

Abstract
Integrins connect the extracellular matrix with the cell interior, and transduce signals through interactions of their cytoplasmic tails with cytoskeletal and signaling proteins. Using the yeast two-hybrid system, we isolated a novel splice variant (filamin-Bvar-1) of the filamentous actin cross-linking protein, filamin-B, that interacts with the cytoplasmic domain of the integrin β1A and β1D subunits. RT-PCR analysis showed weak, but wide, expression of filamin-Bvar-1 and a similar splice variant of filamin-A (filamin-Avar-1) in human tissues. Furthermore, alternative splice variants of filamin-B and filamin-C, from which the flexible hinge-1 region is deleted (ΔH1), were induced during in vitro differentiation of C2C12 mouse myoblasts. We show that both filamin-Avar-1 and filamin-Bvar-1 bind more strongly than their wild-type isoforms to different integrin β subunits. The mere presence of the high-affinity binding site for β1A is not sufficient for targeting the filamin-Bvar-1 construct to focal contacts. Interestingly, the simultaneous deletion of the H1 region is required for the localization of filamin-B at the tips of actin stress fibers. When expressed in C2C12 cells, filamin-Bvar-1(ΔH1) accelerates their differentiation into myotubes. Furthermore, filamin-B variants lacking the H1 region induce the formation of thinner myotubes than those in cells containing variants with this region. These findings suggest that specific combinations of filamin mRNA splicing events modulate the organization of the actin cytoskeleton and the binding affinity for integrins.
Keywords: filamin isoforms; alternative splicing; β1D integrin; myogenesis; cytoskeleton
Introduction
Filamins are ∼560-kD dimeric proteins that, based on their conserved actin-binding domain (ABD)* and actin cross-linking activities, belong, similar to α-actinin, spectrin, and dystrophin, to the family of actin-binding proteins. Filamins organize filamentous actin (F-actin) into either orthogonal networks or parallel bundles, depending on the relative actin/filamin ratio and the source of purified filamin. In humans three filamin isogenes have been identified: FLNA, FLNB, and FLNC (Stossel et al., 2001; van der Flier and Sonnenberg, 2001a). Filamin-A is also named ABP-280 (Gorlin et al., 1990) or filamin-1, and filamin-B is also named ABP-278/276 (Xu et al., 1998), β-filamin (Takafuta et al., 1998), or filamin-3. Filamin-C has been cloned as γ-filamin, ABPL (Xie et al., 1998), or filamin-2 (Thompson et al., 2000).
The first ∼275 NH2-terminal amino acids of filamin contain an ABD, composed of two calponin homology domains. This domain is followed by 24 repetitive, 100-residue segments, interrupted by two ∼30–amino acid flexible loops (hinge-1 [H1] and -2), which show little homology among the products of the filamin isogenes. The H1 loop, between repeats 15 and 16, is lacking in some splice variants of filamin-B and filamin-C (Xie et al., 1998; Xu et al., 1998). It has been suggested that in the dimeric filamin molecule, the flexible H1 regions are essential for the separation of its ABDs and thus for its ability to promote orthogonal cross-linking of actin filaments (Gorlin et al., 1990). The H2 loop between repeats 23 and 24 is present in all filamin isoforms, and the COOH-terminal repeat 24 is essential in the tail–tail dimerization of the filamin molecule (Gorlin et al., 1990). In addition to its ability to cross-link actin filaments, filamin-A serves as a docking site for various transmembrane cell surface molecules such as GP-Ibα, Fc-γRI, tissue factor, and the β2 and β1A integrin subunits (Ohta et al., 1991, 1999; Sharma et al., 1995; Meyer et al., 1997; Loo et al., 1998; Pfaff et al.,1998; Calderwood et al., 1999). Filamin-B binds to GP-Ibα (Takafuta et al., 1998; Xu et al., 1998), whereas the muscle-associated filamin-C binds to sarcoglycans (Thompson et al., 2000) and a number of muscle-specific proteins (Faulkner et al., 2000; van der Ven et al., 2000; Takada et al., 2001). Furthermore, intracellular signal transduction proteins, such as the small GTPases RalA, RhoA, Rac-1, Cdc42 (Ohta et al., 1999), the guanidine-exchange factor Trio (Bellanger et al., 2000), the tumor necrosis factor receptor–associated factor-2 (Leonardi et al., 2000), and the membrane-associated proteases, furin and presenilin, bind to filamin (Liu et al., 1997; Zhang et al., 1998). These interactions point to a scaffolding function of filamin, rather than a distinct role as an actin filament–organizing molecule. Analyses of melanocytic cell lines deficient in filamin-A (Cunningham et al., 1992), as well as of patients with the neuronal migration disorder periventricular heterotopia (Fox et al., 1998; Sheen et al., 2001), indicate a role for filamin-A in cell motility during stabilization of the cell membrane. However, relatively little is known about the specific roles of filamin variants in the properties of cells and the regulation of cellular processes.
Integrins are heterodimeric adhesion receptors that provide a structural link between proteins of the extracellular matrix (e.g., fibronectin, laminin, and collagen) and the intracellular cytoskeleton (Hynes, 1992; van der Flier and Sonnenberg, 2001b). In addition, integrins play a role in several different signaling events (Giancotti and Ruoslahti, 1999). Although the small (20–50 amino acids) cytoplasmic tails of the integrin α and β subunits have no intrinsic catalytic domains or obvious protein–protein interaction domains, studies combining biochemical and genetic approaches, have led to the identification of a range of cytoskeletal, adaptor, and signaling molecules, which interact with the integrin cytoplasmic tails and are therefore implicated in integrin function (Liu et al., 2000; van der Flier and Sonnenberg, 2001b). Cytoskeletal proteins, such as the actin cross-linking proteins talin, α-actinin, and filamin-A (Critchley, 2000), interact with integrin β subunits. These molecules have been implicated in the linking of actin stress fibers to the cell membrane at specialized structures, known as focal adhesions or focal contacts, which are formed at cell–substrate contact sites. Binding to β subunits of this class of proteins was originally shown in biochemical experiments and was recently confirmed in yeast two-hybrid assays (Loo et al., 1998). Another category of proteins that interact with integrins are signaling molecules, including the ARF-GEF protein, cytohesin-1, the WD-repeat proteins Rack1 and WAIT-1, ICAP-1, and integrin-linked kinase (Hannigan et al., 1996; Kolanus et al., 1996; Chang et al., 1997; Liliental and Chang, 1998; Rietzler et al., 1998).
This study originated with the desire to identify cytoplasmic proteins that bind to the integrin β1D subunit, specifically in cardiac and skeletal muscle. Using the yeast two-hybrid technique, we isolated a new splice variant of filamin-B. Characterization of the interactions in yeast two-hybrid analyses and biochemical assays indicated that this filamin variant has a high binding activity and a broad specificity for integrin β subunits. Subsequent analysis of filamin mRNA splicing during in vitro myogenesis revealed that the H1 region of filamin-B and filamin-C is removed during the differentiation of myoblasts. Transfection studies with different filamin-B variants tagged with green fluorescent protein (GFP) revealed that the interaction with integrins and the cellular localization of the various filamin variants is different, and they affect myotube morphology and the pace of in vitro myogenesis of C2C12 cells in a variant-specific manner.
Results
A novel filamin-B splice variant interacts with the β1D integrin subunit in yeast
To identify proteins that interact with the cytoplasmic domain of β1D, specific for cardiac and skeletal muscle, we screened a human skeletal muscle cDNA library for interacting clones, using the β1D cytoplasmic domain as bait in a yeast two-hybrid screen. Approximately 6 × 106 clones were screened. From 67 positive clones, two identical clones encoding the 5 COOH-terminal repeats of filamin-B (amino acids 2027–2602) were isolated. Strikingly, in both filamin-B clones, 41 amino acids were deleted from the region between filamin repeats 19 and 20 (residues 2082–2122). This filamin variant was named filamin-Bvar-1. In a yeast two-hybrid screen for clones interacting with β1A, using a human keratinocyte cDNA library (29 × 106 clones), we isolated, from 16 positive clones, 4 identical filamin-Bvar-1 clones. The other isolated clones encoded either false positives or will be described elsewhere. Additionally, a filamin-Bvar-1 clone encoding EST was published under GenBank/EMBL/DDBJ accession no. W40525 (pancreatic islets). These results show that filamin-Bvar-1 binds to the cytoplasmic domains of both β1A and β1D, and the expression of filamin-Bvar-1 is not restricted to skeletal muscle.
Expression and genomic determination of novel filamin-B splice variants
To explore the expression pattern of the transcript for filamin-Bvar-1, cDNAs of multiple human tissues were analyzed in a PCR reaction, using primers that flank repeats 19 and 20 of filamin-B. Fig. 1 A shows that the cDNA encoding the previously reported filamin-B wild-type sequence was amplified from all tissues tested (683-bp product). In addition, a smaller PCR product of 560 bp, corresponding to the filamin-Bvar-1 specific part, was detectable in heart, lung, and skeletal muscle. Nested PCR analysis revealed a weak expression of filamin-Bvar-1 in all tissues tested (unpublished data). A similar splice variant lacking the corresponding region (amino acids 2127–2167) in human filamin-A, filamin-Avar-1, was also detected by (nested) PCR analysis (unpublished data). Two additional filamin-B–specific PCR products of 830 (filamin-Bvar-2) and 753 bp (filamin-Bvar-3) were detected in cardiac tissue (Fig. 1 A). Cloning and sequencing of these PCR fragments revealed that they represent two partially overlapping cardiac filamin-B cDNAs (Fig. 1 B). Intriguingly, in filamin-Bvar-2, the insertion of a 147-bp sequence results in a truncated protein with a unique COOH-terminal sequence of 24 amino acids. The insertion in filamin-Bvar-3 of only the first 70 of the 147 bp inserted in filamin-Bvar-2 leads to a frameshift, and as a result, four more amino acids are encoded COOH-terminally in this protein. Hence, both cardiac-specific filamin-B transcripts encode truncated filamin-B proteins that lack the four COOH-terminal repeats, including the 24th dimerization domain.
Figure 1.

Identification of three human filamin-B variants and analysis of the genomic organization of the FLN-B gene. (A) Tissue expression of filamin-B variants. PCR was performed on a human multiple tissue cDNA panel, using primers BV1 and BV2 designed to amplify repeats 19 and 20 of filamin-B. The 683-bp band, representing wild-type filamin-B, is present in all samples tested. The 560-bp product marked by one asterisk represents filamin-Bvar-1. The cardiac splice variants (filamin-Bvar-2, 830 bp, and filamin-Bvar-3, 753 bp) are indicated by two asterisks. (B) The partial cDNA sequences and deduced amino acid sequence of the human filamin-B variants. The translation of the two cardiac variants (Bvar-2 and Bvar-3), as well as wild-type and filamin-Bvar-1 is indicated in single letter code. Dots indicate the presence of nucleotides, whereas slashes indicate deletion of the corresponding nucleotides. The putative cAMP-kinase sequence is underlined. The nucleotide sequence data are available from GenBank/EMBL/DDBJ under accession no. AF353666. (C) Schematic diagram representing the genomic organization of the region of FLN-B encoding the repeats 19 and 20 and the composition of the cDNA splice variants. Arrows indicate positions of primers used for cloning. (D) Boundaries at the exon–intron junctions of the FLN-B gene segment. Sizes of exons and introns are indicated as are the consensus splice donor and acceptor sequences; GT/AG of each exon–intron border are underlined. Exons are numbered according to Chakarova et al. (2000). (Footnotes a and b) Nucleotides are numbered according to GenBank/EMBL/DDBJ accession nos. NM_001457 and AF353667 (Fig. 1 D), respectively.
