

Abstract
Glycogen synthase kinase (GSK)-3 is a serine/threonine kinase that has been implicated in several aspects in embryonic development and several growth factor signaling cascades. We now report that an inactive phosphorylated pool of the enzyme colocalizes with F-actin in both neuronal and nonneuronal cells. Semaphorin 3A (Sema 3A), a molecule that inhibits axonal growth, activates GSK-3 at the leading edge of neuronal growth cones and in Sema 3A–responsive human breast cancer cells, suggesting that GSK-3 activity might play a role in coupling Sema 3A signaling to changes in cell motility. We show that three different GSK-3 antagonists (LiCl, SB-216763, and SB-415286) can inhibit the growth cone collapse response induced by Sema 3A. These studies reveal a novel compartmentalization of inactive GSK-3 in cells and demonstrate for the first time a requirement for GSK-3 activity in the Sema 3A signal transduction pathway.
Keywords: GSK-3; axon guidance; actin; Semaphorin 3A; growth cone
Introduction
Axonal pathfinding is mediated by a specialized structure at the leading tip of an extending axon, the growth cone, which is able to recognize guidance cues and translate this information into directed motility. Depending on their nature, guidance cues can either stabilize or destabilize discrete areas of the growth cone. Several signals have been isolated and based on in vitro and/or in vivo studies characterized as positive/attractive or inhibitory/repulsive cues (Tessier-Lavigne and Goodman, 1996). Although we know a considerable amount concerning the coupling of positive/attractive cues to a motile response (Doherty et al., 2000), our knowledge about the signaling cascades that couple the inhibitory cues to growth cone repulsion remains rudimentary.
Glycogen synthase kinase (GSK)*-3 is an evolutionary conserved serine/threonine kinase that has been implicated in several aspects in embryonic development and several growth factor signaling cascades (Siegfried et al., 1992; He et al., 1995; Pierce and Kimelman, 1995; Pap and Cooper, 1998). Two genes encode the closely related proteins GSK-3α and GSK-3β, and both forms are widely distributed with highest levels found in the developing brain (Woodgett, 1990; Lau et al., 1999). A function for GSK-3 in the establishment of functional circuits in the nervous system can be postulated based on its developmental expression in axonal tracts. The steady increase in expression from embryonic stages to early postnatal development in rat brains with a decrease in expression at P20 to low levels broadly correlates with the period of axonal extension and dendritic plasticity (Takahashi et al., 1994, 2000; Leroy and Brion, 1999).
Our understanding of the function of GSK-3 in relationship to axonal growth and guidance is poor and has been limited by the lack of specific antagonists of the enzyme. In the present study, we have used phosphospecific antibodies to reveal the presence of an inactive pool of GSK-3 at the leading edge of the navigating growth cone and migratory cells. We have used LiCl and two specific GSK-3 inhibitors to investigate the function of the enzyme in the growth cone (Coghlan et al., 2000; Cross et al., 2001). Our results provide evidence that GSK-3 plays a crucial role in allowing the growth cone to respond to an inhibitory guidance cue. We show that after treatment with the inhibitory guidance molecule Semaphorin 3A (Sema 3A; Luo et al., 1993) the inactive pool of GSK-3 is rapidly lost from the leading edge of the growth cone, and that the three different GSK-3 antagonists can inhibit the growth cone collapse response induced by Sema 3A. These studies reveal a novel compartmentalization of inactive GSK-3 in cells and demonstrate for the first time a requirement for GSK-3 activity in the Sema 3A signal transduction pathway.
Results and discussion
GSK-3 is maintained inactive in association with F-actin in different cell types
GSK-3 is a unusual protein kinase in that it shows a high basal activity, and growth factors, including insulin, insulin-like growth factor, Wnt, and EGF, induce responses in cells by inhibiting the catalytic activity of the enzyme (Saito et al., 1994; Cook et al., 1996; Eldar-Finkelman et al., 1995; Waltzer and Bienz, 1999; Ding et al., 2000). Inactivation of GSK-3 by insulin is achieved by phosphorylation of Ser9 and Ser21 in GSK-3β and GSK-3α, respectively (Sutherland et al., 1993; Stambolic and Woodgett, 1994; Cross et al., 1995), via a mechanism that involves the binding of a small loop containing the phosphorylated serine back to the substrate binding site on the enzyme (Frame et al., 2001). Although not absolutely required for activity, phosphorylation of a key tyrosine (Tyr279 and Tyr216 in GSK-3α and GSK-3β, respectively) enhances activity (Hughes et al., 1993; Murai et al., 1996), conceivably by increasing substrate access to the enzyme (Dajani et al., 2001).
We have used antibodies that specifically recognize phosphorylated Ser9 and phosphorylated Ser21 in GSK-3β and GSK-3α to examine the distribution of the inactive pools of GSK-3 in cells. In highly migratory MDA-MB-231 breast cancer cells, an inactive pool of GSK-3β is found localized at the leading edge of the cells alongside F-actin (Fig. 1 A). Likewise, inactive GSK-3β colocalizes with F-actin in primary chick fibroblasts (Fig. 1 B). In both cases, inactive GSK-3α showed a similar distribution (unpublished data). In the developing embryo, GSK-3 is highly enriched in the nervous system. Interestingly, inactive pools of both GSK-3α and GSK-3β are found enriched in the filopodia and at the leading edge of the lamellipodia of growth cone extending from an E7 chick dorsal root ganglion (DRG) explants where they again colocalize with F-actin (Fig. 1, C and D). These compartmentalizations were apparent in essentially all growth cones (92 + 2.8% and 94.8 + 2% of growth cones for P-GSK-3α and P-GSK-3β, respectively, n = 4 explants, mean + SEM) and demonstrate that inactive pools of GSK-3 are preferentially enriched in motile regions of growth cones and possibly other cells.
Figure 1.
