

Abstract
Multistep carcinogenesis involves more than six discrete events also important in normal development and cell behavior. Of these, local invasion and metastasis cause most cancer deaths but are the least well understood molecularly. We employed a combined in vitro/in vivo carcinogenesis model, that is, polarized Ha-Ras–transformed mammary epithelial cells (EpRas), to dissect the role of Ras downstream signaling pathways in epithelial cell plasticity, tumorigenesis, and metastasis. Ha-Ras cooperates with transforming growth factor β (TGFβ) to cause epithelial mesenchymal transition (EMT) characterized by spindle-like cell morphology, loss of epithelial markers, and induction of mesenchymal markers. EMT requires continuous TGFβ receptor (TGFβ-R) and oncogenic Ras signaling and is stabilized by autocrine TGFβ production. In contrast, fibroblast growth factors, hepatocyte growth factor/scatter factor, or TGFβ alone induce scattering, a spindle-like cell phenotype fully reversible after factor withdrawal, which does not involve sustained marker changes. Using specific inhibitors and effector-specific Ras mutants, we show that a hyperactive Raf/mitogen-activated protein kinase (MAPK) is required for EMT, whereas activation of phosphatidylinositol 3-kinase (PI3K) causes scattering and protects from TGFβ-induced apoptosis. Hyperactivation of the PI3K pathway or the Raf/MAPK pathway are sufficient for tumorigenesis, whereas EMT in vivo and metastasis required a hyperactive Raf/MAPK pathway. Thus, EMT seems to be a close in vitro correlate of metastasis, both requiring synergism between TGFβ-R and Raf/MAPK signaling.
Keywords: PI3K; MAPK; TGFβ; EMT; metastasis
Introduction
Carcinomas are the most frequent type of malignancies in humans. They arise by multistep mechanisms, involving five to eight genetic alterations in dominant oncogenes or tumor suppressor genes. These mutated genes cause alterations in at least six types of cellular mechanisms, which cooperate to generate a fully malignant carcinoma cell (for review see Hanahan and Weinberg, 2000). Of these events, local invasion and metastasis of tumor cells are the least well understood at the molecular level, although metastases cause ∼90% of human cancer deaths. Particularly, it is unknown which and how oncogenes or tumor suppressors cause changes typical for invasive/metastatic cells such as loss or altered functionality of cadherins (Christofori and Semb, 1999), CAMs (Perl et al., 1999), integrins, and their extracellular matrix ligands (Lukashev and Werb, 1998), and extracellular proteases and their inhibitors (Bergers and Coussens, 2000). Except for E-cadherin (Christofori and Semb, 1999), mutations in these genes themselves have not been observed.
During normal development, tissue reorganization, wound healing, and carcinogenesis, epithelial cells may transiently or even stably lose epithelial polarity and acquire a mesenchymal phenotype. Depending on the cell model and type of exogenous signal applied, these phenotypical changes have been termed transformation, dedifferentiation, scattering, or epithelial mesenchymal transition (EMT). EMT is relatively ill defined, always involving a change to a fibroblastoid spindle-shaped cell morphology, whereas changes in epithelial/mesenchymal markers or gene expression may differ widely (for reviews see Hay, 1995; Thiery and Chopin, 1999). EMT is also still little understood in mechanistic terms and controversially discussed as to its importance for carcinogenesis and metastasis (Boyer et al., 1996; Cui et al., 1996; Bakin et al., 2000).
Transforming growth factor β (TGFβ),* TGFβ receptor (TGFβ-R), and oncogenic Ras have been identified as important molecular players in EMT and metastasis (Oft et al., 1996, 1998) using well-polarized mammary epithelial cells (EpH4) (Reichmann et al., 1992) as a combined in vitro/in vivo model. EpH4 cells are nontumorigenic and require low amounts of TGFβ for organotypic tubular morphogenesis in type I collagen gels, whereas higher TGFβ concentrations induce cell cycle arrest and apoptosis (Oft et al., 1996). In contrast, EpH4 cells transformed by oncogenic Ha-Ras (EpRas) form rapidly growing tumors in mice and undergo EMT in response to TGFβ both in tumors and in collagen gels, giving rise to mesenchymal-like cells (EpXT) in both cases. These EpXT cells are characterized by a spindle-like morphology and loss of epithelial and gain of mesenchymal marker proteins, a phenotype stabilized by an autocrine TGFβ loop in vitro and in vivo (Oft et al., 1996).
Further work revealed that TGFβ-R signaling (Massague and Wotton, 2000) was required for EMT, invasion in vitro, and metastasis in vivo, using diverse murine and human cell systems (Oft et al., 1998). This tumor-promoting role of TGFβ did not agree with either the known growth-inhibitory tumor-suppressive function of TGFβ or inactivating mutations of Smad4 or the TGFβ-R in certain cancers, suggesting a function of these genes as tumor suppressors (Parsons et al., 1995; Schutte et al., 1996). However, more recent findings argue against TGFβ playing only a tumor-suppressive role and suggest an additional tumor-promoting role of TGFβ in late stage carcinogenesis (Cui et al., 1996; Takaku et al., 1998; Onichtchouk et al., 1999). These discrepancies may be explained by the fact that oncogenic Ras renders cells insensitive to TGFβ-induced cell cycle arrest and apoptosis (Filmus et al., 1992; Oft et al., 1996; Lehmann et al., 2000). However, it remains controversial by which mechanisms Ras reverses TGFβ-induced growth inhibition (Kretzschmar et al., 1999; Liu et al., 2000).
Similarly, the signaling pathways by which oncogenic Ras contributes to EMT are discussed controversially. Different reports implicated the mitogen-activated protein kinase (MAPK) pathway (Chen et al., 2000), the phosphatidylinositol 3-kinase (P13K) pathway (Bakin et al., 2000), or both (Potempa and Ridley, 1998) in EMT-like processes, which, however, can also be induced by TGFβ alone (Piek et al., 1999).
In this paper, we use the EpH4/EpRas cell system (Oft et al., 1998) to dissect signaling pathways downstream of oncogenic Ras with respect to their ability to contribute to EMT, tumorigenesis, and metastasis. Analysis of organotypic epithelial structures grown in collagen gels revealed that EMT, a metastable process involving sustained loss of epithelial and gain of mesenchymal markers (Reichmann et al., 1992; Oft et al., 1996), can be distinguished from “scattering,” which is fully reversible and does not involve major epithelial/mesenchymal marker changes. Using both low molecular weight inhibitors and expression of effector-specific Ras mutants (Rodriguez-Viciana et al., 1997), we show that a hyperactive Raf/MAPK pathway is required for TGFβ-induced EMT, tumorigenesis, and metastasis. In contrast, activation of the phosphatidylinositol 3-kinase (PI3K) pathway is required for protection from TGFβ-induced apoptosis, allowing scattering but not EMT and causing tumors but not metastasis. Thus, EMT and metastasis are closely linked processes, both relying on the synergism between constitutive hyperactivation of the Raf/MAPK signaling module and TGFβ-R signaling.
Results
EMT and scattering: two distinct processes in epithelial plasticity induced by growth factors or oncogenes
In addition to oncogenic Ras (Oft et al., 1996), hepatocyte growth factor (HGF)/scatter factor (SF), fibroblast growth factor (FGF), and TGFβ alone induce mesenchymal features in diverse epithelial cell systems (Brinkmann et al., 1995; Piek et al., 1999; Thiery and Chopin, 1999). We investigated whether these different signals would induce similar or different types of mesenchymal phenotypes in EpRas cells. These represent Ha-Ras–transformed fully polarized EpH4 cells (Oft et al., 1996), spontaneously immortalized from mammary glands of midpregnant mice (Reichmann et al., 1992). EpRas cells were seeded in serum-free collagen gels and subjected to different factor treatments. HGF/SF induced EpRas cells to invade collagen gels and grow as disordered chords of nonpolarized cells (Fig. 1 A; true for >95% of >100 structures counted in two to three separate collagen gels; see Materials and methods). Upon HGF/SF removal, >90% of these factor-induced disordered structures reverted to hollow tubular structures after an additional 5 d (Fig. 1 B). The disordered collagen gel structures formed by the HGF/SF-treated cells continued to express β4-integrin, E-cadherin, and ZO-1 at the entire cell surface (indicating loss of polarity) and failed to express vimentin (Fig. 1 C, left; unpublished data). Upon factor withdrawal, β4-integrin regained polarized basal expression in the hollow tubules formed (Fig. 1 C, right). Likewise, E-cadherin and ZO-1 were again expressed basolaterally (Fig. 1 C, inset; unpublished data). Similar results were obtained using FGF (unpublished data).
Figure 1.