Genomic PCR mapping with primers designed from exons 39 and 41, encoding the filamin repeats 19 and 20 (Fig. 1, C and D), revealed that the stretch of 41 amino acids, deleted in filamin-Bvar-1, is encoded by a single exon of 123 bp. This exon is preceded by a 102-bp intron and followed by an intron of ∼2.5 kb. Furthermore, we found that the cDNA sequence of filamin-Bvar-2, specific for heart, is encoded by a 147-bp exon, exon 40B/C. Alternative RNA splicing at the internal splice site, located in exon 40B/C, produces the filamin-Bvar-3 transcript.
Filamin isoforms and their variants determine specificity for association with β subunits
We next tested the interaction of the COOH-terminal domain, i.e., repeats 19–24 of filamin-B and filamin-Bvar-1 with different β subunits, using the yeast two-hybrid system. In addition, we tested the homologous regions of filamin-A and filamin-Avar-1. The results (Fig. 2 A) show that the COOH-terminal domain of wild-type filamin-B(19–24), containing repeats 19–24, interacts only with β1A. In contrast, an equivalent construct encoding filamin-Bvar-1(19–24), which lacks amino acids 2082–2122 that span repeats 19 and 20, and an NH2-terminal deletion mutant of filamin-B, truncated at amino acid 2123 (filamin-B, 20*–24), bind not only to β1A but also to the β1D, β3, and β6 subunits. Similar results were obtained with proteins from the original isolated filamin-Bvar-1 clones that contain amino acids 2027–2602 (unpublished data). Quantitative β-galactosidase activity assays indicated that filamin-Bvar-1(19–24) bound two to three times more strongly to β1A than did wild-type filamin-B(19–24) (Fig. 2 B). The binding of filamin-Bvar-1(19–24) to β3 was weaker and comparable to that of wild-type filamin-B(19–24) to β1A, and that of filamin-Bvar-1(19–24) to β1D was of intermediate strength.
Figure 2.

Characterization of the interaction of filamin isoforms and variants with the cytoplasmic domains of different integrin β subunits in yeast. (A) Cotransformation of yeast host strain PJ69–4A with the listed combinations of β integrin cytoplasmic domains (pAS2.1) and filamin-A and filamin-B deletion constructs (pACT). The NH2- and COOH-terminal positions of the different filamin constructs are indicated. Numbers in parentheses refer to the repeats present in the different constructs. The partially truncated repeat 20 is indicated by an asterisk. Interactions were scored (+) when the plating efficiencies on selective SC-LTHA plates were greater than 80% of those on nonselective SC-LT plates at 5 d of growth, (±) when they were greater than 80% at 10 d of growth, and (−) when no colonies were detected after 10 d of growth. (B) β-Galactosidase activity of the indicated cotransformants was measured by a liquid culture assay with O-nitrophenyl B-d-galactopyranoside as substrate. Controls were used: negative interaction control, empty vectors pAS/pACT; positive interaction control, p53/pSV-40 large T and the complete Gal4 domain pCL1/pAS (171 ± 6 β-galactosidase units; unpublished data). Data are shown as mean ± SD (n = 3).
In contrast to filamin-B(19–24), the corresponding filamin-A(19–24) construct did not bind to β1A or any other β subunit. However, filamin-Avar-1(19–24) and the shorter filamin-A(20*–24) construct (amino acids 2168–2647) interacted strongly with β1A and weakly with β3 and β6, but neither of them interacted with β1D. None of the aforementioned filamin constructs interacted with the β2 cytoplasmic domain (unpublished data). Taken together, these data show that the affinity of filamin-B for β1A is strongly increased when the stretch of 41 amino acids is deleted from it. Similarly, the removal of the same region from filamin-A induces binding to β1A. Furthermore, both variants become capable of binding to other integrin β subunits.
Analysis of a series of filamin-B constructs truncated at the NH2 or COOH terminus indicates that repeat 21 is necessary, but not sufficient, for interaction with β1A (Fig. 2 A). It is possible that the presence of repeat 24 facilitates the dimerization of filamin, and thus of repeat 21, thereby greatly increasing the strength of the binding to β1A. The interaction of filamin-Bvar-1 with β3 and β6 was abolished by the deletion of repeats 23–24, whereas it did not affect binding to β1A or β1D. Interactions of both β1A and β1D with filamin-Bvar-1(19 and 20) could still be demonstrated, although the binding appeared to be weaker than that of a construct containing repeat 21, filamin-Bvar-1(19–21). These data suggest that deletion of the 41–amino acid region in filamin-B leads to either the removal of inhibitory sequences in repeat 19 or to the introduction of a new binding site for β1A and β1D in the remaining part of repeat 20 (amino acids 1995–2185). The presence of repeat 21 increases binding activity, which is consistent with the data showing that this repeat is necessary for efficient binding of filamin-B to β1A and contributes to the binding activity of filamin-Bvar-1 (19–21) to β1 integrins. As may be the case for the binding of filamin-B to β1A, binding of filamin-Bvar-1 to β3 and β6 probably requires dimerization mediated by repeat 24.
Biochemical interaction of filamin variants with integrins
To confirm the interactions between the different filamin splice variants and the β1A and β1D subunits observed in yeast, we expressed the regions containing repeats 19–24 of filamin-Avar-1 or filamin-Bvar-1, or the corresponding regions of wild-type filamin-A(19–24) or filamin-B(19–24), in COS-7 cells. The proteins were tagged at their NH2 terminus with hemagglutinin A (HA) and expression of equivalent amounts of proteins in COS-7 cells was confirmed by immunoblotting with anti-HA antibody (Fig. 3 A). Binding of the filamin constructs to β1A and β1D was tested in a pull-down assay using glutathione-S-transferase (GST) fusion proteins containing the cytoplasmic domains of these integrin subunits, immobilized on glutathione-Sepharose beads (Fig. 3 B). As shown in Fig. 3 C, GST–β1A bound to filamin-Avar-1(19–24) and filamin-Bvar-1(19–24), but not to the corresponding fragments of the wild-type filamin isoforms (filamin-A, 19–24, and filamin-B, 19–24). Binding of GST–β1D to the different filamin-A and filamin-B constructs was either undetectable or very weak. The interaction between β1A and filamin-B(19–24) detected in yeast could not be confirmed in the pull-down assay, probably because it is too weak. Interestingly, we found that a truncated filamin-B construct that lacks the first 14 amino acids of repeat 19 (filamin-B, 2009–2602) could be efficiently precipitated with GST–β1A (unpublished data). Thus, it appears that not only the deletion of COOH-terminal residues of repeat 19, as in the variant-1 protein, but also the deletion of NH2-terminal residues of repeat 19 results in stronger binding of filamin-B to β1A. Together, these data suggest that repeat 19 contains an inhibitory element for binding of filamin-B to β subunits.
Figure 3.
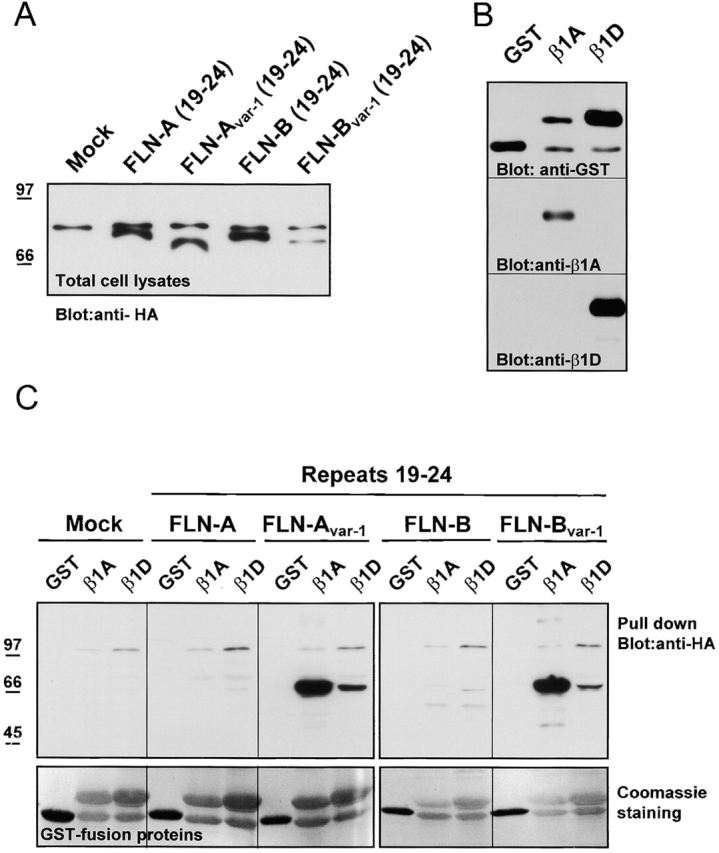
Binding of filamin isoforms and variants to GST–β1A and –β1D fusion proteins. (A) Detection of the expressed products from HA-tagged filamin-B cDNA constructs (filamin repeats 19–24) in lysates of COS-7 cells, transiently transfected with the indicated constructs. The upper band represents nonspecific reactivity of the antibody with an endogenous protein. (B) Expression of recombinant GST–β1A and –β1D fusion proteins. The GST fusion proteins containing the cytoplasmic domains of β1A and β1D were purified on glutathione-Sepharose 4B beads and analyzed by immunoblotting with antibodies against GST, β1A, and β1D subunits. (C) Pull-down assay with GST or GST–β1A and –β1D fusion proteins, immobilized on glutathione beads (A) containing HA-tagged fusion proteins as indicated. (Top) Immunoblot detection of bound filamin-Avar-1 and filamin-Bvar-1 to GST–β1A and very weak binding to GST–β1D. No binding was detected to filamin-A and filamin-B. (Bottom) Coomassie blue–stained polyvinylidene difluoride membrane showing the amount of GST fusion proteins used for each pull-down assay.
Identification of the binding sites for filamins on β1A and β1D
Next, we determined the binding sites for filamin-B and the splice variants of filamin-A and filamin-B on the cytoplasmic domains of the β1A and β1D subunits (Fig. 4) . Swapping the COOH-terminal residues of β1A and β1D in β1A/D796 (EGK to GRKAGL) and β1A/D792 (NPKYEGK to NPNYGRAGL) had no effect on their binding to filamin-B(19–24), filamin-Bvar-1(19–24), and filamin-A(20–24). A swap involving the membrane-proximal region of β1D, where the G778 and A786 residues in β1A are replaced by the corresponding Q778 and P786 residues from β1D, leads to complete abolition of the interaction with filamin-B(19–24) and filamin-A(20*–24). Binding of filamin-Bvar-1 (19–24) could still be detected, although it was weaker. These results indicate that the specificity of the binding of filamin-B and filamin-Avar-1 with β1A depends on the two residues, G778 and A786, that have been substituted in β1D by Q778 and P786. Although a role for Q778 cannot be excluded, we propose that P786, by its ability to create a bend or a rigid kink in polypeptide chains, changes the conformation of the β1D cytoplasmic domain in such a way that it can no longer bind to filamin-B or filamin-Avar-1.
Figure 4.

Characterization of the interaction between mutants of the β1A and β1D cytoplasmic domain with filamin-A and filamin-B splice variants by yeast two-hybrid analysis. Cotransformation of yeast host strain PJ69–4A with the listed combinations of β integrin cytoplasmic domains (in pAS) and filamin (in pACT). Interaction was scored as in Fig. 2. The numbers represent the amino acid positions in β1A and β1D. The conserved NPXY motifs are underlined and the amino acids in β1D that were swapped in the different chimeras are indicated by shading. The common membrane proximal region, which is shared between β1A and β1D, is boxed.
