

Abstract
Duplications and overexpression of the proteolipid protein (PLP) gene are known to cause the dysmyelinating disorder Pelizaeus-Merzbacher disease (PMD). To understand the cellular response to overexpressed PLP in PMD, we have overexpressed PLP in BHK cells and primary cultures of oligodendrocytes with the Semliki Forest virus expression system. Overexpressed PLP was routed to late endosomes/lysosomes and caused a sequestration of cholesterol in these compartments. Similar results were seen in transgenic mice overexpressing PLP. With time, the endosomal/lysosomal accumulation of cholesterol and PLP led to an increase in the amount of detergent-insoluble cellular cholesterol and PLP. In addition, two fluorescent sphingolipids, BODIPY–lactosylceramide and –galactosylceramide, which under normal conditions are sorted to the Golgi apparatus, were missorted to perinuclear structures. This was also the case for the lipid raft marker glucosylphosphatidylinositol–yellow fluorescence protein, which under normal steady-state conditions is localized on the plasma membrane and to the Golgi complex. Taken together, we show that overexpression of PLP leads to the formation of endosomal/lysosomal accumulations of cholesterol and PLP, accompanied by the mistrafficking of raft components. We propose that these accumulations perturb the process of myelination and impair the viability of oligodendrocytes.
Keywords: proteolipid protein; cholesterol; myelin; rafts; Pelizaeus-Merzbacher disease
Introduction
Myelin proteolipid protein (PLP)* is the major membrane protein of central nervous system (CNS) myelin and its expression is largely limited to oligodendrocytes. This protein has several interesting features. It spans the membrane four times and is highly hydrophobic with ∼50% hydrophobic amino acids. The protein is covalently attached to several acyl chains and interacts with cholesterol (Weimbs and Stoffel, 1992; Simons et al., 2000). The association of PLP with cholesterol and galactosylceramide-enriched membrane domains, so-called myelin rafts, during biosynthetic transport in primary cultures of oligodendrocytes may be critical for the correct sorting of PLP and the assembly of myelin in the CNS (Simons et al., 2000). Lipid rafts are cholesterol- and sphingolipid-rich membrane domains that form platforms for specific proteins and regulate, for example, intracellular membrane transport and cell signaling (Simons and Ikonen, 1997; Brown and London, 2000 ). We have previously proposed that PLP-containing myelin rafts represent specialized forms of lipid rafts, characterized by their unique subset of sphingolipids, galactosylceramide and sulfatide (Simons et al., 2000). Whereas cholesterol–sphingolipid rafts have been isolated based on their insolubility in the detergents Triton X-100 and CHAPS, myelin rafts containing PLP can only be recovered from the CHAPS-insoluble membrane fraction (Pereyra et al., 1988; van der Haar et al., 1998; Brown and Rose, 1992; Kim et al., 1995; Krämer et al., 1997; Simons et al., 2000). Although the exact role of PLP in myelin remains undefined, it is known to be associated with human disease. In humans, a variety of mutations, including missense mutations, deletions, and duplications, of the PLP gene are known to cause the dysmyelinating disorders Pelizaeus-Merzbacher disease (PMD) and spastic paraplegia (Garbern et al., 1999). Gene duplications of the human PLP locus are responsible for the majority of cases of PMD and lead to enhanced expression of the PLP protein (Hodes and Dlouhy, 1996). The cellular mechanism of dysmyelination caused by mutations of the PLP gene has only been studied in detail for the missense mutations (Gow and Lazzarini, 1996). This work has shown that most point mutations lead to accumulation of the protein in the ER, presumably due to its misfolding (Gow and Lazzarini, 1996; Gow et al., 1998).
Transgenic mice harboring additional copies of the wild-type PLP gene have demonstrated a premature arrest of myelination and, at higher PLP gene dosage, abnormal oligodendrocyte cell death (Kagawa et al., 1994; Readhead et al., 1994). However, the molecular pathology of PLP overexpression has remained obscure. In the present study, we have studied the overexpression of PLP in BHK cells, cultured oligodendrocytes, and transgenic mice. We demonstrate the abnormal accumulation of PLP, cholesterol, and other raft components in the late endosomal/lysosomal compartment and suggest that the mistrafficking of these myelin components are involved in the pathology associated with PLP overexpression.
Results
Overexpression of PLP leads to endosomal/lysosomal cholesterol accumulation in BHK cells
To analyze the general trafficking behavior of newly synthesized PLP, we used BHK cells, which do not contain endogenous PLP but are often used in trafficking studies. We transiently transfected the cDNA encoding for PLP or PLP containing a myc tag at its COOH terminus into BHK cells and analyzed its distribution by immunofluorescence microscopy at various times after transfection. We found that the protein was exported from the ER (unpublished data) and, at time points early after transfection (up to ∼14 h), was mainly found in the Golgi region and at the plasma membrane (unpublished data). At later time points after transfection, PLP accumulated in the endosomal–lysosomal system, as shown by colocalization with the fluid-phase marker rhodamine–dextran (Fig. 1 a). Cells were labeled with different markers of the endosomal–lysosomal system to determine the exact subcellular localization of PLP. We found no overlap between PLP–myc and early endosomal antigen 1 (EEA1), a marker of early endosomes (Fig. 1 b), or between PLP and transferrin–rhodamine, a marker of recycling endosomes (Fig. 1 c). In contrast, PLP colocalized with lysobisphosphatidic acid (LBPA), which localizes to late endosomes (Fig. 1 d). The presence of PLP within late endosomes/lysosomes is consistent with earlier reports (Gow et al., 1994; Sinoway et al., 1994; Kalway et al., 1997; Krämer et al., 2001).
Figure 1.