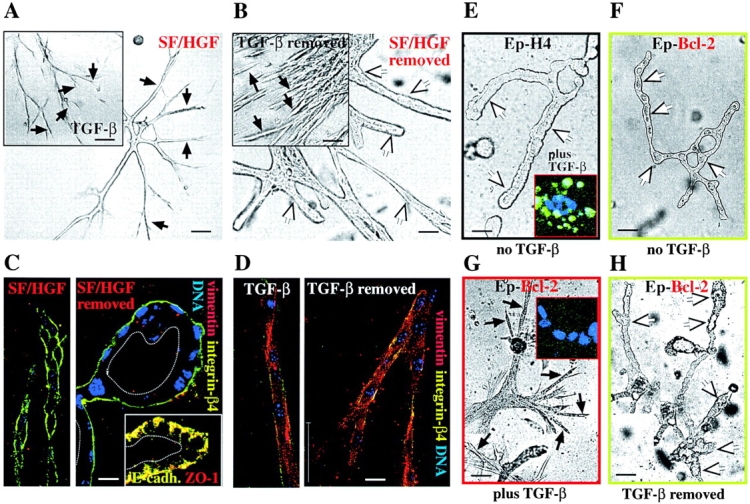
Scattering induced by scatter factor (HGF/SF) and TGFβ can be distinguished from EMT by reversibility and lack of marker changes. (A–D) EpRas collagen gel structures were treated with SF/HGF and TGFβ. (A) EpRas cells seeded in collagen gels for 3 d were treated with HGF/SF or TGFβ (insets) for an additional 5 d and photographed (B). After factor removal, (as described in Materials and methods) cells were maintained for an additional 4 d and photographed again. White arrows indicate lumina of tubular structures; black arrows indicate unordered strands of spindle-like invasive cells. (C and D). Structures treated as in A and B were fixed, stained with antibodies to the epithelial markers β4-integrin, E-cadherin, and ZO-1 (C, insets) and the mesenchymal marker vimentin, and analyzed by confocal immunofluorescence microscopy (as described in Materials and methods). (C) Structures transiently treated with HGF/SF. (D) Structures transiently treated with TGFβ. Note that HGF/SF-treated cells retain nonpolar distributed β4-integrin (C, left), whereas cells completely repolarize after HGF/SF removal (basolateral localized E-cadherin, and ZO-1) (C, left and inset). In contrast, the TGFβ-treated control structures undergo EMT (loss of β4-integrin, and vimentin expression) (D, left), persisting after TGFβ withdrawal (D, right). (E–H) Bcl-2–expressing EpH4 cells (Ep–Bcl-2) and control EpH4 cells were treated with TGFβ, which induced apoptosis (revealed by in situ TUNEL staining) in the EpH4 cells (E, inset) but a spindle cell-like morphology (G) and no apoptosis in the Ep–Bcl-2 cells (G, inset). Untreated cells of both types form normal hollow tubules (E and F). After withdrawal of TGFβ, the EpH4–Bcl-2 cells revert to tubular structures (H). Bars: (A, B, and E–H) 50 μm; (C and D) 10 μm.
Controls using EpRas cells undergoing EMT in response to TGFβ showed that the spindle-like phenotype induced by TGFβ treatment (Fig. 1 A, inset) persisted after factor removal (Fig. 1 B, inset) and involved loss of E-cadherin/β4-integrin, whereas vimentin was strongly induced (Fig. 1 D, left). Again, these marker changes persisted after TGFβ removal (Fig. 1 D, right). These results show that HGF and FGF reversibly induce a spindle-like migratory phenotype in EpRas cells, which is characterized by loss of polarity but involves no lasting changes in epithelial or mesenchymal marker expression. We refer to this phenotype as scattering, since FGF and HGF also induce “classical” scattering on plastic (unpublished data), and distinguish it from EMT, which is not reversed upon factor removal and involves stable loss of epithelial markers and upregulation of mesenchymal markers.
To answer the question if and how TGFβ alone (Piek et al., 1999) would alter the phenotype of nontransformed EpH4 cells, we used EpH4 cells expressing a retrovirally transduced antiapoptotic Bcl-2 protein. This was necessary since doses of TGFβ (2–5 ng/ml) causing EMT in EpRas cells induced growth arrest and apoptosis in EpH4 cells. In the absence of TGFβ, the Bcl-2–expressing EpH4 cells formed tubular structures in collagen gels similar to EpH4 control cells (Fig. 1, E compared with F). When treated with TGFβ, the EpH4–Bcl-2 cells were apoptosis protected as expected (8% TUNEL-positive cells; Fig. 1 G, inset) and formed disordered structures consisting of migratory cells (Fig. 1 G), whereas control EpH4 cells underwent apoptosis (48% TUNEL-positive cells; Fig. 1 E, inset). However, in contrast to EpRas cells the TGFβ-treated Bcl-2–expressing EpH4 cells reverted to hollow tubular structures 4 d after TGFβ removal (Fig. 1 H). Likewise, they maintained E-cadherin expression throughout the experiment, whereas vimentin expression was not detectable (unpublished data). In conclusion, TGFβ causes reversible scattering in EpH4 cells apoptosis protected by Bcl-2 but fails to induce EMT in the absence of oncogenic Ras, suggesting that Ras induces an EMT competent state in addition to prevent TGFβ-induced apoptosis.
Active oncogenic Ras is required for both induction and maintenance of EMT
Using a kinase-dead dominant negative mutant of the TGFβ-RII, we showed previously that maintenance of TGFβ-R signaling is required for EMT and metastasis (Oft et al., 1998). To address if EMT also required maintenance of oncogenic Ras activity, we used a specific nontoxic inhibitor of Ras-farnesylation (L739749 [Kohl et al., 1994]). L739749 reversed EMT in mesenchymal EpXT cells, causing reformation of hollow tubular structures (Fig. 2 B) from the unordered cords and strands of EpXT cells (Fig. 2 A) but did not affect EpH4 (Fig. 2 B, inset). After inhibitor withdrawal, the reverted cells maintained their polarized phenotype as indicated by persistence of tubular structures (Fig. 2 C). However, these structures could be reinduced to undergo EMT when again treated with TGFβ (unpublished data). This indicates that Ras inhibition reversed EMT completely, converting EpXT cells into cells with EpH4 cell properties. In line with this, reactivation of Ras in the reverted cells was insufficient for EMT, again requiring TGFβ as in the original EpRas cells (Oft et al., 1996). Furthermore, cotreatment of the reverted cells with L739749 and TGFβ resulted in cell death (Fig. 2 D), whereas their treatment with Ras inhibitor alone caused no phenotypical changes (Fig. 2 D, inset).
Figure 2.
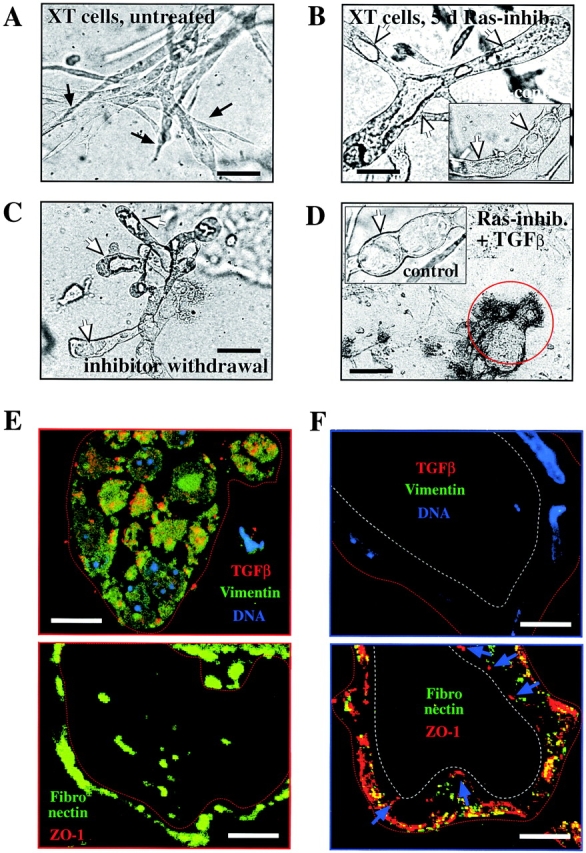
Ras activity is required for induction and maintenance of EMT. (A–D) EpXT cells were seeded into collagen gels in the absence (A) or presence (B) of 10 μM Ras farnesylation inhibitor (L739749), and resulting structures were photographed after 6 d (as described in Materials and methods). Note the complete reversal of EpXT cells to tubular structures with lumina (white arrows), which persisted after removal of the inhibitor for 4 d (C). The inhibitor alone did not affect tubular structures formed by EpRas cells (B, inset) or after reversal (D, inset, control), whereas treatment of the reverted structures with TGFβ plus L739749 caused cell disintegration. (E and F) Immunostaining of frozen ultrathin sections of EpXT collagen gel structures before (E) or after treatment (F) with L739749 for 5 d. Sections were stained with antibodies to TGFβ (red) (Oft et al., 1996) and vimentin (green) plus DAPI (top, DNA). Parallel sections (bottom) were stained with antibodies to ZO-1 (red) and fibronectin (green). L739749 causes loss of intracellular TGFβ and vimentin (top) and extracellular fibronectin (bottom) and reexpression of ZO-1 at apicolateral sites of tight junctions (blue arrows, bottom left). Bars: (A–D) 50 μm; (E and F) 20 μm.
These results were confirmed by analyzing marker expression, using ultrathin cryosections stained with respective fluorescent antibodies (see Materials and methods) from similar collagen gel structures as shown in Fig. 2, A and B. As expected, the mesenchymal chords of untreated EpXT cells expressed vimentin and intracellular TGFβ (Fig. 2 E, top) and deposited fibronectin extracellularly (periphery of structures and intercellular spaces; Fig. 2 E, bottom). After treatment with L739749 for 5 d (Fig. 2 F), vimentin and TGFβ expression was no longer detectable in the hollow structures obtained (Fig. 2 F, top). Two additional mesenchymal markers, CD68 (macrosialin) and calgranulin A, were also downregulated (unpublished data). Furthermore, the L739749-treated cells reexpressed the tight junction marker ZO-1 (Fig. 2 F, bottom) and E-cadherin (unpublished data) in a polarized basolateral fashion. The presence of tight junctions in the epithelial tubules induced by L739749 were confirmed by EM, using immunogold staining for ZO-1, although the mesenchymal chords obtained in the absence of inhibitor failed to show any junction-like structures (unpublished data).
In conclusion, inhibition of Ras in EpXT cells completely restores a normal polarized epithelial phenotype, indicating that sustained oncogenic Ras and TGFβ-R signaling are required for induction and maintenance of EMT in EpRas cells.
Ras downstream signaling required for EMT: analysis by low molecular weight inhibitors
Oncogenic Ha-Ras activates multiple downstream signal transduction pathways, including the Raf/MAPK module and the PI3K-PKB/Akt pathway (for review see Rommel and Hafen, 1998). Both pathways were implied in cell transformation in vitro (Rodriguez-Viciana et al., 1997) and tumorigenesis (Webb et al., 1998). To dissect Ras-activated downstream pathways required for different aspects of Ha-Ras transformation in epithelial cells (EMT, scattering, and apoptosis protection), we employed two specific low molecular weight inhibitors selectively blocking either the MAPK pathway (PD98059) or PI3K signaling (LY294002).