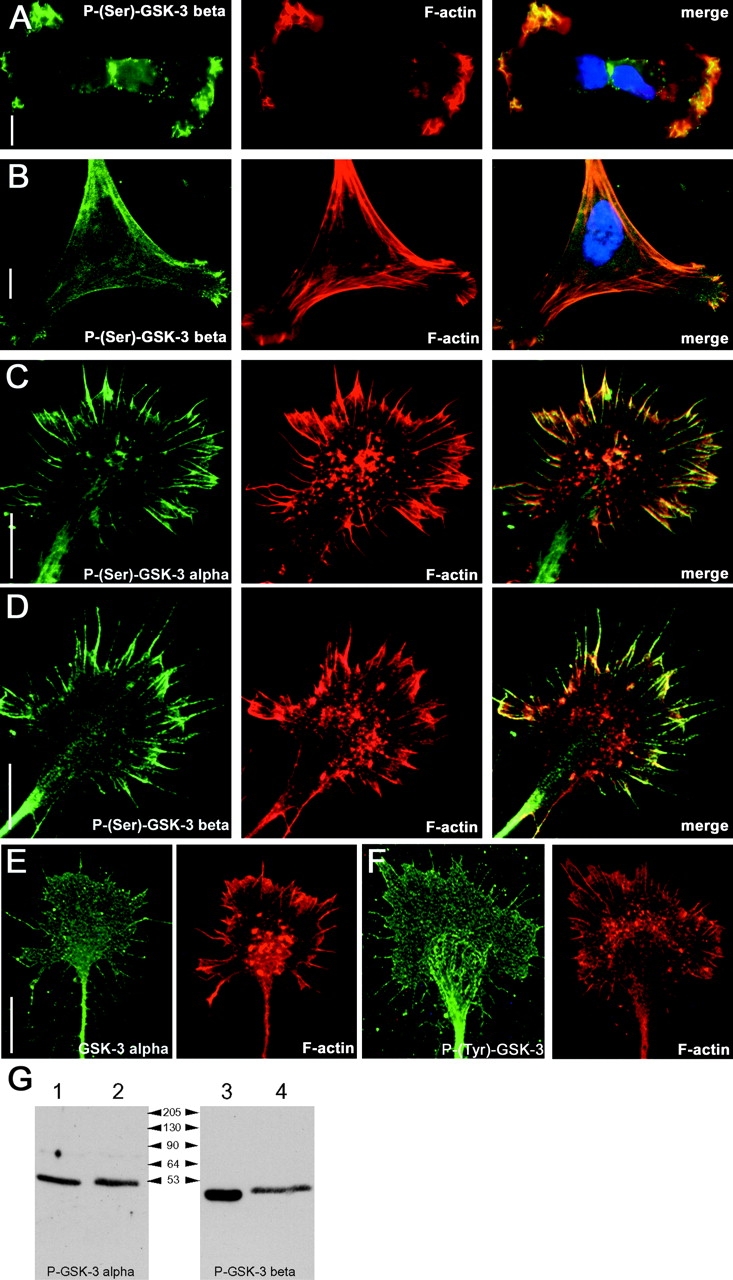
GSK-3 is maintained inactive at the leading edge of migratory cells and colocalizes with F-actin. Distribution of P-(Ser9)-GSK-3β immunoreactivity in MDA-MB-231 breast carcinoma cells (A, left) and in primary chick fibroblast (B, left). A parallel-performed phalloidin staining (middle) reveals a great overlap of inactive serine-phosphorylated GSK-3 with F-actin (right, merge). Likewise, in DRG growth cones the signals detected using an anti–P-(Ser21)-GSK-3α antibody (C) and an anti–P-(Ser9)-GSK-3β (D) are found colocalized with F-actin in the filopodia and at the leading edge of the lamellipodia. Stainings performed using a pan GSK-3α (E) and a P-(Y)-GSK-3 (F) antibody reveal that GSK-3 in present throughout the entire growth cone structure. Bars, 15 μm. (G) Western blots probed with indicated P-(Ser)–specific antibodies: lanes 1 and 3 show chick brain lysate, and lanes 2 and 4 show Cos-7 cell lysate that have been transfected with GSK-3α and GSK-3β, respectively.
The above data might simply reveal differential cellular localization of the GSK-3 protein per se, or it might reflect differential localization of active versus inactive pools of GSK-3. In neuronal growth cones, the latter appears to be the case, since antibodies to the protein backbone of GSK-3 or to the phosphorylated tyrosine residue found in the active enzyme clearly label growth cones in a uniform manner (Fig. 1, E and F). The specificity of the P-(Ser21)-GSK-3α and P-(Ser9)-GSK-3β antibodies was confirmed by Western blotting chick brain lysates and lysates obtained from Cos-7 cells transfected with GSK-3α or GSK-3β, and as expected the antibodies detected single bands of ∼51 and 47 kD (Fig. 1 G).
The phosphatidylinositol (PI) 3-kinase pathway is one of the major pathways that inactivates GSK-3 by stimulating a PKB/Akt-dependent phosphorylation of Ser21 and/or Ser9 (Cross et al., 1995). In primary DRG neurons, treatment with two selective PI 3-kinase inhibitors (wortmannin and LY294002) induces a dramatic reduction in the phosphorylation of GSK-3α on Ser21 and GSK-3β on Ser9 as determined by Western blotting (Fig. 2 A) and immunocytochemistry (Fig. 2 B). These results demonstrate that under our culture conditions PI 3-kinase activity is required for inactivating GSK-3 in the growth cones of primary neurons. It is also interesting to note that although PI 3-kinase inhibition by wortmannin does not result in a collapse of the growth cone, it reduces its outspread morphology and appears to alter the appearance of the actin filaments (Fig. 2 C).
Figure 2.
