

Abstract
Using two-hybrid screening, we isolated the integrin cytoplasmic domain-associated protein (ICAP-1), an interactor for the COOH terminal region of the β1A integrin cytoplasmic domain. To investigate the role of ICAP-1 in integrin-mediated adhesive function, we expressed the full-length molecule in NIH3T3 cells. ICAP-1 expression strongly prevents NIH3T3 cell spreading on extracellular matrix. This inhibition is transient and can be counteracted by coexpression of a constitutively activated mutant of Cdc42, suggesting that ICAP-1 acts upstream of this GTPase. In addition, we found that ICAP-1 binds both to Cdc42 and Rac1 in vitro, and its expression markedly inhibits activation of these GTPases during integrin-mediated cell adhesion to fibronectin as detected by PAK binding assay. In the attempt to define the molecular mechanism of this inhibition, we show that ICAP-1 reduces both the intrinsic and the exchange factor–induced dissociation of GDP from Cdc42; moreover, purified ICAP-1 displaces this GTPase from cellular membranes. Together, these data show for the first time that ICAP-1 regulates Rho family GTPases during integrin-mediated cell matrix adhesion, acting as guanine dissociation inhibitor.
Keywords: integrins; Cdc42; Rac; ICAP-1; GDI; cytoskeleton
Introduction
Rho family GTPases are cytoplasmic proteins controlling several crucial cellular functions such as organization of the cytoskeleton in cell motility, gene expression, and cell cycle progression. Three of these GTPases, Cdc42, Rac1, and RhoA, have been studied in some detail and shown to play a major role in regulating actin cytoskeleton in response to soluble extracellular stimuli known to induce cell motility, such as PDGF and lysophosphatidic acid (LPA)* (Nobes and Hall, 1995; Hall, 1998). In particular Cdc42 and Rac1 regulate the ability of the cell to elongate membrane protrusions, such as filopodia and lamellipodia, whereas RhoA controls the organization of actin in stress fibers (Hall, 1998) ending at “focal adhesions,” sites of cell matrix contact where integrins are clustered.
Filopodia, lamellipodia, and stress fibers are also formed during integrin-mediated cell adhesion to and spreading on insoluble extracellular matrix proteins in the absence of soluble motogenic factors. Integrins are major membrane receptors involved in cell matrix adhesion that generate a cascade of intracellular signals, leading to the organization of the actomyosin filaments (Schoenwaelder and Burridge, 1999; Schwartz and Shattil, 2000). Analysis of such signals indicated that integrins can trigger activation of Rho family GTPases (Clark et al., 1998; Price et al., 1998; Olivo et al., 2000). In particular, it has been shown that integrins can activate p21PAK and ACK2, two known effectors of Cdc42 and Rac1, very shortly after binding to fibronectin (Price et al., 1998; Yang et al., 1999). Activation of these effectors is inhibited by expression of a dominant negative form of Cdc42 (Price et al., 1998; Yang et al., 1999), which also strongly inhibited cell spreading (Clark et al., 1998; Price et al., 1998; Olivo et al., 2000). On the other hand, only modest effect on cell spreading was observed with Rac1 dominant negative mutants or by inhibiting RhoA with C3 exoenzyme (Clark et al., 1998; Price et al., 1998; Olivo et al., 2000). These data clearly indicate that activation of Cdc42 is crucial for integrin-mediated cell spreading on matrix proteins. However, the molecular mechanisms leading to regulation of Cdc42 upon integrin binding to their ligands are still poorly defined.
Activation of monomeric GTPases is determined by cycling between the GDP- and GTP-bound states. This process is controlled by three major classes of molecules known as guanine nucleotide exchange factors (GEFs), GTPase-activating proteins (GAPs), and guanine nucleotide dissociation inhibitors (GDIs) (Van Aelst and D'Souza-Schorey, 1997). GEFs induce the exchange of GDP for GTP, thus promoting the activation of the GTPases. Over 30 different GEFs have been described, all of which contain a Dbl homology (DH) domain flanked at the COOH-terminal by a pleckstrin homology (PH) domain (Hart et al., 1994). Although the DH domain is responsible for the GEF catalytic activity, the PH domain localizes the GEF protein to the plasma membrane where their GTPase substrates reside. Numerous GAPs have also been identified, which increase the intrinsic rate of GTP hydrolysis of the GTPase, thus promoting the transition to the inactive GDP-bound state. The third class of GTPase regulators is represented by the GDIs. Only a few of these proteins have been identified to date, and their structure has been solved recently. GDIs form a complex with GTPases sequestering these molecules in the cytoplasm and preventing their membrane association (Leonard et al., 1992). The formation of this complex is mediated by the COOH-terminal region of the GDI molecule, which forms a hydrophobic pocket accommodating the GTPase-bound isoprenyl lipid necessary for membrane localization, of the GTPase (Gosser et al., 1997; Keep et al., 1997; Hoffman et al., 2000; Scheffzek et al., 2000). The NH2-terminal region of the GDI molecule consists of a flexible arm capable of inhibiting GDP dissociation by binding to the switch I and II domains of the GTPase (Keep et al., 1997; Hoffman et al., 2000; Scheffzek et al., 2000).Thus, activation of a Rho family GTPase likely involves activation of GEFs and the simultaneous inhibition of GDIs and/or GAPs.
Using the two-hybrid system, we identified a group of proteins capable of binding to the cytoplasmic domain of β1-integrin (Brancaccio et al., 1999). Among these, we isolated the integrin cytoplasmic domain associated protein (ICAP-1), which was also isolated by other laboratories using similar baits but different cDNA libraries (Chang et al., 1997; Zhang and Hemler, 1999). The data presented here show that overexpression of ICAP-1 strongly reduces the integrin-mediated cell spreading on extracellular matrix and inhibits both Cdc42 and Rac1. In addition, ICAP-1 induces release of Cdc42 from cellular membranes and prevents the dissociation of GDP from this GTPase. Thus, ICAP-1 is a new GTPase regulatory protein endowed with GDI activity and involved in the integrin-mediated actin cytoskeleton regulation during cell matrix adhesion.
Results
Isolation of ICAP-1 by two-hybrid screening and binding specificity
To identify proteins able to interact with the β1-integrin cytoplasmic domain, we performed an interaction trap approach (Gyuris et al., 1993). Using the entire β1A cytoplasmic region as a bait to screen a cDNA heart neonatal rat library, we isolated three different proteins (Brancaccio et al., 1999). One of these proteins was found to be the integrin cytoplasmic domain-associated protein, ICAP-1, isolated recently also in other laboratories by screening of different cDNA libraries (Chang et al., 1997; Zhang and Hemler, 1999). ICAP-1 was found to specifically bind to β1-integrin but not to β2, β3, and α5. In addition, no binding was observed with the β1B and β1D isoforms, which differ from β1A in the COOH terminus of the protein (Balzac et al., 1994; Belkin et al., 1996; Table I). To map the amino acid sequences of β1 required for binding to ICAP-1, swapping mutants were generated between the β1A and β1D molecules and tested in the two-hybrid system. The results of these experiments indicate that, although important, the NPXY sequences alone (Chang et al., 1997) are not sufficient for ICAP-1 binding (bait β1A/D/A in Table I). In fact, the six amino acid region AVTTVV (878–884) of β1A located between the two NPXY motives is required for ICAP-1 binding (bait β1D/A/D in Table I).