Analysis of deletions of the cytoplasmic domain of β1A shows that for binding to filamin-B, almost the complete cytoplasmic domain of β1A is required. Only the last 10 COOH-terminal amino acids of β1A can be deleted without causing a loss of interaction. The binding site for filamin-Bvar-1 on β1A comprises both the NPXY motifs and the intervening sequence, whereas on β1D additional membrane proximal sequences are needed. Similarly, the minimal binding site for filamin-A(20*–24) on β1A comprises the two NPXY motifs and the intervening sequence, which resembles the binding site for filamin-Bvar-1. Thus, it appears that the binding site on β1A for the low-affinity interaction with filamin-B is distinct from the one mediating high-affinity binding to filamin-Avar-1 and filamin-Bvar-1. The sequence requirements for the binding of β1D to filamin-Bvar-1 resemble those for the low affinity interaction of β1A to filamin-B (Fig. 2).
Alternative splicing of filamins during in vitro myogenesis
During differentiation of mouse C2C12 myoblasts into myotubes, the expression of β1D is induced, whereas that of β1A is downregulated (Belkin et al., 1997; van der Flier et al., 1997). We investigated whether this switch is paralleled by changes in the expression of filamin isoforms and/or their variants. Total RNA was isolated at different time points of myogenic differentiation, and the expression of filamin isoforms was analyzed by RT-PCR using appropriate primers. We studied the splicing of the region encoding the 41 amino acids in filamin-A and filamin-B, as well that of the H1 region of filamin-A, filamin-B, and filamin-C (Fig. 5 A). The latter were included because variants of human filamin-B and filamin-C, lacking the H1 region, have been described previously (Xie et al., 1998; Xu et al., 1998). Fig. 5, B–D, shows that C2C12 myoblasts express all three murine filamin isoforms. In addition, whereas the H1 region is present in filamin-A throughout differentiation, this region is absent from the filamin-B and filamin-C isoforms. This deletion appears to precede the switch from β1A to β1D that occurs during myogenic differentiation. Interestingly, we detected in C2C12 cells and among several murine and human cDNAs (unpublished data) a third filamin-B transcript that encodes a variant with a shorter H1 (H1s) region (Fig. 5 D). This transcript arises as a result of the usage of intrinsic splice-donor and acceptor sites that are present in the murine and human filamin-B genes (position 5280 and 5312, respectively; GenBank/EMBL/DDBJ accession no. NM_001457). We did not detect the filamin-Avar-1 and filamin-Bvar-1 transcripts.
Figure 5.




Expression of filamin isoforms and variants during in vitro myogenesis. (A) Schematic representation of the organization of the filamin domains and the regions analyzed by RT-PCR. (B) RT-PCR analysis of alternative splicing of the H1 region of filamin-A, filamin-B, and filamin-C in mouse C2C12 myoblasts and murine tissues, as indicated. Splicing is analyzed at the indicated time points after induction of differentiation. The H1 region of filamin-A is not spliced, whereas in filamin-B and filamin-C the H1 region is removed during myogenesis. Filamin-B(H1s) is detected in C2C12 cells and adult tissues. The switching from the β1A to the β1D variant during C2C12 differentiation is shown. (C) Immunoblot of the protein expression levels of filamin-B β1D, and MHC during myogenesis. Filamin-B expression decreases, while the myogenic differentiation markers β1D and MHC are both induced. (D) Amino acid alignment of the human and murine H1 and variable-1 region. The partial murine filamin sequences are available from GenBank/EMBL/DDBJ under accession nos. AF353668–353670.
The expression of filamin-B proteins in C2C12 cells was assessed by immunoblotting with an antibody specific for the H1 region of filamin-B (Fig. 5 C). In agreement with the PCR data, the level of filamin-B protein containing the H1 region decreased during the differentiation of C2C12 cells. In contrast, the expression of β1D and sarcomeric myosin heavy chain (MHC) was induced in differentiating C2C12 cells.
GFP COOH-terminal tags do not interfere with filamin dimerization
Before initiating studies to define the cellular localization of the different splice variants of filamin-B, we examined whether the addition of GFP at the COOH terminus of filamin influences the ability of this protein to form dimers. To this end, a filamin-B(19–24) construct with GFP at the COOH-terminal end, and a control construct, tagged with HA at the NH2-terminal end, were transiently expressed in CHO cells. After 2 d, cell lysates and intact cells were treated with the chemical cross-linking reagent dithiobis-succinimidyl propionate (DSP) at two concentrations. As shown in Fig. 6 , the addition of increasing amounts of cross-linker led to a shift from monomeric to dimeric tagged filamins, as visualized by immunoblotting with antibodies against HA or GFP. The similar dimerization capacity of HA- and GFP-tagged filamin-B(19–24) indicates that the GFP tag had no effect on dimerization. The same samples under reducing conditions, which disrupt the disulfide bond, only contained filamin monomers. Specificity of the cross-linking reaction was checked using the NH2-terminal HA-tagged filamin-Bvar-1(19–23) construct, which did not form dimers due to truncation of the COOH-terminal repeat 24, which is required for dimerization.
Figure 6.

The GFP tag on the COOH-terminal part of filamin-B does not interfere with dimerization in vivo. CHO cells were transfected with either the NH2-terminal HA- or the COOH-terminal GFP-tagged FLN-B(19–24) or with the NH2-terminal HA-tagged FLN-Bvar-1(19–23) construct lacking the last repeat. 2 d after transfection, filamin dimers in either cell lysates (lysate) or intact cells (cells) were stabilized by chemical cross-linking at two different concentrations of DSP for 1 h, and dimerization of the epitope-tagged filamins was analyzed by immunoblotting under nonreducing conditions (NR) using anti-HA and anti-GFP antibodies. The specificity of the cross-linkage was checked by including NH2-terminal HA-tagged FLN-Bvar-1(19–23), in which the truncation of COOH-terminal repeat 24 abolished dimerization. In addition, cross-linked samples were separated under reducing (R), DSP disrupting conditions resulting in the presence of filamin-B monomers. Closed arrowheads indicate HA-tagged products, and open arrowheads indicate GFP-tagged fusion proteins. A single arrow indicates a filamin monomer, and an arrow doublet indicates dimers.
Subcellular distribution of full-length filamin-B variants and their interaction with β1 subunits
After excluding potential disadvantageous effects of the GFP tag on the dimerization of filamin, we generated full-length cDNAs encoding four different filamin-B splice variants and tagged them with GFP (Fig. 7 A). These include the previously reported filamin-B and filamin-B lacking H1 (filamin-B[ΔH1]), as well as the novel identified filamin-B lacking the 41 residues between repeats 19 and 20 (filamin-Bvar-1), and filamin-Bvar-1 without H1 (filamin-Bvar-1[ΔH1]). As shown in Fig. 7 B, the latter two filamin-B variants are expressed in a variety of tissues and cell types, including heart, lung, testis, spleen, thymus, and leukocytes. Several cell types were retrovirally transduced, and after fluorescence-activated cell sorting for filamin-GFP–expressing cells, the stable expression of the different filamin fusion proteins was verified by immunoblotting. In all cell lines, full-length GFP-tagged filamin-B variants migrating at ∼300 kD could be detected (shown for C2C12, Fig. 7 C). Filamin-B expression levels were consistently lower than those of the other variants, which were comparable to each other. Occasionally, smaller protein degradation products were detected, which varied in size and quantity, depending on the transduced cell line and the filamin variant (Fig. 7 C). GST pull-down assays confirmed that deletion of the variant-1 region from full-length filamin-B increases binding to β1 integrins (Fig. 7 D). Only the two filamin-B constructs, filamin-Bvar-1 and filamin-Bvar-1 (ΔH1), in which the variant-1 region had been deleted, but not filamin-B or filamin-B(ΔH1), were precipitated by GST–β1A (Fig. 7 D). Binding of filamin-Bvar-1 and filamin-Bvar-1(ΔH1) to β1D again proved to be weak and could only be demonstrated after long exposures of the film. None of the filamin-B constructs interacted with GST.
Figure 7.

Expression of filamin-B variants in human tissues, and characterization of the binding of full-length filamin-B variants to integrin cytoplasmic domains. (A) Schematic presentation of COOH-terminal GFP-tagged filamin-B variant constructs. The internal deletions of filamin-B are indicated by a single line. (B) Detection by PCR amplification of filamin-Bvar-1 and filamin-Bvar-1(ΔH1) transcripts. PCR was performed on cDNAs from different human tissues, using primers BV12 and BV13 (A) designed to specifically amplify filamin-Bvar-1(±ΔH1) cDNAs. cDNAs encoding filamin-B variants with and without the var-1 region were used as positive and negative controls, respectively. As a further negative control, cDNA for filamin-A was used. A PCR product for filamin-Bvar-1 was amplified from most tissues. Heart, spleen, and thymus contained transcripts for filamin-Bvar-1(ΔH1). A third PCR product (indicated by an asterisk) that migrated slightly faster than the filamin-Bvar-1 variant and is present in heart, kidney, liver, lung, and colon, corresponds to filamin-Bvar-1(H1s). The authenticity of the PCR products has been confirmed by sequencing. (C) Expression of GFP-tagged filamin-B variants in C2C12 myoblasts. Stably transduced C2C12 cells were obtained as described in the Materials and methods. Equal amounts of total cells lysed in boiling sample buffer were analyzed by immunoblotting. Filamin-B–GFP was detected by mouse anti-GFP antibody. The upper bands represent the full-length filamin–GFP fusion proteins whereas some fainter lower bands represent COOH-terminal proteolytic products. (D) Pull-down assay of full-length filamin-B variants with GST or GST–β1A and –β1D fusion proteins immobilized on glutathione beads containing GFP-tagged fusion proteins as indicated. Immunoblotting showed binding of filamin-Bvar-1 and filamin-Bvar-1(ΔH1) to GST–β1A, weak binding to GST–β1D, whereas filamin-B and filamin-B(ΔH1) did not bind.
The subcellular distribution of the different GFP-tagged filamin-B variants was examined in GD25-β1A mouse fibroblasts stably expressing these proteins. The localization of GFP-tagged filamin-B (Fig. 8, C and D) was identical to that of endogenous filamin-B, as revealed by immunostaining using an antibody against the H1 region of filamin-B (Fig. 8, A and B). Thus, the GFP tag did not interfere with the normal localization of filamin-B. Filamin-B was localized at actin stress fibers but was not appreciably concentrated in focal contacts. The distribution of filamin-B(ΔH1) and filamin-Bvar-1 variants was similar to that of filamin-B (Fig. 8, E and F). Interestingly, filamin-Bvar-1(ΔH1) was associated with a proportion of the peripheral focal contacts positive for vinculin (Fig. 8, G–O, and Fig. 9 E). There was also GFP fluorescence in the nucleus in many of these cells, the significance of which is unknown. As anticipated, the filamin-Bvar-1(ΔH1) variant did not react with anti–filamin-B H1 antibody. However, its presence in focal contacts was revealed by GFP fluorescence, whereas endogenous filamin-B, which does react with this antibody, was found associated with actin stress fibers (Fig. 8, M–O). Neither endogenous filamin-B (Fig. 8 B) nor any of the filamin-B variants (unpublished data) were enriched in lamellipodia. The expression of filamin-Bvar-1(ΔH1) in focal contacts did not have an apparent effect on the composition and localization of these structures. They were confined at the end of actin stress fibers (Fig. 8, J–L, and Fig. 9 F) and in addition to vinculin, they contained talin, phospho-tyrosine, and paxillin (Fig. 9, A, B, and D). We did not detect α-actinin in focal contacts (Fig. 9 C). Lastly, in GD25-β1D cells expressing filamin-Bvar-1(ΔH1) there was also a prominent staining of some focal contacts (unpublished data).
Figure 8.
