

Abstract
We have previously shown that during early Caenorhabditis elegans embryogenesis PKC-3, a C. elegans atypical PKC (aPKC), plays critical roles in the establishment of cell polarity required for subsequent asymmetric cleavage by interacting with PAR-3 [Tabuse, Y., Y. Izumi, F. Piano, K.J. Kemphues, J. Miwa, and S. Ohno. 1998. Development (Camb.). 125:3607–3614]. Together with the fact that aPKC and a mammalian PAR-3 homologue, aPKC-specific interacting protein (ASIP), colocalize at the tight junctions of polarized epithelial cells (Izumi, Y., H. Hirose, Y. Tamai, S.-I. Hirai, Y. Nagashima, T. Fujimoto, Y. Tabuse, K.J. Kemphues, and S. Ohno. 1998. J. Cell Biol. 143:95–106), this suggests a ubiquitous role for aPKC in establishing cell polarity in multicellular organisms. Here, we show that the overexpression of a dominant-negative mutant of aPKC (aPKCkn) in MDCK II cells causes mislocalization of ASIP/PAR-3. Immunocytochemical analyses, as well as measurements of paracellular diffusion of ions or nonionic solutes, demonstrate that the biogenesis of the tight junction structure itself is severely affected in aPKCkn-expressing cells. Furthermore, these cells show increased interdomain diffusion of fluorescent lipid and disruption of the polarized distribution of Na+,K+-ATPase, suggesting that epithelial cell surface polarity is severely impaired in these cells. On the other hand, we also found that aPKC associates not only with ASIP/PAR-3, but also with a mammalian homologue of C. elegans PAR-6 (mPAR-6), and thereby mediates the formation of an aPKC-ASIP/PAR-3–PAR-6 ternary complex that localizes to the apical junctional region of MDCK cells. These results indicate that aPKC is involved in the evolutionarily conserved PAR protein complex, and plays critical roles in the development of the junctional structures and apico-basal polarization of mammalian epithelial cells.
Keywords: atypical PKC, tight junction, epithelial cell polarity, PAR-3, PAR-6
Introduction
Protein kinase C comprises a family of serine/threonine kinases with NH2-terminal autoregulatory regions containing unique cysteine-rich Zn-finger motifs (for reviews, see Hug and Sarre 1993). Among them, atypical PKC (aPKC) represents a subgroup of this family comprising the λ/ι and ζ isoforms whose activities are affected by neither calcium nor diacylglycerol/phorbol esters. Several works including ours have shown that aPKCs are activated in vivo through a pathway involving phosphatidylinositol 3-kinase (Akimoto et al. 1996; Toker and Cantley 1997), and are involved in several signal transduction pathways directing mitogen-stimulated cell growth (Berra et al. 1993; Akimoto et al. 1996), insulin-stimulated GLUT4 translocation (Kotani et al. 1998), and apoptosis (Diaz-Meco et al. 1996).
Recently, we identified a mammalian homologue of Caenorhabditis elegans PAR-3, one of the six par gene products indispensable for the establishment of the cell polarity of the one-cell embryo (Kirby et al. 1990; Guo and Kemphues 1996), as an aPKC-specific interacting protein (ASIP) (Izumi et al. 1998). Then, we demonstrated that C. elegans aPKC, PKC3, also binds directly to PAR-3 and shows an asymmetric distribution to the anterior periphery in the one-cell embryo, as in the case of PAR-3 (Tabuse et al. 1998). The functional interaction between PKC-3 and PAR-3 was further demonstrated by showing that RNAi of PKC-3 results in the disruption of the asymmetric distribution of PAR-3, as well as defective phenotypes similar to those found in par-3 mutants (Tabuse et al. 1998). These results indicate an unexpected physiological function of aPKC: its requirement for establishing or maintaining the cell polarity of the C. elegans early embryo. Very interestingly, our previous work also demonstrates that, in mammalian epithelial cells that exhibit well-developed apico-basal cell polarity, ASIP/PAR-3 concentrate at the tight junction (TJ) together with aPKC (Drubin and Nelson 1996; Izumi et al. 1998). Considering that TJ is one of the epithelia-specific junctional structures that is thought to be important for maintaining epithelial cell surface polarity, the result suggested a very intriguing possibility that the aPKC-PAR system plays a fundamental role in the establishment of cell polarity not only in the C. elegans embryo, but also in mammalian epithelial cells.
In this work, we report the introduction of a dominant-negative mutant of aPKC (aPKCkn) into MDCK II cells using an adenovirus expression vector to directly examine the functional importance of aPKC in epithelial cell polarity. We present evidence that aPKCkn blocks the completion of tight junction formation after calcium switch or during normal cell growth. Impairment of cell surface polarity of aPKCkn-expressing cells is also demonstrated by increased interdomain diffusion of fluorescent membrane lipids and the disrupted asymmetric distribution of Na+,K+-ATPase. On the other hand, we also demonstrate that aPKC associates with not only ASIP/PAR-3, but also with a mammalian homologue of another par-gene product, PAR-6, which colocalizes and functions interdependently with PKC-3 and PAR-3 in the C. elegans embryo (Watts et al. 1996; Tabuse et al. 1998; Hung and Kemphues 1999). As expected, mammalian PAR-6 localizes to the apical junctional region together with aPKC and ASIP/PAR-3. These results suggest that aPKC is critically involved in the development of the epithelial junctional structures and controls the cell polarity of mammalian epithelial cells, probably by forming a ternary complex with ASIP/PAR-3 and PAR-6.
Materials and Methods
cDNA Cloning of Human PAR-6
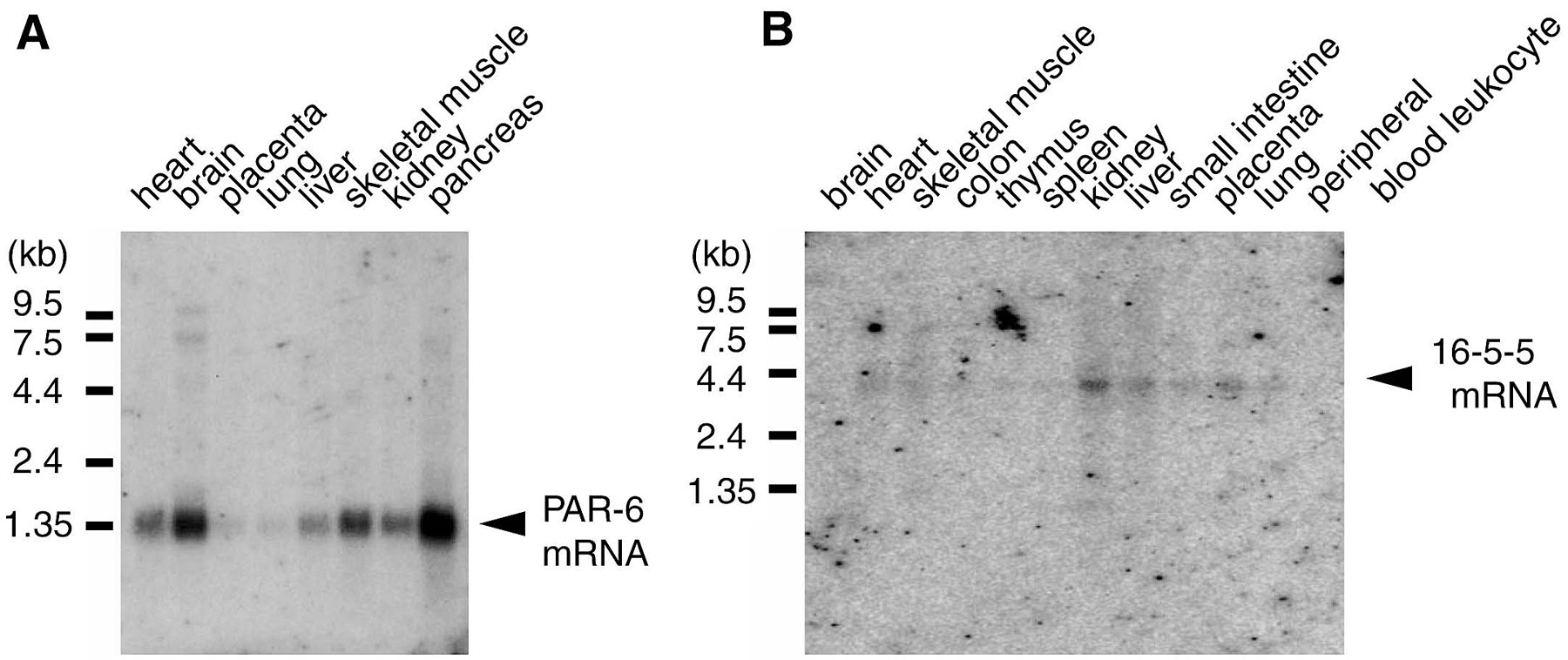
A database search of human EST clones in GenBank identified a human EST clone (AA609625) encoding a peptide closely similar to a part of the C. elegans PAR-6 protein. To obtain cDNA clone(s) covering the entire human PAR-6 protein coding region, we performed backscreening of a human kidney cDNA library (CLONTECH Laboratories, Inc.) using this EST clone as a probe, and identified five cDNA clones, each encoding human PAR-6. The longest clone, n32, carries a 1,269-pb insert containing a 1,041-bp open reading frame encoding a protein of 346 amino acid residues with a calculated molecular weight of 37,388.32. Screening of a HeLa cDNA library (CLONTECH Laboratories, Inc.) with the same probe led to the identification of a different class of cDNA clones encoding a human PAR-6 isoform. The longest clone, 16-5-5, contains 1,162 bp and encodes a 276 amino acid residue, corresponding to a part of the protein (see Supplemental Fig. S1).
Northern Blot Analysis
Northern blot analysis was performed following a standard procedure using human multiple tissue Northern blot (CLONTECH Laboratories, Inc.). Radio-labeled cDNA inserts of clone n32 (1,269 bp) and clone 16-5-5 (1,162 bp) were used as probes to detect human PAR-6 and 16-5-5 mRNA, respectively (see Supplemental Fig. S2).
