

Abstract
β-Spectrin and ankyrin are major components of the membrane cytoskeleton. We have generated mice carrying a null mutation in the βIV-spectrin gene using gene trapping in embryonic stem cells. Mice homozygous for the mutation exhibit tremors and contraction of hindlimbs. βIV-spectrin expression is mostly restricted to neurons, where it colocalizes with and binds to ankyrin-G at axon initial segments (AISs) and nodes of Ranvier (NR). In βIV-spectrin–null neurons, neither ankyrin-G nor voltage-gated sodium channels (VGSC) are correctly clustered at these sites, suggesting that impaired action potential caused by mislocalization of VGSC leads to the phenotype. Conversely, in ankyrin-G–null neurons, βIV-spectrin is not localized to these sites. These results indicate that βIV-spectrin and ankyrin-G mutually stabilize the membrane protein cluster and the linked membrane cytoskeleton at AIS and NR.
Keywords: βIV-spectrin; ankyrin-G; voltage-gated sodium channel; axon initial segment; node of Ranvier
Introduction
The axons of myelinated neurons are wrapped with myelin sheaths produced by oligodendrocytes in the central nervous system, and by Schwann cells in the peripheral nervous system (for review see Arroyo and Scherer, 2000). The insulating property of the sheath facilitates a rapid and efficient propagation of the action potential through the axon (for review see Vabnick and Shrager, 1998). Two sites not wrapped by myelin are the axon initial segment (AIS)* and the node of Ranvier (NR). The AIS is the part of the axon between the cell body and the beginning of the myelin sheath, and the NR is a site of discontinuity between successive myelin sheaths along the axon (for review see Arroyo and Scherer, 2000; Peles and Salzer, 2000). Both of these sites are polarized structures where the same membrane proteins are clustered. These proteins include voltage-gated sodium channels (VGSCs), the cell adhesion molecules neurofascin and NrCAM, and the membrane skeletal protein ankyrin-G (for review see Bennett and Lambert, 1999). VGSC, neurofascin, and NrCAM all bind through their cytoplasmic domains to ankyrin-G (Srinivasan et al., 1988; Davis et al., 1996). VGSCs are responsible for producing the inward ionic flow to generate the action potential at AIS and to maintain the action potential at NR. In addition, these clustered proteins at AIS, together with the linked spectrin-actin membrane cytoskeleton, may function as a diffusion barrier that prevents axonal proteins from leaking out to the cell body (Winckler et al., 1999).
The mechanisms by which these specific membrane proteins are concentrated at AIS and NR are not yet understood. A number of in vivo and in vitro studies have implicated myelinating glial cells in localizing axonal membrane proteins to NR both in the central and peripheral nervous systems (for review see Peles and Salzer, 2000; Rasband and Trimmer, 2001). Several axonal proteins have also been implicated in this process. It has been suggested that neurofascin and NrCAM may have a role in defining the initial sites for clustering in rat sciatic nerves, because clustering of these cell adhesion molecules precedes that of ankyrin-G and VGSC as well as myelination of the axon (Lambert et al., 1997). However, in cerebellar Purkinje cells of ankyrin-G knockout mice, clustering of neurofascin as well as VGSC at AIS is disrupted, leading to a failure in normal action potential firing (Zhou et al., 1998). These results indicate that ankyrin-G is an essential component for localizing neurofascin and VGSC at these sites. Taken together, these observations suggest that the membrane protein clustering is a sequential process that includes glial and axonal proteins, and that the factors defining the site of protein clustering and the factors stabilizing the once-formed cluster are distinct.
Ankyrins are linked to the spectrin-actin membrane cytoskeleton through binding to β-spectrins (Davis and Bennett, 1990; Kennedy et al., 1991). Therefore, it is possible that the membrane cytoskeleton at AIS and NR binds ankyrin-G and defines the localization of associated membrane proteins. Indeed, it has been shown that the putative β-spectrin–binding domain of ankyrin-G alone can be targeted to AIS in cultured neurons (Zhang and Bennett, 1998). As βII-spectrin is localized to axons (whereas βIΣ2- and βIII-spectrins are localized to the cell body and dendrites in neurons) (Riederer et al., 1986; Ohara et al., 1998), βII-spectrin has been believed to bind ankyrin-G at AIS and NR. However, it has not been thought to be involved in defining the localization of ankyrin-G, as it is present throughout the axon (Riederer et al., 1986). Recently, a new member of the β-spectrin family, βIV-spectrin, has been identified (Berghs et al., 2000; Tse et al., 2001). This protein was shown to colocalize with ankyrin-G at AIS and NR (Berghs et al., 2000), raising the possibility that this novel β-spectrin might bind ankyrin-G and thus be involved in clustering ankyrin-G and associated membrane proteins.
Using gene trap mutagenesis to identify and mutate genes regulated by retinoic acid (Komada et al., 2000), we have identified the ROSA62 mouse strain that harbors a null mutation in the βIV-spectrin gene. ROSA62 mice exhibit tremors and hindlimb contraction, and the phenotype increases in severity with age. We demonstrate that βIV-spectrin plays an essential role in localizing ankyrin-G and VGSC at AIS and NR in neurons. Conversely, we show that βIV-spectrin localization to AIS requires ankyrin-G, indicating a mutual role for βIV-spectrin and ankyrin-G in stabilizing the membrane protein cluster and the linked membrane skeleton at these sites. Very recently, Parkinson et al. (2001) showed that the spontaneous mouse mutant, quivering (qv) (Yoon and Les, 1957), carries a mutation in the βIV-spectrin gene. We propose that the phenotype of ROSA62 and qv mice is primarily due to mislocalization of VGSC at AIS and NR.
Results
We have screened for retinoic acid–inducible gene trapping in mouse embryonic stem (ES) cells using the retroviral vector ROSAβgeo*, which transduces a promoterless βgeo* (a fusion gene of β-galactosidase and neomycin phosphotransferase II) as a reporter (Komada et al., 2000; http://www. fhcrc.org/labs/soriano/trap.html). Northern blot analysis confirmed that one of the lines, ROSA62, exhibited a 17-fold induction of the reporter gene expression after 6-h treatment with 1 μM all-trans retinoic acid (unpublished data). This strong induction led us to characterize the gene trap mutation in detail. As the expression of the ROSA62 gene is mostly restricted to neurons in mice (see below), this induction might reflect the ability of retinoic acid to induce neuronal differentiation of ES cells.