Table I. Binding of ICAP-1 to different integrin subunit cytopasmic domain constructs.
| Bait sequences | ICAP-1a binding | |
|---|---|---|
| β1A | KLLMIIHDRREFAKFEKEKMNAKWDTGENPIYKSAVTTVVNPKYEGK | +++ |
| β1D | KLLMIIHDRREFAKFEKEKMNAKWDTQENPIYKSPINNFKNPNYGRKAGL | − |
| β1A/D | KLLMIIHDRREFAKFEKEKMNAKWDTGENPIYKSAVTTVVNPKYGRKAGL | +++ |
| β1D/A/D | KLLMIIHDRREFAKFEKEKMNAKWDTQENPIYKSAVTTVVNPNYGRKAGL | ++ |
| β1A/D/A | KLLMIIHDRREFAKFEKEKMNAKWDTGENPIYKSPINNFKNPKYEGK | − |
| β1D/A | KLLMIIHDRREFAKFEKEKMNAKWDTQENPIYKSPINNFKNPNYEGK | − |
| β1B | KLLMIIHDRREFAKFEKEKMNAKWDTSYKTSKKQSGL | − |
| β2 | KALIHLSDLREYRRFEKEKLKSQWNNDNPLFKSATTTVMNPKFAES | − |
| β3 | KLLITIHDRKEFAKFEEERARAKWDTVRDGAGRFLKSLV | − |
| α5 | KLGFFKRSLPYGTAMEKAQLQPPATSDA | − |
The bolded sequences are specific to the β1A variable region.
Number of + indicates the strength of the interaction judged by intensity of blue after growing colonies on X-gal indicator plates lacking leucine.
ICAP-1 inhibits the initial phase of cell spreading
To test the possible function of ICAP-1, we transiently transfected this molecule in NIH3T3 cells. As detected with our polyclonal antibody, the endogenous level of ICAP-1 was very low in these cells and raised ∼20 times after transfection. Transfected cells were detached from the dish and replated on fibronectin-coated glass to test their ability to adhere and spread. As shown in Fig. 1 , cells expressing ICAP-1 attach to fibronectin, but only 35% ± 6 spread after 60 min of plating on the coated substratum, whereas 84% ± 4 of control untransfected cells present in the same sample spread under the same plating conditions. The inhibition of cell spreading by ICAP-1 expression was not the result of a toxic or non specific effect, since cells recover from inhibition and spread normally after overnight incubation on fibronectin (Fig. 1 A, b). Moreover, expression of a different β1 integrin interactor, such as melusin (Brancaccio et al., 1999), did not alter cell adhesion or spreading (unpublished data).
Figure 1.
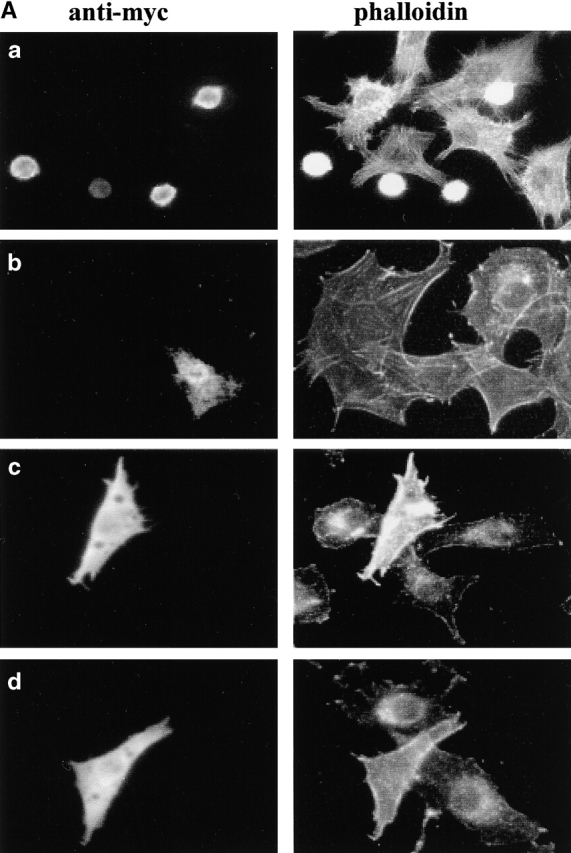

ICAP-1 inhibition of cell spreading and rescue by V12Cdc42. Cells were transiently transfected, detached with EDTA, plated on fibronectin-coated coverslips (5 μg/ml) in serum-free medium, fixed, and stained with different antibodies. (A) NIH3T3 cells transfected with myc–ICAP-1 and plated on fibronectin for 1 h (a) and for 24 h (b). NIH3T3 cotransfected with myc–ICAP-1 and myc-V12Cdc42 plated for 1 h on fibronectin (c). V12Cdc42-NIH3T3 transiently transfected with myc–ICAP-1 and plated for 1 h on fibronectin (d). Transfected cells were visualized by anti-myc monoclonal antibody (left), whereas actin organization was detected in the same sample with FITC phalloidin (right). (B) The percentage of spread cells was calculated as described in Materials and methods. Values are means ± SD from three independent experiments in which >20 cells per condition were scored.
Interestingly, ICAP-1 effect can be appreciated only when cells were detached and replated on fibronectin. In fact, transient transfection of cell monolayer or microinjection of adherent cells with ICAP-1 constructs did not significantly alter the cell morphology (unpublished data). These data show that inhibition of cell spreading by ICAP-1 is specific and occurs only when cells are replated on fibronectin, suggesting a crucial role of ICAP-1 during the initial phases of cell–matrix interaction.
Given that ICAP-1 binds to β1A but not to β3 and β5 integrin subunits (unpublished data; Chang et al., 1997), we wondered whether the ICAP-1 antispreading effect was dependent on integrin β1A binding. To this purpose, we analyzed spreading of ICAP-1–expressing NIH3T3 cells on vitronectin, a substrate for αVβ3 and αvβ5. As shown in Fig. 2 , ICAP-1–expressing cells spread poorly on vitronectin after 1 h of adhesion. This ICAP-1 inhibitory effect was comparable to that observed on fibronectin (Fig. 1, A and B, compared with Fig. 2, A and B). Taken together, these results suggest that in our experimental condition ICAP-1 effect seems not to be dependent on integrin binding.
Figure 2.


ICAP-1 inhibition of cell spreading on vitronectin. (A) NIH3T3 cells were transiently transfected with myc–ICAP-1, detached with EDTA, and allowed to attach on vitronectin-coated coverslips (10 μg/ml) in serum-free medium for 1 h. Transfected cells were visualized by anti-myc monoclonal antibody, whereas actin organization was detected in the same sample with FITC phalloidin. (B) The percentage of spread cells was calculated as described in Materials and methods. Values are means ± SD from three independent experiments in which >20 cells per condition were scored.
ICAP-1 inhibition of cell spreading is reversed by an activated form of Cdc42
Rho family GTPases regulate the actin organization required for cell adhesion and migration (Nobes and Hall, 1999). Previous work has shown that transient expression of a Cdc42 dominant negative mutant strongly inhibits cell spreading during adhesion on extracellular matrix (Clark et al., 1998; Price et al., 1998; Olivo et al., 2000). The fact that expression of ICAP-1 and a dominant negative form of Cdc42 induce a similar phenotype raises the possibility that the two proteins act on the same pathway. To test this hypothesis, we cotransfected ICAP-1 with the constitutively activated form of Cdc42, V12Cdc42. As shown in Fig. 1, ICAP-1 induced only a modest inhibitions of cell spreading in cells transiently transfected with V12Cdc42. Similar results were observed in cells stably expressing V12Cdc42 (Fig. 1). In these cells, ICAP-1 induced a 1.8–2.7-fold decrease of cell spreading, whereas in control cells it induced much strong response (4-fold decrease; Fig. 1 B). These results suggest that Cdc42 acts downstream of ICAP-1 during cell spreading on extracellular matrix.
ICAP-1 binds and inhibits Cdc42 and Rac1
Based on the results described above, we tested the possibility that ICAP-1 could regulate the activity of Rho family GTPases. To measure the activation state of Cdc42 and Rac1, the GTP-bound form of these proteins was isolated using the CRIB domain of the effector protein PAK (Sander et al., 1998). Cells transfected with control vector or ICAP-1 were either maintained in suspension or plated on fibronectin, and cell extracts were processed as described in Materials and methods. COS cells were used in these experiments in order to achieve high level of expression. As shown in Fig. 3 A and Fig. 4 A, the amount of GTP-bound GTPases is increased upon cell adhesion to fibronectin, indicating that both Cdc42 and Rac1 are activated during cell spreading on fibronectin. However, expression of ICAP-1 in these cells strongly reduced the level of GTP-bound form of both GTPases (Fig. 3 and Fig. 4, A and B).