Antibodies
The antibodies used in this study were: rabbit anti–nPKCδ(δ5), anti–aPKCλ(λ1, λ2), and anti–ASIP(C2-3) polyclonal antibodies, previously raised in our laboratory (Mizuno et al. 1991; Akimoto et al. 1994; Izumi et al. 1998); mouse anti–ZO-1 and rat anti–claudin-1 monoclonal antibodies (obtained from Dr. S. Tsukita, Kyoto University, Kyoto, Japan); rabbit anti–Na+,K+-ATPase polyclonal and mouse anti–gp135 monoclonal antibodies (obtained from Dr. J.D. Nelson, Stanford University, Stanford, CA); mouse anti–aPKCι, E-cadherin, and β-catenin monoclonal antibodies (Transduction Laboratories); rabbit anti–occludin and ZO-1 polyclonal antibodies (Zymed Laboratories); rabbit anti–aPKCζ polyclonal antibody (C-20; Santa Cruz Biotechnology, Inc.); mouse anti–T7 monoclonal antibody (Novagen); mouse anti–Flag monoclonal antibody (M2; Sigma-Aldrich). Anti–human PAR-6 polyclonal antibodies, GW2, GC2, and N12, were generated in rabbits. GW2AP was raised against glutathione-S-transferase (GST)–human PAR-6 and purified on a maltose binding protein (MalE)–PAR-6 affinity column. GC2AP was raised against GST–PAR-6 (amino acids 126–346) and purified as above. N12AP was raised against MalE–PAR-6 (amino acids 1-125) and affinity purified on MalE and MalE–PAR-6 columns.
Expression Vectors
Complementary DNAs encoding wild-type mouse aPKCλ and its kinase-deficient mutant (aPKCλkn) were obtained as described previously (Akimoto et al. 1994, Akimoto et al. 1996). Adenovirus vectors encoding these cDNAs were generated using a cosmid vector, pAxCAwt, as described previously (Miyake et al. 1996). The adenovirus vectors for the kinase-deficient mutant of mouse nPKCδ (lysine 376 replaced by alanine; Hirai et al. 1994) and mouse aPKCζ (lysine 281 replaced by tryptophan) were generous gifts from Dr.Oka (Yamaguchi University, Yamaguchi, Japan); the vector for LacZ was obtained from the Riken Gene Bank (Miyake et al. 1996). T7-tagged ASIP expression vectors were constructed on SRHis vector (Izumi et al. 1998); ASIPwt encodes full-length rat ASIP (amino acids 1–1337), while ASIP Δ30 encodes an ASIP splicing isoform lacking amino acid residues 741–743 and 827–856. Flag-tagged human PAR-6 expression vectors were constructed on pME18S-flag vector; PAR-6wt encodes a full-length PAR-6 (amino acids 1–346), PAR-6 ΔaPKCBD lacks amino acids 1–125, corresponding to the aPKC-binding domain, and PAR-6 ΔCRIB/PDZ lacks amino acids 126–258, corresponding to CRIB and PDZ domain. aPKCλ expression vectors encoding wild type or a mutant lacking amino acids 1–47 (aPKCλ ΔN47) have been described previously (Akimoto et al. 1994).
Cell Culture and Adenovirus Infection
MDCK II cells were grown in DMEM containing 10% FCS, penicillin, and streptomycin on 12-mm round coverslips or 12-mm diameter Transwell™ filters (Corning Coaster Corp.) with a pore size of 0.4 μm. For adenovirus infection, cells were seeded on 24-well plates (1.25 × 105 cells/cm2) 1 d before infection, or on filter inserts (1.5 × 105 cells/cm2) 2 d before infection. To enhance the efficiency of viral infection, cell–cell adhesion was disrupted by preincubating cells in low Ca2+ (LC) medium containing 5% FCS and 3 μM Ca2+ (Stuart et al. 1994) for 2 h, and the cells were then incubated for 1 h with 150 μl of the appropriate virus solution diluted to 3 × 108 pfu/ml in LC medium. When subjected to calcium switch (Gumbiner and Simons 1986), the cells were incubated in fresh LC medium for >20 h, and then the medium was replaced with normal Ca2+ (NC) growth medium. At appropriate times after calcium switch, the cells were processed for immunofluorescent analysis. When skipping calcium switch, the cells were incubated in NC medium immediately after virus infection to allow the cells to complete TJ formation before the expression of ectopic proteins.
Immunocytochemistry
Cells grown on coverslips or filters were washed twice with PBS containing 0.9 mM CaCl2 and 0.49 mM MgCl2, and fixed with 2% paraformaldehyde in PBS for 15 min at room temperature. Cells were then permeabilized with PBS containing 0.5%(vol/vol) Triton X-100 for 10 min and blocked in PBS containing 10% calf serum for 1 h at room temperature. Only in the case of claudin-1 immunostaining, fixation and permeabilization procedures were performed simultaneously using PBS containing 0.3% paraformaldehyde and 0.1% Triton X-100. Antibody incubations were performed at 37°C for 45 min in buffer containing 10 mM Tris/HCl, pH7.5, 150 mM NaCl, 0.01% (vol/vol) Tween 20, and 0.1% (wt/vol) BSA. The secondary antibodies used were: BODIPY, or Alexa488-conjugated goat anti–rabbit IgG (Molecular Probes Inc.), Cy3-conjugated goat anti–mouse IgG, Cy3-conjugated goat anti–rabbit IgG (Amersham Pharmacia Biotech), and FITC-conjugated goat anti–mouse IgG antibodies (EY Laboratories). To stain F-actin, rhodamine-phalloidin (Molecular Probes Inc.) was used in place of the secondary antibodies. Coverslips were mounted using Vectashield (Vector Laboratories), and examined under a fluorescence microscope equipped with a confocal system (μRadiance; Bio-Rad Laboratories). For immunohistochemistry, a piece of small intestine from a mouse (12-wk old) was fixed in 2% paraformaldehyde/PBS at 4°C for 30 min, rinsed with 50 mM NH4Cl/PBS, treated with PBS containing 1.0 M sucrose at 4°C for 24 h, and embedded in Tissue Tek OTC compound. The frozen tissue was cut in a cryostat. The sections (∼7 μm) were mounted on glass slides and stained as described above.
Evaluation of the Barrier Functions of TJ in MDCK Cells
Transepithelial electrical resistance (TER) of MDCK II monolayers grown on filters was measured using an ERS electrical resistance system (Millipore) as described elsewhere (Balda et al. 1996). TER values were obtained by subtracting the contribution of the filter and bathing solution and expressed in Ohm × cm2. Paracellular flux measurement was performed on confluent monolayers of adenovirally infected MDCK II cells that had been subjected to calcium switch 2 d before. A stock solution of FITC-conjugated dextrans 40K and 500K (40 and 500 kD, respectively; Molecular Probes, Inc.) was prepared as described previously (Balda et al. 1996). The assay was started by replacing the apical compartment medium with 200 μl DMEM/FBS containing 0.5 mg/ml FITC-conjugated dextran, and the basal compartment medium with 600 μl DMEM/FBS without tracer. The cells were incubated at 37°C for 3 h, and then the basal medium was collected and diffusible FITC-dextran was measured with a fluorometer (excitation, 492 nm; emission, 520 nm). The amount of dextran was calculated from a titration curve of known concentrations of the tracer.
Fluorescent Lipid Labeling of the Plasma Membrane
BODIPY-FL-C5 sphingomyelin (Molecular Probes, Inc.) and defatted BSA (Sigma-Aldrich) were used to prepare sphingomyelin/BSA complexes (5 nmol/ml) in P-buffer (10 mM Hepes, pH 7.4, 1 mM sodium pyruvate, 10 mM glucose, 3 mM CaCl2, and 145 mM NaCl; Pagano and Martin 1994). Adenovirally infected MDCK cells subjected to calcium switch were washed twice with ice-cold P-buffer, and then 200 μl sphingomyelin/BSA complex solution was added to the apical compartment while 600 μl ice-cold P-buffer was added to the basal compartment. After 10-min labeling, cells were washed twice in ice-cold P-buffer and further incubated in P-buffer for 1 h on ice or prepared directly for microscopy. The distribution of the fluorescent lipid was analyzed as described previously using a confocal laser scanning microscope (Balda et al. 1996).
Immunoprecipitation Analysis
COS1 cells were transfected with appropriate cDNA expression plasmids by electroporation (Gene Pulser; Bio-Rad Laboratories). These cells cultured in 10-cm dishes were suspended in 200 μl of lysis buffer containing 20 mM Hepes, pH 7.5, 150 mM NaCl, 1 mM EDTA, 50 mM NaF, 1 mM Na3VO4, 10 μg/ml leupeptin, 1 PMSF, 1.8 μg/ml aprotinin, and 1% Triton X-100. After 30-min incubation on ice, the lysates were clarified by centrifugation at 14,000 rpm for 30 min and incubated with antibodies preabsorbed on Protein G-sepharose (Amersham Pharmacia Biotech) for 1 h at 4°C. After washing four times with lysis buffer, the immunocomplexes were denatured in SDS sample buffer and separated by SDS-PAGE.
Electrophoresis and Western Blot Analysis
MDCK II cell extracts prepared by adding 200 μl of SDS-sample buffer to each well or immunocomplexes described above were subjected to SDS-PAGE in 8% gels. Proteins were transferred onto polyvinylidene difluoride membranes (Millipore), which were then soaked in 5% skimmed milk. Blotted membranes were processed for immunoreactions as described previously (Suzuki et al. 1995), and visualized by a chemiluminescence ECL system (Amersham Pharmacia Biotech). Chemiluminescent signals were quantified using a Luminescent Image Analyzer, LAS-1000 (Fuji film, Tokyo, Japan).
Yeast Two-Hybrid Analysis
Various deletion mutants of PAR-6 and aPKCλ, subcloned into pAS2-1C and pGAD424 (CLONTECH Laboratories, Inc.), respectively, were simultaneously transformed into the yeast strain HF7C (CLONTECH Laboratories, Inc.) by a standard method (Gietz et al. 1992). After 4 d of growth at 30°C on selective culture plates lacking tryptophan and leucine, or lacking tryptophan, leucine, and histidine, the interaction was evaluated by the formation of colonies.
Online Supplemental Material
Supplemental Fig. S1: comparison of human PAR-6 with other PAR-6 homologues. Supplemental Fig. S2: Northern blot analysis of PAR-6 mRNA in human tissues. Online supplemental materials can be found at http://www.jcb.org/cgi/content/full/152/6/1183/DC1.