Neuromuscular defects in ROSA62 mutant mice
Germline chimeric mice were generated by injecting ROSA62 mutant ES cells into blastocysts, and were crossed to 129/S4 and C57BL/6J mice to derive mutants. Heterozygotes exhibited no overt phenotype. Viable homozygous mutant mice were recovered from intercrossing of heterozygotes according to Mendelian expectations and were mostly of normal size. However, by 3 wk of age, homozygous mutants were distinguishable from wild-type and heterozygous mutant littermates by fine tremors. Until 2–3 mo of age, the homozygotes were grossly normal except for the tremor. They also exhibited clasping of the hindlimbs when held by the tail, a hallmark of ataxia. As they grew older, the phenotype became more severe. By 6–10 mo of age, they exhibited continuous contraction of hindlimb skeletal muscle and were not able to walk. The same phenotype was observed on congenic 129/S4, mixed 129/S4 x C57BL/6J, and congenic C57BL/6J (>10-generation backcross) genetic background. The following analyses were performed on mixed 129/S4x C57BL/6J genetic background.
Expression pattern of the ROSA62 gene in mice
Expression of β-galactosidase activity in gene trap mutant mice is driven by the promoter of the trapped gene (Friedrich and Soriano, 1991). Therefore, ROSA62 heterozygous mutant embryos and adult tissues were isolated and stained with X-gal to determine the expression pattern of the trapped gene.
In embryonic day 10.5 embryos, expression was restricted to cranial and dorsal root ganglia (Fig. 1 A). In adult mice, expression was detected in many regions of the brain, with the highest level in the hippocampus and cerebellum (Fig. 1 B). Expression was also detected in the spinal cord (Fig. 1 C). In contrast, no β-galactosidase activity was detected in skeletal muscle (unpublished data), suggesting that the phenotype of ROSA62 mutant mice is primarily due to a defect in neurons but not skeletal muscle.
Figure 1.

Expression pattern of the ROSA62 gene in mice. Expression pattern of the ROSA62 gene was assessed by X-gal staining of heterozygous mutant mice. (A) Embryonic day 10.5 embryo. Expression is detected in the dorsal root ganglia (DRG) and trigeminal ganglia (TG). (B) Adult brain. High level of expression is detected in the hippocampus (HC) and cerebellum (CL). (C) Adult spinal cord. GM, gray matter; WM, white matter. (D) Adult pancreas. Expression is restricted to the islets of Langerhans (L). (E) Adult testis. Expression is restricted to the round spermatids (RS) and elongated spermatids (ES) facing the lumen (lu) of the seminiferous tubule. Bars: (A and C) 500 μm; (B) 1 mm ; (D) 100 μm; (E) 30 μm .
Other than the neuronal tissues, ROSA62 expression was detected in the pancreas (Fig. 1 D) and testis (Fig. 1 E), but not in the heart, lung, liver, kidney, spleen, stomach, small intestine, colon, ovary, and uterus (unpublished data). In the pancreas, expression was restricted to the islet of Langerhans (Fig. 1 D), whereas in the testis, it was detected in round and elongated spermatids (Fig. 1 E). However, no histological abnormalities were observed in any of these tissues of the homozygotes (unpublished data). The homozygous mutant males reproduced very inefficiently when mated with wild-type females. However, this is likely to reflect a mating problem as litter sizes were normal.
Mutation in βIV-spectrin in ROSA62 mutant mice
To identify the gene trapped in this mutant, the ROSA62–βgeo* fusion cDNA was cloned from ROSA62 mutant ES cells by 5′-RACE, and full-length cDNAs were subsequently cloned by screening cDNA libraries. Sequencing of the cDNAs revealed that the trapped gene encodes βIV-spectrin, for which the human homologue has been recently identified (Berghs et al., 2000; Tse et al., 2001). Two βIV-spectrin splice isoforms, Σ1 and Σ6, were cloned (Fig. 2 A). βIVΣ1-spectrin was 2561 amino acids long, consisted of an NH2-terminal actin-binding domain, 17 spectrin repeats, a variable region, and a COOH-terminal plecstrin homology (PH) domain, and had 95% overall sequence identity to its human homologue. The actin-binding domain, the entire spectrin repeats, and the PH domain of βIVΣ1-spectrin were ∼70, 40, and 55% identical in amino acid sequences, respectively, to those of βI-, βII-, and βIII-spectrins, with the highest sequence identity to βII-spectrin.
Figure 2.

Characterization of βIV-spectrin and its gene products. (A) Schematic diagram of the βIVΣ1- and Σ6-spectrin isoforms. The gene trap vector was inserted between exons encoding the spectrin repeat 12 of the βIV-spectrin gene. (B) Sequence of the 5′ region of βIVΣ6-spectrin cDNA and its comparison with the corresponding region of βIVΣ1-spectrin cDNA. The sequence of Σ6 diverges upstream of the nucleotide 4079 of Σ1 (arrow). (C) Northern blot analysis of βIV-spectrin expression in brains from wild-type (+/+), heterozygous (+/−), and homozygous (−/−) mutant mice. The regions of the 5′- and 3′-probes are indicated in (A). Positions of the 28S and 18S rRNAs are indicated. (D) Western blot analysis of βIV-spectrin in brain lysates from wild-type (+/+), heterozygous (+/−), and homozygous (−/−) mutant mice. Positions of size markers are indicated in kD on the left. The sequence data are available from Genbank/EMBL/DDBJ accession no. AB055618 for βIVΣ1-spectrin and AB055619, AB055620, AB055621 for βIVΣ6-spectrin.
The other isoform has not been identified in human and was termed βIVΣ6-spectrin in this study (Σ2–5 isoforms have been reported in humans) (Berghs et al., 2000; Tse et al., 2001). This novel isoform lacked the NH2-terminal actin-binding domain, the spectrin repeats 1–9, and part of spectrin repeat 10 (Fig. 2 A). The βIVΣ6-spectrin cDNA has a distinct 81-bp sequence upstream of nucleotide 4079 of βIVΣ1-spectrin, which is derived from an alternative exon (Fig. 2 B). An in-frame translation stop codon is located 72 bp upstream of the ATG encoding methionine 1321 of the Σ1 isoform, suggesting that this ATG is used as an translation initiation codon for βIVΣ6-spectrin (Fig. 2 B). Two more distinct βIVΣ6-spectrin cDNAs with a different 5′ noncoding region diverging upstream of nucleotide 4079 of βIVΣ1-spectrin were also cloned (unpublished data), indicating that the second exon for βIVΣ6-spectrin can be alternatively spliced from three distinct first exons. The original 5′-RACE product cloned from ROSA62 mutant ES cells encoded the Σ6 isoform, and its sequence indicated that the gene trap vector was inserted between exons that encode the spectrin repeat 12 of βIVΣ1-spectrin (Fig. 2 A). We did not isolate cDNAs encoding the Σ2, Σ3, Σ4, and Σ5 isoforms identified in human.