Figure 3.

ICAP-1 inhibits and binds Cdc42. (A) COS cells transiently transfected with myc-Cdc42 together with control vector or myc–ICAP-1 were suspended by trypsin treatment, kept in suspension 2 h 30 min in serum-free DME (Su), and plated on fibronectin (5 μg/ml) for 1 h (Ad). Cdc42 activity assay was performed as described in Materials and methods using a GST-PAK-CD fusion protein that selectively binds to GTP-Cdc42. The bound GTP-Cdc42 was analyzed by Western blotting with a polyclonal antibody to Cdc42. To probe for Cdc42 and ICAP-1 expression, total cell lysates were blotted with the corresponding antibodies. (B) The amount of GTP-bound and total Cdc42 (A, bottom band of the doublets) was quantified by densitometric analysis, and the activation level of Cdc42 was expressed as a ratio between the values of GTP-bound and total Cdc42. Comparable results were obtained in three independent experiments. (C) Total protein extract of COS cells transfected with GST-Cdc42 was loaded on Sepharose coupled with MBP–ICAP-1 fusion protein or MBP as control. The proteins eluted with glycine-HCl, pH 3.0, buffer (fractions numbers 1-2-3-4) were analyzed by Western blotting with polyclonal antibody against Cdc42. Note that in A, Cdc42 is detected as a doublet of bands; comparison of the electrophoretic mobility indicates that the bottom band of the doublet in A comigrates with the band in C. The top band of the doublet is likely to represent an incompletely processed form of the GTPase. (D) Western blotting with an antibody against Cdc42 of the pooled fractions eluted from MBP–ICAP-1-Sepharose columns (right). The columns were loaded with equal amounts of GST-Cdc42 fusion protein obtained from either the membrane fraction of pCEFL-GST-Cdc42–transfected COS cells (a), the cytoplasmic fraction of the same cells (b), or bacterial lysate of E. coli producing the GST-Cdc42 fusion protein (c). The amount of fusion proteins loaded on the column are shown on the left as control for equal loading.
Figure 4.



ICAP-1 inhibits and binds Rac1. (A) COS cells transiently transfected with control vector or myc–ICAP-1 were suspended by trypsin treatment, kept in suspension 2 h 30 min in serum-free DME (Su), and plated on fibronectin (5 μg/ml) for 1 h (Ad). Rac1 activation level was evaluated as described in Materials and methods using a GST-PAK-CD fusion protein that selectively binds to GTP-Rac1. The bound GTP-Rac1 was analyzed by Western blotting with a polyclonal antibody to Rac1. To probe for Rac1 and ICAP-1 expression, total cell lysates were blotted with the corresponding antibodies. (B) The amount of GTP-bound and total Rac1 (A) was quantified by densitometric analysis; the activation level of Rac1 is expressed as a ratio between the values of GTP-bound and total Rac1. Comparable results were obtained in three independent experiments. (C) Total lysate of COS cells transfected with myc-Rac1 was loaded on Sepharose coupled with MBP–ICAP-1 fusion protein or MBP as control. The proteins eluted with glycine-HCl, pH 3, buffer (fractions numbers 1-2-3-4) were analyzed by Western blotting with a monoclonal antibody against Rac1.
In the experimental condition used to analyze the activation level of Cdc42 and Rac1 during cell spreading, we were not able to detect any significant activation of RhoA, as measured by pull-down assay with the Rho effector protein mDia (Kimura et al., 2000), in accordance with previous reports (Ren et al., 1999; Arthur et al., 2000). Given that the adhesive stimulus was not sufficient to induce RhoA activation, we then activated RhoA by either treating COS cells with LPA or transfecting them with pCEFL-GST-DH/PH coding for a GST fusion protein containing the DH/PH domains of the GEF protein Dbl. However, in both of these conditions ICAP-1 expression did not affect RhoA activity (unpublished data; Fig. 5 A).
Figure 5.

ICAP-1 does neither inhibit nor bind RhoA. (A) RhoA activity assay was performed as described in Materials and methods using COS cells either untransfected or transfected as indicated. The same blot was reprobed for ICAP-1 expression with a polyclonal antibody against ICAP-1. (B) Total protein extract of COS cells was loaded on Sepharose coupled with MBP–ICAP-1 fusion protein or MBP as control. The eluted proteins were analyzed by Western blotting with a monoclonal antibody against RhoA. The same filter was stripped and tested for the presence of Rac1 using a specific monoclonal antibody (B'). Total extract is shown as control.
To better understand the inhibitory action of ICAP-1 on Cdc42 and Rac1, we performed affinity chromatography experiments to test the ability of these proteins to interact with each other. COS cell extracts were incubated with Sepharose coupled to purified MBP–ICAP-1 fusion protein. As shown in Fig. 3 C and Fig. 4 C, Cdc42 and Rac1 bind to MBP–ICAP-1 but not to MBP carrier protein.
On the other hand, affinity chromatography experiments showed that RhoA did not appreciably bind to MBP–ICAP-1 (Fig. 5 B). Moreover, we tested the binding of transfected molecules unrelated to GTPases, such as melusin (Brancaccio et al., 1999), or endogenous COS proteins such as p125Fak and paxillin. None of these proteins bind to ICAP-1 (unpublished data), demonstrating the specificity of the interaction with Cdc42 and Rac1 GTPases.
Rho family GTPases are posttranslationally modified in eukaryotic cells by covalent binding of an isoprenyl chain that allows association with cellular membranes to exert their biological activity. To test whether this modification affects binding to ICAP-1, Cdc42-containing extracts were prepared from membrane and cytosol fractions of COS and prokaryotic cells. Maximal binding was observed for Cdc42 extracted from the membrane fraction, whereas binding of Cdc42 from prokaryotic cells was eightfold less efficient, suggesting that isoprenylation is important for this interaction (Fig. 3 D). In addition, the reduced binding of cytosolic Cdc42 to ICAP-1 compared with the membrane Cdc42 fraction is likely due to a preferential binding of cytoplasmic Cdc42 to endogenous Rho-GDI.
In addition we found that ICAP-1 binds with similar intensity to the GTP or GDP form of Cdc42 (unpublished data).
ICAP-1 inhibits GDP dissociation
GAPs and GDIs are important negative regulators of monomeric GTPases. Comparative sequence analysis indicated a partial degree of homology between ICAP-1 and known GDI molecules (see Fig. 8 and Discussion). To test whether ICAP-1 can exert GDI activity, Cdc42 was expressed in COS cells as a GST fusion protein, and the membrane-bound fraction was affinity purified. After loading of Cdc42 with radioactive GTP, the release of the nucleotide induced either by chelating Mg2+ ions or by the Dbl GEF was measured in the presence of purified ICAP-1, unrelated molecule, such as the carrier protein MBP, or a known GDI such as RhoGDI. As shown in Fig. 6 , ICAP-1 inhibits the dissociation of GDP induced either by EDTA (see Fig. 6 A) or by Dbl GEF (see Fig. 6 B). MBP control protein does not alter the release of GDP with respect to buffer control (see Fig. 6 C).
Figure 8.

Sequence alignment of bovine RhoGDI (RhoGDIBt) and ICAP-1 proteins. Sequences of human ICAP-1 (sequence data available from GenBank/EMBL/DDBJ under accession no. NP_004754) and bovine RhoGDI (accession no. CAA36916) were aligned using CLUSTAL W (1.81). Lines above RhoGDIBt sequence indicate secondary structure elements as reported by Hoffman et al. (2000). Lines under ICAP-1A sequence indicate a prediction of its secondary structure elements as obtained using a Biology Work Bench protein analysis tool based on the DSC algorithm (King and Sternberg, 1990). Shaded grey areas indicate residues of the RhoGDIBt that interact with Cdc42 geranylgeranyl moiety. Asterisks indicate single fully conserved residue. Colons indicate conservation of strong groups. Periods indicate conservation of weak groups.