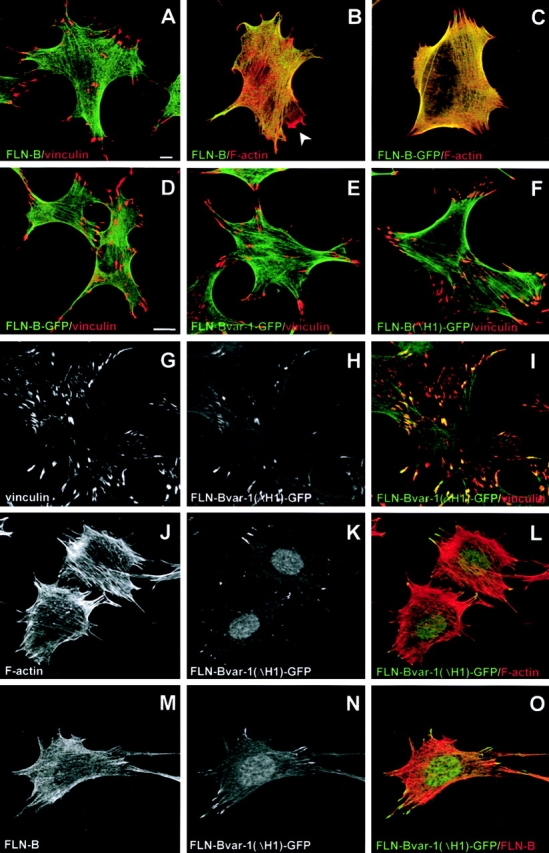
Expression and localization of filamin-B variants in GD25–β1A mouse fibroblasts. GD25–β1A cells (A and B) and GD25–β1A cells stably expressing GFP-tagged filamin-B (C and D), filamin-Bvar-1 (E), or filamin-Bvar-1(ΔH1) (F–O), were analyzed for endogenous filamin-B (A, B, and M) and vinculin (A, D–G, and I) expression using indirect immunofluorescence microscopy, staining of F-actin (B, C, J, and L) with Alexa®568-phalloidin, and GFP fluorescence (C–F, H, I, K, and N). Filamin-B and the variants filamin-Bvar-1 and filamin-B(ΔH1) are associated with actin stress fibers: they did not accumulate in focal contacts. In contrast, filamin-Bvar-1(ΔH1) is concentrated in focal contacts (G–I) at the end of actin stress fibers (J–L, N, and O). Note that the fluorescence pattern of filamin-B–GFP is similar to that of endogenous filamin-B detected with a polyclonal antibody specific for the H1 region of this filamin isoform. This antibody does not react with filamin-B(ΔH1) and therefore in cells that express this variant, it does not react with focal contacts. Filamin-B is not present in membrane ruffles (B, arrow). Bars, 10 μm.
Figure 9.

Localization of filamin-Bvar-1(ΔH1) in focal contacts. Confocal microscopy of GD25-β1A cells showing the green fluorescence of filamin-Bvar-1(ΔH1) compared with red talin (A), phosphotyrosine (B), α-actinin (C), paxillin (D), vinculin (E), and F-actin (F). Insets in C and D are at higher magnifications. Filamin-Bvar-1(ΔH1) is found at the end of actin stress fibers and is colocalized (yellow) with talin, phosphotyrosine, paxillin, and vinculin in focal contacts. Bars: (A–D) 10 μm; (E and F) 5 μm.
We conclude that the localization of filamin-Bvar-1(ΔH1) in focal contacts requires, in addition to the characterized variant-1 high-affinity binding site for β1 integrins, a function that is induced by the loss of the H1 region.
Filamin-B variants affect myoblast differentiation in vitro
The functional significance of the developmentally regulated splicing of the H1 region in filamin-B, as well as the deletion of the 41–amino acid region, was explored by analyzing the effects of ectopic expression of the four different filamin-B splice variants on myogenesis of C2C12 cells. Myogenic differentiation was induced by switching the culture to a medium containing 2% horse serum (differentiation medium). Interestingly, C2C12 cells expressing filamin-Bvar-1(ΔH1) fused into myotubes within 2 to 3 d after the medium switch, which is 1–2 d earlier than the fusing of cells from the other transduced cell lines (Fig. 10 A). Furthermore, the myotubes formed by the cells expressing the filamin-B variants lacking the H1 region (filamin-B[ΔH1] and filamin-Bvar-1[ΔH1]) were thinner than those formed by the other transduced cell lines or GFP control cells. This difference in morphology was more obvious when the myotubes were stained for MHC (Figs. 11 and 12) .
Figure 10.

Effects of the ectopic expression of filamin-B variants on myogenesis. C2C12 myoblasts stably expressing GFP and GFP fusions of the indicated filamin-B variants were grown to confluence and then switched to differentiation medium. (A) The phase contrast micrographs show the morphological appearance of the cells in culture, 4 d after the switch to differentiation medium. (B) Western blot analysis of the expression of sarcomeric MHC in differentiating C2C12 cells. Cell lysates were prepared, separated by SDS-PAGE (5%), and subjected to immunoblot- ting with antibodies against sarco- meric MHC.
Figure 11.

Localization of GFP or GFP fusions of filamin-B or filamin- Bvar-1(ΔH1) in undifferentiated and differentiated C2C12 cells. (A–C) Confocal microscopy of undifferentiated C2C12 myoblasts showing the green fluorescence of GFP and two GFP fusion proteins (filamin-B and filamin-Bvar-1[ΔH1]) compared with red F-actin. (D–I) Triple stainings of differentiated myotubes. (D–F) Localization of GFP and GFP fusions (green). (G–I) Same field showing the distribution of sarcomeric MHC (red) and nuclei (blue).
Figure 12.

Effects of filamin-B variants on myogenesis. Confocal microscopy of C2C12 myoblasts stably expressing GFP fusions of filamin-B variants, 6 d after induction of differentiation. Sarcomeric MHC was immunolabeled in cells to facilitate myotube identification (A–D). The pouch-like myotubes of filamin-B and filamin-Bvar-1 expressing cells and the thinner myotubes in cells expressing the ΔH1 variants are clearly detectable. (E) Staining of differentiated myotubes with anti-sarcomeric α-actinin. (F) Same field showing GFP fluorescence of filamin-B at Z- and M-lines (F). Arrowheads indicate Z-lines. Bars: (A–D) 100 μm; (E and F) 10 μm.
In the differentiating myotubes, filamin-B, filamin-Bvar-1, and filamin-B(ΔH1) were localized diffusely throughout the cytoplasm, with regions of enrichment at the longitudinal actin stress fibers (Fig. 11, B and E). In contrast, the localization of filamin-Bvar-1(ΔH1) was typically polarized and dotted at the periphery of tubes (Fig. 11, C and F). GFP alone was mainly localized in the nucleus (Fig. 11, A and D). Occasionally, depending on the culture conditions, in well-differentiated tubes with clear sarcomeric organization, filamin-B was localized at the Z-lines, as shown by costaining for sarcomeric α-actinin, and at intermediate M-bands (Fig. 12, E and F).
Immunoblot analysis of MHC demonstrated that the morphological differentiation is accompanied by biochemical changes. The induction of MHC in the filamin-Bvar-1(ΔH1)–expressing C2C12 cells was faster than in the other cells, where the induction was similar to that in GFP control cells (Fig. 10 B). Taken together, these results show that expression of the filamin-double variant, filamin-Bvar-1(ΔH1), accelerates muscle differentiation in vitro.
Discussion
In the present study, we have identified novel distinct splice variants produced by the filamin-A and filamin-B genes, and report on functional differences between filamin isoforms and their variants. We found that the deletion of 41 amino acids encoded by exon 40 from filamin-A and filamin-B, which generates the variants filamin-Avar-1 and filamin-Bvar-1, greatly influences their interaction with integrin β subunits. The role of two additional heart-specific filamin-B variants, filamin-Bvar-2 and filamin-Bvar-3, which lack the last four repeated sequences of filamin-B, including the putative dimerization domain, remains elusive. COOH-terminal truncated filamin variants of Drosophila filamin-240 have also been reported (Li et al., 1999b; Sokol and Cooley, 1999; Guo et al., 2000). Furthermore, the recent identification of truncated variants of several other high molecular weight proteins that cross-link actin: i.e., dystropin/utrophin, dystonin, and plectin (Fuchs and Yang, 1999; Blake and Kroger, 2000; Zuellig et al., 2000), raises the interesting possibility that these variants play an unprecedented role in the regulation of actin dynamics and the modification of F-actin organization.
Finally, analysis of the splicing of mRNAs for filamins during in vitro myogenesis of C2C12 cells revealed that the H1 region of filamin-B and filamin-C is removed during myotube formation. The biological consequences of these modifications are discussed below.
Specific interactions of filamin variants
The novel filamin-B variant, filamin-Bvar-1, is the fourth reported β1D-binding protein identified in a yeast two-hybrid scree; the other three are MIBP, melusin, and skelemin (Reddy et al., 1998; Brancaccio et al., 1999; Li et al., 1999a). The yeast two-hybrid interaction assays indicated that filamin-A does not bind to any integrin β subunit, whereas filamin-B only interacts with β1A. On the contrary, the splice variants filamin-Avar-1 and filamin-Bvar-1 bind strongly to β1A and weakly to β3 and β6. Additionally, a moderate binding of filamin-Bvar-1 to β1D was observed. These results provide evidence that isoforms and variants of filamins specifically bind to different integrin β subunits, which render the interpretation of biochemical studies with filamins more complicated. Furthermore, subcellular localization and morphological and myogenic effects were found to be determined by the type of the expressed filamin variant.
Previously, it has been shown by coimmunoprecipitation and pull-down assays that various integrin β subunits have different capacities to bind cytoskeletal proteins. The results of these assays showed that the affinity of β1D for talin is greater than that of β1A, which could result in β1D having a stronger link with the cytoskeleton (Belkin et al., 1997; Pfaff et al., 1998; Zent et al., 2000). In addition, the affinity of the β7 integrin subunit for filamin appeared to be high, whereas that of β1A and β1D was found to be intermediate and low, respectively (Pfaff et al., 1998; Zent et al., 2000). Our data suggest that β1 integrins hardly interact with filamin-A or filamin-B and that most of their binding activity toward these filamin isoforms is with the variant-1 forms. In agreement with previous findings, we observed that the binding of β1A to filamin-Avar-1 and filamin-Bvar-1 is stronger than that of β1D. Loo et al. (1998) has previously reported that filamin-A can interact with the cytoplasmic domain of β1A. However, their conclusion was based on the binding of β1A to a construct containing the COOH-terminal 478 amino acids of filamin-A. This fragment corresponds to filamin-A(20*–24), whose binding properties resemble those of filamin-Avar-1. However, a longer filamin-A protein, extending 176 amino acids at the NH2 terminus, does not bind to integrin β subunits. Thus, the apparent discrepancies between these reported data and ours might be due to the fact that those studies were concerned with the interaction of β1A with a splice variant of filamin-A rather than with wild-type filamin-A. Similarly, the filamin proteins previously identified in pull-down experiments using dimerized integrin cytoplasmic domains presumably do not represent wild-type filamin, but filamin splice variants (Pfaff et al., 1998; Calderwood et al., 1999).
The discrepancy between our pull-down interaction results (Fig. 3) and those of yeast two-hybrid assays (Fig. 2) on the interaction of filamin-B variants with β1D probably can be reduced to a difference in sensitivity between these two assays; the yeast two-hybrid assay being more sensitive than the pull-down assay. Indeed, we found in the β-galactosidase assays that binding of β1D to filamin-Bvar-1 is weaker than that of β1A. An alternative explanation might be that in the GST–β1D fusion protein, the β1D cytoplasmic sequences are not readily accessible for binding.