PLP localizes to late endosomes/lysosomes in BHK cells. (a) BHK cells were transfected with PLP or myc-tagged PLP for 24 h and incubated with rhodamine–dextran for 2 h at 37°C during the last 2 h of transfection. Cells were fixed and labeled with monoclonal anti-myc or polyclonal anti-PLP antibodies (left) to visualize PLP. Rhodamine–dextran is shown in the middle. In the merged image (right), yellow indicates colocalization of PLP and rhodamine–dextran. (b) BHK cells were transfected with myc-tagged PLP, fixed, and double labeled with monoclonal anti-myc antibody to visualize PLP (left) and polyclonal anti-EEA1 to resolve early endosomes (middle). (c) PLP-transfected BHK cells were incubated with rhodamine–transferrin (middle) for 1 h at 37°C as a marker for recycling endosomes and analyzed by immunofluorescence for PLP (left). (d) PLP-transfected BHK cells were fixed and double labeled with polyclonal anti-PLP antibody to visualize PLP (left) and with a monoclonal antibody against LBPA to resolve late endosomes (middle). In the merged image (right), yellow indicates colocalization of PLP and LBPA. Bars, 10 μm.
Cholesterol is not uniformly distributed within the endosomal–lysosomal system. It is enriched within early endosomes and recycling endosomes and relatively depleted from late endosomes and lysosomes (Brotherus and Renkonen, 1977; Hornick et al., 1997; Mukherjee et al., 1998; Gagescu et al., 2000; Kobayashi et al., 2001; Lusa et al., 2001; Nichols et al., 2001). We have previously shown that PLP specifically associates with cholesterol (Simons et al., 2000). We therefore examined whether the accumulation of PLP within late endosomes/lysosomes causes a redistribution of cellular cholesterol. To test this hypothesis, BHK cells were transfected with PLP–myc and the subcellular distribution of cholesterol was monitored with filipin, an antibiotic that specifically binds to free cholesterol. We found that in BHK cells, PLP–myc showed a remarkable colocalization with cholesterol (Fig. 2 a). Co-localization of PLP–myc and cholesterol was also observed in cells that had been incubated during the time of transfection with medium containing lipoprotein-deficient serum, indicating that the cholesterol was from a cellular source (unpublished data). After a 2-h uptake of dextran, the cholesterol-loaded vesicles were identified as endosomes/lysosomes (Fig. 2 a). To analyze whether the redistribution of cellular cholesterol correlates with PLP expression levels, we took advantage of the Semliki Forest virus (SFV) vector. The SFV expression system is ideally suited for this purpose, as infected cells synthesize a synchronized wave of protein that can be followed at various times after infection (Liljeström and Garoff, 1991). Infection of BHK cells with SFV-PLP–myc or SFV-PLP resulted in the infection of >80% of cells. When cells infected with SFV-PLP–myc were fixed 5 h after infection, PLP was observed within the Golgi region and at the plasma membrane in most cells (unpublished data). 10 h after infection, PLP was distributed in intracellular vesicles throughout the cell (Fig. 2 b). 20 h after infection, the vesicles appeared enlarged and many of them exhibited a perinuclear localization (Fig. 2 c). These vesicles were labeled with rhodamine–dextran that was allowed to endocytose for 2 h, but did not colocalize with dextran that had been internalized for only 10 min, thus identifying the labeled structures as late endosomal/lysosomal organelles (unpublished data). Filipin staining demonstrated a striking colocalization of PLP with cholesterol (Fig. 2 c). Quantitative analysis indicated that 78% of the cells that were infected with SFV-PLP–myc for 20 h exhibited intracellular accumulation of cholesterol and PLP (>100 cells examined). Similar results were observed in cells that had been infected with SFV-PLP lacking a myc tag (Fig. 2 d). In contrast, cells that had been infected by a control virus expressing green fluorescence protein (SFV-GFP) or a virus expressing the myelin oligodendrocyte glycoprotein (SFV-MOG) did not accumulate cholesterol (unpublished data). This result shows that overexpression of PLP induces an accumulation of cholesterol in late endosomes/lysosomes of BHK cells.
Figure 2.
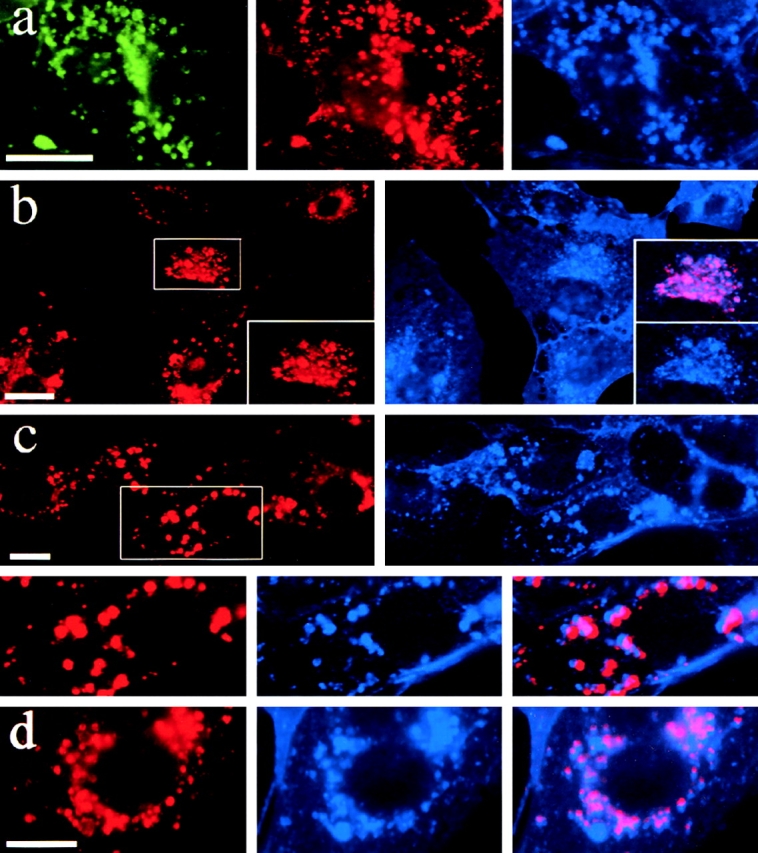
Targeting of PLP to late endosomes/lysosomes leads to cholesterol accumulation. (a) BHK cells were transfected with PLP–myc and, after 24 h, incubated with rhodamine–dextran (red) for 2 h at 37°C. Immunofluorescence was performed to analyze for PLP (green). Cholesterol was labeled with filipin (blue). (b–d) BHK cells were infected with SFV-PLP–myc (b and c) or SFV-PLP (d), fixed 10 (b) or 20 h (c and d) after infection, and analyzed by immunofluorescence for PLP (red) and cholesterol (blue). Insets represent a magnification of the region indicated. In the merged image, pink indicates colocalization of PLP and cholesterol. Bars, 10 μm.