Figure 6.



Inhibitory effect of ICAP-1 on the dissociation of [α-32P]GDP from Cdc42. GST-Cdc42 was expressed in COS cells, and the protein was purified from the membranous fraction and captured on glutathione-Sepharose. The Cdc42 immobilized on the beads was loaded with guanosine nucleotide by incubation with [α-32P]GTP 25 min at room temperature. The dissociation of the GDP induced by chelating Mg2+ with EDTA (A) or by the GEF activity of Dbl (B) was measured in the presence of either the buffer alone, MBP–ICAP-1(10 μg), or the same amount of GST-Rho GDI fusion proteins after 10 min at room temperature. (C) Time course for [α-32P]GDP dissociation from Cdc42 induced by chelating Mg2+ with EDTA in the presence of either 10 μg of MBP–ICAP-1, GST-Rho GDI, MBP carrier protein, or buffer alone. Results shown are means ± SD of values from three experiments. [α-32P]GDP remaining is expressed as the percentage of [α-32P]GDP bound to Cdc42 after loading.
A known activity of GDIs is their ability to induce the release of the GTPase from cellular membranes (Leonard et al., 1992). To test for this activity, membrane fractions from COS cells transfected with Cdc42 constructs were incubated with purified MBP–ICAP-1 fusion protein or MBP alone. As shown in Fig. 7 , increasing amounts of purified ICAP-1 caused an increasing level of Cdc42 to be released into a soluble supernatant fraction. The result demonstrates that ICAP-1, besides being able to inhibit GDP dissociation, is also able to specifically elicit the release of Cdc42 from the membrane.
Figure 7.

ICAP-1 stimulated release of Cdc42 from the membranes of COS cells. Membranes from COS cells transfected with myc-Cdc42 were prepared as described in Materials and methods. The membranes were incubated with the indicated amounts of purified MBP–ICAP-1 or MBP alone as a control for 25 min at room temperature. After centrifugation, the supernatant fractions were examined for the presence of Cdc42 using SDS-PAGE and Western blotting with a specific antibody (A). The relative amount of Cdc42 released into supernatant fraction was determined by densitometric scanning analysis (B).
Discussion
Here we report that ICAP-1, a protein which binds to the β1 integrin cytoplasmic domain, inhibits cell spreading on extracellular matrix substratum by interfering with Cdc42 and Rac1 activation.
Hemler and coworkers have shown that ICAP-1 can promote β1-integrin–dependent cell migration (Zhang and Hemler, 1999). These authors have not analyzed the effect of ICAP-1 on cell spreading on extracellular matrix proteins, but their observation is consistent with our finding that ICAP-1 reduces cell spreading.
Based on tests detecting effector protein activation, such as p21PAK and ACK2, it has been proposed that cell matrix adhesion causes rapid and transient activation of Cdc42 and Rac1 (Price et al., 1998; Yang et al., 1999). On the other hand, during the initial phase of cell spreading RhoA activity is not increased (Ren et al., 1999) and might be even inhibited via a src-p190RhoGAP pathway (Arthur et al., 2000). Although Cdc42 and Rac1 stimulate actin polymerization and assembly of protrusive membrane structures, such as filopodia and lamellipodia, necessary for cell spreading and locomotion on the substratum, RhoA is known to promote actin filament assembly in stress fibers and to increase the contractile forces exerted by the actomyosin filaments (Schoenwaelder and Burridge, 1999). Thus, it has been proposed that inhibition of RhoA during initial phases of cell spreading is necessary to prevent contractile forces that would interfere with the highly dynamic actin-based activity occurring at membrane protrusions during cell adhesion or migration.
Here, by directly measuring GTP-bound forms we show that adhesion to fibronectin induces activation of Cdc42 and Rac1. Moreover, we report that ICAP-1 binds to both Cdc42 and Rac1 and prevents their activation along with cell spreading. Interestingly, RhoA binds very poorly to ICAP-1 and is not significantly activated during cell adhesion to fibronectin under the experimental conditions used. In addition, the activation state of RhoA triggered by the GEF protein Dbl or LPA treatment is not affected by ICAP-1 expression. Together, these data suggest that ICAP-1 acts as specific regulator of Cdc42 and Rac1 GTPases during the initial phases of cell spreading on extracellular matrix protein.
Rho family GTPases are covalently modified by isoprenylation that is required for their translocation from the cytosol to the cellular membrane where they perform their function (Van Aelst and D'Souza-Schorey, 1997). On the other hand, through their isoprenoid moiety these GTPases can interact with GDI molecules that keep them in an inactive cytoplasmic pool (Regazzi et al., 1992). Interestingly, we found that addition of purified ICAP-1 to a membrane fraction induces the release of membrane-bound Cdc42 and that ICAP-1 is able to reduce the release of the guanine nucleotide from Cdc42 in the presence of a known exchange factor such as Dbl. In addition, ICAP-1 was found to bind more strongly to Cdc42 isolated from the membrane fraction than to cytosolic Cdc42 or Cdc42 synthesized in prokaryotic cells where the GTPases cannot be isoprenylated. These properties support the idea that ICAP-1 exert a GDI activity. In our assay conditions, this activity was lower than that of RhoGDI, suggesting that ICAP-1 has distinct and unique properties from a previously characterized GDI. We can hypothesize that ICAP-1 acts only on the limited pool of Cdc42 and Rac1 involved in the turnover of new contact sites, spatially restricted to filopodia and lamellipodia during spreading or migration processes, whereas the remaining pool of Cdc42 could be regulated by other GDI molecules such as RhoGDI.
Interestingly, comparative sequence analysis indicated an overall homology of 27% between ICAP-1 and RhoGDI (Fig. 8) and several other members of the GDI family (unpublished data). The homology was particularly striking (34%) in the COOH terminal region spanning from residues 80 to 200. As shown by Hoffman et al. (2000), this region of RhoGDI consists of an immunoglobulin-like domain that binds to the isoprenyl moiety of the GTPase. More interestingly, 60% of the RhoGDI residues shown to be crucial for this interaction (Fig. 8, gray shaded residues) are conserved in the corresponding region of ICAP-1. Moreover, the predicted secondary structure of this region of ICAP-1 is significantly similar to the one determined by crystallography for the RhoGDI as shown in Fig. 8 (Hoffman et al., 2000). Thus, these structural homologies are consistent with the ability of ICAP-1 to bind Cdc42 and release it from the membrane. In addition, the amino acid residues 45 and 47 in RhoGDI (Fig. 8) crucial for the GDI activity and conserved in several GDI molecules are also conserved in ICAP-1 (Fig. 8).
At present, we do not know whether ICAP-1 has a GDI activity on Rac1, and we cannot definitively conclude whether the inhibition of Rac1 observed with the pull-down PAK assay is due to a direct GDI activity on this GTPase. It is also possible that Rac1 is inhibited as a consequence of Cdc42 inhibition by ICAP-1. It has been proposed that these GTPases can act in cascade, Cdc42 being able to activate Rac1 (Nobes and Hall, 1995). However, the ability of ICAP-1 to bind Rac1 suggests that ICAP-1 should directly affect Rac1 activity. Further studies are necessary to precisely define the detailed mechanisms of ICAP-1 function on Rac.
Previous work has shown that integrins can regulate GEF function. GEFs are proteins capable of stimulating the exchange of GDP with GTP, thus increasing the active GTP-bound form of the GTPases. In particular, two GEF molecules, Vav1 and cytohesin1, can be regulated in response to cell matrix adhesion. Vav1 is an exchange factor for Rac1 that can be activated by tyrosine phosphorylation (Crespo et al., 1997). Indeed, integrin-mediated cell adhesion was shown to induce Vav1 tyrosine phosphorylation in different cell types (Cichowski et al., 1996; Gotoh et al., 1997; Miranti et al., 1998; Yron et al., 1999). Cytohesin1 is a GEF for ARF, a GTPase known to be involved in the regulation of vesicle traffic in the secretory/endocytic pathway. Recent results have shown that cytohesin1 is crucial in regulating integrin affinity state for the extracellular ligand, a process known as inside/out signaling (Geiger et al., 2000). Both Vav1 and cytohesin1 are expressed preferentially in hemopoietic cell lineages and are likely to play a major role in this system. Another GEF protein that is likely to play a role in integrin-mediated activation of Cdc42 and Rac1 is PIX (Turner et al., 1999). This protein is part of a large complex that includes PAK, NCK, p95PKL, and paxillin. The important role played by paxillin in cell adhesion, and the localization of this complex at focal adhesions (Turner et al., 1999) strongly argues for an important role of PIX as a downstream effector in integrin-mediated actin cytoskeleton organization.