Based on the immunoglobulin-like folding of several filamin repeats that are present in the gelation factor of Dictyostelium (Fucini et al., 1997; McCoy et al., 1999), it is likely that as a result of the 41–amino acid deletion, three β-strands of the two adjacent repeats 19 and 20 (strand G from repeat 19 and strands A and B from repeat 20) were lacking in the filamin-A and filamin-B variants. Removal of this stretch of 41 amino acids probably led to the exposure of one or more cryptic binding sites for integrin β subunits in the remaining part of repeat 20 of filamin-A and filamin-B. Alternatively, the disruption of repeat 19 may have removed elements inhibiting the binding of filamin-B to β1A. Finally, together with the deleted 41 amino acids in human filamin-Bvar-1, a conserved cAMP-kinase consensus site (serine 2150) was removed that, when phosphorylated in filamin-A, confers an increased resistance to calpain cleavage at residues 1761–1762 in the H1 region (Gorlin et al., 1990; Jay et al., 2000). This suggests long-range conformational effects by the 19 and 20 repeat region of filamin and, although serine 2150 is not conserved in murine filamin-B, deletion of this region may have additional effects beyond the modulation of the strength and specificity of filamin-β–integrin interactions. Interestingly, filamin-C contains an insertion of 82 amino acids, exactly juxtaposed to the homologous variant-1 region, which could give specific features to this filamin variant associated with striated muscle (Thompson et al., 2000). Evidence of alternative splicing of mRNA has not yet been reported for this region (Xie et al., 1998).
Specialized cellular roles and specific cellular localization of filamin-B variants
Several previous reports have indicated complex expression patterns of filamin variants during myogenesis (Gomer and Lazarides, 1981, 1983a,b; Chiang et al., 2000). In this study, we extended those data by the identification of additional filamin-Bvar and H1 splice variants. We demonstrate that removal of the H1 region from filamin-B and filamin-C is induced during myogenesis in vitro. Furthermore, the presence or absence of the H1 region in filamin-B affects the morphology of the formed myotubes. Filamin-B variants lacking H1 form thinner myotubes in contrast to the more pouch-like tubes formed by H1-containing filamins.
Deletion of the H1 region from both filamin-B and filamin-C might determine the type of actin filaments with which these filamin isoforms interact. This would explain why during differentiation of myoblasts into myotubes, a process that is associated with a dramatic reorganization of the cytoskeleton, the splicing of the mRNA for this region is regulated and why forced expression of it has such a dramatic effect on the morphology of the formed myotubes. Parallel bundles of actin filaments might become more tightly packed by the loss of the H1 region of filamin, because the flexibility of the filamin dimer lacking the H1 region is reduced, as has been anticipated by Gorlin et al. (1990). Our observation that filamin-B variants lacking the H1 region show the tendency to be more polarized at the ends of actin stress fibers supports this hypothesis. The identification of shorter filamin-B(ΔH1s) alternative splice variants in several murine and human tissues provides additional possibilities for the modulation of the H1 region, and subsequent effects on the organization of the F-actin network.
Indeed, several studies have shown the effects of differences in the organization of the actin cytoskeleton on myoblast morphology and differentiation. For example, expression of dystrophin, or its homologue utrophin, in dystrophin-deficient mdx mice affects the type of fibers present in muscle (Rafael et al., 2000). Also, RhoA-mediated induction of stress fibers increases myoblast differentiation, in contrast to the inhibitory effects on myogenesis by Rac-1 and Ras (Wei et al., 1998; Gallo et al., 1999).
By transfecting filamin-B variants into cells, it was shown that the colocalization of filamin with β1-integrin–containing focal adhesions is not solely dependent on the presence of an integrin-binding site on filamin-Bvar-1. Only the filamin-Bvar-1(ΔH1) variant, in which a high affinity binding site for β1A and β1D is combined with the absence of the H1 region, is concentrated at the tips of actin stress fibers, where it is colocalized with β1 integrins. Two previously described chicken filamin variants could resemble filamin-Bvar-1(ΔH1) (Pavalko et al., 1989; Tachikawa et al., 1997), because of their localization at the ends of stress fibers in focal adhesions or at the dense plaques in smooth muscle cells. It is possible that the effect induced by the loss of the H1 region permits the linkage of filamin-Bvar-1(ΔH1) with special organized actin stress fibers in focal contacts, thereby facilitating binding of the variant-1 site to integrins. However, there are other possible explanations, e.g., similar to the deletion of the var-1 region, the deletion of the H1 region may have induced a binding site for one or more focal contact proteins that, together with β1, mediate the localization of filamin-Bvar-1(ΔH1) in focal contacts.
Our finding that filamin-Bvar-1(ΔH1) is present together with talin in focal contacts raises the question of which of these two proteins is actually associated with β1. Filamin-Bvar-1(ΔH1) and talin bind to overlapping sites on the β1 cytoplasmic domain and, therefore, it seems unlikely that these two proteins simultaneously bind to β1. We believe it is reasonable to assume that a proportion of β1 is associated with talin, whereas other β1 subunits interact with filamin-Bvar-1 (ΔH1). An alternative possibility, which we consider to be less likely, but nevertheless cannot formally be excluded, is that only filamin-Bvar-1(ΔH1) is associated with β1 and talin is retained in focal contacts by an interaction with vinculin.
Previous reports have indicated a critical role for integrins in myogenic differentiation (Menko and Boettiger, 1987; Sastry et al., 1996) and muscle integrity (Volk et al., 1990; Mayer et al., 1997; Hayashi et al., 1998). Conceivably, the acceleration of myogenesis observed in C2C12 cells expressing filamin-Bvar-1(ΔH1) might be due to an altered interaction of this filamin variant with the β1 integrins. An increased interaction may help to stabilize the expression of integrins at the cell surface by firmly anchoring them to the actin cytoskeleton. For example, the reexpression of filamin-A, in filamin-A–deficient melanoma cells, has been associated with an increase in the surface levels of both the GP-Ibα and β1 integrins (Meyer et al., 1998). However, we did not detect, by FACS® analysis, any changes in surface expression levels of β1 integrins upon the ectopic expression of filamin-B variants (unpublished data). This discrepancy could be due to the complete lack of filamin-A in the M2 cells used by Meyer et al. (1998). If cells contain endogenous filamin, the ectopic expression of filamin has no detectable effects. The effects of filamin-Bvar-1(ΔH1) on myogenesis may also be the consequence of a different localization and/or function of integrins in C2C12 cells. Indeed, as already mentioned, the distribution of filamin-Bvar-1(ΔH1) is typically polarized in differentiating C2C12 cells and is localized together with β1 in focal contacts in GD25-β1A cells. However, whatever the underlying mechanism for the effect of filamin-Bvar-1(ΔH1) on myogenesis is, the physiological significance of it remains uncertain, because it could not be demonstrated that this variant is expressed in C2C12 cells.
In conclusion, our study provides the first evidence that alternative mRNA splicing controls the cellular localization of filamins and their interaction with integrins. This indicates that individual filamin variants could be specialized in linking integrins or other transmembrane proteins to the actin cytoskeleton, comparable to the filamin-C–specific (Thompson et al., 2000) and dystrophin-mediated connection with the sarcoglycan adhesion complex in muscle cells. Ultimately, these specific activities of filamin variants modulate cell morphology and differentiation.
Materials and methods
Cell lines and antibodies
CHO cells were grown in DME supplemented with 10% FCS and 1% nonessential amino acids. Murine GD25 fibroblasts, lacking the endogenous β1 integrin subunit, were a gift of Dr. R. Fässler (Lund University Hospital, Lund, Sweden; Fässler et al., 1995). GD25-β1A or GD25-β1D cell lines were generated by retroviral transduction of GD25 cells with cDNA constructs for human β1A or β1D, and maintained in DME supplemented with 10% FCS, as described previously (Gimond et al., 1999). The murine myoblast cell line C2C12 (American Type Culture Collection; accession no. CRL 1772) was propagated in DME supplemented with 20% FCS and high glucose (4.5 g/liter). Terminal differentiation of confluent C2C12 monolayers to multinuclear myotubes was induced by changing the culture medium to DME containing 2% horse serum (Yaffe and Saxel, 1977). All media were supplemented with penicillin and streptomycin. Retrovirally transduced cells were selected with 0.2 mg/ml zeocin (Invitrogen). All cell lines were grown at 37°C in a humidified, 5% CO2 atmosphere.
The mouse anti-vinculin (V11F9) (Glukhova et al., 1990) was provided by Dr. M. Glukhova (Institut Curie, Paris, France). The mouse anti-β1D cytoplasmic domain (2B1) and rabbit anti–filamin-B (H1) antibodies have been described previously (van der Flier et al., 1997; Takafuta et al., 1998). Mouse anti-sarcomeric α-actinin (EA-53), anti-talin (8d4), and α-actinin (BM-75.2) were purchased from Sigma-Aldrich; mouse anti-paxillin (349) and anti-phosphotyrosine (PY20) were from Transduction Laboratories; mouse anti–Gal4 binding domain (BD), mouse anti–Gal4 activation domain (AD), and rabbit anti-HA antibodies (sc-510, sc-1663, and sc-805, respectively) were from Santa Cruz Biotechnology, Inc.; mouse anti-HA (12CA5) were from Boehringer; mouse anti-sarcomeric MHC (MF20) were from the Developmental Studies Hybridoma Bank; and rabbit and mouse anti-GFP (B34) were from CLONTECH Laboratories, Inc. and BabCO. The sheep anti–mouse and donkey anti–rabbit HRP-conjugated antibodies were obtained from Amersham Pharmacia Biotech. TO-PRO-3 iodide, Alexa®-568–coupled phalloidin, and Texas red–conjugated goat anti–mouse and goat anti–rabbit were from Molecular Probes.
Yeast two-hybrid screen and plasmid constructs
The β1D (amino acids 752–798) or β1A cytoplasmic domain (amino acids 752–801) were fused to the Gal4 DNA BD in pAS2.1 (CLONTECH Laboratories, Inc.). These constructs were used as bait in yeast two-hybrid screens of a human skeletal muscle and a human keratinocyte library (CLONTECH Laboratories, Inc.; HL4010AB and HL4024AB, respectively) in the yeast Gal4 transcriptional AD expression vector pGAD10 (CLONTECH Laboratories, Inc.). Plasmids were introduced into the yeast host strains PJ69–4A (a gift from Dr. P. James, University of Wisconsin, Madison, WI; James et al., 1996) or Y190 (CLONTECH Laboratories, Inc.) by transformation. The yeast two-hybrid library screen was performed essentially according to the CLONTECH Laboratory, Inc. two-hybrid manual.
To analyze the protein–protein interactions of truncated filamin-A and filamin-B with several integrin cytoplasmic tails, mutants, and truncated filamins, pACT (-derived) and pAS (-derived) constructs were cotransformed into the yeast strain PJ69–4A. Interaction was assayed by selection for growth on plates containing SC-LTHA, a yeast synthetic complete medium lacking the vector markers Leu and Trp, as well as the interaction markers His and Ade, and containing 2 mM 3-aminotriazole (Sigma-Aldrich; A8056) to suppress residual histidine synthesis in the strain PJ69–4A, as described previously (Geerts et al., 1999). Interaction was scored as the percentage of the plating efficiency on SC-LTHA containing 2 mM 3-aminotriazole compared with the plating efficiency on SC-LT, a medium lacking only the vector markers Leu and Trp, when grown for 5 or 10 d at 30°C. Quantitative determination of β-galactosidase activities was performed in triplicate using of three independent clones grown in SC-LT with O-nitrophenyl B-d-galactopyranoside (Sigma-Aldrich) as the substrate, according to the manufacturer's recommendations.