Accumulation of PLP and cholesterol in the endosomal system is accompanied by increased insolubility in CHAPS
We have previously shown that PLP associates in the Golgi complex with a detergent-insoluble membrane fraction (myelin rafts) in primary cultures of oligodendrocytes, but not in BHK cells (Simons et al., 2000). We analyzed whether PLP becomes detergent insoluble when it accumulates with cholesterol in late endosomes/lysosomes. BHK cells were infected with SFV containing PLP–myc for 5, 10, or 20 h, and then cells were extracted for 30 min on ice with 20 mM CHAPS. Detergent-soluble and -insoluble material was separated by centrifugation at 14,000 g for 30 min. As shown previously, 5 h after infection, when PLP was found in the Golgi complex and at the plasma membrane, it was largely soluble in CHAPS (Fig. 3 a). However, 20 h after infection, when PLP accumulates with cholesterol in late endosomes/lysosomes, at least 50% of PLP was recovered from the CHAPS-insoluble membrane fraction (Fig. 3 a). To exclude that detergent insolubility was due to aggregation of the protein rather then association with lipid rafts, we analyzed the distribution of PLP on a flotation gradient. The CHAPS extract was adjusted to 40% Optiprep and loaded at the bottom of a step gradient (40, 30, and 0%). After centrifugation, six fractions were collected and the localization of PLP was determined. The majority of PLP was recovered from the top of the gradient in the low-density, detergent-insoluble membrane fraction (Fig. 3 b). To analyze whether this was dependent on the association with cholesterol, the cells were extracted with 10 mM methyl-β-cyclodextrin (mβCd) for 30 min at 37°C, a procedure that reduces cholesterol levels of BHK cells to ∼40% of normal levels (Keller and Simons, 1998), before detergent extraction. Depletion of cellular cholesterol caused a shift of a significant amount of PLP toward fractions of higher density (Fig. 3 b). To rule out the possibility that raft association at late postinfection times was due to an artifact caused by the viral expression system, we determined the raft association of the viral spike protein E2, 5 and 20 h after infection. In contrast to the observation with PLP, the amount of E2 recovered from the CHAPS-insoluble membrane fraction did not increase 20 h after infection, as compared with 5 h (Fig. 3 c).
Figure 3.
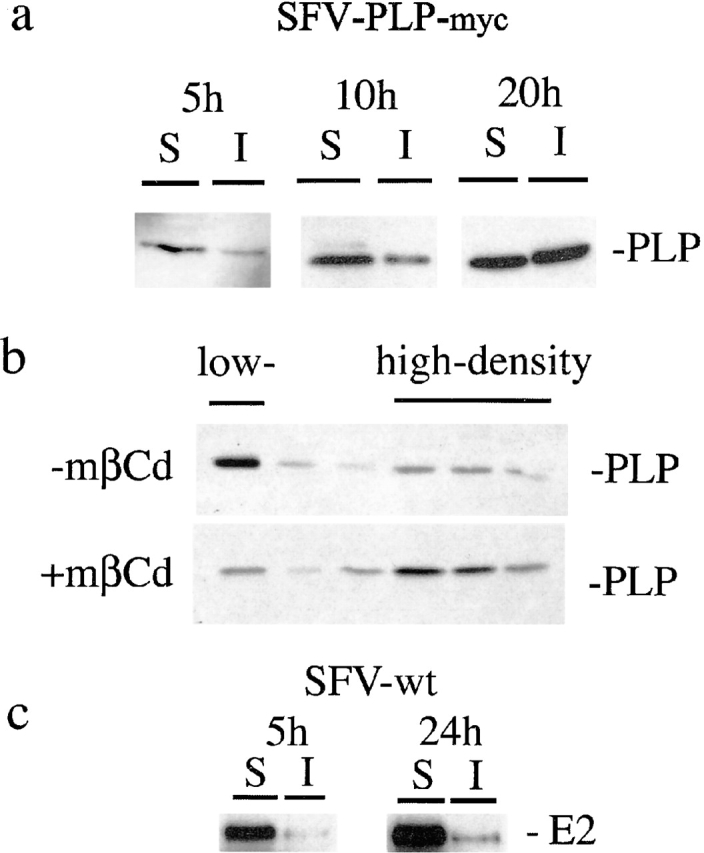
PLP associates with lipid rafts in BHK cells after increasing expression times. (a) BHK cells were infected with SFV-PLP–myc. At 5, 10, or 20 h after infection, cells were extracted with 20 mM CHAPS for 30 min at 4°C and centrifuged for 30 min at 14,000 g. Detergent-insoluble proteins in the pellet (I) and the detergent-soluble proteins in the supernatant (S) were resolved on SDS-PAGE followed by Western blotting. (b) BHK cells were infected with SFV-PLP–myc. 20 h after infection, the cells were treated with 10 mM mβCd or left untreated, extracted with CHAPS, and floated in a density gradient. Six fractions were collected from the top. The top fraction represents the low-density, CHAPS-insoluble membrane fraction, whereas the fractions of high density contain CHAPS-soluble membrane. (c) BHK cells were infected with wild-type SFV. 5 or 20 h after infection, cells were processed as described in a. The pellet (I) and supernatant (S) were analyzed by Western blotting for the viral spike protein E2.