The existence of a GDI protein, like ICAP-1, in the pathway of integrin signaling suggests that during cell matrix adhesion not only GEF should be activated in order to increase the GTP-bound form of the GTPases, but the inhibitory action of the GDI molecule should be released. A hypothetical model can be envisaged in which in cells detached from the matrix, ICAP-1 forms an inhibitory complex with Cdc42 by displacing it from the plasma membrane and preventing the exchange of the GDP for the GTP. Upon interaction with extracellular matrix, ICAP-1 is modified to neutralize its GDI activity. Indeed, it has been shown that ICAP-1 is phosphorylated upon integrin-mediated cell adhesion to fibronectin (Chang et al., 1997; Zhang and Hemler, 1999), and it can thus be postulated that this event affects the GDI activity of the protein. In the experimental system used here, ICAP-1 levels are increased several fold over the basal level in consequence of transfection, and thus its inhibition of the Rho family GTPases persists for longer times, causing the delay in cell spreading. The functional significance of ICAP-1 binding to the β1-integrin cytoplasmic domain is still to be defined. It is possible that ICAP-1 binds to the integrin cytoplasmic domain to perform other functions such as regulation of integrin affinity as proposed by Block and coworkers (Bouvard and Block, 1998). Indeed, we did not observe any difference in ICAP-1 effect on cell spreading using two distinct extracellular matrix substrates, such as fibronectin and vitronectin, suggesting that the potential β1A integrin-mediated control of ICAP-1 function was overcome due to ICAP-1 overexpression levels. However, overexpression approach led us to bring to light the important function of ICAP-1 as a GDI molecule.
Furthermore, ICAP-1 specifically binds to the β1A isoform in the COOH terminal region known to differ among the splicing isoforms (Balzac et al., 1994; Fornaro et al., 1995; Belkin et al., 1996). Binding involves the more COOH terminal NPXY motive present in β1A integrin cytoplasmic domain (Chang et al., 1997) and residues immediately upstream (Table I). Interestingly, two isoforms of the β1 integrin, β1B and β1D, that do not bind ICAP-1 are incompetent to support cell spreading and cell migration on fibronectin (Balzac et al., 1994; Belkin et al., 1997). These properties are consistent with the hypothesis that binding of ICAP-1 to β1 integrin is required to release the inhibitory effect of this protein on Cdc42 and Rac1, thus promoting cell spreading and migration. However, further work is necessary to test this hypothesis.
In conclusion, the data reported here show that ICAP-1, a protein isolated as an interactor of the β1-integrin cytoplasmic domain, binds and negatively regulates Cdc42 and Rac1, acting as a guanine nucleotide dissociation inhibitor. Thus, this protein is a new element, potentially connecting integrin-mediated cell matrix adhesion with regulation of the actin cytoskeleton.
Materials and methods
Interaction trap
Screening for proteins that interact with cytoplasmic tails of β1A was performed as described (Gyuris et al.,1993). To construct the bait plasmid, DNA sequence encoding amino acids 752–798 of β1A was amplified by PCR using primers containing EcoRI and BamHI sites on either ends (β1A, 5′-GGAATTCAAGCTTTTAATGA4TAATT-3′ and 5′-CGGGATCCTCATTTTCCCTCATACTT-3′) and cloned in pEG 202 vector in frame with LexA coding sequence. This plasmid was unable to activate transcription when cotransformed in EGY48 yeast strain with pSH 18–34 reporter plasmid. The expression of this fusion protein in yeast total protein extracts was confirmed by Western blot analysis using an anti-LexA antibody (a gift from A. Zervos, University of Central Florida, Orlando, FL). A cDNA library from heart neonatal rat fused to a galactose-inducible activation domain (a gift from A. Zervos) was transformed in yeast strain EGY48 that already contained pSH 18–34 reporter plasmid and pEG202-β1A using a LiAc high efficiency transformation protocol (Gietz et al., 1992). Primary transformants (2 × 106) were screened by plating 107 colonies on selectable medium lacking Ura, His, Trp, and Leu. Several positive clones were isolated, and their specific binding to integrins was tested by assaying the interaction with control baits such as bicoid, bFGF, cyclin A, and c-Myc (a gift from A. Zervos).
Other baits were produced to test the ability of ICAP-1 to interact with other integrin cytoplasmic tails. These baits were prepared by PCR amplification using the following primers: (5′-catgccatggaaggctctgatccacctg-3′ and 5′ccgctcgagctaactctcagcaaactt-3′) coding for amino acid residues 724–769 of β2; (5′-gcatgccatggaaactcctcatcaccatc-3′ and 5′-ccgctcgagttaagtgccccggtacgt-3′) coding for amino acid residues 716–762 of human β3; (5′-ggaattcaagcttttaatgataatt-3′ and 5′-cgggatccttataagccactttgctt-3′) coding for amino acid residues 752–789 of β1B; (5′-ggaattcaagcttttaatgataatt-3′ and 5′-cgggatcctcagagaccagctttacg-3′) coding for amino acid residues 752–801 of β1D; and (5′-ggaattcaagcttggattcttcaaa-3′ and 5′-cgggatcctcaggcatcagaggtggc-3′) coding for amino acid residues 1022–1049 of α5.
To map the ICAP-1 binding site in β1A, four swapping mutant constructs of β1A and β1D cytoplasmic regions were generated by PCR amplification and cloned in frame with LexA sequence in pEG 202. The following mutants were generated: β1A/D (A778–795/D796–801), β1D/A (D778–795/A796–798), β1A/D/A (A778–785/D786–791/A792–798), and β1D/A/D (D778–785/A786–791/D792–801). Numbers indicate the amino acid residues in the cytoplasmic region of β1A and β1D (Table I).
Constructs and antibodies
Positive clones selected with the two-hybrid screening were sequenced using ABI PRISM Big Dye Terminator Cycle Sequencing Ready Reaction kit (PerkinElmer). A10-1 cDNA fragment contained the sequence of ICAP-1 lacking a 5′ fragment coding for the first 12 amino acids of the protein. The full length of ICAP-1 was constructed by PCR amplification. A10.1 clone was used as template and annealed to a synthetic oligonucleotide coding for the first 12 missing amino acids of the protein. The coding sequence was obtained from the mouse ICAP-1 cDNA (Faisst and Gruss, 1998). A second cycle of PCR amplification was made to introduce a myc tag and KpnI restriction site. The PCR products were digested with KpnI and XhoI and inserted into pcDNA3 eukaryotic expression vector.
To produce large amounts of soluble protein, the full-length ICAP-1 was fused to maltose binding protein for expression in prokaryotic cells. Using pcDNA3-myc–ICAP-1 as template, EcoRI restriction enzyme site was introduced by PCR amplification in ICAP-1 full-length cDNA. The fragment was cloned into the EcoRI/XbaI sites of pMAL-p2 vector. All constructs were sequenced using ABI PRISM Big Dye Terminator Cycle Sequencing Ready Reaction kit.
The following prokaryotic expression constructs were used to express GST fusion proteins: pGEX PAK-CD, encoding the PAK-CRIB, was provided by J. Collard (Netherland Cancer Institute, Amsterdam, Netherlands) (Sander et al., 1998). pGEX RhoGDI was provided by Y. Zheng (University of Tennessee, Memphis, TN) (Leonard et al., 1992). pGEX-Cdc42 was provided by R. Cerione (Cornell University, Ithaca, NY). PGEX-mDIA was provided by S. Narumiya (Kyoto University, Kyoto, Japan).