All filamin-A and filamin-B deletion constructs (starting at the amino acids as indicated in Figs. 2 and 4; for amino acid alignment see van der Flier and Sonnenberg, 2001a) were generated by PCR using Pwo DNA polymerase (Roche Molecular Biochemicals). Filamin-A and filamin-B deletion constructs were cloned in the modified yeast expression vector, pACT2.4 (Gal-4[AD], a derivative of pACT2.1) or pAS2.1 (Gal4[BD]). The Gal4(BD) β1A, β1D, β2, β3A cytoplasmic domain and β1A truncation constructs in the pAS2.1 have been described previously (Wixler et al., 2000). The β6 cytoplasmic domain construct in pAS2.1 was a gift of S. Spong (Lung Biology Center, University of California San Francisco, San Francisco, CA). The β1 chimeric cytoplasmic domains β1A/D792, β1A/D796, and β1D/A787 were generated by PCR and cloned into pAS2.1. All plasmid constructs were checked by sequencing, and the expression of fusion proteins in yeast was confirmed by immunoblotting with Gal4(BD)- and (AD)-specific antibodies. None of the filamin constructs used activated the reporter genes autonomously in yeast, nor did they bind to any of several different integrin α-subunits tested.
Genomic PCR and RT-PCR analysis
RT-PCR analysis was performed on Human Multiple Tissue cDNA Panel I and II (CLONTECH Laboratories, Inc.) using Taq DNA polymerase and primers deduced from the human filamin-B sequence.
The following primer sets were used to detect the human filamin-Bvar-1 cDNA: either BV1/BV2 or nested PCR using primers BV9/BV11 on the BV9/BV10 PCR products. BV11 specifically amplifies the filamin-Bvar-1 cDNA because it spans the 123-nt deleted sequence with one basepair at its 3′ end. Similarly, human filamin-Avar-1 was detected using the nested primers AV1/AV3 on the AV1/AV2 PCR product. Human filamin-Bvar-1 (ΔH1) transcripts with or without the H1 region were detected using primers BV12/BV13. Genomic sequences encoding repeats 19 and 20 of human filamin-B were determined by PCR using Taq DNA polymerase on human genomic DNA isolated from two different cell lines, using a standard procedure and combinations of the primers BV1–BV8 as indicated in Fig. 1 C. All BV primers are numbered according to the filamin-B sequence data available from GenBank/EMBL/DDBJ under accession no. NM_001457, except primers BV7 and BV8, which are numbered according to sequence data submitted under accession no. AF353666. Numbering of AV primers is according to the filamin-A sequence available under accession no. NM_001456. BV1: GAAGATGGCACCTGCAAAGTC (6288–6308); BV2: GACCGGCACCAGGTAGGG (6971–6954); BV3: GTGCCTGGGGTTTATATCGTC (6324–6344); BV4: GGTGCGGGTGATGCTCTCT (6437–6419); BV5: GGAGGGAAGAGTCAAAGAGAG (6404–6424); BV6: GATCTGGGCAGAGCAGTTC (177–159); BV7: GAACTGCTCTGCCCAGATC (159–177); BV8: CACGGTGAACTGGAAGGGG (6686–6668); BV9: TCCAGTCGGAGATTGGTGA (6115–6133); BV10: GACCGGCACCAGGTAGGG (6971–6954); BV11: ATATCACTGCTGTTGATTTC/A (6517–6498/6374); BV12: GTGACCTGCACGGTTCTGA (5071–5089); BV13: ATCACTGCTGTTGATTTC/AGGC (6513–6496/6372–6369); AV1: ACCCGCGATGCAGGCTATG (6386–6404); AV2: CACGGTGAACTGGAAGGGG (6690–6672); AV3: CCGACCAGCACGTGCCTG/A (6543–6771/6675).
Total RNA from C2C12 cells was isolated at various time points after myotube induction, using RNAzol™B (Tel-Testine), as recommended by the manufacturer. Subsequently, cDNA was prepared from 10 μg RNA using an oligo d(T)15 primer and SuperScript™ (Life Technologies). The H1 regions of murine filamin-A, filamin-B, and filamin-C (partial sequences are deposited under GenBank/EMBL/DDBJ accession nos. AF353668, AF353669, and AF354670, respectively) were amplified using the primer sets mAH1/mAH2, mBH1/mBH2, and mCH1/mCH2, respectively. mAH1: CCTGATGGCTCAGAGGTAGA (1–20); mAH2: GTGATCTCCCCTTCTTGATA (344–324); mBH1: CTGACGTCATTGAAAATGAAGAT (1–23); mBH2: AATGGAATCACCAAGTCAAAGG (274–253); mCH1: GATGGGGCAGAGCTCGATG (1–19); mCH2: GTCAGCTCCCCTTTCTGCAC (341–322).
For the detection of potential murine filamin-A and filamin-B variant-1 mRNAs (partial sequences are deposited under GenBank/EMBL/DDBJ accession nos. AF353671 and AF353672, respectively), RT-PCR was performed using mAV1/mAV2 and mBV1/mBV2, respectively. The variant-1 products were further amplified by nested PCR using the mAV1/mAV3 and mBV1/mBV3 primer sets on 1 μl of the RT-PCR products. mAV1: GGCTACGGTGGGCTTAGTC (1–19); mAV2: GAAGGGGCTCCCAGGTACA (453–435); mAV3: TGACCAGCATGTGCCTG/A (138–154/278); mBV1: AGGCTATGGTGGCATATCCT (1–20); mBV2: GAACGGGCTTCCTGTTACG (454–436); mBV3: GCTGACGAGCATGTGCCTG/A (137–155/278). Murine β1A and β1D integrin (GenBank/EMBL/DDBJ accession no. Y00769) were analyzed using GGCAACAATGAAGCTATCG (2219–2237) and CCCTCATTCGGATTGAC (2484–2465) as a primer set. PCR products were analyzed on 2% agarose gels and the identity of all PCR products was confirmed by cloning and subsequent sequencing.
Cloning of HA-tagged and GFP fusion proteins
Truncated forms of filamin-A and filamin-B were fused to the NH2-terminal HA epitope tag in pcDNA3-HANII, a modified pcDNA3 vector (Invitrogen). pCI-puro-eGFPc was generated by inserting an eGFP PCR product, using pEGFP-N1 (CLONTECH Laboratories, Inc.) as a template, into pCI-puro(ΔXbaI/NotI), a pCI-neo–derived (Promega) vector in which the aminoglycoside phosphotransferase gene was replaced by the gene for puromycin-N-acetyl transferase, which induces puromycin resistance. LZRS-eGFPc-IRES-zeo was constructed by subcloning the eGFP fragment from pCI-puro-eGFPc into the retroviral expression vector LZRS-ms-IRES-zeo (Kinsella and Nolan, 1996; van Leeuwen et al., 1997). The four fusion proteins of full-length filamin-B variants and GFP as depicted in Fig. 7 A were generated by a three point ligation into the XbaI/NotI sites of pCI-puro-eGFPc. All four possible combinations between one 5′ filamin-B fragment (XbaI/SacII), either containing or not containing the H1 region, and one 3′ filamin-B fragment (SacII/NotI), either wild-type or variant-1, were prepared. Retroviral expression constructs were obtained by recloning the above filamin-B variants from pCI-puro-eGFP into the retroviral vector LZRS-eGFPc-ms-IRES-zeo. The 5′ filamin-B fragment containing the H1 region was obtained by XbaI/SacII digestion of wild-type filamin-B (Takafuta et al., 1998). A 3′ filamin-B fragment containing a 3′ NotI site for in-frame fusion with GFP was generated by PCR, using primer set D4/U5 and full-length filamin-B as template. A 5′ filamin-B fragment lacking H1 (amino acids 1704–1727) was generated by fusion PCR. In brief, two PCR products were obtained using D1/U1 and D2/U2 primer pairs. Products were annealed through overlapping sequences (underlined) and reamplified with D1/U2 to yield the H1 deletion. This PCR product was then used to replace the MunI/SacII fragment of wild-type filamin-B. A 3′ fragment of filamin-Bvar-1, containing an intrinsic SacII and an in-frame 3′ NotI site was generated by fusion PCR. One PCR product was obtained using primer set D4/U4 and wild-type filamin-B as template. The other PCR product was obtained using primer set D5/U5 and filamin-Bvar-1 cDNA as template. The final fusion PCR product was obtained after reannealling the two obtained products and reamplification using primer set D4/U5. The primers used are under GenBank/EMBL/DDBJ accession no. NM_001457. D1: TGCAATTGATGCCCGAGATGC (4751–4771); U1: ACATAGGCCTCTTCGG TCAC/CATGACAGTGA (5332–5313/5240–5230); D2: ACAGCCCCTTCACTGTCATG/GTGACCGAAGAGG (5221–5240/5313–5325); U2: GGCCGCGGCCATAGACTTT (6165–6147); D4: GTCCAGTCGGAGATTGGTGA (6114–6133); U4: CCACCGCCAAGGATATGCC (6249–6231); D5: GGCATATCCTTGGCGGTGG (6231–6249); U5: gcggcggccgcgAGGCACTGTGACATGAAAAGG (7937–7917) (NotI site is in bold).
Binding to GST fusion proteins
For pull-down assays, β1A and β1D cytoplasmic domains were fused with GST in the bacterial expression vector pRP261, a derivative of the pGEX-3X vector. The recombinant proteins were expressed in Escherichia coli strain BL21 (DE3) and purified from bacterial lysates by the use of glutathione-Sepharose 4B beads. Constructs encoding NH2-terminally truncated filamins were transfected in COS-7 cells using the DEAE-dextran method (two 10-cm Petri dishes with 106 seeded cells were used for three pull-down assays). 48 h after transfection, cells were lysed for 20 min in 1 ml ice cold lysis buffer (10 mM Pipes, pH 6.8, 50 mM NaCl, 150 mM sucrose, 0.5% Triton X-100, 1 mM EDTA, 10 μg/ml calpeptin, 1 mM PMSF, 10 μg/ml soybean trypsin inhibitor, 10 μg/ml leupeptin, and 0.1 U/ml aprotinin). Lysates were cleared by centrifugation at 15,000 g for 10 min at 4°C, and subsequently diluted 10 times in dilution buffer (10 mM Pipes, pH 6.8, 50 mM NaCl, 150 mM sucrose, 3 mM MgCl2). Diluted lysates were incubated overnight at 4°C with 15 μl glutathione-Sepharose 4B beads containing 50 μg GST fusion proteins. The beads were washed with the same lysis buffer and centrifuged through a sucrose cushion (10 mM Pipes, pH 6.8, 50 mM NaCl, 800 mM sucrose, 1 mM MgCl2), and proteins were resolved by SDS-PAGE and immunoblotted with anti-HA antibody. For GST precipitations of full-length COOH-terminally GFP-tagged filamins, stable retrovirally infected C2C12 or GD25-β1A cells were lysed and incubated with the various GST fusion proteins. Expression of the full-length GFP-tagged filamin-B variants was increased by preincubating the cells overnight with 5 mM sodium butyrate. GST fusion protein loading of the beads was checked by Coomassie brilliant blue staining, and the filamin fusion proteins were visualized by immunoblotting, as described previously (Geerts et al., 1999).