Next, we analyzed whether the formation of rafts in late endosomes/lysosomes changes the amount of raft-associated cellular cholesterol in BHK cells. BHK cells were infected with SFV-PLP–myc or were left uninfected, and 20 h after infection, the raft association of cholesterol was determined by extracting with 20 mM CHAPS. Detergent-soluble and -insoluble lipids were quantified from TLC plates. 25% (±6%, n = 3) of cellular cholesterol was CHAPS-insoluble in uninfected cells. A dramatic increase of CHAPS-insoluble cholesterol was observed in cells that had been infected with SFV-PLP–myc (50 ± 7%, n = 3). Cells infected by a control virus expressing GFP (SFV-GFP) contained only 28% (±4%, n = 2) of CHAPS-insoluble cholesterol, arguing against an effect mediated by the viral expression system.
Effect of endosomal accumulation of cholesterol and PLP on cellular cholesterol homeostasis
The redistribution of cellular cholesterol to the degradative compartment of the endosomal system may have important consequences for cellular cholesterol homeostasis. We therefore analyzed the effect of PLP expression on total cholesterol levels, cholesterol efflux, and de novo cholesterol synthesis in BHK cells. We found that total cholesterol levels were not changed (unpublished data). Cholesterol biosynthesis was analyzed by incubating cells infected with SFV-PLP–myc or SFV-GFP 14 h after infection for 4 h with [14C]acetate. After a 2-h chase, [14C]cholesterol levels were quantified from TLC plates. We did not observe a difference between cells infected with SFV-PLP–myc and cells infected with SFV-GFP (unpublished data). To evaluate the cholesterol efflux from the plasma membrane, we labeled cells for 24 h with [14C]cholesterol in lipoprotein-deficient medium, infected the cells with SFV-PLP–myc or SFV-GFP for 20 h in the absence of label, and extracted with mβCd. We used a 30-min incubation with 10 mM mβCd on ice, which extracted 26.8 ± 3% (n = 9) of the cholesterol in uninfected cells. 27.1 ± 2% (n = 3) of the cholesterol was removed from cells that had been infected with SFV-GFP. In cells expressing PLP, a small reduction in cholesterol efflux was observed (20.2% ± 2.2%, n = 9). Because cholesterol is redistributed to late endosomes/lysosomes while total cholesterol levels remain unchanged, there may be less cholesterol available for extracellular acceptors at the plasma membrane in this latter case.
Accumulation of PLP and cholesterol in the endosomal system impairs the trafficking of raft components
Having shown that PLP induces redistribution of cellular cholesterol to late endosomes/lysosomes, we tested whether this affects the sorting of other (raft) lipids. To monitor the trafficking of sphingolipids, we added BODIPY-labeled lipids to the cells. It has been shown previously that BODIPY–lactosylceramide or –galactosylceramide are taken up by cells and transferred to the Golgi apparatus (Puri et al., 1999). In several lysosomal storage diseases, however, these lipids are rerouted to the late endosomes/lysosomes (Puri et al., 1999). We therefore analyzed how these lipids are transported in cells that express PLP. BHK cells were infected with SFV-PLP–myc, and 16–20 h after infection, BODIPY-labeled lipids were added to the cells as BSA complexes for 1 h at 37°C, and internalization was allowed to proceed for 90 min. After “back exchange” (see Materials and methods) of the lipids at 12°C, the distribution of the labeled lipid was monitored. As previously shown, both BODIPY–galactosylceramide and –lactosylceramide distributed to the Golgi region in uninfected cells (Fig. 4, a and c). In cells infected with SFV-PLP–myc, however, both lipids were found in a perinuclear location in enlarged vesicles within the cell (Fig. 4, b and d). These results demonstrate that expression of PLP in BHK cells alters the trafficking of both cholesterol and fluorescent sphingolipids. One consequence of this might be an altered trafficking of lipid rafts. As a marker for lipid rafts, we used yellow fluorescence protein (YFP) fused to a glucosylphosphatidylinositol (GPI) anchor (GPI–YFP; Keller et al., 2001). In BHK cells, GPI–YFP is mainly found at the plasma membrane (Fig. 5 a). When GPI–YFP and PLP were coexpressed in BHK cells, the localization of GPI–YFP was completely altered and both proteins showed a complete colocalization (Fig. 5 c). A similar redistribution of GPI–YFP was observed when GPI–YFP-expressing cells were treated with the drug U18666A, a hydrophobic amine, which induces late endosomal/lysosomal cholesterol accumulation by inhibition of cholesterol mobilization (Liscum, 2000; Fig. 5 b). Taken together, we conclude that the trapping of cholesterol by overexpressed PLP in the endosomal–lysosomal system induces the missorting of raft membrane components.
Figure 4.

Increased expression of PLP changes the normal distribution of BODIPY–lactosylceramide and –galactosylceramide. (a–d) BHK cells were left uninfected (a and c) or were infected with SFV-PLP–myc for 16 h (b and d). BODIPY-labeled lipids were added to the cells as BSA complexes for 1 h at 37°C, and then lipids were washed away and internalization was allowed to proceed for 90 min. After “back exchange” (see Materials and methods) of the lipids at 12°C, the distribution of the lipid was monitored. Distribution of BODIPY–lactosylceramide is shown in uninfected cells (a) and infected cells (b). Distribution of BODIPY–galactosylceramide is shown in uninfected (c) and infected cells (d). Bars, 10 μm.
Figure 5.

Expression of PLP in BHK cells affects the sorting of GPI–YFP. (a) BHK cells were transfected with GPI–YFP and, after 24 h, were fixed and analyzed by immunofluorescence. (b) Cells were transfected with GPI–YFP, incubated for 24 h in the presence of 10 μg/ml U18666A, fixed, and analyzed by immunofluorescence. (c) BHK cells were cotransfected with PLP and GPI–YFP and analyzed after 24 h by immunofluorescence for PLP (red) and GPI–YFP (green). In the merged image, yellow indicates colocalization of PLP and GPI–YFP. Bars, 10 μm.