Expression of βIV-spectrin in mice and its absence in ROSA62 mutant
To determine if the gene trap mutation leads to a null allele, Northern and Western blot analyses were performed. By Northern blot analysis with a βIV-spectrin cDNA fragment encoding the 3′ region (3′-probe in Fig. 2 A), four βIV-spectrin transcripts (∼9, 7, 6.5, and 5 kb in size) were detected in the brain of wild-type and heterozygous mutant mice (Fig. 2 C). Using a 5′-probe specific to βIVΣ1-spectrin (Fig. 2 A), only the 9-kb transcript was detected (Fig. 2 C). As the Σ1 and Σ6 cDNAs we cloned are 8.7 and 4.7 kb long, respectively, mRNAs encoding these isoforms likely correspond to the 9- and 5-kb transcripts, respectively. In the homozygous mutant, all of the transcripts detected with the 3′-probe were absent (Fig. 2 C). Using the 5′-probe, a transcript which is ∼9 kb in size was detected (unpublished data). As this transcript did not hybridize with the 3′ probe, it likely represents the fusion transcript between the 5′ end of βIV-spectrin and the reporter. Western blot analysis was performed using a polyclonal antibody raised against the variable region of βIV-spectrin which has no homology to other proteins. In the wild-type brain, the antibody detected a minor ∼300-kD band and a major ∼160-kD band (Fig. 2 D). The faint band of ∼140 kD may be another splice isoform or a degradation product. From the calculated molecular mass (289 kD for Σ1 and 141 kD for Σ6), the ∼300- and ∼160-kD bands most likely correspond to βIVΣ1- and Σ6-spectrins, respectively. Interestingly, the truncated Σ6 isoform was much highly expressed than the full-length Σ1 isoform both at the level of mRNA as well as protein. The same bands were detected in the heterozygotes, but were less abundant, and they were absent from the homozygous mutant extract (Fig. 2 D). A truncated βIV-spectrin containing the NH2-terminal portion might conceivably be produced in the homozygotes. However, the absence of any mRNAs and proteins detected by the 3′ probe and the antibody, respectively, as well as the gene trap insertion site indicates that it lacks the spectrin repeats 12–17, the variable region, and the PH domain. Therefore, the gene trap insertion has most likely created a functionally null allele of the βIV-spectrin gene.
Subcellular localization of βIV-spectrin
Although spectrin was originally identified as the major component of the plasma membrane skeleton, it is also associated with various intracellular organelles and is implicated in maintaining organelle structures and membrane trafficking (for review see De Matteis and Morrow, 2000). Therefore, subcellular localization of endogenous βIV-spectrin was examined by immunofluorescence staining first in ES cells (the only cell line among those tested expressing βIV-spectrin) using the anti–βIV-spectrin antibody. When plated on gelatinized cover glass, ES cells often formed an epithelia-like monolayer of cells that adhered to each other. In these cells, the anti–βIV-spectrin antibody stained subdomains of the adherens junctions which were also stained for F-actin and β-catenin (Fig. 3 , A–A′′; unpublished data). βIV-spectrin completely colocalized with another adherens junction protein, vinculin (Fig. 3, B-B′′). These results suggest that βIV-spectrin functions as a component of the plasma membrane skeleton.
Figure 3.

Subcellular localization of βIV-spectrin. Cultured ES cells (A–A′′ and B–B′′), dentate gyrus (C–C′′) and cerebellum (D–D′′) of the brain, and sciatic nerves (E–E′′) were stained with anti–βIV-spectrin antibody (A, B, C, D, and E) together with phalloidin (A′), anti-vinculin (B'), anti–ankyrin-G (C′ and D′), and anti-Nav1.6 (E′). (A′′, B′′, C′′, D′′, and E′′) Merged images. In D′′, the granular layer (G), Purkinje cell layer (P), and molecular layer (M) of the cerebellum are separated by dotted lines. Bars: (A–A,′′ B–B,′′ and E–E′′) 30 μm; (C–C′′ and D–D′′) 100 μm.
To understand the function of βIV-spectrin in physiological cell types, its subcellular localization was next examined in neurons in sections of mouse brain, spinal cord, and sciatic nerve. In agreement with the X-gal staining pattern of the heterozygous mutant brain (Fig. 1 B), strong anti–βIV-spectrin staining was observed in the dentate gyrus (Fig. 3 C) and cerebellum (Fig. 3 D). This staining pattern coincided with that of 480/270 kD ankyrin-G (Fig. 3, C′ and D′), a marker of AIS and NR (Kordeli et al., 1995). Colocalization of βIV-spectrin with ankyrin-G was also detected in neurons in other regions of the brain and spinal cord (unpublished data). In sciatic nerves, βIV-spectrin colocalized with Nav1.6, a member of the VGSC α-subunit family that is concentrated at NR (Caldwell et al., 2000; Fig. 3, E-E′′). These results indicate that βIV-spectrin localizes to AIS and NR of neurons in both central and peripheral nervous systems.
Binding of βIV-spectrin to ankyrin-G
βI- and βII-spectrins have been shown to bind ankyrin-R and ankyrin-B (Davis and Bennett, 1990; Kennedy et al., 1991). This fact, together with colocalization of βIV-spectrin with ankyrin-G at AIS and NR, suggested that βIV-spectrin might bind ankyrin-G. To test this possibility, Myc-tagged βIVΣ6-spectrin (Myc-βIVΣ6) was cotransfected with green fluorescent protein (GFP), GFP-tagged 270-kD ankyrin-G (AnkG-GFP), or GFP-tagged 220-kD ankyrin-B (AnkB-GFP) into COS-7 cells. Transfected cells were lysed and immunoprecipitated, and then immunoblotted with anti-Myc or anti-GFP antibody. Whereas Myc-βIVΣ6 was expressed at a similar level in the three transfectants (Fig. 4 C), GFP and AnkB-GFP were much more highly expressed than AnkG-GFP (Fig. 4 A). Consistent with the presence in βIVΣ6-spectrin of the spectrin repeat 15, which has been mapped as an ankyrin-binding domain in βI- and βII-spectrins (Kennedy et al., 1991), the anti-Myc antibody coprecipitated both AnkG-GFP and AnkB-GFP but not GFP alone (Fig. 4 B). However, significantly more AnkG-GFP was brought down than AnkB-GFP (Fig. 4 B). Similarly, AnkG-GFP coprecipitated Myc-βIVΣ6 more efficiently than AnkB-GFP (Fig. 4 D). These results indicate that βIV-spectrin binds to ankyrin-G with high affinity, and to ankyrin-B to a lesser extent.
Figure 4.