The activity of the Mek1/MAPK- and PI3K-PKB/Akt pathways in EpRas and EpH4 cells and the ability of the two inhibitors to selectively suppress either pathway was analyzed by Western blots using phospho-specific antibodies to MAPK/extracellular signal–regulated kinase (Erk)1/2 and PKB/Akt. EpRas cells exhibited strongly elevated phospho-Erk/MAPK and phospho-PKB/Akt levels compared with EpH4 cells (Fig. 3 A, lanes E and R). These elevated levels were largely independent of cell density (unpublished data). Testing of the inhibitors in EpRas cells revealed that 10 μM PD98059 reduced MAPK phosphorylation to basal levels, whereas 40 μM completely abolished MAPK phosphorylation (unpublished data). Conversely, PKB/Akt phosphorylation levels were not affected by PD98059 (Fig. 3 A, left, lanes R and iR; unpublished data). Conversely, 5 μM LY294002 added at 12-h intervals stably reduced elevated PKB/Akt phosphorylation in EpRas cells to basal levels (Fig. 3 A, middle; unpublished data), whereas 30 μM LY294002 essentially abolished PKB/Akt phosphorylation (Fig. 3 A, right). In contrast, even high levels of LY294002 had no effect on MAPK phosphorylation in EpRas cells (Fig. 3 A).
Figure 3.

Inhibitors of Mek-1 prevent and reverse EMT, and PI3K inhibitors abolish protection from TGFβ-induced apoptosis. (A) EpH4 cells (lanes marked E) or EpRas cells mock treated (lanes marked R) or treated with the inhibitors indicated (lanes marked iR) were lysed after 6 h of inhibitor treatment and analyzed for serine phosphorylation of Erk1/2 (MAPK) and PKB/Akt in Western blots (as described in Materials and methods). (B) EpRas cells seeded into collagen gels in the presence or absence (insets) of 5 ng/ml TGFβ. Both types of cultures were then left untreated (top, control) or treated with PD98059 (top right) and LY294002 (bottom) for another 5 d (as described in Materials and methods). Neither inhibitor was toxic to EpRas cells without TGFβ (insets; LY294002 slowed down structure growth). PD98059 (10 μM) completely reverted EMT (white arrows, top right), 5 μM LY294002 had no effect on the mesenchymal structures (black arrows, bottom left), whereas 30 μM caused cell death (bottom right, red circle). (C) Confocal analysis of in situ TUNEL staining (green) performed on collagen gel structures from EpRas cells treated for 4 d as indicated on the microphotographs (counterstaining for DNA in blue). (D) Quantitative analysis of cell death caused by inhibition of MEK-1 and PI3K in the absence or presence of TGFβ using trypan blue dye exclusion assay. The percentages of structures consisting of 25–60% (hatched bars, partially dead) or >60% trypan blue–positive cells (black bars, dead) are shown (as described in Materials and methods). Red asterisks indicate statistically significant induction of apoptosis by TGFβ. Bars: (B and C) 50 μm.
Next, we analyzed the effects of the two inhibitors on TGFβ-induced EMT in collagen gels (Fig. 3 B, top). 10 μM PD98059 prevented EMT in >95% of the structures, giving rise to hollow tubules (Fig. 3 B, top) typical for control EpRas cells (Fig. 3 A, inset). These inhibitor-treated structures failed to express vimentin while retaining E-cadherin (unpublished data). In contrast, 5 μM LY294002 did not affect TGFβ-induced EMT in EpRas cells (Fig. 3 B, bottom left; unpublished data), whereas 30 μM LY294002 (almost completely suppressing PI3K signaling; Fig. 3 A) caused cell disintegration (Fig. 3 B, bottom right). None of the inhibitors strongly altered formation of hollow structures in the absence of TGFβ, showing that they were nontoxic at the respective concentrations used (Fig. 3, A–D, insets).
The disintegration of EpRas cells observed after treatment with 30 μM LY294002 plus TGFβ was clearly due to strongly increased apoptosis as shown by TUNEL staining or dye exclusion (Fig. 3, C and D; see Materials and methods). In contrast, 10 μM PD98059 or 5 μM LY294002 plus TGFβ and LY294004, PD98059, or TGFβ added alone failed to significantly increase apoptosis (Fig. 3, C and D; unpublished data). In conclusion, high levels of Mek1/MAPK activity are necessary for induction and maintenance of EMT. Similar elevated levels of PI3K pathway activity are required for protection from TGFβ-induced apoptosis but not for EMT.
Effector-specific Ras mutants in vitro: MAPK hyperactivation is required for EMT, whereas PI3K signaling causes scattering
To verify these results by an independent approach, two well-characterized Ras mutants were used, which selectively signal along the MAPK pathway, S35-V12–Ras (S35-Ras), or the PI3K pathway, C40-V12–Ras (C40-Ras), due to specific amino acid changes in the effector loop of the Ras protein (Rodriguez-Viciana et al., 1997; Downward, 1998). These mutant proteins and the parental oncogenic Ras protein (V12-Ras) were overexpressed in EpH4 cells using respective retroviral constructs (see Materials and methods). Since V12-Ras did not reach similar expression levels in mass cultures as seen for v-H-Ras in EpRas cells (unpublished data), clones were selected expressing particularly high levels of V12-Ras, S35-Ras, and C40-Ras. In Western blots, S35-Ras–expressing clones showed elevated phosphorylation of Erk1 but only basal levels of PKB/Akt phosphorylation compared with empty vector controls. In contrast, the C40-Ras clones showed only basal level phosphorylation of Erks but elevated levels of PKB/Akt phosphorylation (Fig. 4 A). As expected, the V12-Ras control cells showed elevated levels of both phospho-Erk and phospho-PKB/Akt. Similar results were obtained with pools of >10 clones also selected for high level expression of V12-Ras, S35-Ras, and C40-Ras proteins (Fig. 4 B; unpublished data; see Materials and methods).
Figure 4.
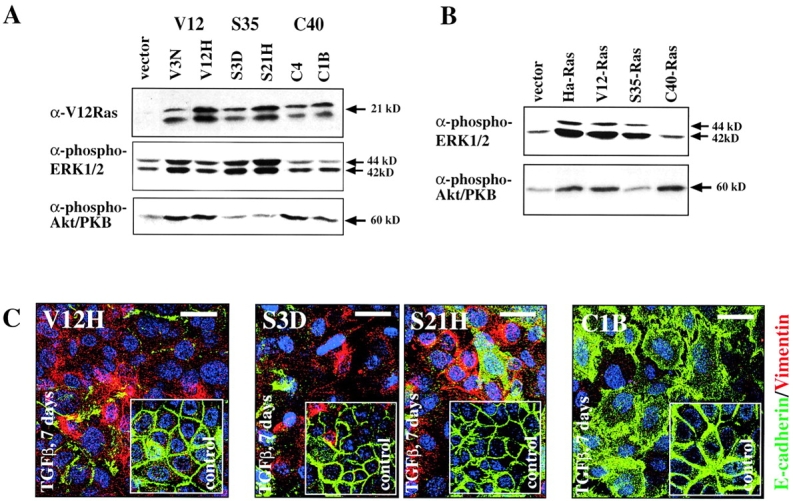
Biochemical and biological characterization of EpH4 cells expressing Ras effector-specific mutants lacking signaling along the MAPK or PI3K pathways. (A) Clones of EpH4 cells expressing the Ras effector mutants S35-Ras, C40-Ras, or V12-Ras were tested for overexpression of Ras proteins in Western blots using V12-Ras–specific antibodies. Clones highly overexpressing the exogenous Ras proteins (top) were then tested for levels of phospho-Erk (middle) or phospho-Akt (bottom) by respective phospho-specific antibodies (as described in Materials and methods). (B) Mass cultures (pools of multiple clones) expressing lower but still clearly elevated levels of H-V12–Ras or Ras effector mutant proteins S35 or C40 (unpublished data) were analyzed for phospho-Erk and phospho-Akt expression as in A. (C) Representative clones expressing V12-Ras (left), S35-Ras (middle), and C40-Ras (right) were cultivated on porous supports for 7 d in the presence (panels) or absence of TGFβ (insets). Cells were immunostained for E-cadherin (green) and vimentin (red) plus DAPI counterstaining for DNA (blue; as described in Materials and methods) and analyzed by confocal immunofluorescence microscopy. Bars, 20 μm.
The Ras mutant-overexpressing clones (Fig. 4 A) were then characterized for their response to TGFβ. On porous supports (filters) allowing epithelial polarization, treatment of control V12-Ras cells and two S35-Ras–overexpressing clones with TGFβ for 7 d resulted in spindle-shaped, vimentin-positive, E-cadherin–negative cells (Fig. 4 C; unpublished data). In contrast, C40-Ras–overexpressing cells failed to lose E-cadherin or to upregulate vimentin but showed loss of polarity (redistribution of E-cadherin; Fig. 4 C). As expected, control cells not treated with TGFβ always expressed lateral E-cadherin and no vimentin (Fig. 4 C, insets).
In collagen gels, untreated S35-Ras cells resembled V12-Ras cells (e.g., forming distended tubular structures with large lumina in collagen gels), whereas C40 cells more closely resembled EpH4 cells (thin tubules with tiny lumina; Fig. 5 A, top). Marker staining revealed basolateral E-cadherin staining and no expression of mesenchymal markers in all three cell types (Fig. 5 B, insets; unpublished data). Treatment of V12-Ras, S35-Ras, and C40-Ras cells with TGFβ resulted in unordered cell strands and cords with spindle-like cellular morphology (Fig. 5 A, middle). After withdrawal of TGFβ, these lumen-less disordered structures persisted in the V12-Ras and S35-Ras cells, whereas C40-Ras cells reverted to thin hollow structures (Fig. 5 A, bottom). Likewise, TGFβ-treated S35-Ras cells showed loss of E-cadherin/β4-integrin staining and induction of the mesenchymal markers vimentin and CD68 (Fig. 5 B, top), a marker distribution persisting after removal of TGFβ (Fig. 5 B, bottom). In contrast, C40-Ras–expressing cells maintained nonpolar E-cadherin staining in the disordered structures induced by TGFβ, whereas mesenchymal markers remained undetectable (Fig. 5 B, top). Upon withdrawal of TGFβ, the C40-Ras structures regained epithelial polarity as indicated by lumen formation and basolateral E-cadherin staining (Fig. 5 B, bottom).