This could possibly lead to a perturbation of endosomal compartments. To assess whether early and late endosomal compartments mix, cells were infected with SFV-PLP–myc, and 20 h after infection, double immunofluorescence with EEA1 and LBPA was performed. Both markers remained separated, indicating that early and late endosomes did not mix (Fig. 6 a). Next, we analyzed whether the mannose-6-phosphate receptor (M6PR), which mostly resides in the TGN in BHK cells, is displaced into late endosomes, as it is in Niemann-Pick type C (NPC) cells or upon treatment with U18666A (Kobayashi et al., 1999). Cells were infected and the distribution of the M6PR was compared with LBPA. Likewise, immunofluorescence microscopy revealed that M6PR and LBPA remained largely separated (Fig. 6 b). Our results suggest that the separation of late endosomes/lysosomes from early endosomes and the TGN is not altered by the expression of PLP in BHK cells.
Figure 6.

Increased expression of PLP does not disturb endosomal compartmentalization. BHK cells were infected with SFV-PLP–myc. 20 h after infection, cells were fixed and double labeled with a monoclonal antibody against LBPA (green), to label late endosomes, and a polyclonal antibody against EEA1 (a, red) or M6PR (b, red). Neither marker colocalizes with late endosomes. Bars, 10 μm.
Overexpression of PLP leads to endosomal accumulation of PLP and cholesterol in oligodendrocytes
Because the pathology of PLP overexpression is mediated by oligodendrocytes in the CNS, it is important to analyze the trafficking of PLP in these cells. In primary cultures of oligodendrocytes, endogenous PLP is incorporated into cholesterol- and galactosylceramide-enriched membrane domains (myelin rafts) and is transported to the plasma membrane (Simons et al., 2000). As shown previously, a fraction of PLP colocalizes with lysosome-associated membrane protein 1 (LAMP-1), a marker for late endosomes/lysosomes (Krämer et al., 2001; unpublished data). To investigate the fate of overexpressed PLP, primary cultures of oligodendrocytes were infected with SFV-PLP–myc to distinguish it from endogenous protein. We found that 8 h after infection, the myc-tagged PLP was not transported out to the distal part of the processes, but was found in vesicles in the cell body and proximal part of the processes (Fig. 7 a). When oligodendrocytes were double labeled for PLP–myc and calnexin, no colocalization was observed (Fig. 7 b), ruling out the possibility that overexpressed PLP was trapped in the ER. Instead PLP–myc colocalized with rhodamine–dextran that had been internalized for 2 h, identifying the vesicles as part of the endosomal–lysosomal system (Fig. 7 a). Labeling of SFV-PLP–myc-infected oligodendrocytes with filipin demonstrated colocalization of overexpressed PLP and cholesterol, which was drastically pronounced 16 h after infection (Fig. 7 c). Quantitative analysis indicated that 82% of the cells infected with SFV-PLP–myc for 16 h exhibited colocalization of cholesterol and PLP (>100 cells examined). To analyze whether the myc tag interfered with the normal trafficking behavior of PLP, we infected oligodendrocytes with SFV-PLP. To minimize the staining from endogenous protein, we used a 10-fold higher dilution than usual of the PLP antibody for immunofluorescence analysis. 16 h after infection, accumulation of cholesterol and PLP was observed as in the cells expressing PLP–myc (unpublished data). To analyze whether overexpression of any myelin protein leads to endosomal/lysosomal accumulation, oligodendrocytes were infected with SFV-MOG for 16 h. We found that MOG was transported to the distal processes and the cells did not accumulate intracellular cholesterol (Fig. 7 d). These results suggest that overexpression of PLP in oligodendrocytes causes accumulation of PLP and sequestration of cholesterol in the endosomal–lysosomal system as observed in BHK cells.
Figure 7.

Exogenously expressed PLP is not transported to the processes but accumulates cholesterol in primary cultures of oligodendrocytes. (a) Oligodendrocytes were infected with SFV containing myc-tagged PLP and incubated with rhodamine–dextran for 2 h at 37°C during the last 2 h of infection. 8 h after infection, cells were fixed and processed for immunofluorescence. Exogenous PLP (green) is mainly found in the cell body. The panels at the right show the cell body at lower exposure. Colocalization of PLP (green) and rhodamine–dextran (red) is shown in the merged image as yellow. (b) Oligodendrocytes were infected with SFV containing myc-tagged PLP, and 8 h after infection, cells were fixed and processed for immunofluorescence for PLP (green) and calnexin (red). (c) Oligodendrocytes were infected with SFV containing myc-tagged PLP, and 16 h after infection, cells were fixed and processed for immunofluorescence for PLP (red) and cholesterol (blue). The region indicated in the upper panel is shown below at a higher magnification. Colocalization of PLP and cholesterol is shown in the merged image as pink. (d) Oligodendrocytes were infected with SFV containing MOG, and 16 h after infection, cells were fixed and processed for immunofluorescence for MOG (red) and cholesterol (blue). Bars, 10 μm.