To express GST fusion proteins in eukaryotic cells, Cdc42Hs cDNA and cDNA coding for Dbl DH/PH domains (residues 497–875) were cloned into the BamHI site of the pCEFL-GST vector provided by S. Gutkind (National Institutes of Health, Bethesda, MD).
pcEXV-myc-V12Cdc42, pRK5-myc-Cdc42, and pRK5-mycRac1 constructs, a gift from A. Hall (University College of London, London, UK), were used for transient expression in NIH3T3 and COS cells.
Polyclonal rabbit antiserum against ICAP-1 was prepared by immunizing a rabbit with MBP–ICAP-1 fusion protein.
Antibodies against Cdc42 and RhoA were purchased from Santa Cruz Biotechnology, Inc. 9E10 anti-myc monoclonal antibody was purchased from Sigma-Aldrich. Anti-Rac1 monoclonal antibody was purchased from Upstate Biotechnology.
Cell cultures and transfection
COS cells cultured in DME supplemented with 10% FCS were transiently transfected at 80% confluence by the DEAE-dextran method (Kluxen and Lubbert, 1993) and used for experiments 48 h after DNA addition.
NIH3T3 fibroblasts were cultured in DME supplemented with 10% calf serum. For Immunofluorescence experiments, cells were plated at a density of 12×104 into 6-well culture plates and transfected with 10 μg of plasmid DNA using the calcium phosphate procedure (Chen and Okayama, 1987). In cotransfection experiments, the various DNA constructs were used at a 1:1 ratio. Cells were used for experiments 36 h after DNA addition.
To obtain stable expression of V12Cdc42, NIH3T3 were transfected with pZipNeo-V12Cdc42 by the calcium phosphate procedure and grown for 3 wk in the presence of 375 μg/ml G418 as described (Olivo et al., 2000).
Immunofluorescence analysis
Transiently transfected cells were detached gently with 5 mM EDTA in PBS, washed with PBS containing 1 mM CaCl2, 1 mM MgCl2, and plated in serum-free DME for 1 h onto fibronectin-coated coverslips. Cells were fixed in 3% paraformaldehyde for 10 min and permeabilized with 0.5% Triton X-100/TBS (150 mM NaCl, 50 mM Tris-HCl, pH 7.4) for 1 min. F-actin was revealed by staining with fluorescein-FITC-phalloidin (Sigma-Aldrich). Transiently transfected cells were revealed with anti-myc monoclonal antibody 9E10. Cells were observed and counted under an Olympus fluorescence microscope, and pictures were taken with an Olympus DP10 digital photomicrography system. To evaluate the degree of cell spreading, the cell area was measured by a computerized image analysis system. Cells with an area >150 μm2 were considered spread.
Rho GTPases activity assays
The assay described by Sander et al. (1998) based on the interaction of the GTP-bound GTPase to the PAK CRIB domain was used. To prepare the GST-PAK CRIB domain fusion protein, Escherichia coli TOPF1 10 cells transformed with the GST-PAK-CD construct were harvested and resuspended in lysis buffer (50 mM Tris-Hcl, pH 7.5, 1 mM EDTA, 100 mM NaCl, 5% glycerol, 0.1% Triton X-100, 1 mM DTT, 10 μg/ml leupeptin, 0.4 μg/ml pepstatin, 0.1 TIU/ml aprotinin, 1 mM PMSF) and sonicated. After centrifugation, the cell lysate supernatant was incubated with glutathione-coupled Sepharose 4B beads (Amersham Pharmacia Biotech) for 30 min at 4°C. The beads were washed three times in lysis buffer, and the amount of bound fusion protein was estimated after separation by SDS-PAGE and Coomassie blue staining.
COS cells transiently transfected with the indicated constructs were detached with trypsin-EDTA (0.05% trypsin, 0.02% EDTA in PBS), washed twice with serum-free DME, and maintained in suspension for 2 h 30 min at 4°C in serum free DME before plating on coated dishes. 1 h after seeding, the cells were washed in ice-cold PBS, incubated 5 min on ice in lysis buffer (50 mM Tris-HCl, pH 7.4, 2 mM MgCl2, 100 mM NaCl, 10% glycerol, 1% NP-40, 10 μg/ml leupeptin, 0.4 μg/ml pepstatin, 0.1 TIU/ml aprotinin, 1 mM PMSF), and then centrifuged for 5 min at 4°C at 16,000 g. Aliquots were taken from the supernatant to compare protein amounts. The supernatant was incubated with GST-PAK-CD bound to glutathione–Sepharose beads (Amersham Pharmacia Biotech) at 4°C for 30 min. The beads were washed three times with an excess of lysis buffer, and bound proteins were eluted in Laemmli sample buffer and analyzed for the presence of Cdc42 or Rac1 molecules by Western blotting using antibodies specific for Cdc42 and Rac.
Rho activation state was assessed by using GST-mDIA fusion protein to pull down GTP-loaded RhoA accordingly with Kimura et al. (2000).
Affinity chromatography
To prepare the affinity matrix, ICAP-1 was expressed as a MBP fusion protein and purified from E. coli by affinity chromatography on amilose resin (Biolabs). 5 mg of purified MBP–ICAP-1 or MBP alone as control were dialyzed overnight at 4°C in coupling buffer (0.1 M NaHCO3/Na2CO3, pH 9.2) and incubated for 2 h at room temperature with 430 mg of dried CNBr-activated Sepharose 4B (Amersham Pharmacia Biotech).
Cdc42-containing cell extracts were prepared by expressing the GST–Cdc42 fusion protein in E. coli and in COS cells using pGEX-Cdc42 and pCEFL-GST-Cdc42 constructs, respectively. Transfected COS cells were lysed with 50 mM Tris-HCl, pH 7.4, 2 mM MgCl2, 100 mM NaCl, 10% glycerol, 1% NP-40, 10 μg/ml leupeptin, 4 μg/ml pepstatin, 0.1 TIU/ml aprotinin, and 1 mM PMSF. After centrifugation at 10,000 g, total soluble proteins were chromatographed on MBP–ICAP-1 Sepharose. After washing with five volumes of TBS, the proteins were eluted with 0.1 M glycine-HCl, pH 3. The collected fractions (500 μl each) were neutralized by titration with 1.5 M Tris-HCl, pH 8.8, lyophilized, and analyzed by SDS-PAGE and Western blotting with polyclonal rabbit antibody against Cdc42.
To compare the binding capacity to ICAP-1 of the membrane-bound cytosolic or E. coli produced Cdc42 extracts from cytoplasmic and membrane fractions of COS cells (see below for membrane fraction preparation) or bacterial lysates were loaded on the MBP–ICAP-1 Sepharose column. Columns were loaded with equal amounts of Cdc42 as determined by Western blotting. To test if the binding activity of the GTPases to ICAP-1 was affected by the nature of the bound guanine nucleotide, the procedure described by Hart et al. (1994) was followed.
To test ICAP-1 binding to Rac1 and RhoA, total extracts of COS cells were used in the affinity chromatography procedure described above, and the eluted proteins were analyzed by Western blotting with specific monoclonal antibodies.
Assay of GDP dissociation from Cdc42
The release of GDP from Cdc42 was measured as described (Leonard et al., 1992).
To purify the isoprenylated form of Cdc42, a construct encoding for the GST-Cdc42 fusion protein (pCEFL-GST-Cdc42) was transfected in COS cells. 48 h after transfection, 4×107 cells were trypsinized, washed with PBS, and lysed in hypotonic lysis buffer (20 mM Tris-HCl, pH 8, 1 mM EDTA, 1 mM DTT, 1 mM PMSF, 0.1 TIU aprotinin, 10 μg/ml leupeptin, 4 μg/ml pepstatin) with 20 strokes of a 7-ml glass/glass Dounce homogenizer. Homogenates were centrifuged at 10,0000 g for 1 h, the cytosol-containing supernatant was removed, and the crude membrane pellet was solubilized with TBS containing 1% Triton X-100, 10 μg/ml leupeptin, 4 μg/ml pepstatin, 0.1 TIU/ml aprotinin, and 1 mM PMSF for 90 min at 4°C and then clarified by centrifugation at 10,0000 g for 1 h. The solubilized membrane fraction was incubated with glutathione-coupled Sepharose beads at 4°C for 30 min. The beads were then washed three times with PBS, and the amount of bound fusion protein was estimated using Coomassie-stained SDS gels.