Figure 5.
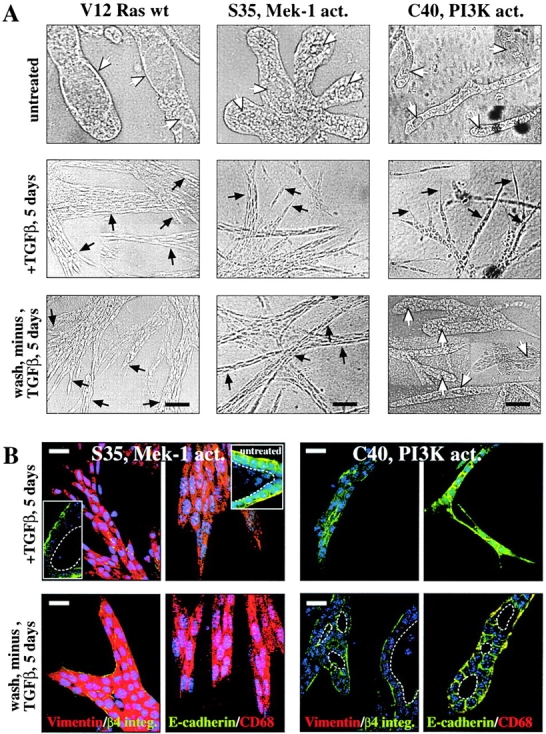
EpH4 cells expressing V12-Ras or S35-Ras undergo EMT upon TGFβ treatment; C40-Ras cells undergo scattering. (A) EpH4 cell clones overexpressing V12-Ras (left) and the Ras effector mutants S35-Ras (middle) and C40-Ras (right; Fig. 4) were seeded in collagen gels and either left untreated (top) or treated (middle) with 5 ng/ml TGFβ for 5 d followed by removal of TGFβ and further cultivation for 5 d (bottom). Photographs of representative tubular structures with lumina (white arrows) or distended chords and strands of invasive cells with mesenchymal morphology (black arrows) are shown. (B) Similar collagen gel structures as in A were stained in situ with antibodies to epithelial markers (green, β4-integrin and E-cadherin) and mesenchymal markers (red, vimentin and CD68; as described in the text) and analyzed by confocal immunofluorescence microscopy (as described in Materials and methods). (Top left, insets) tubular structures formed by untreated S35-Ras cells. (Top) “Invasive” structures formed by S35-Ras and C40-Ras cells after TGFβ treatment (note the persistent nonpolarized expression of epithelial markers in the C40-Ras structures). (Bottom) Structures formed by S35-Ras and C40-Ras after removal of TGFβ (mesenchymal shape and mesenchymal markers persist in the S35-Ras cells; C40-Ras cells revert to tubular structures with lumina [white dotted lines] basolaterally expressed epithelial markers). Bars: (A) 50 μm; (B) 20 μm.
In conclusion, a hyperactive MAPK/Erk pathway plus TGFβ is sufficient to cause EMT, whereas elevated PI3K signaling plus TGFβ induces scattering, that is, spindle-shaped cells without loss of epithelial and gain of mesenchymal markers.
Effector-specific Ras mutants: protection from TGFβ-induced apoptosis requires PI3K signaling
Oncogenic Ras and Raf abolish TGFβ-induced apoptosis in several epithelial cell systems (Oft et al., 1996; Lehmann et al., 2000). V12-Ras, S35-Ras, and C40-Ras cells (from both clones and clone pools; Fig. 4 A) were therefore tested for their apoptotic response upon TGFβ treatment. This was done using cells pregrown in collagen gels for 3–4 d (Fig. 6 A), since detection of apoptosis was much easier in these cultures than on plastic (Lehmann et al., 2000). Collagen structures were either collagenase digested and the cells subjected to TUNEL staining in suspension (Fig. 6 C, top) or TUNEL stained and counted in situ, that is, within the intact collagen gels (Fig. 6, B and C, bottom; see Materials and methods).
Figure 6.

V12-Ras and C40-Ras but not S35-Ras protect EpH4 cells from TGFβ-induced apoptosis. (A) For apoptosis determination in collagen gels, EpH4 cells expressing Ras or Ras effector–specific mutants were allowed to form structures in collagen gels for 3 d and were treated with various concentrations of TGFβ (0.5–40 ng/ml) for 4 d (dotted bar). Cells were either TUNEL stained in suspension (C, top) or in situ (B, and C, bottom panel; as described in Materials and methods). (B) Confocal immunofluorescence analysis of gel structures formed by clones overexpressing S35-Ras (left) and C40-Ras cells (right) treated with moderate (top, 2.5 ng/ml) or high levels of TGFβ (40 ng/ml; see A) and subjected to in situ TUNEL staining (green). DAPI staining (blue) indicates live cells. (C) Quantitation of TUNEL-positive cells from collagen gel structures either stained and counted in suspension after collagenase digestion (top; red asterisks indicate statistically significant increases in apoptosis induced by TGFβ) or stained and counted in situ (bottom; as described in Materials and methods). Mean percentages of apoptotic cells from multiple gel structures plus standard deviations (error bars) from three independent determinations are shown. Bars, 50 μm.
Both approaches yielded similar results. Already at low TGFβ levels (1 ng/ml), apoptosis of EpH4 control cells was enhanced significantly, whereas >95% of these cells were TUNEL positive and thus apoptotic at 5 ng/ml TGFβ (Fig. 6 C). EpH4 cells expressing V12-Ras and C40-Ras showed no significant elevation of apoptotic cells even at high TGFβ concentrations. In contrast, S35-Ras cells were consistently sensitive to TGFβ-induced apoptosis but required higher TGFβ levels for complete apoptosis than EpH4 cells (Fig. 6, B and C). Thus, hyperactivation of the PI3K pathway is required for protection from TGFβ-induced apoptosis. In contrast, hyperactive MAPK signaling in S35-Ras cells failed to induce apoptosis protection at higher TGFβ levels.
Effector-specific Ras mutants in vivo: elevated MAPK and PI3K signaling are each sufficient for tumor formation
To analyze the importance of Ras downstream signaling pathways for tumor formation in vivo, clones strongly overexpressing V12-Ras, S35-Ras, and C40-Ras were injected into the mammary gland fat pads of nude mice. All cell types caused tumors with comparable efficiencies (16/16 injection sites; Fig. 7 A). Similar results were obtained with respective pools of clones overexpressing the different Ras signaling mutants except that tumor formation by cells from S35-Ras clone pools was significantly slower than from respective C40-Ras clone pools (unpublished data). Bcl-2–EpH4 control cells formed similarly small regressing nodules as EpH4 cells, which contained only well-polarized, E-cadherin–positive, vimentin-negative cells (unpublished data). Therefore, protection of EpH4 cells from TGFβ-dependent apoptosis is not sufficient for tumorigenesis.
Figure 7.

S35-Ras– and C40-Ras–overexpressing cells form tumors, but only S35-Ras cells undergo EMT in vivo. (A) Various clones (see also Fig. 4 A) overexpressing V12-Ras (black symbols) S35-Ras (red symbols) and C40-Ras (blue symbols) were injected into the mammary gland fat pads of mice (four mice with a total of 16 injection sites per clone). Growth of the multiple tumors developing in all mice was followed by measuring mean tumor sizes (as described in Materials and methods). (B) Ex-tumor cells recultivated from 4–5-wk-old tumors (as described in Materials and methods) induced by V12-Ras, S35-Ras, and C40-Ras clones (A) were seeded onto porous supports and analyzed by immunostaining for E-cadherin (green) and vimentin (red). Blue, DAPI counterstain for DNA. Bars, 20 μm.
To address which phenotypic changes of the Ras mutant-expressing cells accompanied tumor growth, we recultivated cells from 4–5-wk-old tumors, using G418 to select for ex-tumor cells of donor origin (see Materials and methods) and analyzed them for E-cadherin and vimentin expression by immunofluorescence. These ex-tumor cells closely resembled the respective Ras mutant-expressing cells subjected to TGFβ treatment in vitro (Fig. 4 C compared with Fig. 7 B). V12-Ras and S35-Ras ex-tumor cells showed a typical spindle-shaped morphology (unpublished data) and were vimentin-positive and E-cadherin–negative (Fig. 7 B, left and middle). In contrast, the C40-Ras ex-tumor cells showed epithelioid morphology (unpublished data) and E-cadherin staining, whereas vimentin was undetectable. In conclusion, the separate activation of either the PI3K pathway or the MAPK pathway is sufficient to confer tumorigenic potential to EpH4 cells, whereas EMT in vivo requires the MAPK pathway (see Discussion).
Effector-specific Ras mutants in vivo: only hyperactive MAPK can induce metastasis
Finally, we sought to determine whether or not V12-Ras–, S35-Ras–, or C40-Ras–expressing cells were able to form metastases. In line with metastasis formation in nude mice being a rare event (McClatchey, 1999), EpRas-derived primary tumors rarely progressed to metastasis before killing the animal. Resection of primary tumors to allow for late metastasis formation met with technical difficulties. Therefore, ex-tumor cells derived from two clones of S35-Ras cells and one clone each of C40-Ras and V12-Ras cells were injected into the tail veins of nude mice, an assay testing for survival of injected cells in the circulation and for evasation and colonization of distant organs. Most of the mice injected with V12-Ras or S35-Ras cells died between 2 and 5 wk, whereas all C40-Ras–injected mice survived until the end of the experiments (10 wk; Fig. 8 A). Upon histological analysis, the lungs of dead or moribund animals injected with V12-Ras or S35-Ras cells showed multiple metastases in all cases, often surrounded by blood vessels (Fig. 8 B, left). In contrast, lungs from C40-Ras–EpH4 injected animals were histologically free of metastases (Fig. 8 B, right), and no metastases were found in other organs (unpublished data). Similar results were obtained using pooled clones of V12-Ras–, S35-Ras–, or C40-Ras–EpH4 cells except that the S35-Ras cell-derived metastases appeared much later in line with the slower growth rate of the respective primary tumors (unpublished data).