Overexpressed PLP does not associate with myelin rafts during biosynthetic transport, but forms raft aggregates in the endosomal system
Normally, the majority of PLP is incorporated into myelin rafts and is subsequently transported to and retained in myelin. We therefore determined the association of overexpressed PLP with myelin rafts. Oligodendrocytes were infected with SFV-PLP–myc, and 8 h after infection, the cells were extracted with 20 mM CHAPS followed by density gradient centrifugation. PLP–myc was mainly recovered from the fractions of higher density, indicating that exogenous PLP does not associate with myelin rafts (Fig. 8). In contrast, and as shown previously, endogenous PLP and its smaller isoform DM20 were predominantly found in the low-density, detergent-insoluble membrane fraction (Fig. 8). It is possible that exogenous overexpressed PLP does not associate with rafts during biosynthetic transport, because rafts are saturated with endogenous PLP. To investigate this possibility, we expressed PLP and PLP–myc using the SFV expression system in immortalized oligodendroglial precursor cells. These cells do not synthesize endogenous PLP, but they express significant amounts of myelin lipid (Jung et al., 1995). We found that at 8 h after infection, PLP and PLP–myc were incorporated to the same extent (∼40–50%, n = 3) into myelin rafts in oligodendroglial precursor cells (unpublished data). These data rule out the possibility that the viral expression system or the myc tag interferes with raft incorporation and suggest that raft association is reduced in primary cultures of oligodendrocytes because the protein is overexpressed.
Figure 8.

Exogenously expressed PLP associates with rafts in primary cultures of oligodendrocytes only after increasing expression times. Oligodendrocytes were infected with SFV-PLP–myc or were left uninfected (endogenous). 8 or 16 h after infection, the cells were extracted with 20 mM CHAPS for 30 min at 4°C and floated in a density gradient. Six fractions were collected from the top and proteins were resolved on SDS-PAGE. After Western blotting, proteins were detected with anti-PLP (endogenous) or anti-myc (SFV-PLP–myc, 8 or 16 h) antibodies. The top fraction represents the low-density, CHAPS-insoluble membrane fraction, whereas the fractions of high density contain CHAPS-soluble membrane.
In BHK cells, the continuous accumulation of PLP and cholesterol in late endosomes/lysosomes was accompanied by increased insolubility in CHAPS. To analyze whether the same is true for oligodendrocytes, the cells were analyzed at later time points after SFV-PLP–myc infection, when a substantial fraction of PLP and cholesterol had accumulated (Fig. 7 c). Indeed, a large fraction of PLP–myc was recovered from the low-density, detergent-insoluble membrane fraction 16 h after infection (Fig. 8). Taken together, these data show that exogenously overexpressed PLP does not associate with rafts during biosynthesis; only later, when the protein accumulates in late endosomes/lysosomes, does raft binding ensue.
PLP-overexpressing mice accumulate PLP and cholesterol in late endosomes/lysosomes
Transgenic mice with increased dosage of the PLP gene have been useful models for PMD (Kagawa et al., 1994; Readhead et al., 1994). These mice develop a dys- and demyelinating disease, the severity of which is related to the level of PLP overexpression. Immunocytochemistry with antibodies against PLP/DM20 has shown that PLP/DM20 distributes differently in transgenic mice compared with wild type (Gow et al., 1998). PLP/DM20 is found in myelin tracts that can be followed over a long distance through brain sections in wild-type mice, whereas PLP/DM20 localizes to short and swollen myelin segments in transgenic mice. Furthermore, PLP/DM20 was found to accumulate at a perinuclear location in many oligodendrocytes of the transgenic animal (Macklin et al., 1995; Gow et al., 1998). The subcellular compartment of these perinuclear accumulations has not yet been determined. To analyze whether PLP/DM20 accumulates in late endosomes/lysosomes in PLP-overexpressing oligodendrocytes, we performed immunolocalization of PLP in situ. Sagittal sections of the brain of 3-wk-old homozygous PLP-overexpressing mice (line no. 66 in Readhead et al., 1994) were stained with antibodies against PLP/DM20 and LAMP-1. We found that PLP/DM20 accumulated in the perinuclear region and colocalized with LAMP-1 (Fig. 9 a). Quantitative analysis of the sections indicated that colocalization of PLP and LAMP-1 was increased >10-fold in PLP-overexpressing mice (line no. 66) compared with age-matched wild-type control mice. When tissue sections were labeled with filipin, we found that a significant amount (∼40%) of LAMP-1– and PLP-positive structures were also labeled with filipin (Fig. 9, b and c). These results strongly suggest that PLP overexpression in vivo also causes the proteins to accumulate in late endosomes/lysosomes; thus, our in vitro results are also relevant for the pathogenesis of PMD.
Figure 9.

Perinuclear PLP/DM20 localizes to late endosomes/lysosomes and colocalizes with cholesterol in PLP-overexpressing transgenic mice. 3-wk-old homozygous PLP-overexpressing transgenic mice were perfused, and sagittal sections of the brain were prepared and stained with antibodies against PLP/DM20 and LAMP-1. Immunofluorescence staining is shown in red for PLP/DM20 and green for LAMP-1 (a–c). Nuclei are visualized with DAPI and shown in blue in the merged image (a). In the merged image (a), yellow indicates colocalization of PLP/DM20 and LAMP-1. The inset represents a magnification of the region indicated. Cholesterol was labeled with filipin and shown in blue (b and c). Several cells are shown in the overview (b), whereas a single cell is shown in the image of higher magnification (c). Bars, 8 μm.