Results
Functional Depletion of aPKC Induces the Mislocalization of ASIP/PAR-3 as Well as ZO-1
It has been shown that a kinase-deficient mutant of aPKCλ (aPKCλkn), in which a conserved lysine residue in the ATP-binding site is replaced by glutamate, exerts dominant-negative effects on aPKC-dependent TRE (TPA-response element) activation in HepG2 cells, as well as on insulin-stimulated glucose uptake and GLUT4 translocation in 3T3-L1 adipocytes (Akimoto et al. 1996; Kotani et al. 1998). As a first step to evaluate the role of aPKC in epithelial cell polarity, we overexpressed this aPKCλ mutant in MDCK II cells using an adenovirus-mediated gene transfer approach, and analyzed its effects on the junctional localization of ASIP/PAR-3. As shown in Fig. 1, a and c, adenoviral infection of confluent monolayers of MDCK II cells resulted in the heterogenous expression of aPKCλkn and aPKCλwt ranging from an extreme high level giving saturated immunofluorescent signals to the lowest level, with signals slightly higher than background. Infection efficiencies in the present conditions estimated from the immunostaining results of aPKCλwt and aPKCλkn were both ∼40% (Fig. 1, a and c, and Table ). SDS-PAGE analysis revealed an average fivefold overexpression of both proteins compared with endogenous aPKCλ (data not shown). Overexpressed aPKCλkn as well as aPKCλwt distributed diffusely in the cytosol and did not show dominant junctional localization as reported for the endogenous protein (Dodane and Kachar 1996; Izumi et al. 1998), suggesting that few membrane-anchoring sites for ectopic aPKC remain (note that the anti–aPKCλ antibody used here could not detect endogenous aPKCλ clearly; see Fig. 1 c, top left). This appears to be consistent with the fact that substantial amounts of endogenous aPKC also distribute in the cytosol (Izumi et al. 1998).
Figure 1.

Overexpression of the kinase-deficient mutant of aPKCλ (aPKCkn) in confluent MDCK monolayers disrupts the junctional localization of ASIP and ZO-1 only when the cells are subjected to calcium switch. (a) Confluent MDCK cells seeded on cover slips were infected with adenovirus vector encoding aPKCλkn. 20 h after viral infection, the cells were subjected to calcium switch (+CS) or left untreated (−CS) (see Materials and Methods). After another 20 h, the cells were doubly labeled with anti–aPKCλ and anti–ASIP as indicated at the top (left and middle). Each photograph represents the projected view of optical sections (0.4 μm) obtained from the apical to basal membrane using a laser scanning confocal microscope (unless otherwise noted, the following data were obtained in the same way). Note that fluorescent signals for endogenous aPKCλ are not observed in the photographs because the anti–aPKCλ antibody used (λ2) is not sensitive enough to clearly detect the endogenous protein in MDCK II cells and because the exposure times were adjusted to cover the highly heterogenous level of aPKCλkn expression. (Right) Independent specimens stained by the anti–ZO-1 antibody. Bar, 25 μm. (b) Correlation between the expression level of aPKCλkn and the severity of ZO-1 mislocalization. Based on the fluorescent intensity of aPKCλ, cells were categorized as showing undetectable (−/+), low (+), medium (++), or high (+++) expression, and the ZO-1 staining pattern of the corresponding cell was estimated as complete (white), partially disappeared (hatched), or completely disappeared (black). Original statistical data are presented in Table . (c) LacZ- or aPKCλwt-expressing cells subjected to calcium switch were doubly stained as in a, with anti–aPKCλ and anti–ZO-1 antibodies. Bar, 25 μm.
Table 1.
Correlation between the Expression Level of Ectopic Proteins and the Severity of ZO-1 Mislocalization
| ZO-1 staining | Small ring structure | Cells expressing detectable levels of ectopic aPKCs | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Completely disappeared | Fragmentary | Complete | |||||||
| % | % | % | % | ||||||
| LacZ | 0 | 0 | 6 | 1.2 | 504 | 98.8 | 0 | ND | |
| aPKCλwt | 0 | 0 | 0 | 0 | 564 | 100 | 0 | 38.8 | |
| PKCλkn | 86 | 10.4 | 426 | 51.4 | 316 | 38.2 | 43.1 | ||
| Expression level | −/+ | 9 | 1.9 | 198 | 42.0 | 264 | 56.1 | 37 | |
| + | 24 | 10.8 | 152 | 68.5 | 46 | 20.7 | 8 | 26.8 | |
| ++ | 30 | 33.7 | 54 | 60.7 | 5 | 5.6 | 1 | 10.7 | |
| +++ | 23 | 50.0 | 22 | 47.8 | 1 | 2.1 | 0 | 5.65 | |
When aPKCλkn was expressed in MDCK II cells maintained under normal calcium conditions, the junctional localization of ASIP/PAR-3 was not impaired even in cells showing extremely high levels of aPKCλkn expression (Fig. 1 a, −CS). However, if cells expressing aPKCλkn were subjected to calcium switch to induce a disruption–regeneration process of cell–cell adhesion, then the junctional staining of ASIP/PAR-3 was significantly affected (Fig. 1 a, +CS). Despite the apparently normal restoration of a confluent monolayer as indicated by phase contrast observations (data not shown), ASIP/PAR-3 did not develop a complete junctional distribution over the entire cell circumference, even 6 h after calcium switch (see Fig. 4 a), which is achieved <2 h after calcium switch in control cells. This fragmentary staining of the cell–cell boundaries remains unchanged until 20 h after calcium switch (Fig. 1 a, +CS), suggesting that the junctional localization of ASIP/PAR-3 is significantly inhibited by aPKCλkn. Interestingly, the junctional localization of ZO-1, a TJ marker, in cells was similarly disturbed in a calcium switch-dependent manner, suggesting that TJ formation itself is severely affected (Fig. 1 a, right). Table summarizes the statistical results of ZO-1 mislocalization observed 20 h after calcium switch. While most (>99%) of the cells infected with adenovirus vectors carrying LacZ as well as aPKCλwt display normal ZO-1 staining (Fig. 1 c, and Table ), >60% infected with an aPKCλkn-encoding adenovirus vector exhibit partial or complete mislocalization of ZO-1. The severity of ZO-1 mislocalization correlates well with the level of aPKCλkn expression (Table , and Fig. 1 b); for example, >50% of cells with a high fluorescent signal (+++) exhibit a complete disappearance of ZO-1, whereas the rate is <11% in cells with a low fluorescent signal (+). On the other hand, the data also indicate that >40% of cells apparently negative for an aPKCλ fluorescent signal (−/+) also show fragmentary ZO-1 distribution. Considering the low sensitivity of the anti–aPKCλ antibody used, and the proportional correlation between fluorescent signal intensity and phenotype severity (Fig. 1 b), we infer that the actual infection efficiency is higher than estimated and some cells negative for a fluorescent signal also express levels of aPKCλkn that are undetectable but still sufficient to affect ZO-1 distribution.
Figure 4.

Distribution of various junctional components and F-actin in aPKCλkn-expressing cells. Confluent monolayers of aPKCλkn-expressing MDCK II cells were subjected to calcium switch. 6 h later, ZO-1 distribution was compared with that of other TJ components (a) or adherens junction components (b) by double staining as indicated. For E-cadherin and rhodamine-phalloidin staining, the data for LacZ-expressing cells are also presented for reference. Arrows indicate the positions of aberrant small ring junctional structures, while arrowheads indicate the regions where discrepant staining is observed between ZO-1 and the compared TJ component. Bar, 25 μm. (c and d) Western blot analysis of several junctional proteins in adenovirally infected MDCK II cells. Cells were subjected to calcium switch, lysed 6 h later, and processed for Western blot analysis using the antibodies indicated (left). (c) Typical results of Western blot analysis are presented. The infected viruses are indicated (top). The total amount of protein in each lane was finely adjusted based on the quantitative results of Coomassie brilliant blue staining of the blot membrane (data not shown). Quantified results based on three independent experiments examining the amounts of the indicated proteins in MDCK cells expressing LacZ (white bar), aPKCλwt (hatched bar), or aPKCλkn (black bar) are presented in d. Chemiluminescent signals from the immunostained bands for each junctional protein were quantified using a luminescence Image Analyzer.
Fig. 2 a demonstrates that coinfection with increasing amounts of aPKCλwt and aPKCλkn gradually restores the network-like staining of ZO-1 in a dose-dependent manner. In addition, Fig. 2 b shows that the same phenotypes were observed after the introduction of aPKCζkn, but not nPKCδkn. Because of the significant sequence similarity between aPKCλ and aPKCζ, we cannot exclude the possibility that aPKCλkn exerts its effect not only on endogenous aPKCλ, but also on aPKCζ, and vice versa. In fact, we observed that coinfection with aPKCζwt can rescue the defective phenotype of TJ formation caused by aPKCλkn (data not shown). Therefore, we conclude that the observed mislocalizations of ASIP/PAR-3 and ZO-1 are caused specifically by the dominant negative effects of the aPKC kinase-deficient mutants on endogenous aPKC (λ and/or ζ) activity.
Figure 2.

aPKCkn specifically disturbs the junctional localization of ZO-1 by its dominant negative effect on endogenous aPKC activity. (a) MDCK II cells coinfected with adenovirus vectors encoding aPKCλwt and aPKCλkn were subjected to calcium switch as described in Fig. 1 and, 20 h later, stained with anti–ZO-1 antibody. Total multiplicity of infection was normalized to 120 by mixing with a LacZ-encoding virus vector. The mixed ratios of the each vector are as follows: aPKCλkn:aPKCλwt:LacZ = 2:0:2 (top), 2:1:1 (middle), and 2:2:0 (bottom). Note that increasing amounts of coexpressed aPKCλwt increasingly rescue the phenotype caused by aPKCλkn. (b) MDCK II cells overexpressing aPKCζwt, ζkn, or δkn were similarly prepared as described in Fig. 1 and, 20 h later, doubly stained with anti–PKC antibodies corresponding to each PKC subtype (left) and anti–ZO-1 (right). Bar, 25 μm.
The calcium switch dependence of the effects of aPKCλkn suggests that this dominant-negative mutant is effective only when the cells develop junctional structures to establish epithelial cell polarity. In fact, we further observed the similar effects of aPKCλkn when cells form de novo cell–cell contacts and develop TJ under normal growth conditions. As shown in Fig. 3, where cells were sparsely reseeded immediately after viral infection and cultured for 40 h before immunofluorescent analysis, cells expressing LacZ or aPKCλwt show a normal appearance while aPKCλkn-expressing cells exhibit a flattened shape with prominent lamellipodia (Fig. 3 c) with many cell–cell boundaries negative for ASIP/PAR-3 and ZO-1 staining (Fig. 3, a and b). However, it should be noted that aPKCλkn-expressing cells still form islands and remain close to each other through cell–cell adhesions even in the absence of ASIP/PAR-3 or ZO-1 staining at the cell–cell boundary (Fig. 3 c). These results suggest that the effect of aPKCλkn on TJ formation is not the result of the complete disruption of cell–cell adhesions.
Figure 3.