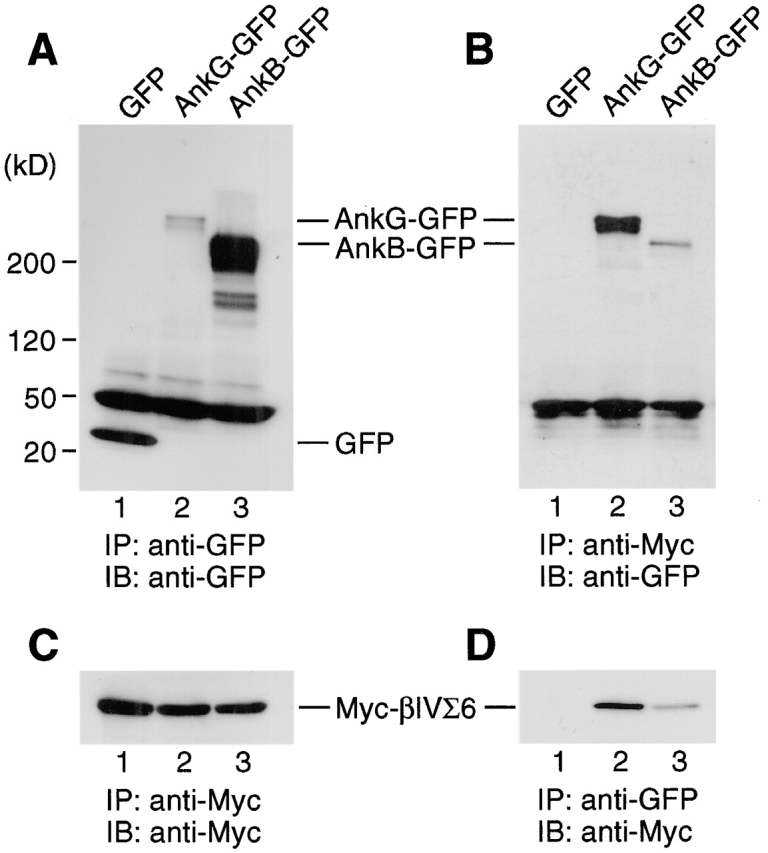
Binding of βIV-spectrin to ankyrin. COS-7 cells were transfected with Myc-tagged βIVΣ6-spectrin (Myc-βIVΣ6) together with GFP (lanes 1), GFP-tagged 270 kD ankyrin-G (AnkG-GFP; lanes 2), or GFP-tagged 220 kD ankyrin-B (AnkB-GFP; lanes 3). Cell lysates were immunoprecipitated with anti-GFP (A and D) or anti-Myc (B and C). Immunoprecipitates were separated by SDS-PAGE and immunoblotted with anti-GFP (A and B) or anti-Myc (C and D). Positions of size markers are indicated in kD on the left.
Clustering of ankyrin-G and VGSC at AIS and NR in βIV-spectrin–null neurons
The subcellular localization of βIV-spectrin as well as binding to ankyrin-G raised the possibility that it is involved in regulating the localization of ankyrin-G and thus ankyrin-G-binding membrane proteins to AIS and NR. To test this possibility, we examined ankyrin-G and VGSC localization in βIV-spectrin–null neurons.
As expected from the Northern and Western blot analyses (Fig. 2, C and D), no anti–βIV-spectrin staining was detected above background level in the βIV-spectrin mutant cerebellar Purkinje and hippocampal pyramidal neurons (Fig. 5, B and D) . Compared with wild-type neurons where ankyrin-G colocalized with βIV-spectrin at AIS (Fig. 5, A, A′, C, C′, and E), staining for ankyrin-G was undetectable or very faint at these sites in most βIV-spectrin–null neurons (Fig. 5, B′, D′, and F). In mutant Purkinje cells with faint anti–ankyrin-G staining, staining was not restricted to the AIS but was spread over the rest of the axon (unpublished data). Although anti–ankyrin-G staining was still restricted to the AIS in some neurons of the mutant, it was much weaker than in wild-type neurons (Fig. 5, E and F).
Figure 5.

Clustering of ankyrin-G at AIS of βIV-spectrin –null cerebellar and hippocampal neurons. Cerebellum (A, A′, B, and B′) and hippocampus (C, C′, D, and D′) of wild-type (A, A′, C, and C') and βIV-spectrin–null (B, B′, D, and D′) mice were stained for βIV-spectrin and ankyrin-G as indicated. In E and F, Purkinje cells from wild-type (E) and βIV-spectrin–null (F) mice were double stained for ankyrin-G (red) and calbindin D-28K to visualize their cell bodies (green), and are shown at higher magnification. The granular layer (G), Purkinje cell layer (P), and molecular layer (M) of the cerebellum are separated by dotted lines (A, A′, B, and B′), and Purkinje cell bodies (Pu) are indicated in E and F. AIS of Purkinje cells are indicated by arrowheads (A, A′, E, and F). Arrows in D and D′ indicate the pyramidal cell layer of the hippocampus, where AIS of pyramidal neurons are detected in wild-type mice (arrowheads in C and C′). Mice were killed at 3 mo of age. Bars, 50 μm.
The localization of VGSC was examined using an anti–pan-sodium channel antibody that recognizes all the known vertebrate sodium channel isoforms (Rasband et al., 1999). In wild-type mice, VGSC colocalized with βIV-spectrin at AIS of cerebral cortex (Fig. 6, A and A′) and hippocampal pyramidal (Fig. 6, C and C′) neurons. However, in βIV-spectrin mutant mice, VGSC staining was very faint in the cerebral cortex (Fig. 6, B and B′) and mostly undetectable in the hippocampus (Fig. 6, D and D′). Similar mislocalization was also observed for a specific sodium channel isoform Nav1.6 (Fig. 6, F and F′), which normally localizes to AIS in Purkinje cells (Fig. 6, E and E′; S. Jenkins and V. Bennett, personal communication).
Figure 6.

Clustering of VGSC at AIS of βIV-spectrin –null cerebral, hippocampal, and cerebellar neurons. Cerebral cortex (A, A′, B, and B′), hippocampus (C, C′, D, and D′), and cerebellum (E, E′, F, and F′) of wild-type (A, A′, C, C′, E, and E′) and βIV-spectrin–null (B, B′, D, D′, F, and F′) mice were stained with anti–βIV-spectrin (A, B, C, D, E, and F) together with anti–pan-sodium channel (pan-SC; A′, B′, C′, and D′) or anti-Nav1.6 (E′ and F′). Arrowheads indicate AIS of cerebral (A, A′, and B′), hippocampal pyramidal (C and C′), and Purkinje (E and E') neurons. The granular layer (G), Purkinje cell layer (P), and molecular layer (M) of the cerebellum are separated by dotted lines (E, E′, F, and F′). Mice were killed at 3 mo of age. Bars, 20 μm.