Discussion
We found that overexpressed PLP is routed to late endosomes/lysosomes and causes a sequestration of cholesterol in these compartments. This result is surprising, as unesterified cholesterol is normally found mainly in the plasma membrane, Golgi apparatus, and early or recycling endosomes (Brotherus and Renkonen, 1977; Hornick et al., 1997; Mukherjee et al., 1998; Gagescu et al., 2000; Kobayashi et al., 2001; Lusa et al., 2001). Under normal conditions, internalized cholesterol taken up by the LDL receptor is rapidly deesterified, released into the lumen of the late endosome, and translocated through the activity of the NPC1 protein (Brown and Goldstein, 1986; Liscum, 2000). Internalized cholesterol present in lipid rafts is distributed to early and recycling endosomes from where it can recycle back to the plasma membrane. It is largely restricted from entering the degradative compartment of the endosomal system (Brotherus and Renkonen 1977; Hornick et al., 1997; Mukherjee et al., 1998; Gagescu et al., 2000; Kobayashi et al., 2001; Lusa et al., 2001; Nichols et al., 2001). Taken together, these mechanisms ensure that cholesterol levels are kept low in late endosomes. Our results raise the question as to how PLP causes accumulation of cholesterol in late endosomes/lysosomes. The direct interaction of PLP with cholesterol (Simons et al., 2000) supports the view of a cotransport of PLP and cholesterol to this compartment. Alternatively, it may be that accumulations of PLP in the late endosomal–lysosomal system perturb the translocation of cholesterol from these compartments. Normally, the majority of PLP is incorporated into cholesterol- and galactosylceramide-enriched domains (myelin rafts) and is subsequently transported to and retained in myelin. Because the entry of rafts into the degradative compartment of the endosomal system is restricted, PLP might normally recycle back to the plasma membrane when entering the endocytic system. However, in BHK cells, possibly due to the absence of sufficient amounts of myelin lipids such as galactosylceramide and sulfatide, PLP is not incorporated into rafts. Furthermore in oligodendrocytes, exogenously overexpressed PLP also does not associate with rafts. The increased expression of PLP most likely saturates the available myelin raft pathway. As a result, PLP may be routed to the degradative compartment of the endosomal system. With time, the accumulated PLP is increasingly insoluble in CHAPS and thus seems to become raft associated. This might be due to the trapping of other raft lipids. It is known that in lipid storage diseases, the accumulation of one class of raft lipid within late endosomes/lysosomes leads to the accumulation of other raft lipids. For example, in NPA and -B cells, which have a defect in sphingomyelinase, sphingomyelin accumulation is accompanied by cholesterol accumulation in late endosomes/lysosomes. Conversely, NPC cells, which accumulate cholesterol due to a defect in the NPC1 protein, also accumulate sphingomyelin (Pentchev et al., 1995; Puri et al., 1999; Simons and Gruenberg, 2000). Thus, the primary accumulation of one raft lipid can lead to the secondary accumulation of another lipid. We suggest that insolubility of PLP at later time points of infection reflects the formation of lipid rafts in the degradative compartment of the endosomal system. This conclusion is supported by our finding that increased insolubility of PLP is accompanied by the increased insolubility of cholesterol in BHK cells. In addition, we show that two sphingolipids, BODIPY–lactosylceramide and –galactosylceramide, which normally associate with the Golgi complex, are redistributed to perinuclear vesicles in cells expressing PLP, supporting our conclusion that sphingolipids follow the transport of PLP and cholesterol. Furthermore, the lipid raft marker GPI–YFP also exhibits an abnormal localization in cells that express PLP.
If PLP is able to form and associate with rafts in the endosomal–lysosomal system of BHK cells, why is the protein not incorporated into rafts in the Golgi complex in the first place? The affinity of PLP to BHK rafts may be low, and therefore PLP might not be incorporated into rafts during the biosynthetic transport. Only after accumulation of PLP in late endosomes and cumulative sequestration of cholesterol and other raft lipids is association to rafts possible. It could be that the accumulation leads to clustering of raft lipids, and this in turn increases PLP association with rafts. For example, the NPC protein is detergent soluble in normal fibroblasts, but in NPC cells, when lipid-rafts accumulate in late endosomes/lysosomes, it becomes increasingly detergent insoluble (Lusa et al., 2001). Likewise, the clustering of rafts might also be involved in the activation of lymphoid cells and is known to enhance the affinity of the T cell receptor to rafts (Viola, 2001). More work will be required to elucidate the interaction of PLP with the different raft lipids and to define how these interactions influence the trafficking behavior of PLP.
What is the significance of this finding for the pathogenesis of PMD? There are now several lines of evidence that PMD is a result of toxic “gain of function” of PLP, rather than a loss of functional protein. For example, PLP knockout mice have a remarkably mild phenotype (Klugmann et al., 1997), whereas missense mutations or increased gene dosage of PLP lead to severe forms of PMD (Garbern et al., 1999). For the missense mutations, it has been shown that the underlying pathogenic mechanism is the accumulation of misfolded protein in the ER (Gow and Lazzarini, 1996). Misfolded PLP is harmful to the cell and provokes an “unfolded protein response,” which leads to subsequent cell death by apoptosis (Gow et al., 1998). However, for the majority of patients with PMD due to gene duplication followed by an increased gene dosage of PLP, the molecular nature of the toxic “gain of function” has remained obscure. As a disease mechanism, we and others have proposed that PLP overexpression results in an arrest of oligodendrocyte and myelin development (Kagawa et al., 1994; Readhead et al., 1994). However, these studies did not show whether arrested myelin development is a direct effect of PLP expression or possibly a secondary effect due to abnormal accumulation of PLP. It is unlikely that overexpression of PLP leads to accumulation of misfolded protein in the ER, as observed for the missense mutations. When the SFV expression system was used, PLP was not retained in the ER of BHK cells or oligodendrocytes. Instead, we found that overexpression of PLP leads to the formation of detergent-insoluble accumulation of cholesterol and PLP within late endosomes/lysosomes. Because these raft aggregates seem to disturb raft membrane trafficking, it is possible that they also impair the process of myelination. Other myelin lipids may be trapped along with PLP in late endosomes/lysosomes. The capacity of late endosomes/lysosomes to recycle and/or degrade myelin membrane might also be disturbed and induce or accelerate the disease process. Furthermore, important signaling molecules that are required to initiate myelination, such as fyn, are found in rafts in oligodendrocytes (Krämer et al., 1997). Myelination is significantly reduced in fyn-deficient transgenic mice (Umemori et al., 1994; Sperber et al., 2001), and morphological differentiation of oligodendrocytes requires activation of fyn tyrosine kinase (Osterhout et al., 1999). It is tempting to speculate that the impairment of raft membrane trafficking induced by overexpression of PLP disturbs fyn signaling.