Figure 8.

S35-Ras– but not C40-Ras–overexpressing EpH4 cells form lung metastases upon tail vein injection. (A) Ex-tumor cells from the V12-Ras (black symbols), S35-Ras (red symbols), and C40-Ras clones (blue symbols) indicated were injected intravenously into four nude mice per cell type. Moribund mice were killed and analyzed for the presence of lung metastases. The two surviving mice injected with S35-Ras clones were killed at day 55 and also contained lung metastases. None of the four mice injected with C40-Ras cells (killed after 10 wk) showed any lung metastases. (B) Histological stainings of sections from lungs of uninjected animals (left) and S35-Ras (middle) and C40-Ras–injected mice (two animals each). Note large metastases (also around blood vessels) only in the S35-Ras–injected mice. Bar, 100 μm.
These results indicate that metastasis formation seems to depend entirely on hyperactivity of the Raf/MAPK pathway, whereas a hyperactive PI3K pathway (in C40-Ras cells) is not sufficient for metastasis formation.
Discussion
In this paper, we utilize a well-characterized combined in vitro/in vivo model of mammary carcinogenesis (EpRas) to address the role of distinct Ras downstream signaling pathways in epithelial plasticity, tumorigenesis, and metastasis. We show that Ras-dependent hyperactive signaling of the Raf/MAPK pathway is required for EMT, whereas Ras activation of the PI3K pathway causes scattering and is required for protection from TGFβ-induced apoptosis. In vivo, a hyperactive PI3K pathway correlated with tumor formation, whereas a hyperactive MAPK pathway caused both tumors and metastasis. Thus, activation of two different Ras downstream signaling pathways during tumorigenesis and metastasis cause two clearly distinct changes toward a motile phenotype, only one of which (EMT) is associated with metastatic potential.
EMT and scattering, two distinct aspects of epithelial plasticity
Our work and work by others have established several operational criteria for EMT. These are (a) a requirement for cooperation of a hyperactive MAPK–Erk1/2 pathway with TGFβ-R signaling, (b) metastability, that is, persistence of the EMT phenotype after removal of TGFβ, and (c) drastic long-lasting molecular changes such as loss of E-cadherin and a switch to a mesenchymal gene expression pattern (Oft et al., 1996; Eger et al., 2000; Lehmann et al., 2000). These criteria apply only partially to epithelial plasticity changes (often also termed EMT) observed by others in different cell systems in response to Ras alone (Chen et al., 2000), Ras or Raf plus TGFβ (Oft et al., 1998; Lehmann et al., 2000), FGF, EGF, and SF/HGF (Thiery and Chopin, 1999; Boyer et al., 2000), and finally TGFβ alone (Piek et al., 1999; Bakin et al., 2000). These systems differ widely with respect to reversibility and switch from an epithelial to a mesenchymal gene expression program, their common denominator being morphological changes to a spindle-like phenotype.
These discrepancies may be explained by one major finding of this paper, showing that different signals can cause qualitatively different alterations of epithelial plasticity in the same cell type. In EpH4 cells, Ras or upstream receptor tyrosine kinases (e.g., activated HER2; unpublished data) plus TGFβ caused EMT. In contrast, HGF, FGF, or TGFβ alone caused scattering in apoptosis-protected EpH4 cells. Scattering involves a similar spindle-like migratory phenotype as EMT but involves only transient gene expression changes and is fully reversible. Thus, it is possible that some of the various types of epithelial plasticity termed EMT by others (see above) may correspond to scattering rather than EMT or represent hybrid phenotypes. Scattering may also occur during the initial stages of organotypic tubule formation in collagen gels (unpublished data), perhaps corresponding to tissue remodeling during outgrowth of glandular epithelial cells apparently dependent on PI3K plus Mek1/Erk signaling (Khwaja et al., 1998).
Multiple functions of Ras during EMT and tumorigenesis
Oncogenic Ras, the second player in EMT, displays diverse functions in EpH4 cells. First, Ras protects EpH4 cells from apoptosis, both by upregulation of PI3K and by downregulation of Fas receptor, rendering the cells refractory to Fas ligand (Peli et al., 1999). Second, oncogenic Ras or Raf alone cause more plastic cell–cell contacts, increased migration, and distended tubular or alveolar structures with large lumina in collagen gels (Oft et al., 1996; Lehmann et al., 2000). These changes may be due to Ras-dependent upregulation of N-cadherin expression in EpH4 cells, which functionally substituted for E-cadherin and contributed to the altered behavior of EpRas cells (unpublished data). In line with this, N-cadherin regulates cell motility and invasiveness during development and in various carcinoma cell lines (Radice et al., 1997; Nieman et al., 1999).
Importantly, even strongly overexpressed oncogenic Ras does not abolish epithelial polarity and organotypic structure formation on its own in several epithelial cell types (Lehmann et al., 2000; Gotzmann et al., 2002). The evidence from others with respect to this issue is controversial. In MDCK cells, Ras either caused loss of E-cadherin and ZO-1 expression and change to a fibroblastic morphology (Chen et al., 2000) or a partial loss of epithelial polarity, maintaining intact, tight, and adherent junctions (Schoenenberger et al., 1991). These discrepancies, probably due to heterogeneity of the MDCK cells studied or effects by TGFβ present in serum, underline the importance of using cell systems, which form organotypic epithelial structures under serum-free conditions where levels of TGFβ and other factors can be strictly controlled.
On the other hand, TGFβ-dependent EMT required strong overexpression of oncogenic Ras. This was shown in cells expressing low or intermediate Ras levels, which are protected from apoptosis and undergo scattering but not EMT in response to TGFβ (unpublished data). The same was true for saturating doses of TGFα (1 μg/ml) when added to EpH4 cells together with standard doses of TGFβ (unpublished data). These findings raise the question whether EMT requires sustained nuclear translocation of MAPK, occurring during neuronal differentiation of PC12 cells induced by overexpressed receptor tyrosine kinases (Traverse et al., 1994). It is also unclear which endogenous pathways cooperate with TGFβ family ligands when EMT is induced during development (Sun et al., 2000).
Dissection of Ras downstream signaling
A second important finding of our paper was that hyperactivation of the Raf/MAPK pathway caused EMT in vitro, and in vivo tumor formation and metastasis. Respective activation of the PI3K pathway was sufficient for tumorigenesis but not metastasis, causing both scattering and protection from TGFβ-induced apoptosis. These findings were obtained using both low molecular weight inhibitors of the two pathways and Ras effector mutants selectively activating these pathways. However, these mutants might signal through additional pathways, which we did not assay for. To study whether or not activation of the MAPK and PI3K pathways was sufficient for EMT and tumor formation, respectively, we are analyzing EpH4 cells expressing constitutively active PI3K variants and Mek1–Erk fusion proteins and an estrogen-inducible RafER fusion protein. Preliminary results suggest that RafER is not sufficient for EMT of EpH4 cells in serum-free collagen gels (unpublished data) in contrast to RafER-expressing MDCK cells, which underwent EMT in serum-containing cultures upon RafER activation (Lehmann et al., 2000). We currently analyze whether RafER-EpH4 cells require both TGFβ and basal PI3K signaling induced by endogenous RTK ligands, both obviously provided by serum.
So far, our studies did not address the role of the MAPK and PI3K pathways in controlling proliferation of EpRas cells either in collagen gels or in tumors. Our recent work revealed that a moderate upregulation of the PI3K pathway causes hyperproliferation of EpH4 cells in collagen gels and rapid tumor growth but not EMT. In contrast, cells showing a similarly upregulated MAPK pathway grow less rapid in collagen gels and form only slowly growing tumors, which nevertheless contained islands of mesenchymal tumor cells having undergone EMT (unpublished data). This indicates that tumor growth and EMT are controlled by independent pathways and makes it very unlikely that the lack of in vivo EMT in the C40-Ras–induced tumors is due to their slightly slower growth rate as compared with respective S35-Ras clones (Fig. 7 A).
Prevention of TGFβ-dependent growth arrest/apoptosis without abolishing TGFβ-R signaling?
TGFβ blocks cell cycle progression and causes apoptosis in most normal epithelial cells, suggesting that an important step in carcinogenesis is to overcome this antiproliferative effect of TGFβ (Fynan and Reiss, 1993). Multiple human tumors indeed carry inactivating mutations in Smad4 (Schutte et al., 1996) or even TGFβ-RII (Parsons et al., 1995). However, complete suppression of TGFβ-R signaling by such tumor suppressive mutations would also abolish the dominant oncogenic effects of TGFβ, for example, EMT and metastasis. This discrepancy is not yet fully explained by available data. However, the rare tumors with TGFβ-RII mutations have a low or absent ability to metastasize (Oft et al., 1998). Furthermore, TGFβ stimulation still induced a subset of TGFβ-specific promoters in MEF's from Smad4−/− mice (Sirard et al., 1998), suggesting that mammalian cells might contain a second Smad4 family member already described in frogs (Smad4β/Smad10) (Howell et al., 1999). Finally, TGFβ-induced cell cycle arrest is also lost frequently by mutations in downstream effectors like p16ink4A/p15ink4B (Hall and Peters, 1996).
These results suggest that most carcinoma cells are selected for resistance to TGFβ cell cycle arrest while remaining sensitive for induction of migration/invasion by TGFβ. In line with this, murine and human cell lines completely resistant to cell cycle arrest by TGFβ retain apparently normal TGFβ-R signaling (Oft et al., 1998; McEarchern et al., 2001).