Next, we examined the localization of Nav1.6 at NR of the peripheral nervous system. In the βIV-spectrin–null sciatic nerves, Nav1.6-positive but βIV-spectrin–negative NR were detected (Fig. 7 , B-B′′). However, the number of Nav1.6-positive NR was less than in the wild-type littermate in which the NR were also positive for βIV-spectrin (Fig. 7, A–A′′). The number of Nav1.6-positive NR was quantified in sciatic nerve sections of wild-type and the mutant mice. Counting of nine randomly chosen 0.1-mm2 fields from three mice of each genotype revealed that the numbers were 98 ± 23 and 44 ± 15 (mean ± SD) per field in the wild-type and the mutant, respectively (55% reduction in the mutant; Fig. 7 C). In addition, anti-Nav1.6 staining of the NR was often fainter in the mutant than in the wild-type control (Fig. 7, A′ and B′, insets). Phase contrast microscopy showed that the number as well as the morphology of NR was unaffected in the mutant (unpublished data).
Figure 7.

Clustering of Nav1.6 at NR of βIV-spectrin –null sciatic nerves. Double staining of wild-type (A, A′, and A′′) and βIV-spectrin–null (B, B′, and B′′) sciatic nerves with anti–βIV-spectrin (A and B) and anti-Nav1.6 (A′ and B′) antibodies. (A′′) and (B′′) are merged images. Arrowheads in A′, B′, A′′, and B′′ indicate some of the Nav1.6-positive NR which are also positive for βIV-spectrin in the wild-type (A). Insets in A′ and B′ show enlarged images of typical NR. In C, the number of Nav1.6-positive NR in wild-type (+/+) and βIV-spectrin–null (−/−) sciatic nerve sections were quantified. Mean numbers per 0.1 mm2 field are shown with SD (n = 9 for each genotype). Mice were killed at 3 mo of age. Bars, 100 μm.
Taken together, these results indicate that normal localization of ankyrin-G and VGSC to AIS and NR requires βIV-spectrin in its membrane skeleton.
Localization of βIV-spectrin at AIS of ankyrin-G–null neurons
Next, we examined whether localization of βIV-spectrin to AIS requires ankyrin-G. To test this, we examined the localization of βIV-spectrin in cerebellar neurons of mutant mice generated by Zhou et al. (1998) in which the cerebellum-specific form of ankyrin-G is knocked out. Compared with wild-type cerebellum where AIS of both Purkinje and granular neurons were positive for ankyrin-G and βIV-spectrin (Fig. 8, A , A′, and D), βIV-spectrin staining as well as ankyrin-G staining was mostly lost in the knockout mice (Fig. 8, B, B′, and E). βIV-spectrin localization was normal in the mutant in other regions than the cerebellum, such as the hippocampus, where ankyrin-G was expressed (Fig. 8, C and C′; unpublished data), indicating that ankyrin-G is a prerequisite for the correct βIV-spectrin localization in AIS.
Figure 8.

Localization of βIV-spectrin at AIS of ankyrin-G–null neurons. Cerebellum (A, A′, B, and B′) and hippocampus (C and C′) of wild-type (A and A′) and cerebellum-specific ankyrin-G– null (B, B′, C, and C′) mice were stained with anti–ankyrin-G (A, B, and C) and anti–βIV-spectrin (A′, B′, and C′) antibodies. In D and E, Purkinje cells from wild-type (D) and ankyrin-G–null (E) mice were double stained for bIV-spectrin (red) and calbindin D-28K (green), and are shown at higher magnification. The granular layer (G), Purkinje cell layer (P), and molecular layer (M) of the cerebellum are separated by dotted lines (A, A′, B, and B′). Purkinje cell bodies (Pu) are indicated in (D) and (E). Arrowheads indicate AIS of Purkinje cells (A, A′, and D). In B and B′, the contours of Purkinje cell bodies were observed in the absence of specific staining in the mutant due to background staining. Mice were killed at 5 wk of age. Bars, 50 μm.
Discussion
Cell polarization involves the localization of individual proteins to specific subdomains in the cell and requires additional protein components to maintain this localization. The spectrin family is believed to be involved in localizing specific membrane proteins at specific submembrane domains (for review, see Drubin and Nelson, 1996). Here, we demonstrate that a new member of the β-spectrin family, βIV, plays an essential role in maintaining localization of specific membrane proteins to polarized regions of neurons, and that this regulated localization is essential for normal neurological function.
Molecular mechanisms underlying the βIV-spectrin–null phenotype
Mislocalization of VGSC to AIS and NR in the βIV-spectrin mutant suggested that the neurological phenotypes of the mutant are, at least in part, due to impaired firing and propagation of the action potential in the central and peripheral nervous systems. This is supported by mouse mutants in which expression or localization of VGSC is affected. Nav1.6, also called Scn8a, is the major isoform of VGSC α-subunit localized at AIS and NR of cerebellar neurons as well as the sciatic nerve (S. Jenkins and V. Bennett, personal communication; Caldwell et al., 2000). It has been shown that in mice with mutations in the Nav1.6 gene, action potential firing is impaired in Purkinje cells (Raman et al., 1997). Similarly in cerebellum-specific ankyrin-G knockout mice, mislocalization of VGSC to AIS is associated with the impaired action potential firing in Purkinje cells (Zhou et al., 1998). As ankyrin-G is not correctly localized to AIS in the βIV-spectrin mutant, localization of other ankyrin-G-binding membrane proteins such as neurofascin and NrCAM is also likely to be affected. Therefore, the possibility that mislocalization of these proteins also contributes to the phenotype cannot be excluded. In this context, it will be interesting to compare the βIV-spectrin mutant phenotype with that of the NrCAM knockout mice, in which a possible defect in AIS and NR has not been examined yet (Moré et al., 2001).
Although disruption of the ankyrin-G gene is restricted to the cerebellum in the mutant mice generated by Zhou et al. (1998), the resulting phenotype is more severe than that observed in our βIV-spectrin mutant. The ankyrin-G mutant mice frequently fall down when prodded to walk, and in some cases exhibit premature death following uncontrollable jumping and convulsions (Zhou et al., 1998). These phenotypes were not observed in the βIV-spectrin mutant. Faint ankyrin-G and Nav1.6 staining at AIS in some Purkinje cells of the βIV-spectrin mutant might indicate that the milder phenotype is due to the trace amount of VGSC remaining in cerebellar neurons. Taking into account the fact that the Na v1.6-null mice die before 4 wk of age due to muscular atrophy (Duchen and Stefani, 1971; Burgess et al., 1995), incomplete mislocalization of VGSC associated with relatively mild phenotype in the βIV-spectrin mutant is likely.