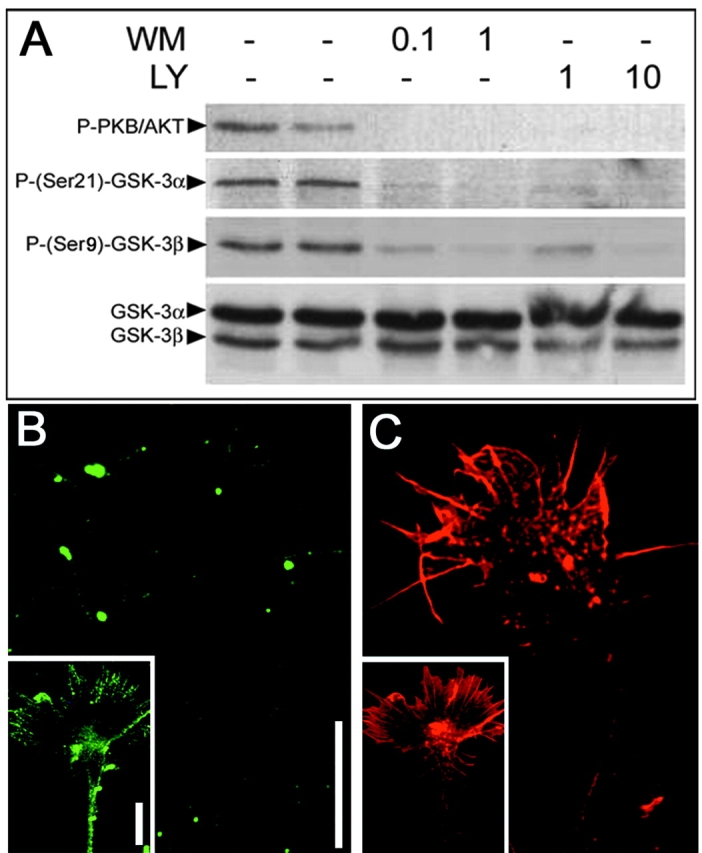
Dephosphosphorylation of GSK-3 by inhibition of PI 3-kinase. (A) In primary DRG neurons, treatment with wortmannin (WM) or LY294002 (LY) at given μM concentrations for 1 h reduces the phosphorylation of PKB/AKT and both GSK-3α and GSK-3β. (B) Treatment of DRG explant with wortmannin at 0.1 μM results in a loss of the P-Ser(9)-GSK-3β signal seen in untreated control cultures (insert). (C) In a parallel-performed phalloidin staining, the growth cone is clearly visible. Bars, 15 μm.
Sema 3A activates GSK-3 at the leading edge of the growth cone
The specific localization of an inactive pool of GSK-3 at the leading edge of the growth cone suggests a function in the control of growth cones motility. However it seems highly unlikely GSK-3 activity is required for responsiveness to guidance cues that promote growth. For example, several factors that promote axonal growth (e.g., the neurotrophins and the fibroblast growth factors) do so by activating tyrosine kinase receptors that have been shown to couple to PI 3-kinase–dependent pathways (Torres et al., 1999; Hadari et al., 2001; Huang and Reichardt, 2001; Ong et al., 2001) and would thereby be expected to inhibit GSK-3 activity. An alternative possibility is that growth cone responsiveness to inhibitory guidance cues might depend on GSK-3 activity. Sema 3A is an inhibitory guidance cue that restricts axonal extension to permissive areas by demarcating inhibitory territories (Luo et al., 1993; Messersmith et al., 1995). A “hallmark” of Sema 3A activity is its ability to induce a very rapid collapse of growth cones, a response that initially involves depolymerization and/or redistribution of F-actin at the leading edge of the growth cone (Fan et al., 1993; Fournier et al., 2000). However, if growth cones are treated with Sema 3A for a relatively short time (2 min) the redistribution of signaling components within growth cones can be assessed in the absence of a full collapse response (Fournier et al., 2000). Treatment with a Sema 3A–Fc chimera (Eickholt et al., 1997) under these conditions has no obvious effect on the distribution of GSK-3 protein within the partially collapsed growth cones (Fig. 3 A). In contrast, the short treatment with Sema 3A induces a dramatic loss of phosphorylation of both GSK-3α and GSK-β at the periphery of the growth cone (Fig. 3, B and C). Western blotting using the phospho–GSK-3 antibodies on cell lysates obtained from untreated primary DRG cultures and Sema 3A–treated cultures revealed that the total level of P–GSK-3 was in some experiments decreased (unpublished data). However, these results were not robust, and this might be due to the effects on the small pool of GSK-3 at the leading edge of the growth cone being masked by little or no change in the larger pool present in other neuronal compartments including, for example, the axon.
Figure 3.
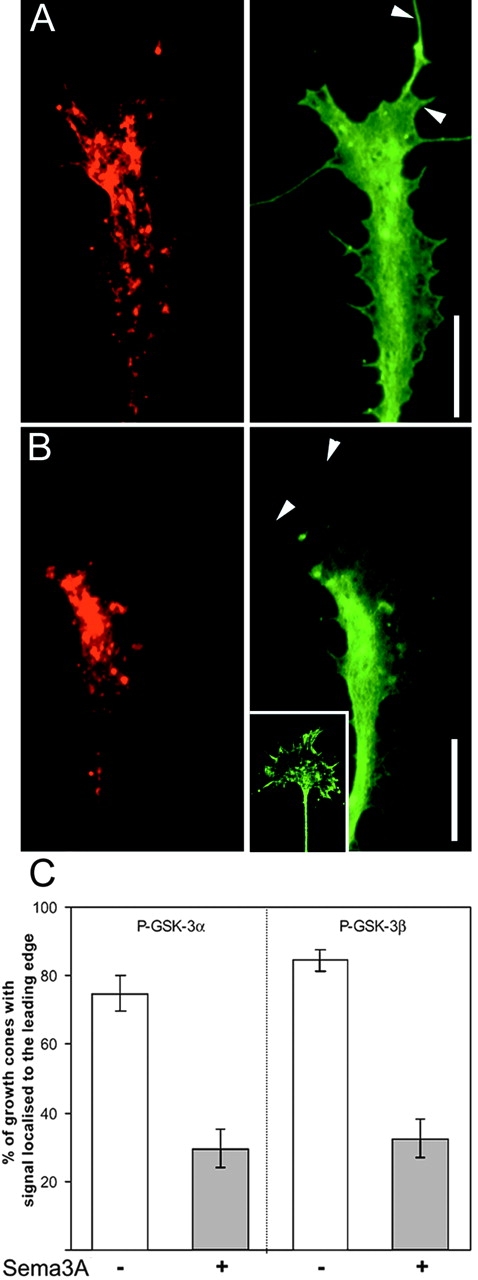
Sema 3A activates GSK-3 at the leading edge of sensory growth cones. DRG cultures grown overnight were treated with a Sema 3A–Fc chimera for 2 min before fixation. Growth cones were stained with phalloidin–Texas red (left) and anti–GSK-3α (A, right) and with phalloidin–Texas red (left) and anti–P-(Ser21)-GSK-3α (B, right). After exposure to Sema 3A, the entire GSK-3α pool remains homogeneously distributed throughout the growth cone and is still detectable in filopodia, whereas the inactive P-(Ser21)-GSK-3α is depleted at the leading edge of the growth cone and filopodia (arrowheads). The insert shows the parallel performed P-(Ser21)-GSK-3α staining in untreated control cultures. Bars, 15 μm. (C) Percentage of growth cones showing enriched staining at the leading edge before and after treatment with Sema 3A–Fc. Bars show ± SEM.