Obviously, oncogenic Ras completely substitutes for this type of tumor cell selection in the various epithelial cell models described above, including also strong protection from TGFβ-induced apoptosis. However, possible mechanisms for how Ras may modulate TGFβ-R signaling are still controversially discussed. Ras signaling was reported to attenuate Smad2/3 signaling via inhibitory phosphorylation, preventing nuclear translocation (Kretzschmar et al., 1999), but we and others failed to observe Ras-dependent inhibition of Smad2 or Smad3 nuclear translocation by elevated Ras or Raf activity in several systems (Lehmann et al., 2000; Liu et al., 2000). Therefore, the MAPK pathway might attenuate TGFβ responses via the induction and/or activation of new distinct transcription factors, corepressors, or coactivators recruited by Smads into a variety of transcriptional modules, which determine the diversity of responses to similar TGFβ signals in different cell types (Massague and Wotton, 2000).
Metastasis: which EMT-related changes in gene expression are important?
The results presented here strongly correlate metastasis to EMT, requiring cooperation of TGFβ-R–signaling with a hyperactive MAPK pathway and thus arguing for a dominant oncogenic role of the TGFβ-R pathway in addition to possible tumor suppressive functions. This positive role of TGFβ in EMT and metastasis correlates best with the defect of TGFβ-3−/− mice in palate development (Taya et al., 1999), which involves EMT, and by the defects in mesoderm induction in mice lacking Smad4 or Smad2 (Weinstein et al., 1998; Yang et al., 1998).
Which TGFβ-induced gene expression changes are important for EMT? One clear example is the loss of E-cadherin typical for EMT, which has been implicated in tumorigenesis and metastasis (Christofori and Semb, 1999). In line with this, the transcriptional repressor Snail blocks E-cadherin expression and causes a fibroblastoid phenotype and enhanced tumor progression in weakly tumorigenic carcinoma cell lines (Cano et al., 2000). However, mesenchymal FosER cells after EMT completely lack E-cadherin but form neither tumors nor metastases (Reichmann et al., 1992), indicating that loss of E-cadherin expression may be necessary but not sufficient for tumor progression.
Materials and methods
Cells and cell culture
The origin and culture conditions for EpH4 mouse mammary epithelial cells and their Has-Ras–transformed derivative EpRas have been described earlier (Reichmann et al., 1992; Oft et al., 1996). V12-Ras–, S35-Ras–, and C40-Ras–expressing EpH4 cells were generated by retroviral gene transfer using retroviral vectors of V12-Ras and its two mutants S35-V12–Ras and C40-V12–Ras in pLXSN (Rodriguez-Viciana et al., 1997). Bcl-2–expressing cells were generated as above by transfer of pBabe-puro Bcl-2 (a gift from M. Busslinger, Institute of Molecular Pathology). Construct DNA was used to transfect cells from the Phoenix™ packaging cell line (grown in DME plus 10% FCS and 2 mM glutamine, after recent hygromycin B/diphteria toxin selection for optimal virus production). After 2–3 d, virus supernatants from the transfected packaging cells were used to infect EpH4, which were then selected with 800 μg/ml G418 (5–6 d) or 2 μg/ml puromycin (3–4 d). Clones were isolated from retrovirally infected and drug-selected cell populations by seeding at low density, marking single adherent cells 8 h later, expansion for 7–9 d, and isolation of colonies using trypsin-soaked filter paper circles.
Cells from mouse tumors were recultivated from finely minced tumors by digestion in 2 mg/ml collagenase type VI (Sigma-Aldrich) plus 5% FCS for 30 min. Dispersed cells were seeded in Eagle's medium plus 15% FCS, selected for 7 d in 800 μg/ml G418 (day 2–8), and subcultured at a ratio of 1:3 every 2 d. During the first 2 d, 2 ng/ml TGFβ were added to facilitate outgrowth of ex-tumor cells.
Reagents and antibodies
10 μg/ml stocks of recombinant TGFβ-1, acidic FGF-1, and HGF/SF (R & D Systems) were prepared according to manufacturer's instructions, that is, FGF-1 solutions containing 1 μg/ml of heparin. The PI3K and Mek-1 inhibitors LY294002 and PD98059 (Calbiochem) were kept as 20–50-mM stocks in DMSO. The Ras farnesylation inhibitor L 739.749 (Kohl et al., 1994; a gift from Dr. F. Himmelsbach, Boehringer, Ingelheim, Germany) was kept as 10-mM stock solutions in DMSO at –70°C and refrozen directly after use. The following primary antibodies were used for immunofluorescence: mouse anti–E-cadherin (C20820; Transduction Laboratories) or polyclonal rabbit anti–E-cadherin (K84; a gift from R. Kemler, Max Planck Institute of Immunobiology, Freiburg, Germany), monoclonal anti–mouse vimentin, Vim-13.4 (V-2258; Sigma-Aldrich) or goat anti–mouse Vimentin, C-20 (sc-7557; Santa Cruz Biotechnology, Inc.), rabbit polyclonal anti–integrin-β4 (sc-9090; Santa Cruz Biotechnology, Inc.), goat polyclonal antimacrosialin/CD68 (sc-7084; Santa Cruz Biotechnology, Inc.), and rabbit polyclonal antifibronectin (F-3648; Sigma-Alddrich), rabbit anti–ZO-1 (61–7300; Zymed Laboratories). The antibodies to detect TGFβ-1, -2, and -3 by immunofluorescence have been described earlier (Oft et al., 1996). Phospho-specific rabbit anti–phospho-Erk and anti–phospho-Akt antibodies and antibodies detecting total Erk1/2 and Akt were from New England Biolabs (9100 and 9270, respectivelty). Anti-pan Ras Val-12 antibody with increased specificity to oncogenic V12-Ras was purchased from Calbiochem (OP38).
Western blot analysis
Cells to be used for Western Blot analysis of either serine-phosphorylated or total Erk and Ras proteins were 80% confluent and cultured in 4% FCS or starved for 48 h (for phospho-Akt and total Akt). Plates were washed in PBS and lysed on ice with kinase buffer (10 mM Tris, pH 7.6, 50 mM NaCl, 1 mM EGTA, 1% Triton X-100, 50 mM NaF, 30 mM sodium pyrophosphate, 10 mM Na3VO4, 2 nM ocadoic acid, 10 mM Pefablock™ and cocktail of protease inhibitors Complete™ both from Boehringer), centrifuged at 12,000 g for 10 min at 4°C, and the pellet was discarded. Freshly prepared lysates were analyzed by 8–10% SDS-PAGE and immunoblotted as described by others (Yu and Sato, 1999). Total V12-Ras expression was analyzed by 12% SDS-PAGE followed by Western Blot analysis as above.
Collagen gel cultures
Serum-free three-dimensional cultures of EpH4 cells, EpRas cells, and their derivatives were performed as described earlier (Oft et al., 1996, 1998) with minor modifications. Cells (in serum-containing medium) and rat tail collagen (3–4 mg/ml) (40236; Becton Dickinson) were mixed rapidly at 0°C (final collagen concentration 1.5 mg/ml), and 100-μl droplets containing between 2,000 and 3,000 cells were dispensed into 17-mm wells. When indicated, collagen solutions gels were supplemented with 10% vol/vol Matrigel solution (GF reduced) (40 230; Becton Dickinson) directly before use. After solidification on a level surface at 23°C for 15–30 min, the gels were incubated at 37°C in a CO2 incubator for another 1–2 h and overlaid with 500 μl of serum-free medium (mammary epithelial basal medium) (C-21010; PromoCell GmbH) supplemented with growth factors according to manufacturer's instructions. The batches of bovine pituitary extract had to be pretested for optimal performance. The medium overlaying the gel was changed 1 d after seeding and then every other day unless stated otherwise.
After allowing structure formation of the cells for 4–7 d, growth factors (20 ng/ml HGF, 20 ng/ml acidic FGF, and 5 ng/ml TGFβ unless stated otherwise) were added upon medium change every other day. Predetermined amounts of pharmacological inhibitors (20% higher final concentrations to correct for collagen gel volume) where added every 12 or 24 h for 5 d unless stated otherwise. Growth factors were withdrawn from collagen gels by switching them to medium without factors, changing medium after 24 h, and further cultivation for 2–5 d. In all experiments, at least 50–100 structures were inspected to quantify lumen-containing structures versus disordered strands or chords. If >90% of the structures were of one type, quantification is not further mentioned in the Results section.
Immunofluorescent staining and confocal microscopy of collagen gel structures
Collagen gels (sometimes split in half by a scalpel) were fixed with 1% formaldehyde in 250 mM Hepes, pH 7.4, freshly diluted from 16% paraformaldehyde stocks stored at –20°C. After 15 min at room temperature, the gels were washed once each with Tris- and phosphate-buffered saline plus 0.2% Tween 20 (TBST and PBST, respectively) and treated for 1 h at 4°C with blocking solution (PBST, 0.1% gelatin plus 10 μg/ml nonimmune bovine IgG). Gels were incubated with primary antibodies plus DAPI in blocking solution for 1 h at 37°C in a wet chamber, washed three times in PBST for 30 min, and postfixed with 4% PFA in PBST for 15 min at 23°C. After 30-min washes in TBST, PBST, and blocking solution, the gels were incubated with appropriate secondary antibody mixtures made up in blocking solution for at least 1 h or overnight at 4°C. Gels were washed three times in PBS, once in distilled water, and mounted in Mowiol (Hoechst). Confocal analysis was performed using a Leica TCS-NT confocal microscope (DAPI visualized by two-photon excitation microscopy using Coherent-Vitesse pulsed NIR laser). From representative gel structures, 5–10 horizontal scans using a 40× (1.3 NA) oil immersion objective were recorded for each channel and used to calculate an extended focus image with the respective software (exported as a TIFF file).
Cryo-sectioning and immunofluorescent staining of collagen gel structures
Cryo-sectioning was performed to visualize TGFβ and ZO-1 by antibody staining not possible using whole-mount gels. Collagen gels were fixed for 15 min at 4% PFA in 250 mM Hepes followed by 8% PFA in 250 mM Hepes for 1 h and then washed in TBS. Individual structures were excised from the gel under a dissection microscope and placed into PVP/sucrose (Sigma-Aldrich) on ice. Blocks were incubated in PVP/sucrose overnight, mounted upright onto a cryo-nail, flash-frozen in liquid nitrogen, and stored in liquid nitrogen until further use. For sectioning, these nails were placed into a Leica Cryo-Microtome and semi-thin sections of 500–800 nm were placed on a gelatin-coated slide. Alternatively, ultrathin sections of 100–130 nm were placed on copper grids for EM analysis. Immunostaining of semi-thin sections was performed in small drops on the slides directly with the same solutions as mentioned above for whole-mount stainings. Samples were analyzed by conventional fluorescent microscopy, recorded with a monochrome digital camera, and the three channel color image was restored from the individual images.