Role for βIV-spectrin in membrane protein clustering at AIS and NR
Mislocalization of ankyrin-G and VGSC in the βIV-spectrin mutant raises the question of whether βIV-spectrin is required for targeting these membrane proteins to AIS and NR or for stabilizing their localization at these sites. The observation that some ankyrin-G and VGSC labeling still occurs in some AIS and NR in the mutant tends to support the latter, although it is also possible that trace amounts of other β-spectrins at AIS and NR partially compensate for βIV-spectrin function. The increased severity of the βIV-spectrin mutant phenotype with age is also consistent with the role for βIV-spectrin in stabilizing the membrane protein cluster. Conversely, βIV-spectrin was also excluded from AIS of Purkinje cells in region-specific ankyrin-G knockout mice, suggesting that ankyrin-G in turn stabilizes the βIV-spectrin localization at these sites. High binding affinity between βIV-spectrin and ankyrin-G, as shown by coimmunoprecipitation experiments, must underlie this mutual stabilization. As βIV-spectrin localization to AIS was almost completely disrupted in the ankyrin-G mutant, ankyrin-G may also play a role in targeting βIV-spectrin to AIS and NR during development of neurons. Initial clustering of ankyrin-G at these sites is thought to be regulated by other axonal proteins such as VGSC, neurofascin, or NrCAM, that are already clustered at these sites by a glial signal (for review see Peles and Salzer, 2000; Rasband and Trimmer, 2001). Therefore, it is possible that ankyrin-G, which is recruited to these sites by other membrane proteins, recruits βIV-spectrin, and once βIV-spectrin forms a complex with the membrane proteins, it in turn serves to stabilize the protein clustering at these sites.
βIV-spectrin has a variable region between the spectrin repeat 17 and the PH domain. This domain was termed ERQES domain because it contains four repeats of those five amino acid residues in human (Berghs et al., 2000). This domain (∼300 amino acids) is much larger than the corresponding regions of other β-spectrins (∼100 amino acids) and it is rich in proline (15%) and positively (20%), and negatively charged (16%) residues. This domain has no sequence homology to any other proteins including other β-spectrins in databases, suggesting that it has a function specific to βIV-spectrin. One possibility is that it serves as an interaction domain with an unknown protein to regulate protein clustering at AIS and NR.
The major βIV-spectrin isoform, Σ6
In this study, we cloned cDNAs encoding two splice isoforms for βIV-spectrin, Σ1 and Σ6, from three independent libraries. The Σ1 isoform is a full-length type consisting of an actin-binding domain, 17 spectrin repeats, a variable region, and a PH domain, and has been reported previously in human (Berghs et al., 2000; Tse et al., 2001). The other isoform, Σ6, is an NH2-terminal truncation lacking the actin-binding domain and the spectrin repeats 1–9 and a part of 10. Although various splice isoforms have been reported for other β-spectrins, this type has not been identified, suggesting that this is a unique isoform for βIV-spectrin. Other groups have identified four additional βIV-spectrin isoforms (Σ2–Σ5) in human (Berghs et al., 2000; Tse et al., 2001). It is noteworthy that among these isoforms, Σ6 appears to be the major isoform at least in the brain, as the size of the Σ6 cDNA (4.7 kb) and its protein product (141 kD) roughly correspond to that of the major bands in Northern blotting (∼5 kb) and in Western blotting (∼160 kD), respectively. On the contrary, the size of mRNAs encoding Σ2, Σ3, and Σ4 are much larger than 5 kb (∼9 kb) (Berghs et al., 2000), whereas the size of mRNA encoding Σ5 is expected to be much smaller than 5 kb (∼2.5 kb) (Tse et al., 2001). By Western blotting with antibodies raised against the variable region and the COOH-terminal region of βIV-spectrin, Berghs et al. (2000) also detected two major truncated isoforms with similar molecular mass (βIV-spectrin 160 and 140) which were not characterized further. One of these proteins is likely to be the Σ6 isoform.
In a classical model of the spectrin-actin membrane skeleton, a tetramer consisting of two α- and two β-spectrin molecules is linked to the actin filament through the actin-binding domain of β-spectrin (for review see Bennett and Gilligan, 1993; Winkelmann and Forget, 1993). As Σ6 lacks the actin-binding domain as well as part of the spectrin repeats, it is unclear how this isoform fits into the model. Although it is impossible to distinguish Σ1 and Σ6 in tissues by immunostaining, as our anti–βIV-spectrin antibody recognizes the variable region, Western blotting suggested that immunostaining in neurons with the antibody mostly reflects the expression of Σ6. These results indicate an unknown structure of the membrane skeleton specific for AIS and NR.
Quivering, a spontaneous mutation in βIV-spectrin in mice
The βIV-spectrin gene has been mapped to the human chromosome 19q13.13 and mouse chromosome 7 near the centromere (Berghs et al., 2000; Tse et al., 2001; unpublished data). Very recently, Parkinson et al. (2001) reported that the spontaneous mouse mutation “quivering” (qv), which has been mapped to chromosome 7 at 14.5 cM from the centromere (Yoon and Les, 1957), carries a mutation in the βIV-spectrin gene. Qv is an autosomal recessive mutation that causes various phenotypes including tremors, progressive ataxia with hindlimb paralysis, and deafness (Yoon and Les, 1957) (MGD; http://www.informatics.jax.org). Although we have not examined deafness in our βIV-spectrin mutant, other neurological phenotypes in the qv/qv mice are consistent with our findings. The allele of βIV-spectrin that we have generated is a null allele, based on Northern and Western blot analyses, whereas at least five of seven known spontaneous qv mutations are the results of single nucleotide changes or a small deletion/insertion (Parkinson et al., 2001). Parkinson et al. (2001) compared three alleles in a test for auditory brainstem function and suggest that various mutations may result in different degrees of severity of the phenotype. Therefore, it will be interesting to perform a molecular analysis of the different qv alleles (for example, the Σ6 isoform can be normally expressed from the qv lnd allele) and to compare the phenotypic severity with the allele presented in this work.
The results presented in our study strongly suggest that the ROSA62 and the qv/qv phenotype, and the impaired action potential firing and propagation in the auditory neurons in the qv/qv mice, are primarily due to mislocalization of VGSC to AIS and NR. Parkinson et al. (2001) also showed mislocalization of the potassium channel KCNA1 in axons of qv/qv mice. However, this is likely to be a secondary defect, as KCNA1 is not localized to AIS and NR but to the juxtaparanodal regions flanking NR by a distinct mechanism involving binding to a cell recognition molecule Caspr2 via an unidentified PDZ domain protein (Wang et al., 1993; Poliak et al., 1999). The demonstration of an essential role for a region-specific spectrin membrane skeleton in neurons might provide new insights into the molecular mechanisms underlying certain neuropathological conditions in human.
Materials and methods
Derivation and genotyping of mice
The ROSA62 mutant ES cell clone was obtained by screening for retinoic acid–inducible gene trapping as described (Komada et al., 2000). Blastocyst injections were performed using standard procedures. Mice were genotyped by Southern blot analysis using DNAs from tail biopsies (Komada et al., 2000). DNAs were digested with BamHI and hybridized with a neo probe. Heterozygotes and homozygotes were distinguished by the intensity of a neo-positive band.