To load Cdc42 with a radiolabeled nucleotide, the beads were first incubated with 20 mM Tris-HCl, pH 8, 50 mM NaCl, 100 μM AMP-PNP, 100 μM DTT, 2 mM MgCl2, and 0.7 μM [α-32P]GTP for 25 min at room temperature and washed extensively in the same buffer to remove unbound radiolabeled nucleotide. Aliquots (5 μl) of guanine nucleotide–loaded Cdc42 bound to the beads (typically 200–300 ng in 5 μl) were incubated with 20 μl of 50 mM Tris-HCl, pH 7.5, 1 mM DTT, 1 mM EDTA, and 2 mM MgCl2, containing purified proteins (MBP, MBP–ICAP-1, GST-RhoGDI, GST-Dbl/DH-PH), or the buffer alone and were allowed to incubate for an additional 10 min at room temperature. To induce the release of the GDP, 32 μl of GDI buffer (55 mM Tris-HCl, pH 7.5, 45 mM NaCl, 1 mM AMP-PNP, 100 μM DTT, 5 mM EDTA, 1 mM GDP, 0.55 mg/ml BSA) were added to each sample. After the indicated time of incubation at room temperature, the reaction was stopped by diluting the samples in 1.8 ml of ice-cold buffer containing 20 mM Tris-HCl, pH 8, 100 mM NaCl, and 10 mM MgCl2, washed three times with the same buffer, and analyzed in the β counter. When the dissociation of the GDP was measured in the presence of 100 ng of the GEF GST-Dbl, the assays were performed essentially as outlined above except that EDTA was omitted and 2 mM MgCl2 was added to the GDI buffer.
RhoGDI was expressed as a GST fusion protein from pGEX vector and purified from E. coli using glutathione–Sepharose 4B column. GST-Dbl (DH-PH domain) was transfected in COS cells and purified using glutathione–Sepharose 4B column.
Release of Cdc42 protein from COS cell membranes
Adherent COS cells were transfected with pRK5-myc-Cdc42 construct as described above. Transfected cells were frozen in liquid nitrogen and thawed in 10 ml hypotonic buffer consisting of 20 mM Tris-HCl, pH 8, 6 mM EDTA, 1 mM DTT, 1 mM PMSF, 0.1 TIU aprotinin, 10 μg/ml leupeptin, 4 μg/ml pepstatin, and lysed with 10 strokes of a 7-ml glass/glass Dounce homogenizer. The nuclei and any unbroken cells were removed from the sample by centrifugation at 1,000 g for 12 min. The supernatant was centrifuged at 27,000 g for 10 min at 4°C to pellet the particulate fraction. After removal of the supernatant, the membranes were washed twice in hypotonic buffer, resuspended in 1.5 ml of 20 mM Tris-HCl, 1 mM EDTA, 1 mM DTT, pH 8, and aliquoted for long-term storage at –80°C.
A 10-μl aliquot of membrane fraction was mixed with 8 μl of reaction buffer (100 mM Tris-HCl, 250 mM NaCl, 25 mM MgCl2, pH 8) and 22 μl of distilled H2O containing increasing amounts of purified MBP–ICAP-1 or MBP as negative control. This mixture was incubated at room temperature for 25 min and then centrifuged at 27,000 g for 10 min at 4°C to separate the membranes from the soluble fraction. The supernatant and the pellet were then analyzed by SDS-PAGE and Western blotting with polyclonal rabbit antibody against Cdc42. The relative amount of Cdc42 was determined by densitometric scanning analysis.
Acknowledgments
We are indebted to A. Zervos, J. Collard, Y. Zheng, R. Cerione, S. Gutkind, S. Narumiya, and A. Hall for shearing reagents and to C. Brancolini for the microinjection analysis. We also thank C. Olivo, P. Defilippi, and G. Topley for helpful discussion and suggestions.
This work was supported by grants from the Italian Association for Cancer Research to G. Tarone and A. Eva and MURST (Cofinanziamento Programmi di Ricerca).
S. Degani and F. Balzac contributed equally to this work.
Footnotes
Abbreviations used in this paper: DH, Dbl homology; GAP, GTPase-activating protein; GDI, guanine nucleotide dissociation inhibitor; GEF, guanine nucleotide exchange factor; ICAP, integrin cytoplasmic domain-associated protein; LPA, lysophosphatidic acid; PH, pleckstrin homology.
References
- Arthur, W.T., L.A. Petch, and K. Burridge. 2000. Integrin engagement suppresses RhoA activity via a c-Src-dependent mechanism. Curr. Biol. 10:719–722. [DOI] [PubMed] [Google Scholar]
- Balzac, F., S.F. Retta, A. Albini, A. Melchiorri, V.E. Koteliansky, M. Geuna, L. Silengo, and G. Tarone. 1994. Expression of beta1B integrin isoform in CHO cells results in a dominant negative effect on cell adhesion and motility. J. Cell Biol. 127:557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkin, A.M., N.I. Zhidkova, F. Balzac, F. Altruda, D. Tomatis, A. Maier, G. Tarone, V.E. Koteliansky, and K. Burridge. 1996. Beta1D integrin displaces the beta 1A isoform in striated muscles: localization at junctional structures and signaling potential in nonmuscle cells. J. Cell Biol. 132:211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkin, A.M., S.F. Retta, O.Y. Pletjushkina, F. Balzac, L. Silengo, R. Fassler, V.E. Koteliansky, K. Burridge, and G. Tarone. 1997. Muscle beta1D integrin reinforces the cytoskeleton-matrix link: modulation of integrin adhesive function by alternative splicing. J. Cell Biol. 139:1583–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvard, D., and M.R. Block. 1998. Calcium/calmodulin-dependent protein kinase II controls integrin alpha5 beta1-mediated cell adhesion through the integrin cytoplasmic domain associated protein-1alpha. Biochem. Biophys. Res. Commun. 252:46–50. [DOI] [PubMed] [Google Scholar]
- Brancaccio, M., S. Guazzone, N. Menini, E. Sibona, E. Hirsch, M. De Andrea, M. Rocchi, F. Altruda, G. Tarone, and L. Silengo. 1999. Melusin is a new muscle-specific interactor for beta1 integrin cytoplasmic domain. J. Biol. Chem. 274:29282–29288. [DOI] [PubMed] [Google Scholar]
- Chang, D.D., C. Wong, H. Smith, and J. Liu. 1997. ICAP-1, a novel beta1 integrin cytoplasmic domain-associated protein, binds to a conserved and functionally important NPXY sequence motif of beta1 integrin. J. Cell Biol. 138:1149–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C., and H. Okayama. 1987. High-efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell. Biol. 7:2745–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichowski, K., J.S. Brugge, and L.F. Brass. 1996. Thrombin receptor activation and integrin engagement stimulate tyrosine phosphorylation of the proto-oncogene product, p95vav, in platelets. J. Biol. Chem. 271:7544–7550. [DOI] [PubMed] [Google Scholar]
- Clark, E.A., W.G. King, J.S. Brugge, M. Symons, and R.O. Hynes. 1998. Integrin-mediated signals regulated by members of the rho family of GTPases. J. Cell Biol. 142:573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo, P., K.E. Schuebel, A.A. Ostrom, J.S. Gutkind, and X.R. Bustelo. 1997. Phosphotyrosine-dependent activation of Rac-1 GDP/GTP exchange by the vav proto-oncogene product. Nature. 385:169–172. [DOI] [PubMed] [Google Scholar]
- Faisst, A.M., and P. Gruss. 1998. A novel murine gene expressed in restricted areas of the brain. Dev. Dyn. 212:293–303. [DOI] [PubMed] [Google Scholar]