Sema 3A can also “collapse” neural crest cells (Eickholt et al., 1999), and it inhibits the migration of several other cell types (Miao et al., 1999; Bagnard et al., 2001). We have observed recently that MDA-MB-231 breast cancer cells show similar responsiveness to Sema 3A (unpublished data). Since inactive GSK-3 is found almost exclusively at the leading edge of these cells (Fig. 1), we reasoned that they might be a good model for examining the effects of Sema 3A on GSK-3 phosphorylation. In these cells, Western blotting consistently revealed that treatment with Sema 3A (applied for 1 h at 1 μg/ml) induces a substantial reduction in serine phosphorylation on GSK-3β in the absence of any detectable change in the level of GSK-3 protein (Fig. 4). As expected, this activation was not apparent when LiCl was included in the cultures (20 mM) (Fig. 4). These data not only confirm that Sema 3A can activate GSK-3, they also extend the observation to nonneuronal cells.
Figure 4.

Decrease in phosphorylation of GSK-3 after Sema 3A treatment in MDA-MB-231 breast carcinoma cell lines. Western blots of MDA-MB 231 cell extracts using anti–P-(Ser9)-GSK-3β, anti–GSK-3β, and antiactin antibodies. Cell lysates were prepared from PBS-treated control and Sema 3A–Fc–treated (1 μg/ml for 1 h) MDA-MB 231 cells in the presence or absence of LiCl (at 20 mM). All experiments were performed in duplicates. Sema 3A treatment notably decreases Ser9 phosphorylation of GSK-3. Treatment with the GSK-3 inhibitor LiCl enhances GSK-3 phosphorylation slightly and antagonizes the Sema 3A–mediated decrease in Ser9 phosphorylation. Blots were reprobed with an anti–GSK-3β antibody to confirm equal loading of samples.
In conclusion, before treatment with Sema 3A growth cones and migratory cells maintain an inactive pool of GSK-3 at their leading edges. Treatment with Sema 3A rapidly activates the GSK-3 pool at their leading edge of the growth cone and substantially activates the total pool of GSK-3 in migratory cells. These data raise the possibility that this activity “switch” of GSK-3 might contribute to the rapid changes in morphology that are seen in both neuronal and nonneuronal cells upon exposure to Sema 3A.
Inhibition of GSK-3 masks the biological activity of Sema 3A
A full collapse response to Sema 3A is much more dramatic than the partial collapse response (Fig. 5). To test whether GSK-3 activity is required for this response, we tested the effect of LiCl in the “collapse assay” since it is an established inhibitor of both GSK-3α and GSK-3β (Stambolic et al., 1996) and can prevent the Sema 3A activation of GSK-3 (Fig. 4). Treatment with LiCl at 20 mM had no effect on the basal level of collapsed growth cones (unpublished data); however, it inhibited the growth cone collapse response induced by Sema 3A from 75.3 ± 4.1% to 22.7 ± 3.2% (n = 8 explants; Fig. 5, A and B). In control experiments, we found that NaCl at 20 mM had no effect on the Sema 3A response (78.5 ± 5.8%, n = 6). LiCl is not a selective inhibitor of GSK-3 and has several additional targets (Sherman et al., 1981; Davies et al., 2000). Therefore, we tested two more specific GSK-3 inhibitors for their ability to inhibit the collapse response. SB-216763 and SB-415286 are structurally distinct maleimides that are potent inhibitors of GSK-3α and GSK-3β in an ATP competitive manner, and the specificity of these antagonists has been established in assays against 25 kinases (Coghlan et al., 2000). In the collapse assay, both compounds were able to inhibit the biological activity of Sema 3A in a dose-dependent fashion (Fig. 5) with SB-216763 being more effective than SB-415286, which correlates with their established efficacy as GSK-3 inhibitors (Coghlan et al., 2000; Cross et al., 2001). These data establish that GSK-3 activity is required for the biological activity of Sema 3A in the collapse assay.
Figure 5.

Inhibition of GSK-3 prevents Sema 3A–induced growth cone collapse. (A) Addition of Sema 3A–Fc to DRG explant cultures for 30 min induces a growth collapse response. The graphs show the percentage of collapsed growth cone ± SEM (n ≥ 4 independent experiments). In each experiment ≥100 growth cones were counted. In the presence of 20 mM LiCl, the Sema 3A response is substantially inhibited, whereas NaCl at 20 mM did not alter the Sema 3A–induced growth cone collapse. Likewise, the two specific GSK-3 inhibitors, SB-216763 and SB-415286 (used at μM concentrations as stated), inhibited the Sema 3A–induced growth cone collapse in a dose-dependent manner. (B) Examples of phalloidin-stained DRG growth cones in order to visualize the distribution of F-actin. First micrograph shows a control untreated (−) growth cone; all subsequent pictures show growth cones that have been treated with Sema 3A (+) in the absence (control) and presence of LiCl (20 mM), SB-216763 (10 μM), and SB-415286 (30 μM). In the presence of the GSK-3 antagonist, growth cone collapse is clearly inhibited. Bar, 15 μm.