The overexpression of aPKCkn also disrupts the junctional localization of ASIP and ZO-1 in MDCK II cells under normal growth conditions. MDCK II cells infected with the adenovirus vectors indicated were reseeded sparsely (1.7 × 104 cells/cm2) on cover slips and, 40 h later, subjected to immunofluorescent analysis. (Top) The antibodies used are indicated. Bar, 25 μm.
Aberrant Localization of TJ Components and F-actin Organization in aPKCλkn-expressing MDCK II Cells
Fig. 4 shows the results of a detailed immunofluorescent analysis of the confluent monolayer of aPKCλkn-expressing cells performed 6 h after calcium switch. Double staining for ZO-1 and ASIP/PAR-3 revealed that these peripheral TJ proteins with three PDZ domains colocalize completely, as indicated by the disrupted junctional staining (Fig. 4 a). Further, the membrane accumulation of the major membrane proteins that comprise TJ strands, occludin and claudin-1 (Furuse et al. 1993, Furuse et al. 1998b), is also inhibited. Since immature junctional structures that show positive staining for ZO-1 but not for occludin or claudin-1 are observed, the effects of aPKCλkn overexpression on these membrane proteins seems to be more severe than the effects on ZO-1 or ASIP/PAR-3 (Fig. 4 a, arrowheads). Recent results (Gow et al. 1999) have shown that claudin-11–deficient mice lack TJ strands in the myelin sheaths of oligodendrocytes and Sertoli cells in which this claudin subtype is primarily expressed (Morita et al. 1999). Thus, the above results suggest that aPKC λkn-expressing cells show defects in the development of the TJ structure itself. The basolateral localization of E-cadherin appears to be less affected, but the fluorescence intensity is rather weaker at contact regions lacking ZO-1 staining (Fig. 4 b). Rhodamine-phalloidin staining revealed that aPKCλkn-expressing cells show defects in the formation of developed cortical F-actin bundles surrounding the epithelial cell circumference (Fig. 4 b). Instead, these cells show remarkable retention of the stress fiber-like structures of F-actin. Furthermore, some cells show characteristic large F-actin aggregates whose location corresponds completely to aberrant small ring structures containing all the TJ components examined here (Fig. 4, a and b, arrows). These results indicate that aPKCλkn interferes with the development of the epithelia-specific junctional structures such as belt-like adherent junction (AJ) and TJ, which is mediated by cooperative interactions between F-actin and junctional components. Since the aberrant small ring structures of TJ are observed only in cells showing low expression of aPKCλkn (Table ), these structures might be produced when the suppression of endogenous aPKC activity by aPKCλkn is not complete.
Fig. 4c and Fig. d, show the results of Western blotting analysis to examine the amounts of several junctional proteins in adenovirally infected MDCK II cells harvested 6 h after calcium switch. Prolonged incubation (20 h) of aPKCλkn-expressing cells after calcium switch results in a decrease in the amounts of ASIP/PAR-3 and ZO-1, probably because they fail to be stabilized by being recruited into the junctional complexes (data not shown). However, at least in the early phase of cell polarization when aPKCλkn-expressing cells clearly exhibit defects in TJ formation (Fig. 4 a), the amounts of the junctional proteins examined do not decrease substantially (Fig. 4c and Fig. d). These results suggest that the effects of the overexpression of aPKCλkn do not result from an enhanced degradation of junctional proteins.
Disrupted TJ Barrier Functions in aPKCλkn-expressing MDCKII Cells
The defects in TJ formation in aPKCλkn-expressing cells was further demonstrated functionally by measuring TER to passive ion flow across a cell monolayer grown on permeable support (Fig. 5 a). Consistent with the immunostaining results shown in Fig. 1 a, none of the ectopically expressed proteins, including LacZ, nPKCδkn, or aPKCλwt, as well as aPKCλkn, affected TER if the experiments were arranged so as to induce the expression of the ectopic proteins after the completion of TJ biogenesis (Fig. 5 a, top, 0–45 h). This also suggests that the overexpression of these proteins, especially aPKCλkn, does not produce any artificial cytotoxic effects on TJ barrier function. On the other hand, if the cells were subsequently subjected to calcium switch (2-h incubation in LC medium followed by switching to NC medium), only cells expressing aPKCλkn showed a large retardation in TER development (Fig. 5 a, top, 45–70 h). If the preincubation in low calcium medium was prolonged to 20 h to ensure the dissociation of cell–cell attachments before calcium switch, the effect of aPKCλkn on TER development was more significant: similar to the immunostaining experiments shown in Fig. 1, the expression of ectopic proteins was induced in the cells cultured in LC medium, and calcium switch was applied 20 h after virus infection (Fig. 5 a, bottom). In this case, TER development of aPKCλkn-expressing cells was substantially suppressed until 48 h after calcium switch compared with LacZ- or aPKCλwt-expressing control cells, suggesting that the development of functional TJ is strongly suppressed in aPKCλkn-expressing cells.
Figure 5.
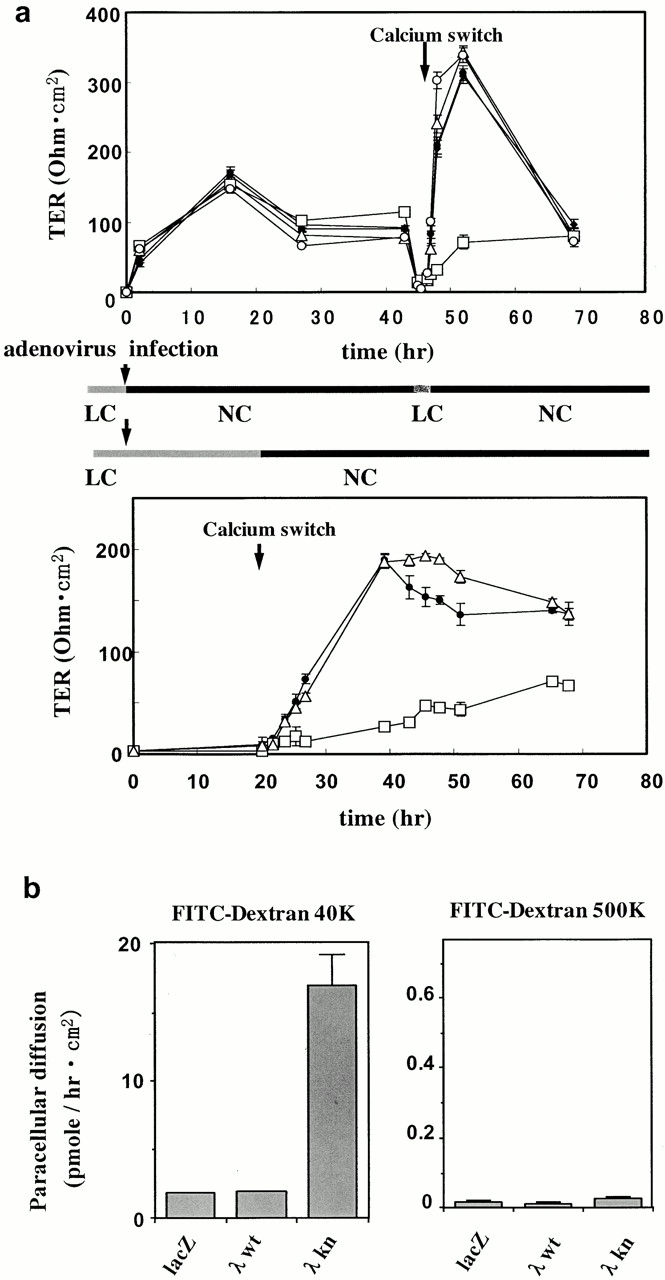
Overexpression of aPKCλkn inhibits the development of TER of MDCK II cells after calcium switch. (a) Confluent MDCK II cells grown on a filter were infected with adenovirus vectors, and TER was measured. (Top) The ectopically expressed proteins are as follows: none (X), LacZ (•), aPKCλkn (□), aPKCλwt (▵), and nPKCδkn (○). 2 d after cell seeding (1.5 × 105 cells/cm2) when the TER values reached a plateau, the cells were processed for adenovirus infection (time 0, left) for 2 h in LC medium. Immediately after virus infection, the medium was changed to NC growth medium to allow the cells to complete junctional structure formation before the expression of ectopic proteins. After TER measurement for 45 h, the cells were subjected to calcium switch by changing the medium to LC for 2 h, and then back to NC. The values given represent mean values (1 ± SD) of three parallel cultures. The background resistance obtained from empty filters was deducted. Note that only cells expressing aPKCλkn show a retardation in TER development after calcium switch. (Bottom) MDCK II cells were infected with adenovirus vectors encoding LacZ (•), aPKCλkn (□), or aPKCλwt (▵) as in a. In this case, the cells were kept in fresh LC medium to induce ectopic protein expression in the absence of cell–cell adhesion. 20 h after virus infection, the medium was changed to NC medium, and the development of TER was monitored for 48 h. Note that aPKCλkn-expressing cells show substantial suppression of TER development even 48 h after calcium switch. (b) Overexpression of aPKCλkn increases the paracellular diffusion of FITC-dextran in a molecular mass-dependent manner. Paracellular diffusion of FITC-dextran 40K (left) and 500K (right) across adenovirus-infected MDCK II cells was evaluated. FITC-dextran was added to the medium on the apical side of cells subjected to calcium switch 2 d before. After 3 h incubation at 37°C, the fluorescence intensity of the medium in the basolateral side was measured with a fluorometer. Values given represent the mean values (±SD) of three parallel cultures.
The impairment of TJ barrier function in aPKCλkn-expressing cells was also demonstrated by measuring the paracellular diffusion of a nonionic solute (Fig. 5 b). In these experiments, the cells were prepared as in Fig. 1, and the diffusion of FITC-dextran across MDCK II monolayers was measured 48 h after calcium switch to ensure the development of epithelial cell polarity. As shown in Fig. 5 b, cells expressing aPKCλkn, but not LacZ or aPKCλwt, still showed fivefold enhanced diffusion of FITC-conjugated dextran 40 K (average 40 kD) over a 3-h period at 37°C. On the other hand, the diffusion of dextran 500 (average 500 kD) showed only a 1.1-fold enhancement, confirming that the enhancement of dextran 40 diffusion is not due to a cytotoxic effect of aPKCλkn expression. These results indicate that aPKCλ activity is required for the development of TJ, which is essential for the barrier function of epithelial cells.