Immuofluorescence of cells grown on porous supports
Cells were cultivated on porous supports (cell culture inserts, pore size 0.4 μm; Becton Dickinson) for 2–7 d and fixed either at ∼70% confluency (or as fully confluent well-polarized epithelial sheets). Filters were rinsed twice with Hank's solution plus glucose or PBS, fixed in acetone/methanol (1:1) for 5 min at –20°C, dried, washed with PBS, and blocked for 1 h in 0.2% gelatin in PBS-containing nonimmune goat or bovine IgGs (20 μg/ml) and 0.05% Tween. Filters were then incubated with first antibody (diluted in blocking solution lacking nonimmune IgGs) for 1 h, washed five times in PBS containing 0.05% Tween, treated with similarly prepared dilutions of secondary antibodies for 30 min, and washed again as above (first wash containing DAPI; 1 mg/ml stock, final dilution 1:10,000). Alexa-conjugated secondary antibodies against rabbit or mouse IgG (Molecular Probes, Inc.) were diluted 1:1,000, whereas Cy3-conjugated goat anti–rabbit or anti–mouse IgG (Jackson ImmunoResearch Laboratories) were diluted 1:300. Confocal microscopy was performed as above, recording only single horizontal scans for each channel.
Apoptosis assays using cells and structures from collagen gels
2–3 d after seeding, cell structures within collagen gels were treated with TGFβ plus or minus other factors/inhibitors for 2.5 or 4 d. After washing in PBS, the structures were either processed to yield cell suspensions or TUNEL stained in situ. To generate and stain cell suspensions, collagen gels were digested with 2 mg/ml collagenase in 0.5 ml PBS for 10 min. Pooled cells from 8 gels were spun down, washed in PBS, transferred into v-shaped wells of a 96-well plate, fixed with 4% PFA for 1 h, treated with 0.1% Triton X-100 in 0.1% sodium citrate and subjected to the TUNEL assay (method for cells in suspension) (1684795; Roche) according to manufacturer's instructions. After staining with DAPI in the last wash, TUNEL-stained cell pellets were suspended in 30 μl Mowiol, spread on glass slides, and apoptotic indices were determined by counting green versus blue nuclei under the fluorescence microscope. For each point, at least 500 cells were scored. Mean values and SD were calculated from three independent determinations.
For TUNEL staining of collagen gels in situ, the collagen gels were fixed with 4% PFA for 2 h, washed in PBS, and subjected to the TUNEL assay (as above) according to the manufacturer's instructions for tissue sections with minor modifications. Briefly, 65 μl of reaction mix diluted 1:1 in PBS was added to each collagen gel in 17-mm wells, and the wells were then covered with parafilm and incubated at 37°C for 2.5 h. Subsequently, labeled collagen gels were washed three times in PBS and once with HBS containing DAPI. Collagen gels were spread on slides, mounted in Mowiol, and TUNEL-positive cells (FITC-labeled) were scored against DAPI-stained nuclei under a fluorescence microscope (counting >1,000 cells from three to six randomly chosen fields in two to three collagen gels). Mean apoptotic indices ± SD are shown.
Trypan blue exclusion assay
4 d after the start of inhibitor treatment, collagen gels were rinsed in PBS, incubated 5 min in 0.5 ml trypan blue solution 0.4% (T 8154; Sigma-Aldrich), and rinsed twice in PBS. Thereafter, the percentage of stained structures was determined under a bright field microscope, counting at least 100 structures (“partially dead” and “dead” structures containing 25–60% or >60% cells of trypan blue–positive cells).
Induction of tumors and metastases in mice
To test the various cell types for tumorigenicity, groups of three to six BalbC athymic nude mice (nu/nu; 5–9 wk old; obtained from Charles River Wiga, GmbH, Sulzfeld, Germany) were used for each cell type per experiment. Confluent cells from several V12-Ras, S35-Ras, and C40-Ras clones were trypsinized and washed three times in PBS. Multiple doses of 105 cells suspended in 25 μl PBS were injected into the 3rd and 4th mammary gland pairs of Metofane anaesthetized mice (shallow injection into the nipple area). Initial tumor nodules were confined to the mammary fat pad in almost all cases. Primary tumor growth was weekly measured with a caliper in mm. Mean tumor diameter was calculated as the sum of diameters of all tumors detected divided by the number of injection sides. For the experimental metastasis assay, 105 ex-tumor cells derived from tumors induced by the above clones (as described in Cells and cell culture) were suspended in 100 μl PBS and injected into the tail veins of MF1 nu/nu mice (Charles River Wiga, GmbH). Lethality of injection due to lung metastasis was assessed daily, and freshly dead or moribund mice were processed for histopathological analysis. For this, lungs were fixed in PFA 3.7% in PBS, postfixed with 70% EtOH, and embedded in paraffin. Sections were cut at 7.5-μm steps and stained with hematoxylin and eosin according to standard protocols. The surviving mice were killed after 10 wk, inspected visually for metastases in major organs, and the lungs were subjected to histological analyses as above.
Acknowledgments
The authors thank Dr. R. Kemler for his gift of anti–E-cadherin antibodies and useful discussions, Drs. G. Christofori, M. Baccarini, and R. Foisner (all Vienna Biocenter, Vienna, Austria) for critically reading the article, and Gabi Litos for excellent technical assistance.
E. Janda, S. Grunert, and H. Beug were supported by grants from the European Union Training and Mobility of Researchers network (ERBFMRXCT-980197), the Austrian Research funding agency (FWF; SFB 006/612), and the Austrian Industrial Research Promotion Fund (FFF project no. 803776).
K. Lehmann's present address is Dept. of Oncology, metaGen GmbH, Oudenarder Str. 16, 13347 Berlin, Germany.
Footnotes
Abbreviations used in this paper: EMT, epithelial mesenchymal transition; Erk, extracellular signal–regulated kinase; FGF, fibroblast growth factor; HGF, hepatocyte growth factor; MAPK, mitogen-activated protein kinase; PI3K, phosphatidylinositol 3-kinase; SF, scatter factor; TGFβ, transforming growth factor β; TGFβ-R, TGFβ receptor.
References
- Bakin, A.V., A.K. Tomlinson, N.A. Bhowmick, H.A. Moses, and C.L. Arteaga. 2000. Phosphatidylinositol 3-kinase function is required for transforming growth factor β-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 275:36803–36810. [DOI] [PubMed] [Google Scholar]
- Bergers, G., and L.M. Coussens. 2000. Extrinsic regulators of epithelial tumor progression: metalloproteinases. Curr. Opin. Genet. Dev. 10:120–127. [DOI] [PubMed] [Google Scholar]
- Boyer, B., A.M. Valles, and J.P. Thiery. 1996. Model systems of epithelium-mesenchyme transitions. Acta Anat. 156:227–239. [DOI] [PubMed] [Google Scholar]
- Boyer, B., A.M. Valles, and N. Edme. 2000. Induction and regulation of epithelial-mesenchymal transitions. Biochem. Pharmacol. 60:1091–1099. [DOI] [PubMed] [Google Scholar]
- Brinkmann, V., H. Foroutan, M. Sachs, K.M. Weidner, and W. Birchmeier. 1995. Hepatocyte growth factor/scatter factor induces a variety of tissue-specific morphogenic programs in epithelial cells. J. Cell Biol. 131:1573–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano, A., M.A. Perez-Moreno, I. Rodrigo, A. Locascio, M.J. Blanco, M.G. del Barrio, F. Portillo, and M.A. Nieto. 2000. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2:76–83. [DOI] [PubMed] [Google Scholar]
- Chen, Y., Q. Lu, E.E. Schneeberger, and D.A. Goodenough. 2000. Restoration of tight junction structure and barrier function by down-regulation of the mitogen-activated protein kinase pathway in ras-transformed Madin-Darby canine kidney cells. Mol. Biol. Cell. 11:849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofori, G., and H. Semb. 1999. The role of the cell adhesion molecule E cadherin as a tumour suppressor gene. Trends Biochem. Sci. 24:73–76. [DOI] [PubMed] [Google Scholar]
- Cui, W., D.J. Fowlis, S. Bryson, E. Duffie, H. Ireland, A. Balmain, and R.J. Akhurst. 1996. TGFbeta1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell. 86:531–542. [DOI] [PubMed] [Google Scholar]
- Downward, J. 1998. Mechanisms and consequences of activation of protein kinase B/Akt. Curr. Opin. Cell Biol. 10:262–267. [DOI] [PubMed] [Google Scholar]
- Eger, A., A. Stockinger, B. Schaffhauser, H. Beug, and R. Foisner. 2000. Epithelial mesenchymal transition by c-Fos estrogen receptor activation involves nuclear translocation of beta-catenin and upregulation of beta-catenin/lymphoid enhancer binding factor-1 transcriptional activity. J. Cell Biol. 148:173–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filmus, J., J. Zhao, and R.N. Buick. 1992. Overexpression of H-ras oncogene induces resistance to the growth-inhibitory action of transforming growth factor beta-1 (TGF-beta 1) and alters the number and type of TGF-beta 1 receptors in rat intestinal epithelial cell clones. Oncogene. 7:521–526. [PubMed] [Google Scholar]