GSK-3 is primarily regarded as an enzyme that is constitutively active in cells with extrinsic agents influencing cell function via mechanisms that depend on inactivation of the enzyme. Here we demonstrate the presence of an inactive pool of GSK-3 that colocalizes with F-actin at the leading edge in migratory cells including neurons. The differential distribution of active and inactive pools of GSK-3 in the neuronal growth cone suggests a function of the enzyme in the control of motility events. This notion is supported by our observation that GSK-3 is activated at the leading edge of growth cones after treatment with the inhibitory axon guidance molecule Sema 3A and the fact that GSK-3 antagonists inhibit the growth cone collapse response induced by Sema 3A. However, activation of GSK-3 by wortmannin does not induce a growth cone collapse, and this may suggest that although required, GSK-3 activation is not sufficient to mimic the biological activity of Sema 3A. At present, several additional signaling molecules have been implicated in the Sema 3A response, and these include the collapse response mediator protein (crmp), the small GTPase rac-1, and LIM kinase (Goshima et al., 1995; Hamajima et al., 1996; Jin and Strittmatter, 1997; Kuhn et al., 1999; Vastrik et al., 1999; Aizawa et al., 2001). It will be of interest to determine if any of these components can cross talk to GSK-3–dependent pathways. The “substrates” that couple GSK-3 activity to the growth cone collapse response are not known. Although it is well established that microtubule dynamics are influenced by GSK-3 activity in neurons (Hanger et al., 1992; Mandelkow et al., 1992; Lovestone et al., 1994; Baum et al., 1995; Sperber et al., 1995; Garcia-Perez et al., 1998; Goold et al., 1999; Sayas et al., 1999; Lucas et al., 2001; Sanchez et al., 2001), the rapid nature of the collapse response together with the established role of F-actin in this process (Fan et al., 1993; Fritsche et al., 1999) suggest that this is unlikely to explain our results. In addition, we also find that stabilization of microtubules with taxol does not interfere with the rapid collapse response (unpublished data). An alternative possibility is that our findings reveal a more direct function for GSK-3 in the control of the actin-based cytoskeleton. It is conceivable that after phosphorylation an as yet to be identified GSK-3 substrate might play a role in the destabilization and/or rearrangement of F-actin that accompanies the growth cone collapse response.
The presence of a pool of GSK-3 that is specifically maintained in an inactive state leads to the immediate issue as to how this phosphorylation is controlled. In principle, this could reflect a relative increase in the activity of the enzymes that phosphorylate GSK-3 within this region of the growth cone or a decrease in the activity of phosphatases. It is also possible that the inactive and active forms of GSK-3 have different binding partners and that the phosphorylated GSK-3 might selectively interact with F-actin or an F-actin binding protein. Our results show that at least in neurons the inactivation of both GSK-3α and GSK-3β is under the control of the PI 3-kinase pathway. Since an increased activity of PI 3-kinase at the leading edge in several cell types has been linked to chemotactic migratory responses (Meili et al., 1999; Parent and Devreotes, 1999; Servant et al., 2000), inactive GSK-3 may be the common link by which PI 3-kinase keeps the growth cone in a “motile” state.
Materials and methods
Antibodies and inhibitors
Mouse monoclonal anti–GSK-3 (clone 4G-1E) and anti–P-(Y279/216)-GSK-3 (clone 5G-2F) antibodies were obtained from Upstate Biotechnology. Rabbit polyclonal antibody against P-(Ser9)-GSK-3β was purchased from Biosource International. Polyclonal sheep antibodies recognizing GSK-3α and P-(Ser21)-GSK-3α (lot 18087) were purchased from Upstate Biotechnology, and the P-AKT antibody was purchased from Cell Signaling Technology. The mouse monoclonal antibody against actin was from Roche. The PI 3-kinase inhibitors LY294002 and wortmannin were purchased from Calbiochem.
Cell culture and collapse assays
Fertile eggs were obtained from a local supplier (Needle farm). DRG explants cultures from E7 chick embryos were prepared as described previously (Eickholt et al., 1997). For immunohistochemistry, DRG explants were plated onto poly-l-Lysine (20 μg/ml)/laminin (20 μg/ml)–coated glass coverslips, and cultures were incubated for 20 h in DME/10% FCS/penicillin/streptomycin supplemented with 20 ng/ml NGF before fixation in 4% paraformaldhyde/10% sucrose. For collapse assays, DRGs were cultured for 24 h on 20 μg/ml laminin-coated Labtec chamberslides (Nunc). LiCl (Sigma-Aldrich) and specific GSK-3 inhibitors were applied at given concentrations and incubated for 1 h before the Sema 3A–Fc was applied (at 1 μg/ml). After 30 min, the cultures were carefully fixed in 4% paraformaldehyde/10% sucrose.
For cultures of isolated neurons, E7 DRGs were incubated in 1 mg/ml trypsin (Worthington) diluted in Hanks' buffer for 10 min at 37°C. The trypsin solution was removed, and explants were triturated in DME/10% FCS using a fire-polished Pasteur pipette. Isolated cells were preplated in DME/10% FCS for 2 h on tissue culture plastic in order to enrich for neurons, which were cultured on laminin (20 μg/ml) as before. MDA-MB-231 cells (American Type Culture Collection) were maintained in DME/10% FCS/PenStrep.
Immunocytochemical procedures
PFA-fixed DRG explants were washed twice with PBS, permeablized in PBS/1% Triton X-100, and blocked in blocking buffer (PBS, 0.5% Triton, 2% BSA). Primary antibody was applied (all antibodies were diluted 1:50 in blocking buffer except anti-P(Y279/216)-GSK-3 antibody that was diluted 1:100) and incubated overnight at 4°C with agitation. Bound antibody was visualized using FITC-conjugated secondary antibodies (Sigma-Aldrich; anti–sheep antibody was from Dako). The distribution of F-actin was visualized using Texas red–conjugated phalloidin (Molecular Probes). All samples were analyzed using volume deconvolution.