Disrupted Cell Surface Polarity in aPKCλkn-expressing MDCK II Cells
TJ have been suggested to contribute to the establishment of epithelial cell surface polarity by acting as fences for the diffusion of lipids in the outer leaflet of the plasma membrane between the apical and basolateral membrane domains (van Meer and Simons 1986). Therefore, the finding that functional depletion of aPKC blocks the development of the epithelia-specific junctional structures, including TJ, suggests the possibility that aPKCkn expression also results in the disruption of epithelial cell surface polarity. To confirm this possibility, we examined the two-dimensional diffusion of ectopically introduced fluorescent lipids in aPKCλkn-expressing cells using confocal microscopy. In Fig. 6 a, the apical membranes were labeled with BODIPY-sphingomyelin for 10 min at 4°C, and left for an additional 60 min on ice. Confocal microscopic analysis of the x–z sections of these cells demonstrated that only aPKCλkn-expressing cells show markedly enhanced labeling of the lateral membrane after a 60-min chase, suggesting a reduced diffusion fence between the apical and basolateral membranes.
Figure 6.
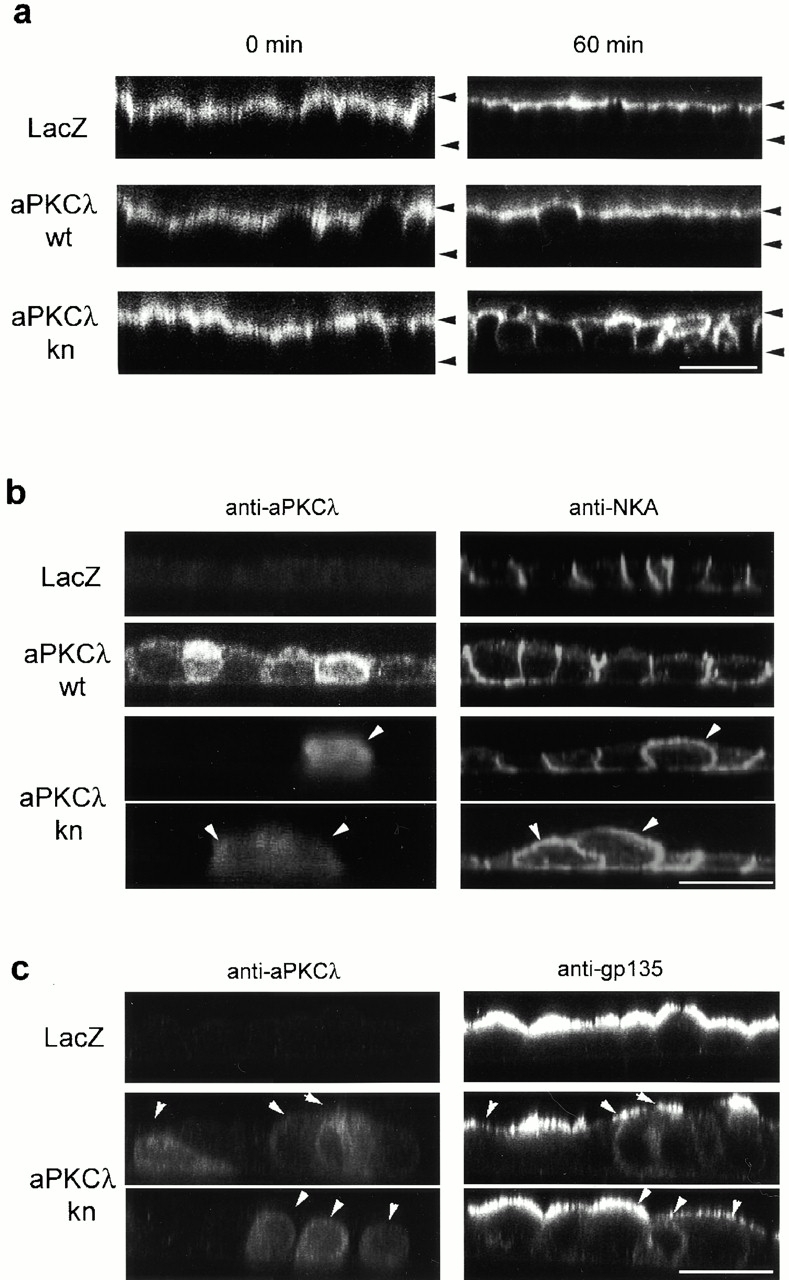
Overexpression of aPKCλkn disrupts apico-basal cell surface polarity of MDCK II cells. (a) Two-dimensional diffusion of ectopically labeled fluorescent lipid from the apical to the basolateral domain. The apical surface of filter-grown MDCK II cell monolayers infected with the indicated adenovirus vectors and subjected to calcium switch were labeled for 10 min on ice with BODIPY-sphingomyelin. The cells were then immediately mounted (0 min) or left for an additional 60 min on ice after extensive washing. The distribution of the fluorescent lipid was then analyzed by taking z-sectional views using a confocal microscope. The upper and lower arrowheads indicate the positions of the apical and basal membranes, respectively. Bar, 25 μm. (b and c) Effect of aPKCλkn on the asymmetric distribution of epithelial polarity markers. Adenovirally infected MDCK II cells were doubly immunostained with anti–aPKCλ and anti–Na+, K+-ATPase (b) or anti–gp135 antibodies (c) 20 h after calcium switch. Representative xz-sectional views obtained by confocal microscopic analysis are presented. Note that aPKCλkn-expressing cells (arrowheads) exhibit disturbed localization of NKA as well as gp135, proteins that show polarized distributions in LacZ-expressing control cells. Bars, 25 μm.
On the other hand, it has been demonstrated that several membrane proteins, such as a basolateral membrane marker, Na+, K+-ATPase (NKA), as well as an apical membrane marker, gp135, retain their polarized localizations even in the absence of TJ via their interaction with domain-specific membrane-skeletal structures (Ojakian and Schwimmer 1988; McNeill et al. 1990). Therefore, to assess the possibility that the overexpression of aPKCλkn affects epithelial cell surface polarity not only by inhibiting TJ biogenesis, but also by interfering with these additional mechanisms for cell polarization, we examined the distribution of endogenous NKA and gp135 in aPKCλkn-expressing cells. NKA is restricted to the lateral membrane in control cells expressing LacZ, whereas its polarized localization in cells expressing aPKCλwt is slightly disturbed to produce a leaky distribution in the apical membrane (Fig. 6 b). Significantly, cells that express aPKCλkn highly show an almost even distribution of NKA in both the apical and basolateral membrane domains (Fig. 6 b, arrowheads), suggesting defects in the machinery required for the maintenance of the polarized distribution of NKA. The apical localization of gp135 was also affected by aPKCλkn overexpression (Fig. 6 c): cells expressing aPKCλkn tend to show a reduced level of gp135 on the apical membrane. Instead, increased cytosolic signals are often detected in these cells.
aPKC and ASIP/PAR-3 Form a Protein Complex Containing a Mammalian Homologue of C. elegans PAR-6 in Polarized Epithelial Cells
Together with the evolutionarily conserved interaction between aPKC and ASIP/PAR-3, the above finding that aPKC activity is required for the establishment of epithelial cell polarity strongly supports a notion that the cell polarization machinery composed of aPKC and PAR proteins found in C. elegans may also be conserved in mammalian epithelial cells. Therefore, we next determined to identify and characterize mammalian homologue of C. elegans PAR-6, which interdependently works with PKC-3 and PAR-3 in C. elegans one-cell embryo (Watts et al. 1996; Tabuse et al. 1998; Hung and Kemphues 1999). Cloned human homologue of PAR-6 shows overall similarity with PAR-6 homologues in other species, sharing several stretches of conserved sequences (supplemental Fig. S1; Fig. 7 c): (a) a sequence at the NH2-terminal region containing two highly conserved regions, CR 1 and CR2; (b) a sequence similar to the CRIB motif (Burbelo et al. 1995); and (c) the single PDZ domain in the COOH-terminal half that has been described previously (Hung and Kemphues 1999). On the other hand, the COOH-terminal region after the PDZ domain is rarely conserved. Northern analysis of human tissues revealed that PAR-6 is expressed in a variety of human tissues (supplemental Fig. S2). To confirm the expression of PAR-6 in MDCK cells, we raised three kinds of anti–PAR-6 antibodies, GW2AP, GC2AP, and N12AP, which were raised against full length amino acids 126–346 and 1–125 of human PAR-6. As shown in Fig. 7 a, immunoprecipitation analysis of MDCK cell lysate revealed the presence of a 43-kD protein reactive with GW2AP as well as N12AP, which is commonly immunoprecipitated with GW2AP and GC2AP. Taken together with the fact that this 43-kD protein comigrates with human PAR-6 (calculated to be 37.4 kD) overexpressed in COS1 cells (Fig. 7 a), we concluded that this 43-kD band represents endogenous PAR-6 protein in epithelial MDCK cells.
Figure 7.

Identification of endogenous PAR-6 and its association with aPKCλ and ASIP in fully polarized epithelial cells. (a) Identification of endogenous PAR-6 protein in MDCK II cells. Semi-confluent MDCK cells were subjected to immunoprecipitation with 1 μg of affinity-purified anti–PAR-6 rabbit polyclonal antibodies (GW2AP, GC2AP) or control normal rabbit IgG. The resultant immunoprecipitates were subjected to Western blot analysis using the antibodies indicated in parentheses (right). GW2AP and GC2AP specifically immunoprecipitate a 43-kD protein comigrating with PAR-6 expressed in COS cells, which was recognized by N12AP as well as GW2AP. (b) Ternary complex formation of PAR-6, aPKCλ, and ASIP/PAR-3 in vivo. Anti–PAR-6 (GW2AP) immunoprecipitate (IP), prepared as in a, was analyzed using anti–aPKCλ monoclonal or anti–ASIP polyclonal antibody. PAR-6 immunoprecipitates specifically contain endogenous aPKCλ, full-length ASIP, and its splicing variant (bottom band). (c and d) Summary of the yeast two-hybrid assays to analyze the PAR-6–aPKCλ interaction. The interaction was examined by growth on culture plates lacking histidine. The NH2-terminal region including CR1 and 2 of PAR-6 is sufficient for the interaction with aPKCλ (c), while NH2-terminal residues 22–113 of aPKCλ are sufficient for the interaction with PAR-6 (d).