X-gal staining and histology
X-gal staining was performed as described (Komada et al., 2000). For staining adult tissues, mice were perfused transcardially with 4% formaldehyde. Brains and spinal cords were sectioned at 100 μm using a vibratome before staining. After staining, pancreas and testis were embedded in paraffin, sectioned at 5 μm, and counterstained with Nuclear Fast Red.
cDNA cloning and Northern blot analysis
Total RNA was prepared as described (Komada et al., 2000), and poly(A)+ RNA was purified using oligo(dT)-cellulose. ROSA62–βgeo* fusion cDNA was amplified from poly(A)+ RNA of ROSA62 mutant ES cells by 5′-RACE as described (Hildebrand and Soriano, 1999). The 5′-RACE product was then used to screen mouse ES cell, embryonic day 15 embryo, and adult brain libraries to obtain full-length cDNAs. Northern blot analysis was performed using standard procedures.
Generation of antibody, Western blotting, and immunoprecipitation
A chicken antibody (Aves Lab) was raised against the variable region of βIV-spectrin (amino acid 2171–2345 of βIVΣ1-spectrin) which was expressed in Escherichia coli as a glutathione S-transferase fusion protein. Purified IgY fraction from immunized chicken was absorbed against the βIV-spectrin–null brain sliced using a vibratome. For Western blotting, a brain was homogenized in 1 ml lysis buffer (20 mM Tris-HCl, pH 7.4, 140 mM NaCl, 1% Nonidet P-40, 1 mM PMSF, and 2 μg/ml aprotinin). After removing insoluble materials by low-speed centrifugation (1,000 g for 5 min), 20 μl of the lysate was run on SDS-PAGE. After transferring proteins to a Trans-Blot polyvinylidene difluoride membrane (Bio-Rad Laboratories), βIV-spectrin was detected using the anti–βIV-spectrin antibody (1:400), peroxidase-conjugated anti–chicken IgY antibody (1:10,000; Jackson ImmunoResearch Laboratories), and the ECL reagent (Amersham Pharmacia Biotech).
For coimmunoprecipitation experiments, COS-7 cells were transfected with Myc-βIVΣ6 together with GFP, AnkG-GFP, or AnkB-GFP, gifts of V. Bennett (Duke University Medical Center, Durham, NC) using the FuGENE 6 Transfection Reagent (Roche). 2 d after transfection, cells were lysed and the lysates were immunoprecipitated with anti-Myc antibody 9E10 or anti-GFP (Molecular Probes). Immunoprecipitates were used for Western blotting with the same antibodies.
Immunofluorescence staining
ES cells were plated on gelatin-coated cover glass, fixed with 4% formaldehyde, permeabilized with 0.2% Triton X-100, and stained with the anti–βIV-spectrin (1:400) together with mouse anti-vinculin antibody (1:400; Sigma-Aldrich) by standard procedures. For staining F-actin, FITC-conjugated phalloidin (Sigma-Aldrich) was included in incubation with secondary antibody. Brains and spinal cords were recovered from mice perfused transcardially with 1% formaldehyde, cryoprotected with 30% sucrose, embedded in the OCT compound (Tissue Tec), and sectioned at 10 μm using a cryostat. The sections were stained with the anti–βIV-spectrin (1:400), mouse anti–ankyrin-G (1:200), a gift of V. Bennett), mouse anti–pan-sodium channel (10 μg/ml; Sigma-Aldrich), rabbit anti-Nav1.6 (Scn8a) (10 μg/ml; Alomone Labs), and rabbit anti-calbindin D-28K (1:1,000; Chemicon) antibodies. Secondary antibodies used were Alexa594-conjugated anti–chicken IgY, Alexa594-conjugated anti–mouse IgG, Alexa488-conjugated anti–mouse IgG, and Alexa488-conjugated anti–rabbit IgG antibodies (Molecular Probes). Fluorescent images were captured using a confocal microscope.
Cerebellum-specific ankyrin-G knockout mice originally generated by Zhou et al. (1998) were provided by H. Kamiguchi and V. Bennett.
Acknowledgments
We thank Vann Bennett for providing antibodies and cDNAs; Hiroyuki Kamiguchi (RIKEN Brain Science Institute, Wako, Japan) and Vann Bennett for cerebellum-specific ankyrin-G knockout mice; Naomi Kitamura and Steve Tapscott for helpful discussion and encouragement; Peter Mueting-Nelsen and Philip Corrin for help with genotyping of mice; and Steve Tapscott and our laboratory colleagues for critical reading of the manuscript.
This work was supported by National Institute of Child Health and Human Development grant HD24875 to P. Soriano.
Footnotes
Abbreviations used in this paper: AIS, axon initial segment; ES, embryonic stem; GFP, green fluorescent protein; NR, node(s) of Ranvier; PH, plecstrin homology; qv, quivering; VGSC, voltage-gated sodium channel.