Cell/tissue lysates and Western blot analysis
Cos-7 cells were transfected using Lipofectamine Plus reagents according to the manufacturer's protocol. Transfected cells were washed with ice-cold PBS and lysed in lysis buffer (20 mM Hepes, 150 mM NaCl, 1% Triton, 5 mM CaCl2, 1 mM MgCl2, protease and phosphatase inhibitors). Brain lysate from E9 chick brains and lysates of primary DRG neurons were prepared using the same lysis conditions. MDA-MB-231 cells were stimulated with Sema 3A–Fc (1 μg/ml) for 1 h and washed on ice with ice-cold PBS. LiCl was applied at 20 mM for 30 min before the Sema 3A–Fc was added. Cells were lysed in lysis buffer (10 mM Tris, pH 8, 0.25 sucrose, 0.5% NP-40, 10 mM MgCl2, 0.1 mM DTT, protease and phosphatase inhibitors). After removing insoluble material by centrifugation, protein concentration was determined using Bio-Rad Laboratories protein assay, and protein extracts (40 μg per lane) were separated by SDS-PAGE (10%) and transferred onto nitrocellulose. Bound proteins were detected by Western blotting. All primary antibodies were used at 1:1,000. Secondary antibodies were purchased from Vector Laboratories and used at 1:5,000.
Acknowledgments
We would like to thank Alistair D. Reith, Darren A. Cross, and Philip Gordon-Weeks for useful discussions during the course of the study, and Adrian Harwood for helpful comments on the article.
The work was supported in part by a King's College London Inter-Disciplinary Research Group fellowship to B.J. Eickholt and the Welcome Trust.
Footnotes
Abbreviations used in this paper: DRG, dorsal root ganglion; GSK, glycogen synthase kinase; PI, phosphatidylinositol; Sema 3A, Semaphorin 3A.
References
- Aizawa, H., S. Wakatsuki, A. Ishii, K. Moriyama, Y. Sasaki, K. Ohashi, Y. Sekine-Aizawa, A. Sehara-Fujisawa, K. Mizuno, Y. Goshima, et al. 2001. Phosphorylation of cofilin by LIM-kinase is necessary for semaphorin 3A-induced growth cone collapse. Nat. Neurosci. 4:367–373. [DOI] [PubMed] [Google Scholar]
- Bagnard, D., C. Vaillant, S.T. Khuth, N. Dufay, M. Lohrum, A.W. Puschel, M.F. Belin, J. Bolz, and N. Thomasset. 2001. Semaphorin 3A-vascular endothelial growth factor-165 balance mediates migration and apoptosis of neural progenitor cells by the recruitment of shared receptor. J. Neurosci. 21:3332–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum, L., R. Seger, J.R. Woodgett, S. Kawabata, K. Maruyama, M. Koyama, J. Silver, and T. Saitoh. 1995. Overexpressed tau protein in cultured cells is phosphorylated without formation of PHF: implication of phosphoprotein phosphatase involvement. Brain Res. Mol. Brain Res. 34:1–17. [DOI] [PubMed] [Google Scholar]
- Coghlan, M.P., A.A. Culbert, D.A. Cross, S.L. Corcoran, J.W. Yates, N.J. Pearce, O.L. Rausch, G.J. Murphy, P.S. Carter, L. Roxbee Cox, et al. 2000. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem. Biol. 7:793–803. [DOI] [PubMed] [Google Scholar]
- Cook, D., M.J. Fry, K. Hughes, R. Sumathipala, J.R. Woodgett, and T.C. Dale. 1996. Wingless inactivates glycogen synthase kinase-3 via an intracellular signalling pathway which involves a protein kinase C. EMBO J. 15:4526–4536. [PMC free article] [PubMed] [Google Scholar]
- Cross, D.A., D.R. Alessi, P. Cohen, M. Andjelkovich, and B.A. Hemmings. 1995. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 378:785–789. [DOI] [PubMed] [Google Scholar]
- Cross, D.A., A.A. Culbert, K.A. Chalmers, L. Facci, S.D. Skaper, and A.D. Reith. 2001. Selective small-molecule inhibitors of glycogen synthase kinase-3 activity protect primary neurones from death. J. Neurochem. 77:94–102. [DOI] [PubMed] [Google Scholar]
- Dajani, R., E. Fraser, S.M. Roe, N. Young, V. Good, T.C. Dale, and L.H. Pearl. 2001. Crystal structure of glycogen synthase kinase 3beta. structural basis for phosphate-primed substrate specificity and autoinhibition. Cell. 105:721–732. [DOI] [PubMed] [Google Scholar]
- Davies, S.P., H. Reddy, M. Caivano, and P. Cohen. 2000. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351:95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, V.W., R.H. Chen, and F. McCormick. 2000. Differential regulation of glycogen synthase kinase 3beta by insulin and Wnt signaling. J. Biol. Chem. 275:32475–32481. [DOI] [PubMed] [Google Scholar]
- Doherty, P., G. Williams, and E.J. Williams. 2000. CAMs and axonal growth: a critical evaluation of the role of calcium and the MAPK cascade. Mol. Cell. Neurosci. 16:283–295. [DOI] [PubMed] [Google Scholar]
- Eickholt, B.J., R. Morrow, F.S. Walsh, and P. Doherty. 1997. Structural features of collapsin required for biological activity and distribution of binding sites in the developing chick. Mol. Cell. Neurosci. 9:358–371. [DOI] [PubMed] [Google Scholar]
- Eickholt, B.J., S.L. Mackenzie, A. Graham, F.S. Walsh, and P. Doherty. 1999. Evidence for collapsin-1 functioning in the control of neural crest migration in both trunk and hindbrain regions. Development. 126:2181–2189. [DOI] [PubMed] [Google Scholar]
- Eldar-Finkelman, H., R. Seger, J.R. Vandenheede, and E.G. Krebs. 1995. Inactivation of glycogen synthase kinase-3 by epidermal growth factor is mediated by mitogen-activated protein kinase/p90 ribosomal protein S6 kinase signaling pathway in NIH/3T3 cells. J. Biol. Chem. 270:987–990. [DOI] [PubMed] [Google Scholar]