Importantly, Western blot analysis further revealed that aPCKλ as well as ASIP/PAR-3 are specifically coimmunoprecipitated with PAR-6 (Fig. 7 b), raising a possibility that PAR-6 interacts with aPKC-ASIP/PAR-3 complex. Consistently, yeast two-hybrid analyses shown in Fig. 7 c confirmed that the NH2-terminal 125 amino acid residues of PAR-6 interact with aPKCλ. Since this NH2-terminal region includes two conserved regions, CR1 and CR2, we tried to narrow the regions required for their interaction further. However, neither amino acids 1–64, including CR1 but not CR2, nor 34–346, including CR2 but not CR1, interacts with aPKCλ (Fig. 7 c), suggesting that the NH2-terminal 125 amino acid sequence including CR1 and CR2 forms a structural domain (termed aPKCBD) required for protein–protein interaction. Another series of analyses also revealed that the NH2-terminal residues 22–113 of aPKCλ are sufficient for the interaction with PAR-6 (Fig. 7 d). Again, NH2- or COOH-terminal deletion of this region results in the disappearance of the interaction (Fig. 7 d), suggesting that this region, corresponding to the D1 region (diversed region 1) of aPKCλ, forms another structural domain for protein–protein interaction.
aPKC Mediates the Interaction between ASIP/PAR-3 and PAR-6 as a Linker
Previously, we demonstrated that aPKCλ directly binds to ASIP/PAR-3 through its kinase domain. Therefore, the above results raise the possibility that aPKCλ binds both ASIP/PAR-3 and PAR-6 simultaneously and mediates the formation of an aPKC-ASIP/PAR-3–PAR-6 ternary complex. To examine this possibility, we next performed a series of immunoprecipitation experiments in COS1 cells (Fig. 8). As shown in Fig. 8 a, when Flag-tagged PAR-6 was overexpressed and immunoprecipitated with an anti–Flag antibody, coexpressed T7-tagged ASIP was coprecipitated together with endogenous aPKCλ. Significantly, when T7-tagged ASIP Δ30, which corresponds to an isoform lacking aPKC-binding region was coexpressed with Flag-tagged PAR-6 instead of wild-type ASIP, endogenous aPKCλ but not this ASIP isoform was coimmunoprecipitated with PAR-6, suggesting that ASIP indirectly associates with PAR-6 by way of aPKCλ. In fact, a T7-tagged PAR-6 mutant lacking NH2-terminal aPKCλ-binding region (ΔaPKCBD) does not show interactions not only with endogenous aPKCλ but also with ASIP (Fig. 8 b), although the other PAR-6 mutant (ΔCRIB/PDZ) lacking the CRIB and PDZ domains but retaining aPKCλ-binding region can interact with both proteins. Furthermore, Fig. 8 c shows that overexpression of aPKCλ enhances the coprecipitation of T7-tagged ASIP with Flag-tagged PAR-6. On the other hand, overexpression of aPKCλ ΔN47 that cannot bind to PAR-6 does not show such enhancement, but rather suppresses the coprecipitation of ASIP/PAR-3 with PAR-6. This can be explained as a dominant negative effect of this aPKCλ mutant on ASIP, inhibiting the indirect association of ASIP with PAR-6 by way of endogenous aPKCλ. Taken together, we conclude that aPKC serves as a linker molecule between PAR-6 and ASIP, and mediates the formation of a ternary protein complex composed of aPKCλ, ASIP/PAR-3, and PAR-6 (Fig. 8 d).
Figure 8.

Ternary complex formation of PAR-6, aPKCλ, and ASIP/PAR-3. (a–c) COS cells were transfected with expression vectors as indicated (top). The cell lysates (Sup) were processed for immunoprecipitation (IP) with anti–Flag (a and c) or anti–T7 (b) monoclonal antibodies, and the coimmunoprecipitated proteins were analyzed using with anti–aPKCλ (ι), anti–T7, anti–Flag, or anti–ASIP (C2-3AP) antibodies. (a) The Flag-PAR-6 immunoprecipitates contained T7-ASIPwt, but not T7-ASIP Δ30 lacking the sequence critical for the interaction with aPKC. (b) ASIPwt was not contained in PAR-6 ΔaPKCBD immunoprecipitates but in PAR-6wt or ΔCRIB/PDZ immunoprecipitates. (c) Coimmunoprecipitation of T7-ASIP with Flag-PAR-6 was enhanced by the coexpression aPKCλwt, but not aPKCλ ΔN47, which lacks the sequences in the PAR-6–binding region. (d) Schematic diagram of the PAR-6–aPKC-ASIP/PAR-3 ternary complex. PAR-6 interacts with ASIP through aPKCλ as a linker.
Colocalization of PAR-6 as Well as aPKCλ and ASIP/PAR-3 to the Epithelial Junctional Complex with ZO-1
To evaluate the physiological significance of the physical interaction between aPKCλ, ASIP/PAR-3, and PAR-6 in epithelial cells, we next examined the intracellular localization of PAR-6 in MDCK cells. As shown in Fig. 9 a, the anti–PAR-6 antibody, GW2AP, clearly stains the cell–cell boundary of confluent MDCK II cells. Since the similar result was obtained with the other independent antibody, GC2AP (Fig. 9 b), we concluded that these junctional stainings represent the genuine localization of endogenous PAR-6 in MDCK II cells. Closer inspection of the localization of endogenous PAR-6 by confocal z-sectioning revealed PAR-6 staining at the most apical end of the cell–cell contact region with ZO-1 (Fig. 9 c). Since, as previously suggested (Izumi et al. 1998), aPKCλ and ASIP/PAR-3 also localize to the corresponding region with ZO-1 (Fig. 9 c), these results strongly suggest that PAR-6 colocalizes with aPKCλ and ASIP/PAR-3 to the apical junctional complex of epithelial cells. To clarify further the localization of PAR-6 in epithelial cells, we next stained mouse intestinal epithelia, a typical tissue containing polarized epithelial cells. Similar to aPKCλ and ASIP/PAR-3, PAR-6 also localizes to the most apical end of the junctional complex with ZO-1 (Fig. 9 d). In addition, like ASIP/PAR-3, the junctional localization of PAR-6 is severely disturbed in aPKCλkn-expressing cells, showing complete colocalization with ZO-1 (Fig. 9 e). Taken together with the physical interactions among these three proteins, the above results provide evidence supporting the idea that PAR-6, aPKCλ, and ASIP/PAR-3 asymmetrically localize in the apical junctional complex in polarized epithelial cells as a ternary protein complex.
Figure 9.
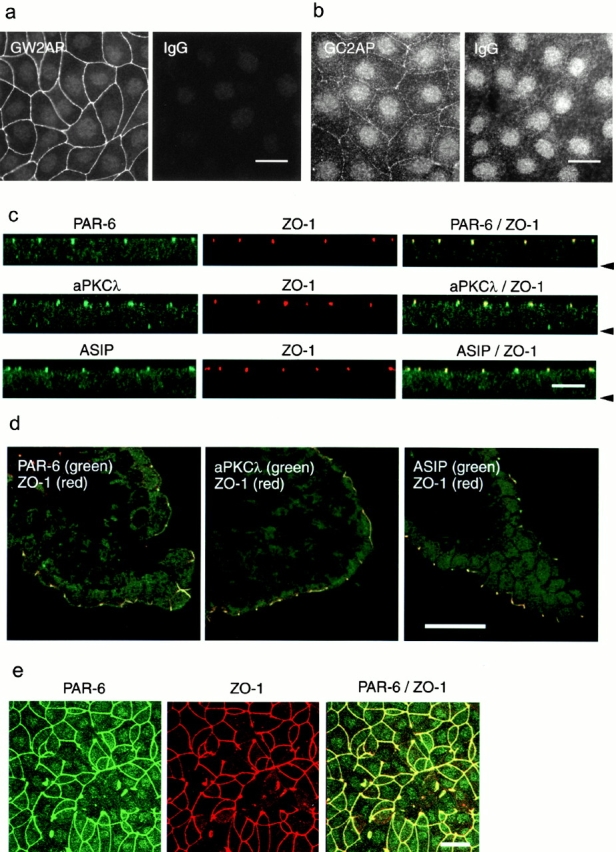
Colocalization of PAR-6, aPKCλ, and ASIP/PAR-3 with ZO-1 at the apical end of cell–cell contact region of epithelial cells. (a and b) Immunofluorescence staining of MDCK cells with anti–PAR-6 polyclonal antibodies, GW2AP (a) and GC2AP (b). IgG means an equal amount of normal rabbit IgG used as a negative control. In b, the photograph was taken at higher sensitivity than in a to visualize weak cell–cell staining by GC2AP. (c) Confocal z-sectional view of MDCK cells doubly stained with anti–PAR-6 (GW2AP), anti–aPKCλ (λ1), or anti–ASIP antibodies (green), together with anti–ZO-1 antibody (red). The arrowhead indicates the position of the basal membrane. All three proteins colocalize with ZO-1 to the apical end of lateral membrane. (d) Immunostaining of a frozen section of mouse intestinal epithelium with anti–PAR-6 (GW2AP), anti–aPKCλ, or anti–ASIP antibodies (green). Only merged views with anti–ZO-1 staining (red) are shown. (e) Overexpression of aPKCλkn affects the junctional localization of PAR-6 as well as ZO-1. Bars, 25 μm (a, b, d, and e) and 10 μm (c).
Discussion
aPKC Kinase Activity Is Required for the Establishment of the Junctional Structures and Epithelial Cell Polarity
Pharmacological studies using PKC activators or inhibitors have indicated the involvement of PKC in TJ biogenesis (Citi 1992; Balda et al. 1993; Stuart and Nigam 1995; Denker et al. 1996). However, one cell type expresses several PKC isotypes with differing sensitivities to activators or inhibitors (Ogita et al. 1991; Mizuno et al. 1995). In fact, MDCK II cells express at least cPKCα, nPKCε, η, and aPKCλ, ζ (Shimizu, M., unpublished results). Therefore, the results obtained so far using these agents should reflect the sum of their effects on several PKCs in MDCK cells, even if it is assumed that their targets are restricted to PKC.