Chemical cross-linking of the HA- and eGFP-tagged filamin-B truncations and immunoblotting
CHO cells were transfected with NH2-terminal HA-tagged or COOH-terminal GFP-tagged truncated constructs of full-length filamin-B (FLN-B[19–24] 2009–2602 or FLN-Bvar-1[19–23] 2027–2047), using a cationic lipid-based DNA delivery protocol (Lipofectamin; Life Technologies). After a 3-h incubation, the lipofectamin reagent was replaced by fresh growth medium. 2 d later, transfected cells were lysed in 0.5 ml lysis buffer (20 mM Hepes, pH 7.4, 150 mM NaCl, 1.5 mM MgCl2, 1% Nonidet P-40) for 5 min on ice and subsequently subjected to cross-linkage for 1 h on ice by adding different concentrations (0, 0.25, or 1 mM) of DSP (Pierce Chemical Co.; 25 mM stock in DMSO). Alternatively, the intact cell monolayer was treated with 1 mM of the cell-permeable cross-linker, dissolved in phosphate-buffered saline, for 1 h. Subsequently, the cross-linking reaction in the cell lysate was stopped by adding 25 μl of 1 M Tris-HCl (pH 6.8), whereas the intact treated cells were washed twice with PBS and lysed in 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Nonidet P-40. All cell lysates were cleared by centrifugation at 14,000 rpm for 10 min and 1/12 portions of the total cell lysates were separated by SDS-PAGE on 7% gels under both nonreducing and reducing (5% β-mercaptoethanol) conditions and analyzed by immunoblotting.
Immunofluorescence and flow cytometry
Bulk populations of sorted cells expressing GFP fusion proteins were grown on coverslips and, in one step, fixed and permeabilized in 3% paraformaldehyde, 2% Triton X-100 in PBS for 10 min at room temperature. Cells were blocked in PBS, 2% BSA for 1 h, and incubated with primary antibodies (optionally in the presence of Alexa®568-labeled phalloidin or TO-PRO-3) in the same buffer for 1 h at room temperature. After washing in PBS, cells were incubated in the presence of FITC- or Texas red–conjugated secondary antibodies for 1 h. Preparations were then washed in PBS, mounted in Vectashield (Vector Laboratories), and analyzed with a confocal Leica TCS NT microscope.
For flow cytometry and cell sorting, cultured cells were trypsinized, washed twice in PBS, 2% FCS, and sorted on a FACStar Plus® (Becton Dickinson) by their level of GFP expression.
Acknowledgments
The authors thank P. Engelfriet, E. Roos, and C. Geuijen for careful reading of the manuscript.
A. van der Flier was supported by a Yamanouchi Studentship (Yamanouchi Research Institute, Oxford, UK) and a grant from The Netherlands Heart Foundation.
Arjan van der Flier's present address is Center for Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139.
Dirk Geerts' present address is Department of Human Genetics, Academic Medical Center, University of Amsterdam, Meibergdreef 9, 1105 AZ Amsterdam, Netherlands.
Footnotes
Abbreviations used in this paper: ABD, actin-binding domain; AD, activation domain; BD, binding domain; DSP, dithiobis-succinimidyl propionate; F-actin, filamentous actin; FLN-B, full-length filamin-B; GFP, green fluorescent protein; GST, glutathione-S-transferase; H1, hinge-1; H1s, shorter H1; HA, hemagglutinin A; MHC, myosin heavy chain.
References
- Belkin, A.M., S.F. Retta, O.Y. Pletjushkina, F. Balzac, L. Silengo, R. Fässler, V.E. Koteliansky, K. Burridge, and G. Tarone. 1997. Muscle β1D integrin reinforces the cytoskeleton–matrix link: modulation of integrin adhesive function by alternative splicing. J. Cell Biol. 139:1583–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellanger, J.M., C. Astier, C. Sardet, Y. Ohta, T.P. Stossel, and A. Debant. 2000. The Rac1- and RhoG-specific GEF domain of trio targets filamin to remodel cytoskeletal actin. Nat. Cell Biol. 2:888–892. [DOI] [PubMed] [Google Scholar]
- Blake, D.J., and S. Kroger. 2000. The neurobiology of Duchenne muscular dystrophy: learning lessons from muscle? Trends Neurosci. 23:92–99. [DOI] [PubMed] [Google Scholar]
- Brancaccio, M., S. Guazzone, N. Menini, E. Sibona, E. Hirsch, M. De Andrea, M. Rocchi, F. Altruda, G. Tarone, and L. Silengo. 1999. Melusin is a new muscle-specific interactor for β1 integrin cytoplasmic domain. J. Biol. Chem. 274:29282–29288. [DOI] [PubMed] [Google Scholar]
- Calderwood, D.A., R. Zent, R. Grant, D.J. Rees, R.O. Hynes, and M.H. Ginsberg. 1999. The Talin head domain binds to integrin beta subunit cytoplasmic tails and regulates integrin activation. J. Biol. Chem. 274:28071–28074. [DOI] [PubMed] [Google Scholar]
- Chakarova, C., M.S. Wehnert, K. Uhl, S. Sakthivel, H.-P. Vosberg, P.F.M. Van der Ven, and D.O. Fürst. 2000. Genomic structure and fine mapping of the two human filamin gene paralogues FLNB and FLNC and comparative analysis of the filamin gene family. Hum. Genet. 107:597–611. [DOI] [PubMed] [Google Scholar]
- Chang, D.D., C. Wong, H. Smith, and J. Liu. 1997. ICAP-1, a novel β1 integrin cytoplasmic domain–associated protein, binds to a conserved and functionally important NPXY sequence motif of β1 integrin. J. Cell Biol. 138:1149–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang, W., M.L. Greaser, and G.E. Lyons. 2000. Filamin isogene expression during mouse myogenesis. Dev. Dyn. 217:99–108. [DOI] [PubMed] [Google Scholar]
- Critchley, D.R. 2000. Focal adhesions - the cytoskeletal connection. Curr. Opin. Cell Biol. 12:133–139. [DOI] [PubMed] [Google Scholar]
- Cunningham, C.C., J.B. Gorlin, D.J. Kwiatkowski, J.H. Hartwig, P.A. Janmey, H.R. Byers, and T.P. Stossel. 1992. Actin-binding protein requirement for cortical stability and efficient locomotion. Science. 255:325–327. [DOI] [PubMed] [Google Scholar]
- Fässler, R., M. Pfaff, J. Murphy, A.A. Noegel, S. Johansson, R. Timpl, and R. Albrecht. 1995. Lack of β1 integrin gene in embryonic stem cells affects morphology, adhesion, and migration but not integration into the inner cell mass of blastocysts. J. Cell Biol. 128:979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner, G., A. Pallavicini, A. Comelli, M. Salamon, G. Bortoletto, C. Ievolella, S. Trevisan, S. Kojic, F. Dalla Vecchia, P. Laveder, et al. 2000. FATZ, a filamin-, actinin-, and telethonin-binding protein of the Z-disc of skeletal muscle. J. Biol. Chem. 275:41234–41242. [DOI] [PubMed] [Google Scholar]
- Fox, J.W., E.D. Lamperti, Y.Z. Eksioglu, S.E. Hong, Y. Feng, D.A. Graham, I.E. Scheffer, W.B. Dobyns, B.A. Hirsch, R.A. Radtke, et al. 1998. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron. 21:1315–1325. [DOI] [PubMed] [Google Scholar]
- Fuchs, E., and Y. Yang. 1999. Crossroads on cytoskeletal highways. Cell. 98:547–550. [DOI] [PubMed] [Google Scholar]
- Fucini, P., C. Renner, C. Herberhold, A.A. Noegel, and T.A. Holak. 1997. The repeating segments of the F-actin cross-linking gelation factor (ABP-120) have an immunoglobulin-like fold. Nat. Struct. Biol. 4:223–230. [DOI] [PubMed] [Google Scholar]
- Gallo, R., M. Serafini, L. Castellani, G. Falcone, and S. Alema. 1999. Distinct effects of Rac1 on differentiation of primary avian myoblasts. Mol. Biol. Cell. 10:3137–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geerts, D., L. Fontao, M.G. Nievers, R.Q. Schaapveld, P.E. Purkis, G.N. Wheeler, E.B. Lane, I.M. Leigh, and A. Sonnenberg. 1999. Binding of integrin α6β4 to plectin prevents plectin association with F-actin but does not interfere with intermediate filament binding. J. Cell Biol. 147:417–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giancotti, F.G., and E. Ruoslahti. 1999. Integrin signaling. Science. 285:1028–1032. [DOI] [PubMed] [Google Scholar]
- Gimond, C., A. van der Flier, S. van Delft, C. Brakebusch, I. Kuikman, J.G. Collard, R. Fässler, and A. Sonnenberg. 1999. Induction of cell scattering by expression of β1 integrins in β1-deficient epithelial cells requires activation of members of the rho family of GTPases and downregulation of cadherin and catenin function. J. Cell Biol. 147:1325–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glukhova, M.A., M.G. Frid, and V.E. Koteliansky. 1990. Developmental changes in expression of contractile and cytoskeletal proteins in human aortic smooth muscle. J. Biol. Chem. 265:13042–13046. [PubMed] [Google Scholar]
- Gomer, R.H., and E. Lazarides. 1981. The synthesis and deployment of filamin in chicken skeletal muscle. Cell. 23:524–532. [DOI] [PubMed] [Google Scholar]
- Gomer, R.H., and E. Lazarides. 1983. a. Highly homologous filamin polypeptides have different distributions in avian slow and fast muscle fibers. J. Cell Biol. 97:818–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomer, R.H., and E. Lazarides. 1983. b. Switching of filamin polypeptides during myogenesis in vitro. J. Cell Biol. 96:321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlin, J.B., R. Yamin, S. Egan, M. Stewart, T.P. Stossel, D.J. Kwiatkowski, and J.H. Hartwig. 1990. Human endothelial actin-binding protein (ABP-280, nonmuscle filamin): a molecular leaf spring. J. Cell Biol. 111:1089–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Y., S.X. Zhang, N. Sokol, L. Cooley, and G.L. Boulianne. 2000. Physical and genetic interaction of filamin with presenilin in Drosophila. J. Cell Sci. 113:3499–3508. [DOI] [PubMed] [Google Scholar]
- Hannigan, G.E., C. Leung-Hagesteijn, L. Fitz-Gibbon, M.G. Coppolino, G. Radeva, J. Filmus, J.C. Bell, and S. Dedhar. 1996. Regulation of cell adhesion and anchorage-dependent growth by a new β1-integrin-linked protein kinase. Nature. 379:91–96. [DOI] [PubMed] [Google Scholar]
- Hayashi, Y.K., F.L. Chou, E. Engvall, M. Ogawa, C. Matsuda, S. Hirabayashi, K. Yokochi, B.L. Ziober, R.H. Kramer, S.J. Kaufman, et al. 1998. Mutations in the integrin α7 gene cause congenital myopathy. Nat. Genet. 19:94–97. [DOI] [PubMed] [Google Scholar]
- Hynes, R.O. 1992. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 69:11–25. [DOI] [PubMed] [Google Scholar]
- James, P., J. Halladay, and E.A. Craig. 1996. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics. 144:1425–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay, D., E.J. Garcia, J.E. Lara, M.A. Medina, and M. de la Luz Ibarra. 2000. Determination of a cAMP-dependent protein kinase phosphorylation site in the C-terminal region of human endothelial actin-binding protein. Arch. Biochem. Biophys. 377:80–84. [DOI] [PubMed] [Google Scholar]
- Kinsella, T.M., and G.P. Nolan. 1996. Episomal vectors rapidly and stably produce high-titer recombinant retrovirus. Hum. Gene Ther. 7:1405–1413. [DOI] [PubMed] [Google Scholar]