- Fynan, T.M., and M. Reiss. 1993. Resistance to inhibition of cell growth by transforming growth factor-beta and its role in oncogenesis. Crit. Rev. Oncog. 4:493–540. [PubMed] [Google Scholar]
- Gotzmann, J., Hottuber, C. Thallinger, M. Wolschek, B. Jansen, R. Schulte-Hermann, H. Beug, and W. Mikulits. 2002. Heptocytes convert to a fibroblastoid phenotype through the cooperation of TGF-β1 and Ha-ras; steps toward invasiveness. J. Cell Sci. In press. [DOI] [PubMed] [Google Scholar]
- Hall, M., and P.G. Peters. 1996. Genetic alterations of cyclins, cyclin-dependent kinases and Cdk inhibitors in human cancer. Adv. Cancer Res. 68:67–108. [DOI] [PubMed] [Google Scholar]
- Hanahan, D., and R.A. Weinberg. 2000. The hallmarks of cancer. Cell. 100:57–70. [DOI] [PubMed] [Google Scholar]
- Hay, E.D. 1995. An overview of epithelia-mesenchymal transition. Acta Anat. 154:8–20. [DOI] [PubMed] [Google Scholar]
- Howell, M., F. Itoh, C.E. Pierreux, S. Valgeirsdottir, S. Itoh, P. ten Dijke, and C.S. Hill. 1999. Xenopus Smad4beta is the co-Smad component of developmentally regulated transcription factor complexes responsible for induction of early mesodermal genes. Dev. Biol. 214:354–369. [DOI] [PubMed] [Google Scholar]
- Khwaja, A., K. Lehmann, B.M. Marte, and J. Downward. 1998. Phosphoinositide 3-kinase induces scattering and tubulogenesis in epithelial cells through a novel pathway. J. Biol. Chem. 273:18793–18801. [DOI] [PubMed] [Google Scholar]
- Kohl, N.E., F.R. Wilson, S.D. Mosser, E. Giuliani, S.J. deSolms, M.W. Conner, N.J. Anthony, W.J. Holtz, R.P. Gomez, T.J. Lee, et al. 1994. Protein farnesyltransferase inhibitors block the growth of ras-dependent tumors in nude mice. Proc. Natl. Acad. Sci. USA. 91:9141–9145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzschmar, M., J. Doody, I. Timokhina, and J. Massague. 1999. A mechanism of repression of TGFbeta/Smad signaling by oncogenic Ras. Genes Dev. 13:804–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann, K., E. Janda, C.E. Pierreux, M. Rytömaa, A. Schulze, M. McMahon, C.S. Hill, H. Beug, and J. Downward. 2000. Raf induces TGFβ production while blocking its apoptotic but not invasive responses: a mechanism leading to increased malignancy in epithelial cells. Genes Dev. 14:2610–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X., Y. Sun, M. Ehrlich, T. Lu, Y. Kloog, R.A. Weinberg, H.F. Lodish, and Y.I. Henis. 2000. Disruption of TGF-beta growth inhibition by oncogenic ras is linked to p27Kip1 mislocalization. Oncogene. 19:5926–5935. [DOI] [PubMed] [Google Scholar]
- Lukashev, M.E., and Z. Werb. 1998. ECM signaling: orchestrating cell behaviour and misbehaviour. Trends Cell Biol. 8:437–441. [DOI] [PubMed] [Google Scholar]
- Massague, J., and D. Wotton. 2000. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 19:1745–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClatchey, A.I. 1999. Modeling metastasis in the mouse. Oncogene. 18:5334–5339. [DOI] [PubMed] [Google Scholar]
- McEarchern, J.A., J.J. Kobie, V. Mack, R.S. Wu, L. Meade-Tollin, C.L. Arteaga, N. Dumont, D. Besselsen, E. Seftor, M.J. Hendrix, et al. 2001. Invasion and metastasis of a mammary tumor involves TGF-beta signaling. Int. J. Cancer. 91:76–82. [DOI] [PubMed] [Google Scholar]
- Nieman, M.T., R.S. Prudoff, K.R. Johnson, and M.J. Wheelock. 1999. N-cadherin promotes motility in human breast cancer cells regardless of their E-cadherin expression. J. Cell Biol. 147:631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oft, M., J. Peli, C. Rudaz, H. Schwarz, H. Beug, and E. Reichmann. 1996. TGFβ1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev. 10:2462–2477. [DOI] [PubMed] [Google Scholar]
- Oft, M., K.H. Heider, and H. Beug. 1998. TGFβ signaling is essential for carcinoma cell invasiveness and metastasis. Curr. Biol. 8:1243–1252. [DOI] [PubMed] [Google Scholar]
- Onichtchouk, D., Y.G. Chen, R. Dosch, V. Gawantka, H. Delius, J. Massague, and C. Niehrs. 1999. Silencing of TGF-beta signaling by the pseudoreceptor BAMBI. Nature. 401:480–485. [DOI] [PubMed] [Google Scholar]
- Parsons, R., L.L. Myeroff, B. Liu, J.K. Willson, S.D. Markowitz, K.W. Kinzler, and B. Vogelstein. 1995. Microsatellite instability and mutations of the transforming growth factor beta type II receptor gene in colorectal cancer. Cancer Res. 55:5548–5550. [PubMed] [Google Scholar]
- Peli, J., M. Schroter, C. Rudaz, M. Hahne, C. Meyer, E. Reichmann, and J. Tschopp. 1999. Oncogenic Ras inhibits Fas ligand-mediated apoptosis by downregulating the expression of Fas. EMBO J. 18:1824–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl, A.-K., U. Dahl, P. Wilgenbus, H. Cremer, H. Semb, and G. Christofori. 1999. Reduced expression of neural cell adhesion molecule indicates metastatic dissemination of pancreatic β tumor cells. Nat. Med. 5:286–291. [DOI] [PubMed] [Google Scholar]
- Piek, E., A. Moustakas, A. Kurisaki, C.H. Heldin, and P. ten Dijke. 1999. TGF-β type I receptor/ALK-5 and Smad proteins mediate epithelial to mesenchymal transdifferentiation in NMuMG breast epithelial cells. J. Cell Sci. 112:4557–4568. [DOI] [PubMed] [Google Scholar]
- Potempa, S., and A.J. Ridley. 1998. Activation of both MAP kinase and phosphatidylinositide 3-kinase by Ras is required for hepatocyte growth factor/scatter factor-induced adherens junction disassembly. Mol. Biol. Cell. 9:2185–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radice, G.L., H. Rayburn, H. Matsunami, K.A. Knudsen, M. Takeichi, and R.O. Hynes. 1997. Developmental defects in mouse embryos lacking N-cadherin. Dev. Biol. 181:64–78. [DOI] [PubMed] [Google Scholar]
- Reichmann, E., H. Schwarz, E.M. Deiner, I. Leitner, M. Eilers, M. Busslinger, and H. Beug. 1992. Activation of an inducible c-fos ER fusion protein causes loss of epithelial polarity and triggers epithelial-fibroblastoid conversion. Cell. 71:1103–1116. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana, P., P.H. Warne, A. Khwaja, B.M. Marte, D. Pappin, P. Das, M.D. Waterfield, A. Ridley, and J. Downward. 1997. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 89:457–467. [DOI] [PubMed] [Google Scholar]
- Rommel, C., and E. Hafen. 1998. Ras—a versatile cellular switch. Curr. Opin. Genet. Dev. 8:412–418. [DOI] [PubMed] [Google Scholar]
- Schoenenberger, C.A., A. Zuk, D. Kendall, and K.S. Matlin. 1991. Multilayering and loss of apical polarity in MDCK cells transformed with viral K-ras. J. Cell Biol. 112:873–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutte, M., R.H. Hruban, L. Hedrick, K.R. Cho, G.M. Nadasdy, C.L. Weinstein, G.S. Bova, W.B. Isaacs, P. Cairns, H. Nawroz, et al. 1996. DPC4 gene in various tumor types. Cancer Res. 56:2527–2530. [PubMed] [Google Scholar]
- Sirard, C., J.L. de la Pompa, A. Elia, A. Itie, C. Mirtsos, A. Cheung, S. Hahn, A. Wakeham, L. Schwartz, S.E. Kern, et al. 1998. The tumor suppressor gene Smad4/Dpc4 is required for gastrulation and later for anterior development of the mouse embryo. Genes Dev. 12:107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, D., S. Baur, and E.D. Hay. 2000. Epithelial-mesenchymal transformation is the mechanism for fusion of the craniofacial primordia involved in morphogenesis of the chicken lip. Dev. Biol. 228:337–349. [DOI] [PubMed] [Google Scholar]
- Takaku, K., M. Oshima, H. Miyoshi, M. Matsui, M.F. Seldin, and M.M. Taketo. 1998. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell. 92:645–656. [DOI] [PubMed] [Google Scholar]
- Taya, Y., S. O'Kane, and M.W. Ferguson. 1999. Pathogenesis of cleft palate in TGF-beta3 knockout mice. Development. 126:3869–3879. [DOI] [PubMed] [Google Scholar]
- Thiery, J.P., and D. Chopin. 1999. Epithelial cell plasticity in development and tumor progression. Cancer Metastasis Rev. 18:31–42. [DOI] [PubMed] [Google Scholar]
- Traverse, S., K. Seedorf, H. Paterson, C.J. Marshall, P. Cohen, and A. Ullrich. 1994. EGF triggers neuronal differentiation of PC12 cell that overexpress the EGF receptor. Curr. Biol. 4:694–701. [DOI] [PubMed] [Google Scholar]
- Webb, C.P., L. Van Aelst, M.H. Wigler, and G.F. Vande Woude. 1998. Signaling pathways in Ras-mediated tumorigenicity and metastasis. Proc. Natl. Acad. Sci. USA. 95:8773–8778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein, M., X. Yang, C. Li, X. Xu, J. Gotay, and C.X. Deng. 1998. Failure of egg cylinder elongation and mesoderm induction in mouse embryos lacking the tumor suppressor smad2. Proc. Natl. Acad. Sci. USA. 95:9378–9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X., C. Li, X. Xu, and C. Deng. 1998. The tumor suppressor SMAD4/DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc. Natl. Acad. Sci. USA. 95:3667–3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, Y., and J.D. Sato. 1999. MAP kinases, phosphatidylinositol 3-kinase, and p70 S6 kinase mediate the mitogenic response of human endothelial cells to vascular endothelial growth factor. J. Cell. Physiol. 178:235–246. [DOI] [PubMed] [Google Scholar]