J., Fages C., Rauvala H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-κB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J. Biol. Chem. 1999;274:19919–19924. doi: 10.1074/jbc.274.28.19919. [DOI] [PubMed] [Google Scholar]
- Lisanti M.P., Scherer P.E., Vidugiriene J., Tang Z., Hermanowski-Vosatka A., Tu Y.H., Cook R.F., Sargiacomo M. Characterization of caveolin-rich membranes domains isolated from an endothelial-rich sourceimplications for human disease. J. Cell Biol. 1994;126:111–126. doi: 10.1083/jcb.126.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merenmies J., Pihlaskari R., Laitinen J., Wartiovaara J., Rauvala H. 30-kD heparin-binding protein of brain (amphoterin) involved in neurite outgrowth. Amino acid sequence and localization in the filopodia of the advancing plasma membrane. J. Biol. Chem. 1991;266:16722–16729. [PubMed] [Google Scholar]
- Mistry A., Falciola L., Monaco L., Tagliabue R., Acerbis G., Knight A., Harbottle R.P., Soria M., Bianchi M.E., Coutelle C., Hart S.L. Recombinant HMG1 protein produced in Pichia pastoris: a non-viral gene delivery agent. Biotechniques. 1997;22:718–729. doi: 10.2144/97224rr01. [DOI] [PubMed] [Google Scholar]
- Murdoch C., Finn A. Chemokine receptors and their role in inflammation and infectious diseases. Blood. 2000;15:3032–3043. [PubMed] [Google Scholar]
- Nair S.M., Jungalwala F.B. Characterization of a sulfoglucuronyl carbohydrate binding protein in the developing nervous system. J. Neurochem. 1997;68:1286–1297. doi: 10.1046/j.1471-4159.1997.68031286.x. [DOI] [PubMed] [Google Scholar]
- Neeper M., Schmidt A.M., Brett J., Yan S.D., Wang F., Pan Y.C., Elliston K., Stern D., Shaw A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992;267:14998–15004. [PubMed] [Google Scholar]
- Neer E.J. Heterotrimeric G proteinsorganizers of transmembrane signals. Cell. 1995;80:249–257. doi: 10.1016/0092-8674(95)90407-7. [DOI] [PubMed] [Google Scholar]
- Park L., Raman K.G., Lee K.J., Ferran L.J.J., Chow W.S., Stern D., Schmidt A.M. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat. Med. 1998;4:1025–1031. doi: 10.1038/2012. [DOI] [PubMed] [Google Scholar]
- Parkkinen J., Raulo E., Merenmies J., Nolo R., Kajander E.O., Baumann M., Rauvala H. Amphoterin, the 30 kDa protein in a family of HMG1-type polypeptides. J. Biol. Chem. 1993;268:19726–19738. [PubMed] [Google Scholar]
- Parkkinen J., Rauvala H. Interactions of plasminogen and tissue plasminogen activator (t-PA) with amphoterin. Enhancement of t-PA-catalyzed plasminogen activation by amphoterin. J. Biol. Chem. 1991;266:16730–16735. [PubMed] [Google Scholar]
- Passalacqua M., Zicca A., Sparatore B., Patrone M., Melloni E., Pontremoli S. Secretion and binding of HMG1 protein to the external surface of the membrane are required for murine erythroleukemia cell differentiation. FEBS Lett. 1997;400:275–279. doi: 10.1016/s0014-5793(96)01402-0. [DOI] [PubMed] [Google Scholar]
- Rauvala H., Merenmies J., Pihlaskari R., Korkolainen M., Huhtala M.-L., Panula P. The adhesive and neurite-promoting molecule p30analysis of the amino-terminal sequence and production of antipeptide antibodies that detect p30 at the surface of neuroblastoma cells and of brain neurons. J. Cell Biol. 1988;107:2293–2305. doi: 10.1083/jcb.107.6.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read C.M., Cary P.D., Crane-Robinson C., Driscoll P.C., Norman D.G. Solution structure of a DNA-binding domain from HMG1. Nucleic Acids Res. 1993;21:3427–3436. doi: 10.1093/nar/21.15.3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley A.J. Membrane ruffling and signal transduction. Bioessays. 1994;16:321–327. doi: 10.1002/bies.950160506. [DOI] [PubMed] [Google Scholar]
- Salmivirta M., Rauvala H., Elenius K., Jalkanen M. Neurite growth-promoting protein (amphoterin, p30) binds syndecan. Exp. Cell Res. 1992;200:444–451. doi: 10.1016/0014-4827(92)90194-d. [DOI] [PubMed] [Google Scholar]
- Schägger H., von Jagow G. Tricine-sodium dodecyl sulfate–polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- Schmidt A.M., Yan S.D., Wautier J.L., Stern D. Activation of receptor for advanced glycation end productsa mechanism for chronic vascular dysfunction in diabetic vasculopathy and atherosclerosis. Circ. Res. 1999;84:489–497. doi: 10.1161/01.res.84.5.489. [DOI] [PubMed] [Google Scholar]
- Schwartz S.M. Smooth muscle migration in atherosclerosis and restenosis. J. Clin. Invest. 1997;99:2814–2816. doi: 10.1172/JCI119472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparatore B., Passalacqua M., Patrone M., Melloni E., Pontremoli S. Extracellular high-mobility group 1 protein is essential for murine erythroleukaemia cell differentiation. Biochem. J. 1996;320:253–256. doi: 10.1042/bj3200253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier F.W., Moffatt B.A. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- Taguchi A., Blood D.C., del Toro G., Canet A., Lee D.C., Qu W., Tanji N., Lu Y., Lalla E., Fu C. Blockage of RAGE-amphoterin signalling suppresses tumour growth and metastasis. Nature. 2000;405:354–360. doi: 10.1038/35012626. [DOI] [PubMed] [Google Scholar]
- van Leeuwen R.T.J. Extracellular proteolysis and the migrating vascular smooth muscle cell. Fibrinolysis. 1996;10:59–74. [Google Scholar]
- Wang H., Bloom O., Zhang M., Vishnubhakat J.M., Ombrellino M., Che J., Frazier A., Yang H., Ivanova S., Borovikova L. HMG-1 as a late mediator of endotoxin lethality in mice Science. 285 1999. 248 251a [DOI] [PubMed] [Google Scholar]
- Wang H., Vishnubhakat J.M., Bloom O., Zhang M., Ombrellino M., Sama A., Tracey K.J. Proinflammatory cytokines (tumor necrosis factor and interleukin 1) stimulate release of high mobility group protein-1 by pituicytes Surgery (St. Louis). 126 1999. 389 392b [PubMed] [Google Scholar]
- Weir H.M., Kraulis P.J., Hill C.S., Raine A.R.C., Laue E.D., Thomas J.O. Structure of the HMG box motif in the B-domain of HMG1. EMBO (Eur. Mol. Biol. Organ.) J. 1993;12:1311–1319. doi: 10.1002/j.1460-2075.1993.tb05776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]