In this paper, we used dominant-negative mutants of aPKC to demonstrate that aPKCλ or ζ is required for the assembly of TJs. This conclusion is based on the following results. (a) Cells expressing dominant–negative point mutants of aPKCλ or ζ (aPKCkn), but not the wild types, show an inhibited junctional localization of ZO-1 even 20 h after calcium switch. (b) The effects are specific to dominant–negative mutants of aPKCs and are not observed when such mutants of nPKCδ and cPKCα (data not shown) are used. (c) The effects of aPKCλkn are rescued by the cointroduction of aPKCλwt in a dose-dependent manner, suggesting that the effects of aPKCλkn are based on its dominant suppression of endogenous aPKC activity. (d) The junctional localizations of not only ZO-1, but all TJ components tested, are similarly affected by aPKCλkn, indicating that the development of the TJ structure itself is blocked in aPKCλkn-expressing cells. (e) Overexpression of aPKCλkn impairs the restoration of the barrier function of MDCK cells after calcium switch, as measured by TER as well as by the paracellular diffusion of dextran. It should be noted that these effects of aPKCλkn are observed only when the ectopic protein is present during the de novo formation of TJ structures. Generally, the dominant–negative effects of kinase-deficient point mutants are thought to arise from competition with the endogenous kinase for interaction with other molecules required for their proper function. The target molecules can be (a) specific substrates of the kinase, (b) activators, or (c) anchoring partners that determine where the kinase should be activated physiologically and efficiently. At this stage, we cannot conclude in which way the aPKCkn mutants we used work within the cells. However, the present results clearly indicate that regulated aPKC kinase activity is required during the dynamic reconstructing of the junctional structures and epithelial cell polarity.
Together with previous pharmacological studies, the features of the effect of aPKCkn reported here agree with those shown for the effects of the PKC-specific inhibitors, H7 and calphostin (Stuart and Nigam 1995): both inhibit TJ assembly as monitored by ZO-1 distribution and TER development, but have lesser effect on the basolateral accumulation of E-cadherin in MDCK cells. Both specifically affect the formation but not the maintenance of TJ. These coincidences may indicate that the main targets of the PKC inhibitors so far examined are aPKCs. In fact, the inhibition constant of H7 and calphostin against aPKCζ measured in vitro (90 and 1 μM for ε-peptide, respectively; Mizuno et al. 1995), are four- to fivefold higher than those for cPKCα, and this may be why the effects of these inhibitors on ZO-1 distribution are rather weak compared with the effect of aPKCkn (Stuart and Nigam 1995, reported that 25 μM H7, as well as 500 nM calphostin, in the medium retards, but does not completely block, the junctional ring formation of ZO-1). However, it should also be noted that aPKCs are atypical PKCs that are insensitive to diacylglycerol (Akimoto et al. 1994). This means that our data do not explain the finding in a previous study that 1,2-dioctanoylglycerol (a diacylglycerol analogue) induces TJ-like structures even in the absence of extracellular calcium without AJ formation (Balda et al. 1993). One working hypothesis that can explain all the above results is that not only aPKC but also diacylglycerol-responsive PKC (conventional and/or novel PKC) plays a positive role in the regulation of TJ formation. We are now directing our efforts to validating this hypothesis.
Molecular Mechanism by which aPKC Regulates the Junctional Structure Formation
The molecular basis of the involvement of aPKC in TJ biogenesis is not yet clear. Considering the fact that endogenous aPKC as well as ASIP/PAR-3, its specific binding protein with three PDZ domains, localize at TJ in polarized epithelial cells (Izumi et al. 1998), it is tempting to speculate that aPKC is recruited into TJ by ASIP/PAR-3, and thereby exerts kinase activity to promote the development of the TJ structures. It may seem that this idea is inconsistent with the present observation that aPKCkn is distributed mainly in the cytosol and not at the cell–cell borders (Fig. 1 a); however, this may suggest that the mutant inhibits the translocation of endogenous aPKC to the cell–cell junctional area by competing for binding to ASIP/PAR-3 or other binding proteins in the cytosol. We do not have direct evidence suggesting that the phosphorylation by aPKC is required for the TJ targeting of ASIP/PAR-3, but it should be remembered that the polarized localizations of aPKC and PAR-3 are mutually dependent in the C. elegans one-cell embryo.
Of course, the possibility remains that the region where aPKC mainly functions is not the cell–cell junction, but the cytosol. In fact, substantial amounts of endogenous aPKC are detected in the cytosol, although some is concentrated at the tip of the lateral membrane of epithelial cells (Dodane and Kachar 1996). In addition, upon disruption of cell–cell adhesions, aPKC as well as ASIP/PAR-3 localizing at cell–cell junctions redistribute to the cytosol, suggesting the dynamic nature of the aPKC-ASIP/PAR-3 complex (Izumi et al. 1998). In this respect, it is interesting that there are several reports suggesting that aPKCs are involved in the regulation of vesicle transport. For example, the dominant–negative mutant of aPKC specifically inhibits insulin-dependent Glut4 translocation to plasma membranes in adipocytes (Kotani et al. 1998). In addition, aPKCs colocalize with p62/ZIP and Rab7 at vesicular-like structures in the cytosol of Hela cells, and its dominant–negative mutant severely impairs the endocytic membrane transport of the EGF receptor with no effect on the transferrin receptor (Sanchez et al. 1998). Therefore, another interesting possibility is that aPKC regulates the polarized membrane targeting of proteins, including junctional proteins, in response to initial cell–cell adhesion. This may explain the present results in which the polarized localization of NKA and gp135, which can be maintained even in the absence of TJ, is disturbed in aPKCλkn-expressing MDCK cells. On the other hand, we cannot exclude the possibility that the primary target of aPKC may not be TJ biogenesis itself, but upstream events triggered by initial cell–cell attachment. In fact, we observed that aPKCλkn-expressing cells also show decreased E-cadherin accumulation to cell–cell borders, and defects in the formation of developed F-actin peripheral bundles remaining stress fiber-like structures instead (Fig. 4 b). Therefore, aPKC might regulate the reorganization of the submembranous and/or intracellular cytoskeleton, which is closely linked with the asymmetric development of epithelia-specific junctional structures including TJ, belt-like AJ, and desmosome. This idea may be consistent with the recent reports that aPKCλ and ζ participate in the Ras-mediated reorganization of the F-actin cytoskeleton in NIH3T3 cells, or that aPKCλ and ζ work downstream of Cdc42 to disrupt stress fibers in NIH3T3 cells (Uberall et al. 1999; Coghlan et al. 2000). The defects in the mislocalization of NKA and gp135 in aPKCλkn-expressing MDCK cells can also be explained by this idea. To address these issues, further studies are needed to identify the physiological substrates of this kinase, whose phosphorylated state and activity change dynamically during the process of epithelial cell polarization.
aPKC–PAR System in Mammalian Epithelial Cells
Here, we present data showing that the overexpression of a dominant–negative mutant of aPKC disturbs epithelial cell surface polarity along the apico-basal axis (Fig. 6). These results correspond well with our previous finding that the C. elegans one-cell embryo lacking aPKC (PKC-3) shows defects in the establishment of the anterior–posterior polarity (Izumi et al. 1998). Consistently, we further demonstrate here that a mammalian homologue of PAR-6 forms a ternary complex with aPKC and ASIP/PAR-3, and accumulates at the apical tip of epithelial cell–cell junctions. Moreover, as in the case of the C. elegans embryo lacking PKC-3 expression, the junctional localizations of ASIP/PAR-3 and PAR-6 are disturbed by the overexpression of aPKCλkn. Although we have not succeeded in directly demonstrating the involvement of ASIP/PAR-3 and PAR-6 in aPKC function in the present paper, the above coincidences increasingly support the hypothesis that the aPKC-PAR system acts as evolutionarily conserved cell polarization machinery not only in the C. elegans early embryo but also in mammalian epithelial cells. Importantly, a Drosophila homologue of aPKC was recently suggested to associate with Bazooka (Drosophila homologue of PAR-3) and control the polarity of epithelia as well as neuroblasts (Wodarz et al. 2000). The present study provides additional insight into this machinery by indicating that aPKC activity is required for the development of the polarized junctional structures to which the aPKC/PAR-3/PAR-6 complex anchors. This might suggest that, in general, the establishment of the asymmetrical membranous/submembranous structures is a primary step for subsequent cell polarization. To fully understand the molecular basis of this evolutionarily conserved role of aPKC, mammalian epithelial cells provide an advantageous system for future biochemical analyses, offering a convenient method to reconstitute the cell polarization process in vitro.
Supplemental Material
Acknowledgments
We thank S. Tsukita, M. Itoh, and M. Furuse for providing us with anti–ZO-1 and anti–claudin-1 antibodies and for helpful discussion, J.D. Nelson for providing anti–Na+,K+-ATPase and anti–gp135 antibodies, and Y. Oka and H. Katagiri for providing adenovirus vectors encoding wild-type and kinase-deficient mutants of PKCζ and δ.
This work was supported by grants from the Japan Society for the Promotion of Science (S. Ohno) and the Ministry of Education, Science, Sports and Culture of Japan (S. Ohno and A. Suzuki).
Footnotes
The online version of this article contains supplemental material.
Drs. Suzuki and Yamanaka contributed equally to this work and should be considered co-first authors.
Dr. Izumi's present address is Department of Developmental Neurobiology, Institute of Development, Aging and Cancer, Tohoku University, Sendai 980-8575, Japan.
Abbreviations used in this paper: AJ, adherent junction; aPKC, atypical PKC; ASIP, aPKC-specific interacting protein; LC, low Ca2+; NC, normal Ca2+; NKA, Na+, K+-ATPase; TER, transepithelial electrical resistance; TJ, tight junction.