- Fan, J., S.G. Mansfield, T. Redmond, P.R. Gordon-Weeks, and J.A. Raper. 1993. The organization of F-actin and microtubules in growth cones exposed to a brain-derived collapsing factor. J. Cell Biol. 121:867–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier, A.E., F. Nakamura, S. Kawamoto, Y. Goshima, R.G. Kalb, and S.M. Strittmatter. 2000. Semaphorin 3A enhances endocytosis at sites of receptor-F-actin colocalization during growth cone collapse. J. Cell Biol. 149:411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame, S., P. Cohen, and R.M. Biondi. 2001. A common phosphate binding site explains the unique substrate specificity of gsk3 and its inactivation by phosphorylation. Mol. Cell. 7:1321–1327. [DOI] [PubMed] [Google Scholar]
- Fritsche, J., B.F. Reber, B. Schindelholz, and C.E. Bandtlow. 1999. Differential cytoskeletal changes during growth cone collapse in response to hSema III and thrombin. Mol. Cell. Neurosci. 14:398–418. [DOI] [PubMed] [Google Scholar]
- Garcia-Perez, J., J. Avila, and J. Diaz-Nido. 1998. Implication of cyclin-dependent kinases and glycogen synthase kinase 3 in the phosphorylation of microtubule-associated protein 1B in developing neuronal cells. J. Neurosci. Res. 52:445–452. [DOI] [PubMed] [Google Scholar]
- Goold, R.G., R. Owen, and P.R. Gordon-Weeks. 1999. Glycogen synthase kinase 3beta phosphorylation of microtubule-associated protein 1B regulates the stability of microtubules in growth cones. J. Cell Sci. 112:3373–3384. [DOI] [PubMed] [Google Scholar]
- Goshima, Y., F. Nakamura, P. Strittmatter, and S.M. Strittmatter. 1995. Collapsin-induced growth cone collapse mediated by an intracellular protein related to UNC-33. Nature. 376:509–514. [DOI] [PubMed] [Google Scholar]
- Hadari, Y.R., N. Gotoh, H. Kouhara, I. Lax, and J. Schlessinger. 2001. Critical role for the docking-protein FRS2 alpha in FGF receptor-mediated signal transduction pathways. Proc. Natl. Acad. Sci. USA. 98:8578–8583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamajima, N., K. Matsuda, S. Sakata, N. Tamaki, M. Sasaki, and M. Nonaka. 1996. A novel gene family defined by human dihydropyrimidinase and three related proteins with differential tissue distribution. Gene. 180:157–163. [DOI] [PubMed] [Google Scholar]
- Hanger, D.P., K. Hughes, J.R. Woodgett, J.P. Brion, and B.H. Anderton. 1992. Glycogen synthase kinase-3 induces Alzheimer's disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 147:58–62. [DOI] [PubMed] [Google Scholar]
- He, X., J.P. Saint-Jeannet, J.R. Woodgett, H.E. Varmus, and I.B. Dawid. 1995. Glycogen synthase kinase-3 and dorsoventral patterning in Xenopus embryos. Nature. 374:617–622. [DOI] [PubMed] [Google Scholar]
- Huang, E.J., and L.F. Reichardt. 2001. Neurotrophins: roles in neuronal development and function. Annu. Rev. Neurosci. 24:677–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, K., E. Nikolakaki, S.E. Plyte, N.F. Totty, and J.R. Woodgett. 1993. Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation. EMBO J. 12:803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, Z., and S.M. Strittmatter. 1997. Rac1 mediates collapsin-1-induced growth cone collapse. J. Neurosci. 17:6256–6263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn, T.B., M.D. Brown, C.L. Wilcox, J.A. Raper, and J.R. Bamburg. 1999. Myelin and collapsin-1 induce motor neuron growth cone collapse through different pathways: inhibition of collapse by opposing mutants of rac1. J. Neurosci. 19:1965–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau, K.F., C.C. Miller, B.H. Anderton, and P.C. Shaw. 1999. Expression analysis of glycogen synthase kinase-3 in human tissues. J. Pept. Res. 54:85–91. [DOI] [PubMed] [Google Scholar]
- Leroy, K., and J.P. Brion. 1999. Developmental expression and localization of glycogen synthase kinase-3beta in rat brain. J. Chem. Neuroanat. 16:279–293. [DOI] [PubMed] [Google Scholar]
- Lovestone, S., C.H. Reynolds, D. Latimer, D.R. Davis, B.H. Anderton, J.M. Gallo, D. Hanger, S. Mulot, B. Marquardt, S. Stabel, et al. 1994. Alzheimer's disease-like phosphorylation of the microtubule-associated protein tau by glycogen synthase kinase-3 in transfected mammalian cells. Curr. Biol. 4:1077–1086. [DOI] [PubMed] [Google Scholar]
- Lucas, J.J., F. Hernandez, P. Gomez-Ramos, M.A. Moran, R. Hen, and J. Avila. 2001. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 20:27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, Y., D. Raible, and J.A. Raper. 1993. Collapsin: a protein in brain that induces the collapse and paralysis of neuronal growth cones. Cell. 75:217–227. [DOI] [PubMed] [Google Scholar]
- Mandelkow, E.M., G. Drewes, J. Biernat, N. Gustke, J. Van Lint, J.R. Vandenheede, and E. Mandelkow. 1992. Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein tau. FEBS Lett. 314:315–321. [DOI] [PubMed] [Google Scholar]