- Kolanus, W., W. Nagel, B. Schiller, L. Zeitlmann, S. Godar, H. Stockinger, and B. Seed. 1996. ALβ2 integrin/LFA-1 binding to ICAM-1 induced by cytohesin-1, a cytoplasmic regulatory molecule. Cell. 86:233–242. [DOI] [PubMed] [Google Scholar]
- Leonardi, A., H. Ellinger-Ziegelbauer, G. Franzoso, K. Brown, and U. Siebenlist. 2000. Physical and functional interaction of filamin (actin-binding protein-280) and tumor necrosis factor receptor-associated factor 2. J. Biol. Chem. 275:271–278. [DOI] [PubMed] [Google Scholar]
- Li, J., R. Mayne, and C. Wu. 1999. a. A novel muscle-specific β1 integrin binding protein (MIBP) that modulates myogenic differentiation. J. Cell Biol. 147:1391–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M.G., M. Serr, K. Edwards, S. Ludmann, D. Yamamoto, L.G. Tilney, C.M. Field, and T.S. Hays. 1999. b. Filamin is required for ring canal assembly and actin organization during Drosophila oogenesis. J. Cell Biol. 146:1061–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liliental, J., and D.D. Chang. 1998. Rack1, a receptor for activated protein kinase C, interacts with integrin β subunit. J. Biol. Chem. 273:2379–2383. [DOI] [PubMed] [Google Scholar]
- Liu, G., L. Thomas, R.A. Warren, C.A. Enns, C.C. Cunningham, J.H. Hartwig, and G. Thomas. 1997. Cytoskeletal protein ABP-280 directs the intracellular trafficking of furin and modulates proprotein processing in the endocytic pathway. J. Cell Biol. 139:1719–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, S., D.A. Calderwood, and M.H. Ginsberg. 2000. Integrin cytoplasmic domain-binding proteins. J. Cell Sci. 113:3563–3571. [DOI] [PubMed] [Google Scholar]
- Loo, D.T., S.B. Kanner, and A. Aruffo. 1998. Filamin binds to the cytoplasmic domain of the β1-integrin. Identification of amino acids responsible for this interaction. J. Biol. Chem. 273:23304–23312. [DOI] [PubMed] [Google Scholar]
- Mayer, U., G. Saher, R. Fässler, A. Bornemann, F. Echtermeyer, H. von der Mark, N. Miosge, E. Pöschl, and K. von der Mark. 1997. Absence of integrin α7 causes a novel form of muscular dystrophy. Nat. Genet. 17:318–323. [DOI] [PubMed] [Google Scholar]
- McCoy, A.J., P. Fucini, A.A. Noegel, and M. Stewart. 1999. Structural basis for dimerization of the Dictyostelium gelation factor (ABP120) rod. Nat. Struct. Biol. 6:836–841. [DOI] [PubMed] [Google Scholar]
- Menko, A.S., and D. Boettiger. 1987. Occupation of the extracellular matrix receptor, integrin, is a control point for myogenic differentiation. Cell. 51:51–57. [DOI] [PubMed] [Google Scholar]
- Meyer, S.C., D.A. Sanan, and J.E. Fox. 1998. Role of actin-binding protein in insertion of adhesion receptors into the membrane. J. Biol. Chem. 273:3013–3020. [DOI] [PubMed] [Google Scholar]
- Meyer, S.C., S. Zuerbig, C.C. Cunningham, J.H. Hartwig, T. Bissell, K. Gardner, and J.E. Fox. 1997. Identification of the region in actin-binding protein that binds to the cytoplasmic domain of glycoprotein Ibα. J. Biol. Chem. 272:2914–2919. [DOI] [PubMed] [Google Scholar]
- Ohta, Y., T.P. Stossel, and J.H. Hartwig. 1991. Ligand-sensitive binding of actin-binding protein to immunoglobulin G Fc receptor I (Fc γ RI). Cell. 67:275–282. [DOI] [PubMed] [Google Scholar]
- Ohta, Y., N. Suzuki, S. Nakamura, J.H. Hartwig, and T.P. Stossel. 1999. The small GTPase RalA targets filamin to induce filopodia. Proc. Natl. Acad. Sci. USA. 96:2122–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott, I., E.G. Fischer, Y. Miyagi, B.M. Mueller, and W. Ruf. 1998. A role for tissue factor in cell adhesion and migration mediated by interaction with actin-binding protein 280. J. Cell Biol. 140:1241–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavalko, F.M., C.A. Otey, and K. Burridge. 1989. Identification of a filamin isoform enriched at the ends of stress fibers in chicken embryo fibroblasts. J. Cell Sci. 94:109–118. [DOI] [PubMed] [Google Scholar]
- Pfaff, M., S. Liu, D.J. Erle, and M.H. Ginsberg. 1998. Integrin β cytoplasmic domains differentially bind to cytoskeletal proteins. J. Biol. Chem. 273:6104–6109. [DOI] [PubMed] [Google Scholar]
- Rafael, J.A., E.R. Townsend, S.E. Squire, A.C. Potter, J.S. Chamberlain, and K.E. Davies. 2000. Dystrophin and utrophin influence fiber type composition and post-synaptic membrane structure. Hum. Mol. Genet. 9:1357–1367. [DOI] [PubMed] [Google Scholar]
- Reddy, K.B., P. Gascard, M.G. Price, E.V. Negrescu, and J.E.B. Fox. 1998. Identification of an interaction between the m-band protein skelemin and β-integrin subunits. Colocalization of a skelemin-like protein with β1- and β3-integrins in non-muscle cells. J. Biol. Chem. 273:35039–35047. [DOI] [PubMed] [Google Scholar]
- Rietzler, M., M. Bittner, W. Kolanus, A. Schuster, and B. Holzmann. 1998. The human WD repeat protein WAIT-1 specifically interacts with the cytoplasmic tails of β7-integrins. J. Biol. Chem. 273:27459–27466. [DOI] [PubMed] [Google Scholar]
- Sastry, S.K., M. Lakonishok, D.A. Thomas, J. Muschler, and A.F. Horwitz. 1996. Integrin α subunit ratios, cytoplasmic domains, and growth factor synergy regulate muscle proliferation and differentiation. J. Cell Biol. 133:169–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, C.P., R.M. Ezzell, and M.A. Arnaout. 1995. Direct interaction of filamin (ABP-280) with the β2-integrin subunit CD18. J. Immunol. 154:3461–3470. [PubMed] [Google Scholar]
- Sheen, V.L., P.H. Dixon, J.W. Fox, S.E. Hong, L. Kinton, S.M. Sisodiya, J.S. Duncan, F. Dubeau, I.E. Scheffer, S.D. Schachter, et al. 2001. Mutations in the X-linked filamin 1 gene cause periventricular nodular heterotopia in males as well as in females. Hum. Mol. Genet. 10:1775–1783. [DOI] [PubMed] [Google Scholar]
- Sokol, N.S., and L. Cooley. 1999. Drosophila filamin encoded by the cheerio locus is a component of ovarian ring canals. Curr. Biol. 9:1221–1230. [DOI] [PubMed] [Google Scholar]
- Stossel, T.P., J. Condeelis, L. Cooley, J.H. Hartwig, A. Noegel, M. Schleicher, and S.S. Shapiro. 2001. Filamins as integrators of cell mechanics and signalling. Nat. Rev. Mol. Cell Biol. 2:138–145. [DOI] [PubMed] [Google Scholar]
- Tachikawa, M., H. Nakagawa, A.G. Terasaki, H. Mori, and K. Ohashi. 1997. A 260-kDa filamin/ABP-related protein in chicken gizzard smooth muscle cells is a new component of the dense plaques and dense bodies of smooth muscle. J. Biochem. 122:314–321. [DOI] [PubMed] [Google Scholar]
- Takada, F., D.L. Woude, H.Q. Tong, T.G. Thompson, S.C. Watkins, L.M. Kunkel, and A.H. Beggs. 2001. Myozenin: an α-actinin- and γ-filamin-binding protein of skeletal muscle Z lines. Proc. Natl. Acad. Sci. USA. 98:1595–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takafuta, T., G. Wu, G.F. Murphy, and S.S. Shapiro. 1998. Human β-filamin is a new protein that interacts with the cytoplasmic tail of glycoprotein Ibα. J. Biol. Chem. 273:17531–17538. [DOI] [PubMed] [Google Scholar]
- Thompson, T.G., Y.M. Chan, A.A. Hack, M. Brosius, M. Rajala, H.G. Lidov, E.M. McNally, S. Watkins, and L.M. Kunkel. 2000. Filamin 2 (FLN2): a muscle-specific sarcoglycan interacting protein. J. Cell Biol. 148:115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier, A., and A. Sonnenberg. 2001. a. Structural and functional aspects of filamins. Biochem. Biophys. Acta. 1538:99–117. [DOI] [PubMed] [Google Scholar]
- van der Flier, A., and A. Sonnenberg. 2001. b. Function and interactions of integrins. Cell Tissue Res. 305:285–298. [DOI] [PubMed] [Google Scholar]
- van der Flier, A., A.C. Gaspar, S. Thorsteinsdóttir, C. Baun, E. Groeneveld, C.L. Mummery, and A. Sonnenberg. 1997. Spatial and temporal expression of the β1D integrin during mouse development. Dev. Dynam. 210:472–486. [DOI] [PubMed] [Google Scholar]
- van der Ven, P.F., S. Wiesner, P. Salmikangas, D. Auerbach, M. Himmel, S. Kempa, K. Hayess, D. Pacholsky, A. Taivainen, R. Schroder, et al. 2000. Indications for a novel muscular dystrophy pathway. γ-Filamin, the muscle-specific filamin isoform, interacts with myotilin. J. Cell Biol. 151:235–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen, F.N., H.E. Kain, R.A. Kammen, F. Michiels, O.W. Kranenburg, and J.G. Collard. 1997. The guanine nucleotide exchange factor Tiam1 affects neuronal morphology: opposing roles for the small GTPases Rac and Rho. J. Cell Biol. 139:797–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk, T., L.I. Fessler, and J.H. Fessler. 1990. A role for integrin in the formation of sarcomeric cytoarchitecture. Cell. 63:525–536. [DOI] [PubMed] [Google Scholar]
- Wei, L., W. Zhou, J.D. Croissant, F.E. Johansen, R. Prywes, A. Balasubramanyam, and R.J. Schwartz. 1998. RhoA signaling via serum response factor plays an obligatory role in myogenic differentiation. J. Biol. Chem. 273:30287–30294. [DOI] [PubMed] [Google Scholar]
- Wixler, V., D. Geerts, E. Laplantine, D. Westhoff, N. Smyth, M. Aumailley, A. Sonnenberg, and M. Paulsson. 2000. The LIM-only protein DRAL/FHL2 binds to the cytoplasmic domain of several α and β integrin chains and is recruited to adhesion complexes. J. Biol. Chem. 275:33669–33678. [DOI] [PubMed] [Google Scholar]
- Xie, Z., W. Xu, E.W. Davie, and D.W. Chung. 1998. Molecular cloning of human ABPL, an actin-binding protein homologue. Biochem. Biophys. Res. Commun. 251:914–919. [DOI] [PubMed] [Google Scholar]
- Xu, W., Z. Xie, D.W. Chung, and E.W. Davie. 1998. A novel human actin-binding protein homologue that binds to platelet glycoprotein Ibα. Blood. 92:1268–1276. [PubMed] [Google Scholar]
- Yaffe, D., and O. Saxel. 1977. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature. 270:725–727. [DOI] [PubMed] [Google Scholar]
- Zent, R., C.A. Fenczik, D.A. Calderwood, S. Liu, M. Dellos, and M.H. Ginsberg. 2000. Class- and splice variant-specific association of CD98 with integrin β cytoplasmic domains. J. Biol. Chem. 275:5059–5064. [DOI] [PubMed] [Google Scholar]
- Zhang, W., S.W. Han, D.W. McKeel, A. Goate, and J.Y. Wu. 1998. Interaction of presenilins with the filamin family of actin-binding proteins. J. Neurosci. 18:914–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuellig, R.A., B.C. Bornhauser, I. Knuesel, F. Heller, J.M. Fritschy, and M.C. Schaub. 2000. Identification and characterization of transcript and protein of a new short N-terminal utrophin isoform. J. Cell Biochem. 77:418–431. [DOI] [PubMed] [Google Scholar]