References
- Akimoto K., Mizuno K., Osada S., Hirai S., Tanuma S., Suzuki K., Ohno S. A new member of the third class in the protein kinase C family, PKC lambda, expressed dominantly in an undifferentiated mouse embryonal carcinoma cell line and also in many tissues and cells. J. Biol. Chem. 1994;269:12677–12683. [PubMed] [Google Scholar]
- Akimoto K., Takahashi R., Moriya S., Nishioka N., Takayanagi J., Kimura K., Fukui Y., Osada S., Mizuno K., Hirai S. EGF or PDGF receptors activate atypical PKClambda through phosphatidylinositol 3-kinase. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:788–798. [PMC free article] [PubMed] [Google Scholar]
- Balda M.S., Gonzalez-Mariscal L., Matter K., Cereijido M., Anderson J.M. Assembly of the tight junctionthe role of diacylglycerol. J. Cell Biol. 1993;123:293–302. doi: 10.1083/jcb.123.2.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balda M.S., Whitney J.A., Flores C., Gonzalez S., Cereijido M., Matter K. Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical-basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J. Cell Biol. 1996;134:1031–1049. doi: 10.1083/jcb.134.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berra E., Diaz-Meco M.T., Dominguez I., Municio M.M., Sanz L., Lozano J., Chapkin R.S., Moscat J. Protein kinase C zeta isoform is critical for mitogenic signal transduction. Cell. 1993;74:555–563. doi: 10.1016/0092-8674(93)80056-k. [DOI] [PubMed] [Google Scholar]
- Burbelo P.D., Drechsel D., Hall A. A conserved binding motif defines numerous candidate target proteins for both Cdc42 and Rac GTPases. J. Biol. Chem. 1995;270:29071–29074. doi: 10.1074/jbc.270.49.29071. [DOI] [PubMed] [Google Scholar]
- Citi S. Protein kinase inhibitors prevent junction dissociation induced by low extracellular calcium in MDCK epithelial cells. J. Cell Biol. 1992;117:169–178. doi: 10.1083/jcb.117.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghlan M.P., Chou M.M., Carpenter C.L. Atypical protein kinases Clambda and -zeta associate with the GTP-binding protein Cdc42 and mediate stress fiber loss. Mol. Cell. Biol. 2000;20:2880–2889. doi: 10.1128/mcb.20.8.2880-2889.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denker B.M., Saha C., Khawaja S., Nigam S.K. Involvement of a heterotrimeric G protein alpha subunit in tight junction biogenesis. J. Biol. Chem. 1996;271:25750–25753. doi: 10.1074/jbc.271.42.25750. [DOI] [PubMed] [Google Scholar]
- Diaz-Meco M.T., Municio M.M., Frutos S., Sanchez P., Lozano J., Sanz L., Moscat J. The product of par-4, a gene induced during apoptosis, interacts selectively with the atypical isoforms of protein kinase C. Cell. 1996;86:777–786. doi: 10.1016/s0092-8674(00)80152-x. [DOI] [PubMed] [Google Scholar]
- Dodane V., Kachar B. Identification of isoforms of G proteins and PKC that colocalize with tight junctions. J. Membr. Biol. 1996;149:199–209. doi: 10.1007/s002329900020. [DOI] [PubMed] [Google Scholar]
- Drubin D.G., Nelson W.J. Origins of cell polarity. Cell. 1996;84:335–344. doi: 10.1016/s0092-8674(00)81278-7. [DOI] [PubMed] [Google Scholar]
- Furuse M., Hirase T., Itoh M., Nagafuchi A., Yonemura S., Tsukita S. Occludina novel integral membrane protein localizing at tight junctions. J. Cell Biol. 1993;123:1777–1788. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuse M., Sasaki H., Fujimoto K., Tsukita S. A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts J. Cell Biol 143 1998. 391 401b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz D., St A., Jean R.A., Woods, Schiestl R.H. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992;20:1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow A., Southwood C.M., Li J.S., Pariali M., Riordan G.P., Brodie S.E., Danias J., Bronstein J.M., Kachar B., Lazzarini R.A. CNS myelin and sertoli cell tight junction strands are absent in Osp/claudin-11 null mice. Cell. 1999;99:649–659. doi: 10.1016/s0092-8674(00)81553-6. [DOI] [PubMed] [Google Scholar]
- Gumbiner B., Simons K. A functional assay for proteins involved in establishing an epithelial occluding barrieridentification of a uvomorulin-like polypeptide. J. Cell Biol. 1986;102:457–468. doi: 10.1083/jcb.102.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S., Kemphues K.J. Molecular genetics of asymmetric cleavage in the early Caenorhabditis elegans embryo. Curr. Opin. Genet. Dev. 1996;6:408–415. doi: 10.1016/s0959-437x(96)80061-x. [DOI] [PubMed] [Google Scholar]
- Hirai S., Izumi Y., Higa K., Kaibuchi K., Mizuno K., Osada S., Suzuki K., Ohno S. Ras-dependent signal transduction is indispensable but not sufficient for the activation of AP1/Jun by PKC delta. EMBO (Eur. Mol. Biol. Organ.) J. 1994;13:2331–2340. doi: 10.1002/j.1460-2075.1994.tb06517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug H., Sarre T.F. Protein kinase C isoenzymesdivergence in signal transduction? Biochem. J. 1993;291:329–343. doi: 10.1042/bj2910329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung T.J., Kemphues K.J. PAR-6 is a conserved PDZ domain-containing protein that colocalizes with PAR-3 in Caenorhabditis elegans embryos. Development (Camb.) 1999;126:127–135. doi: 10.1242/dev.126.1.127. [DOI] [PubMed] [Google Scholar]
- Izumi Y., Hirose T., Tamai Y., Hirai S., Nagashima Y., Fujimoto T., Tabuse Y., Kemphues K.J., Ohno S. An atypical PKC directly associates and colocalizes at the epithelial tight junction with ASIP, a mammalian homologue of Caenorhabditis elegans polarity protein PAR-3. J. Cell Biol. 1998;143:95–106. doi: 10.1083/jcb.143.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby C., Kusch M., Kemphues K. Mutations in the par genes of Caenorhabditis elegans affect cytoplasmic reorganization during the first cell cycle. Dev. Biol. 1990;142:203–215. doi: 10.1016/0012-1606(90)90164-e. [DOI] [PubMed] [Google Scholar]
- Kotani K., Ogawa W., Matsumoto M., Kitamura T., Sakaue H., Hino Y., Miyake K., Sano W., Akimoto K., Ohno S., Kasuga M. Requirement of atypical protein kinase C lambda for insulin stimulation of glucose uptake but not for Akt activation in 3T3-L1 adipocytes. Mol. Cell. Biol. 1998;18:6971–6982. doi: 10.1128/mcb.18.12.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill H., Ozawa M., Kemler R., Nelson W.J. Novel function of the cell adhesion molecule uvomorulin as an inducer of cell surface polarity. Cell. 1990;62:309–316. doi: 10.1016/0092-8674(90)90368-o. [DOI] [PubMed] [Google Scholar]
- Miyake S., Makimura M., Kanegae Y., Harada S., Sato Y., Takamori K., Tokuda C., Saito I. Efficient generation of recombinant adenoviruses using adenovirus DNA-terminal protein complex and a cosmid bearing the full-length virus genome. Proc. Natl. Acad. Sci. USA. 1996;93:1320–1324. doi: 10.1073/pnas.93.3.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno K., Kubo K., Saido T.C., Akita Y., Osada S., Kuroki T., Ohno S., Suzuki K. Structure and properties of a ubiquitously expressed protein kinase C, nPKC delta. Eur. J. Biochem. 1991;202:931–940. doi: 10.1111/j.1432-1033.1991.tb16453.x. [DOI] [PubMed] [Google Scholar]
- Mizuno K., Noda K., Ueda Y., Hanaki H., Saido T.C., Ikuta T., Kuroki T., Tamaoki T., Hirai S., Osada S. UCN-01, an anti-tumor drug, is a selective inhibitor of the conventional PKC subfamily. FEBS Lett. 1995;359:259–261. doi: 10.1016/0014-5793(95)00042-8. [DOI] [PubMed] [Google Scholar]
- Morita K., Sasaki H., Fujimoto K., Furuse M., Tsukita S. Claudin-11/OSP-based tight junctions of myelin sheaths in brain and Sertoli cells in testis. J. Cell Biol. 1999;145:579–588. doi: 10.1083/jcb.145.3.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogita K., Ono Y., Kikkawa U., Nishizuka Y. Expression, separation, and assay of protein kinase C subspecies. Methods Enzymol. 1991;200:228–234. doi: 10.1016/0076-6879(91)00142-j. [DOI] [PubMed] [Google Scholar]
- Ojakian G.K., Schwimmer R. The polarized distribution of an apical cell surface glycoprotein is maintained by interactions with the cytoskeleton of Madin-Darby canine kidney cells. J. Cell Biol. 1988;107:2377–2387. doi: 10.1083/jcb.107.6.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano R.E., Martin O.C. Use of fluorescent analogs of ceramide to study the Golgi apparatus of animal cells. In: Celis J.E., editor. Cell BiologyA Laboratory Handbook. Academic Press, Inc.; San Diego, CA: 1994. pp. 387–393. [Google Scholar]
- Sanchez P., De Carcer G., Sandoval I.V., Moscat J., Diaz-Meco M.T. Localization of atypical protein kinase C isoforms into lysosome-targeted endosomes through interaction with p62. Mol. Cell. Biol. 1998;18:3069–3080. doi: 10.1128/mcb.18.5.3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart R.O., Nigam S.K. Regulated assembly of tight junctions by protein kinase C. Proc. Natl. Acad. Sci. USA. 1995;92:6072–6076. doi: 10.1073/pnas.92.13.6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart R.O., Sun A., Panichas M., Hebert S.C., Brenner B.M., Nigam S.K. Critical role for intracellular calcium in tight junction biogenesis. J. Cell Physiol. 1994;159:423–433. doi: 10.1002/jcp.1041590306. [DOI] [PubMed] [Google Scholar]
- Suzuki A., Yoshida M., Ozawa E. Mammalian alpha 1- and beta 1-syntrophin bind to the alternative splice-prone region of the dystrophin COOH terminus. J. Cell Biol. 1995;128:373–381. doi: 10.1083/jcb.128.3.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabuse Y., Izumi Y., Piano F., Kemphues K.J., Miwa J., Ohno S. Atypical protein kinase C cooperates with PAR-3 to establish embryonic polarity in Caenorhabditis elegans . Development (Camb.) 1998;125:3607–3614. doi: 10.1242/dev.125.18.3607. [DOI] [PubMed] [Google Scholar]
- Toker A., Cantley L.C. Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature. 1997;387:673–676. doi: 10.1038/42648. [DOI] [PubMed] [Google Scholar]
- Uberall F., Hellbert K., Kampfer S., Maly K., Villunger A., Spitaler M., Mwanjewe J., Baier-Bitterlich G., Baier G., Grunicke H.H. Evidence that atypical protein kinase C-lambda and atypical protein kinase C-zeta participate in Ras-mediated reorganization of the F-actin cytoskeleton. J. Cell Biol. 1999;144:413–425. doi: 10.1083/jcb.144.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G., Simons K. The function of tight junctions in maintaining differences in lipid composition between the apical and the basolateral cell surface domains of MDCK cells. EMBO (Eur. Mol. Biol. Organ.) J. 1986;5:1455–1464. doi: 10.1002/j.1460-2075.1986.tb04382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts J.L., Etemad-Moghadam B., Guo S., Boyd L., Draper B.W., Mello C.C., Priess J.R., Kemphues K.J. par-6, a gene involved in the establishment of asymmetry in early C. elegans embryos, mediates the asymmetric localization of PAR-3. Development (Camb.) 1996;122:3133–3140. doi: 10.1242/dev.122.10.3133. [DOI] [PubMed] [Google Scholar]
- Wodarz A., Ramrath A., Grimm A., Knust E. Drosophila atypical protein kinase C associates with Bazooka and controls polarity of epithelia and neuroblasts. J. Cell Biol. 2000;150:1361–1374. doi: 10.1083/jcb.150.6.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}