- Fornaro, M., D.Q. Zheng, and L.R. Languino. 1995. The novel structural motif Gln795-Gln802 in the integrin beta1C cytoplasmic domain regulates cell proliferation. J. Biol. Chem. 270:24666–24669. [DOI] [PubMed] [Google Scholar]
- Geiger, C., W. Nagel, T. Boehm, Y. Van Kooyk, C.G. Figdor, E. Kremmer, N. Hogg, L. Zeitlmann, H. Dierks, K.S. Weber, and W. Kolanus. 2000. Cytohesin-1 regulates beta-2 integrin-mediated adhesion through both ARF-GEF function and interaction with LFA-1. EMBO J. 19:2525–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz, D., A. St Jean, R.A. Woods, and R.H. Schiestl. 1992. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 20:1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyuris, J., E. Golemis, H. Chertkov, and R. Brent. 1993. Cdi1, a human G1 and S phase protein phosphatase that associates with Cdk2. Cell. 75:791–803. [DOI] [PubMed] [Google Scholar]
- Gosser, Y.Q., T.K. Nomanbhoy, B. Aghazadeh, D. Manor, C. Combs, R.A. Cerione, and M.K. Rosen. 1997. C-terminal binding domain of Rho GDP-dissociation inhibitor directs N-terminal inhibitory peptide to GTPases. Nature. 387:814–819. [DOI] [PubMed] [Google Scholar]
- Gotoh, A., H. Takahira, R.L. Geahlen, and H.E. Broxmeyer. 1997. Cross-linking of integrins induces tyrosine phosphorylation of the proto-oncogene product Vav and the protein tyrosine kinase Syk in human factor-dependent myeloid cells. Cell Growth Differ. 8:721–729. [PubMed] [Google Scholar]
- Hall, A. 1998. Rho GTPases and the actin cytoskeleton. Science. 279:509–514. [DOI] [PubMed] [Google Scholar]
- Hart, M.J., A. Eva, D. Zangrilli, S.A. Aaronson, T. Evans, R.A. Cerione, and X. Zheng. 1994. Cellular transformation and guanine nucleotide exchange activity are catalyzed by a common domain on the Dbl oncogene product. J. Biol. Chem. 269:62–65. [PubMed] [Google Scholar]
- Hoffman, G.R., N. Nassar, and R.A. Cerione. 2000. Structure of the Rho family GTP-binding protein Cdc42 in complex with the multifunctional regulator RhoGDI. Cell. 100:345–356. [DOI] [PubMed] [Google Scholar]
- Keep, N.H., M. Barnes, I. Barsukov, R. Badii, L.Y. Lian, A.W. Segal, P.C. Moody, and G.C. Roberts. 1997. A modulator of rho family G proteins, rhoGDI, binds these G proteins via an immunoglobulin-like domain and a flexible N-terminal arm. Structure. 5:623–633. [DOI] [PubMed] [Google Scholar]
- Kimura, K., T. Tsuji, Y. Takada, T. Miki, and S. Narumiya. 2000. Accumulation of GTP-bound RhoA during cytokinesis and a critical role of ECT2 in this accumulation. J. Biol. Chem. 275:17233–17236. [DOI] [PubMed] [Google Scholar]
- King, R.D., and M.J.E. Sternberg. 1990. Machine learning approach for the prediction of protein secondary structure. J. Mol. Biol. 216:441–457. [DOI] [PubMed] [Google Scholar]
- Kluxen, F.W., and H. Lubbert. 1993. Maximal expression of recombinant cDNAs in COS cells for use in expression cloning. Anal. Biochem. 208:352–356. [DOI] [PubMed] [Google Scholar]
- Leonard, D., M.J. Hart, J.V. Platko, A. Eva, W. Henzel, T. Evans, and R.A. Cerione. 1992. The identification and characterization of a GDP-dissociation inhibitor (GDI) for the CDC42Hs protein. J. Biol. Chem. 267:22860–22868. [PubMed] [Google Scholar]
- Miranti, C.K., L. Leng, P. Maschberger, J.S. Brugge, and S.J. Shattil. 1998. Identification of a novel integrin signaling pathway involving the kinase Syk and the guanine nucleotide exchange factor Vav1. Curr. Biol. 8:1289–1299. [DOI] [PubMed] [Google Scholar]
- Nobes, C.D., and A. Hall. 1995. Rho, Rac, and Cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 81:53–62. [DOI] [PubMed] [Google Scholar]
- Nobes, C.D., and A. Hall. 1999. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J. Cell Biol. 144:1235–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivo, C., C. Vanni, P. Mancini, L. Silengo, M.R. Torrisi, G. Tarone, P. Defilippi, and A. Eva. 2000. Distinct involvement of cdc42 and RhoA GTPases in actin organization and cell shape in untransformed and Dbl oncogene transformed NIH3T3 cells. Oncogene. 19:1428–1436. [DOI] [PubMed] [Google Scholar]
- Price, L.S., J. Leng, M.A. Schwartz, and G.M. Bokoch. 1998. Activation of Rac1 and Cdc42 by integrins mediates cell spreading. Mol. Biol. Cell. 9:1863–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regazzi, R., A. Kikuchi, Y. Takai, and C.B. Wollheim. 1992. The small GTP-binding proteins in the cytosol of insulin-secreting cells are complexed to GDP dissociation inhibitor proteins. J. Biol. Chem. 267:17512–17519. [PubMed] [Google Scholar]
- Ren, X.D., W.B. Kiosses, and M.A. Schwartz. 1999. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 18:578–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander, E.E., S. van Delft, J.P. ten Klooster, T. Reid, R.A. van der Kammen, F. Michiels, and J.G. Collard. 1998. Matrix-dependent Tiam1/Rac1 signaling in epithelial cells promotes either cell–cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. J. Cell Biol. 143:1385–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffzek, K., I. Stephan, O.N. Jensen, D. Illenberger, and D. Gierschik. 2000. The Rac-RhoGDI complex and the structural basis for the regulation of Rho proteins by RhoGDI. Nat. Struct. Biol. 7:122–126. [DOI] [PubMed] [Google Scholar]
- Schoenwaelder, S.M., and K. Burridge. 1999. Bidirectional signaling between the cytoskeleton and integrins. Curr. Opin. Cell Biol. 11:274–286. [DOI] [PubMed] [Google Scholar]
- Schwartz, M.A., and S.J. Shattil. 2000. Signaling networks linking integrins and Rho family GTPases. Trends Biochem. Sci. 25:388–391. [DOI] [PubMed] [Google Scholar]
- Turner, C.E., M.C. Brown, J.A. Perrotta, M.C. Riedy, S.N. Nikolopoulos, A.R. McDonald, S. Bagrodia, S. Thomas, and P.S. Leventhal. 1999. Paxillin LD4 motif binds PAK and PIX through a novel 95-kD ankyrin repeat, ARF-GAP protein: a role in cytoskeletal remodeling. J. Cell Biol. 145:851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Aelst, L., and C. D'Souza-Schorey. 1997. Rho GTPases and signaling networks. Genes Dev. 11:2295–2322. [DOI] [PubMed] [Google Scholar]
- Yang, W., Q. Lin, J.L. Guan, and R.A. Cerione. 1999. Activation of the Cdc42-associated tyrosine kinase-2 (ACK-2) by cell adhesion via integrin beta1. J. Biol. Chem. 274: 8524–8530. [DOI] [PubMed] [Google Scholar]
- Yron, I., M. Deckert, M.E. Reff, A. Munshi, M.A. Schwartz, and A. Altman. 1999. Integrin-dependent tyrosine phosphorylation and growth regulation by Vav. Cell Adhes. Commun. 7:1–11. [DOI] [PubMed] [Google Scholar]
- Zhang, X.A., and M.E. Hemler. 1999. Interaction of the integrin beta1 cytoplasmic domain with ICAP-1 protein. J. Biol. Chem. 274:11–19. [DOI] [PubMed] [Google Scholar]