- Miao, H.Q., S. Soker, L. Feiner, J.L. Alonso, J.A. Raper, and M. Klagsbrun. 1999. Neuropilin-1 mediates collapsin-1/semaphorin III inhibition of endothelial cell motility: functional competition of collapsin-1 and vascular endothelial growth factor-165. J. Cell Biol. 146:233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meili, R., C. Ellsworth, S. Lee, T.B. Reddy, H. Ma, and R.A. Firtel. 1999. Chemoattractant-mediated transient activation and membrane localization of Akt/PKB is required for efficient chemotaxis to cAMP in Dictyostelium. EMBO J. 18:2092–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messersmith, E.K., E.D. Leonardo, C.J. Shatz, M. Tessier-Lavigne, C.S. Goodman, and A.L. Kolodkin. 1995. Semaphorin III can function as a selective chemorepellent to pattern sensory projections in the spinal cord. Neuron. 14:949–959. [DOI] [PubMed] [Google Scholar]
- Murai, H., M. Okazaki, and A. Kikuchi. 1996. Tyrosine dephosphorylation of glycogen synthase kinase-3 is involved in its extracellular signal-dependent inactivation. FEBS Lett. 392:153–160. [DOI] [PubMed] [Google Scholar]
- Ong, S.H., Y.R. Hadari, N. Gotoh, G.R. Guy, J. Schlessinger, and I. Lax. 2001. Stimulation of phosphatidylinositol 3-kinase by fibroblast growth factor receptors is mediated by coordinated recruitment of multiple docking proteins. Proc. Natl. Acad. Sci. USA. 98:6074–6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pap, M., and G.M. Cooper. 1998. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-kinase/Akt cell survival pathway. J. Biol. Chem. 273:19929–19932. [DOI] [PubMed] [Google Scholar]
- Parent, C.A., and P.N. Devreotes. 1999. A cell's sense of direction. Science. 284:765–770. [DOI] [PubMed] [Google Scholar]
- Pierce, S.B., and D. Kimelman. 1995. Regulation of Spemann organizer formation by the intracellular kinase Xgsk-3. Development. 121:755–765. [DOI] [PubMed] [Google Scholar]
- Saito, Y., J.R. Vandenheede, and P. Cohen. 1994. The mechanism by which epidermal growth factor inhibits glycogen synthase kinase 3 in A431 cells. Biochem. J. 303:27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez, S., C.L. Sayas, F. Lim, J. Diaz-Nido, J. Avila, and F. Wandosell. 2001. The inhibition of phosphatidylinositol-3-kinase induces neurite retraction and activates GSK3. J. Neurochem. 78:468–481. [DOI] [PubMed] [Google Scholar]
- Sayas, C.L., M.T. Moreno-Flores, J. Avila, and F. Wandosell. 1999. The neurite retraction induced by lysophosphatidic acid increases Alzheimer's disease-like Tau phosphorylation. J. Biol. Chem. 274:37046–37052. [DOI] [PubMed] [Google Scholar]
- Servant, G., O.D. Weiner, P. Herzmark, T. Balla, J.W. Sedat, and H.R. Bourne. 2000. Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science. 287:1037–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman, W.R., A.L. Leavitt, M.P. Honchar, L.M. Hallcher, and B.E. Phillips. 1981. Evidence that lithium alters phosphoinositide metabolism: chronic administration elevates primarily d-myo-inositol-1-phosphate in cerebral cortex of the rat. J. Neurochem. 36:1947–1951. [DOI] [PubMed] [Google Scholar]
- Siegfried, E., T.B. Chou, and N. Perrimon. 1992. wingless signaling acts through zeste-white 3, the Drosophila homolog of glycogen synthase kinase-3, to regulate engrailed and establish cell fate. Cell. 71:1167–1179. [DOI] [PubMed] [Google Scholar]
- Sperber, B.R., S. Leight, M. Goedert, and V.M. Lee. 1995. Glycogen synthase kinase-3 beta phosphorylates tau protein at multiple sites in intact cells. Neurosci. Lett. 197:149–153. [DOI] [PubMed] [Google Scholar]
- Stambolic, V., and J.R. Woodgett. 1994. Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem. J. 303:701–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambolic, V., L. Ruel, and J.R. Woodgett. 1996. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr. Biol. 6:1664–1668. [DOI] [PubMed] [Google Scholar]
- Sutherland, C., I.A. Leighton, and P. Cohen. 1993. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem. J. 296:15–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, M., K. Tomizawa, R. Kato, K. Sato, T. Uchida, S.C. Fujita, and K. Imahori. 1994. Localization and developmental changes of tau protein kinase I/glycogen synthase kinase-3 beta in rat brain. J. Neurochem. 63:245–255. [DOI] [PubMed] [Google Scholar]
- Takahashi, M., K. Tomizawa, and K. Ishiguro. 2000. Distribution of tau protein kinase I/glycogen synthase kinase-3beta, phosphatases 2A and 2B, and phosphorylated tau in the developing rat brain. Brain Res. 857:193–206. [DOI] [PubMed] [Google Scholar]
- Tessier-Lavigne, M., and C.S. Goodman. 1996. The molecular biology of axon guidance. Science. 274:1123–1133. [DOI] [PubMed] [Google Scholar]
- Torres, M.A., H. Eldar-Finkelman, E.G. Krebs, and R.T. Moon. 1999. Regulation of ribosomal S6 protein kinase-p90(rsk), glycogen synthase kinase 3, and beta-catenin in early Xenopus development. Mol. Cell. Biol. 19:1427–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vastrik, I., B.J. Eickholt, F.S. Walsh, A. Ridley, and P. Doherty. 1999. Sema3A-induced growth-cone collapse is mediated by Rac1 amino acids 17-32. Curr. Biol. 9:991–998. [DOI] [PubMed] [Google Scholar]
- Waltzer, L., and M. Bienz. 1999. The control of beta-catenin and TCF during embryonic development and cancer. Cancer Metastasis Rev. 18:231–246. [DOI] [PubMed] [Google Scholar]
- Woodgett, J.R. 1990. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 9:2431–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]