References
- Arroyo, E.J., and S.S. Scherer. 2000. On the molecular architecture of myelinated fibers. Histochem. Cell Biol. 113:1–18. [DOI] [PubMed] [Google Scholar]
- Bennett, V., and D.M. Gilligan. 1993. The spectrin-based membrane skeleton and micron-scale organization of the plasma membrane. Annu. Rev. Cell Biol. 9:27–66. [DOI] [PubMed] [Google Scholar]
- Bennett, V., and S. Lambert. 1999. Physiological roles of axonal ankyrins in survival of premyelinated axons and localization of voltage-gated sodium channels. J. Neurocytol. 28:303–318. [DOI] [PubMed] [Google Scholar]
- Berghs, S., D. Aggujaro, R. Dirkx, Jr., E. Maksimova, P. Stabach, J.-M. Hermel, J.-P. Zhang, W. Philbrick, V. Slepnev, T. Ort, and M. Solimena. 2000. βIV spectrin, a new spectrin localized at axon initial segments and nodes of Ranvier in the central and peripheral nervous system. J. Cell Biol. 151:985–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess, D.L., D.C. Kohrman, J. Galt, N.W. Plummer, J.M. Jones, B. Spear, and M.H. Meisler. 1995. Mutation of a new sodium channel gene Scn8a in the mouse mutant “motor endplate disease.” Nat. Genet. 10:461–465. [DOI] [PubMed] [Google Scholar]
- Caldwell, J.H., K.L. Schaller, R.S. Lasher, E. Peles, and S.R. Levinson. 2000. Sodium channel Nav1.6 is localized at nodes of Ranvier, dendrites, and synapses. Proc. Natl. Acad. Sci. USA. 97:5616–5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, L.H., and V. Bennett. 1990. Mapping the binding sites of human erythrocyte ankyrin for the anion exchanger and spectrin. J. Biol. Chem. 265:10589–10596. [PubMed] [Google Scholar]
- Davis, J.Q., S. Lambert, and V. Bennett. 1996. Molecular composition of the node of Ranvier: identification of ankyrin-binding cell adhesion molecules neurofascin (mucin+/third FNIII domain-) and NrCAM at nodal axon segments. J. Cell Biol. 135:1355–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Matteis, M.A., and J.S. Morrow. 2000. Spectrin tethers and mesh in the biosynthetic pathway. J. Cell Sci. 113:2331–2343. [DOI] [PubMed] [Google Scholar]
- Drubin, D.G., and W.J. Nelson. 1996. Origins of cell polarity. Cell. 84:335–344. [DOI] [PubMed] [Google Scholar]
- Duchen, L.W., and E. Stefani. 1971. Electrophysiological studies of neuromuscular transmission in hereditary “motor end-plate disease” of the mouse. J. Physiol. 212:535–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich, G., and P. Soriano. 1991. Promoter traps in embryonic stem cells: a genetic screen to identify and mutate developmental genes in mice. Genes Dev. 5:1513–1523. [DOI] [PubMed] [Google Scholar]
- Hildebrand, J.D., and P. Soriano. 1999. Shroom, a PDZ domain-containing actin-binding protein, is required for neural tube morphogenesis in mice. Cell. 99:485–497. [DOI] [PubMed] [Google Scholar]
- Kennedy, S.P., S.L. Warren, B.G. Forget, and J.S. Morrow. 1991. Ankyrin binds to the 15th repetitive unit of erythroid and nonerythroid β-spectrin. J. Cell Biol. 115:267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komada, M., D.J. McLean, M.D. Griswold, L.D. Russell, and P. Soriano. 2000. E-MAP-115, encoding a microtubule-associated protein, is a retinoic acid-inducible gene required for spermatogenesis. Genes Dev. 14:1332–1342. [PMC free article] [PubMed] [Google Scholar]
- Kordeli, E., S. Lambert, and V. Bennett. 1995. AnkyrnG: a new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier. J. Biol. Chem. 270:2352–2359. [DOI] [PubMed] [Google Scholar]
- Lambert, S., J.Q. Davis, and V. Bennett. 1997. Morphogenesis of the node of Ranvier: co-clusters of ankyrin and ankyrin-binding integral proteins define early developmental intermediates. J. Neurosci. 17:7025–7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moré, M.I., F.-P. Kirsch, and F.G. Rathjen. 2001. Targeted ablation of NrCAM or ankyrin-B results in disorganized lens fibers leading to cataract formation. J. Cell Biol. 154:187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohara, O., R. Ohara, H. Yamakawa, D. Nakajima, and M. Nakayama. 1998. Characterization of a new β-spectrin gene which is predominantly expressed in brain. Mol. Brain Res. 57:181–192. [DOI] [PubMed] [Google Scholar]
- Parkinson, N.J., C.L. Olsson, J.L. Hallows, J. McKee-Johnson, B.P. Keogh, K. Noben-Trauth, S.G. Kujawa, and B.L. Tempel. 2001. Mutant β-spectrin 4 causes auditory and motor neuropathies in quivering mice. Nat. Genet. 29:61–65. [DOI] [PubMed] [Google Scholar]
- Peles, E., and J.L. Salzer. 2000. Molecular domains of myelinated axons. Curr. Opin. Neurobiol. 10:558–565. [DOI] [PubMed] [Google Scholar]
- Poliak, S., L. Gollan, R. Martinez, A. Custer, S. Einheber, J.L. Salzer, J.S. Trimmer, P. Shrager, and E. Peles. 1999. Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron. 24:1037–1047. [DOI] [PubMed] [Google Scholar]
- Raman, I.M., L.K. Sprunger, M.H. Meisler, and B.P. Bean. 1997. Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron. 19:881–891. [DOI] [PubMed] [Google Scholar]
- Rasband, M.N., and J.S. Trimmer. 2001. Developmental clustering of ion channels at and near the node of Ranvier. Dev. Biol. 236:5–16. [DOI] [PubMed] [Google Scholar]
- Rasband, M.N., E. Peles, J.S. Trimmer, S.R. Levinson, S.E. Lux, and P. Shrager. 1999. Dependence of nodal sodium channel clustering on paranodal axoglial contact in the developing CNS. J. Neurosci. 19:7516–7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riederer, B.M., I.S. Zagon, and S.R. Goodman. 1986. Brain spectrin (240/235) and brain spectrin (240/235E): two distinct spectrin subtypes with different locations within mammalian neural cells. J. Cell Biol. 102:2088–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan, Y., L. Elmer, J. Davis, V. Bennett, and K. Angelides. 1988. Ankyrin and spectrin associate with voltage-dependent sodium channels in brain. Nature. 333:177–180. [DOI] [PubMed] [Google Scholar]
- Tse, W.T., J. Tang, O. Jin, C. Korsgren, K.M. John, A.L. Kung, B. Gwynn, L.L. Peters, and S.E. Lux. 2001. A new spectrin, βIV, has a major truncated isoform that associates with promyelocytic leukemia protein nuclear bodies and the nuclear matrix. J. Biol. Chem. 276: 23974–23985. [DOI] [PubMed] [Google Scholar]
- Vabnick, I., and P. Shrager. 1998. Ion channel redistribution and function during development of the myelinated axon. J. Neurobiol. 37:80–96. [PubMed] [Google Scholar]
- Wang, H., D.D. Kunkel, T.M. Martin, P.A. Schwartzkroin, and B.L. Tempel. 1993. Heteromultimeric K+ channels in terminal and juxtaparanodal regions of neurons. Nature. 365:75–79. [DOI] [PubMed] [Google Scholar]
- Winckler, B., P. Forscher, and I. Mellman. 1999. A diffusion barrier maintains distribution of membrane proteins in polarized neurons. Nature. 397:698–701. [DOI] [PubMed] [Google Scholar]
- Winkelmann, J.C., and B.G. Forget. 1993. Erythroid and nonerythroid spectrins. Blood. 81:3173–3185. [PubMed] [Google Scholar]
- Yoon, C.H., and E.P. Les. 1957. Quivering, a new first chromosome mutation in mice. J. Hered. 48:176–180. [Google Scholar]
- Zhang, X., and V. Bennett. 1998. Restriction of 480/270-kD ankyrinG to axon proximal segments requires multiple ankyrinG-specific domains. J. Cell Biol. 142:1571–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, D., S. Lambert, P.L. Malen, S. Carpenter, L.M. Boland, and V. Bennett. 1998. AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J. Cell Biol. 143:1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]