Interestingly, increased dosage of the peripheral myelin protein 22 is involved in diseases of peripheral myelin (Charcot-Marie tooth disease type 1 and hereditary neuropathy with liability to pressure palsies; Suter and Snipes, 1995). Myelinating cells may be particularly sensitive to an imbalance in the synthesis and turnover of myelin components due to their high rate of myelin synthesis (Pfeiffer et al., 1993). It is likely that oligodendrocytes trigger a death program early in the disease process. Indeed, mouse models of PMD that carry extra copies of the PLP gene show that oligodendrocytes die prematurely, i.e., as early as a few weeks after birth depending on the levels of expression of the PLP transcript (Kagawa et al., 1994; Readhead et al., 1994; Inoue et al., 1996). Furthermore, we have observed that the viability of immortalized oligodendroglial precursor cells is reduced by 24% in cells where PLP was expressed for 20 h with the SFV expression system, compared with cells that expressed MOG (unpublished data). An important issue for future studies is the mechanism of induction and execution of cell death in PLP-overexpressing oligodendrocytes.
Materials and methods
Reagents
Unless otherwise stated, all chemicals were obtained from the sources described previously by Krämer et al. (1997) and Simons et al. (2000). Filipin, U18666A, and lipoprotein-deficient FCS were from Sigma-Aldrich. 70 kD rhodamine–dextran, rhodamine–transferrin, BODIPY–galactosylceramide, and BODIPY–lactosylceramide were from Molecular Probes. The following rabbit pAbs were used: antibodies recognizing the COOH terminus of PLP (Linington and Waehneldt, 1990), EEA1 (Simonsen et al., 1998), M6P receptor, and the SFV protein E2. The following mouse mAbs were used: antibodies recognizing the c-myc epitope (Sigma-Aldrich), LBPA (Kobayashi et al., 1998), LAMP-1 (BD PharMingen), and MOG (clone 818C5). Secondary antibodies were from Dianova.
Cell culture and transfection
Primary cultures of oligodendrocytes were prepared and maintained as described previously (Simons et al., 2000). The immortalized oligodendroglial precursor cell line (Oli-neu) was cultured as described previously (Jung et al., 1995). BHK-21 cells were cultured in G-MEM (GIBCO BRL) containing 5% FCS and supplemented with penicillin (100 U/ml), streptomycin (100 μg/ml), and 2 mM glutamine (all from GIBCO BRL). Transfection of BHK cells was performed with FuGENE transfection reagents (Roche Molecular Biochemicals) according to the manufacturer's protocol.
Preparation of detergent-insoluble membrane fractions
Detergent extraction was performed as described by Simons et al. (2000) and Brown and Rose (1992). Primary oligodendrocytes or BHK cells cultured in a 5-cm dish were washed in PBS, scraped into 300 μl of 50 mM Tris-HCl, pH 7.4, 5 mM EDTA (TE), supplemented with protease inhibitors, and extracted for 30 min by adding CHAPS (20 mM final concentration). For lipid analysis, cells were grown in 10-cm dishes, washed, and scraped in PBS. The cell pellet was extracted with 350 μl of 20 mM CHAPS in TE buffer. The samples were either floated in an Optiprep step gradient (40, 30, 0% Optiprep) as previously described (Simons et al., 2000) or centrifuged at 14,000 g for 30 min at 4°C and processed as described by Brown and Rose (1992). The resulting fractions were processed for SDS-PAGE, followed by immunoblotting, or for TLC as described.
DNA constructs, preparation of recombinant SFV, and infection of cells
For preparation of recombinant SFV, we followed the protocol of Liljeström and Garoff (1991). A c-myc epitope (EQKLISEEDL) was introduced at the COOH terminus of PLP and subcloned into the pCMV vector. The PLP–myc, PLP, MOG, and GFP cDNA were cloned into the SmaI restriction site of pSFV1. pSFV1 was linearized using Nru1, pSFV-helper 1 using Spe1, and runoff transcription was performed with SP6 RNA polymerase. The transcription mix was cotransfected with the helper–transcription mix into BHK cells using electroporation. The culture supernatant was collected after a 24-h incubation (5% CO2, 37°C). The virus-containing supernatant was titrated on BHK cells. For infection, cells were incubated for 1 h with recombinant SFV diluted in conditioned maintenance medium (5% CO2, 37°C). The virus was replaced by maintenance medium and the infection continued for 5–20 h.
Lipid analysis
To analyze de novo cholesterol synthesis, BHK cells grown in 5-cm dishes were infected with SFV-PLP–myc or SFV-GFP or were left uninfected. 16 h after infection, cells were labeled with [14C]acetate (50 μCi/ml) for 4 h in G-MEM containing 5% lipoprotein-deficient FCS and chased for 2 h in normal medium. Cells were scraped into ice-cold PBS, harvested by centrifugation, and resuspended in 2% NaCl. To analyze total cholesterol levels or CHAPS insolubility of cholesterol, cells were infected for 20 h or left uninfected and processed as described above. Lipids were extracted, separated, and quantified as described by Simons et al. (2000). Cholesterol efflux was performed essentially as detailed by Lusa et al. (2001). BHK cells were labeled with 0.5 μCi/ml [14C]cholesterol in G-MEM containing 5% lipoprotein-deficient FCS for 24 h, infected or not, and incubated for 20 h in the absence of label in normal maintenance medium. Efflux to 10 mM mβCd was performed on ice for 30 min in serum-free medium. Medium was removed, cells were harvested in 2% NP-40, 0.2% SDS in TE, and the radioactivity was determined in both fractions by liquid scintillation counting. BODIPY–galactosylceramide and –lactosylceramide were added to cells as a BSA complex, and cells were cultured for 45 min at 37°C in culture medium (Puri et al., 1999). They were then washed and further incubated for 1 h at 37°C in culture medium. Fluorescent lipid present at the plasma membrane was then removed by washing six times with 3% defatted BSA at 12°C (“back exchange”) (Martin and Pagano 1994).
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde in PBS, pH 7.4, permeabilized with 0.1% Triton X-100 at room temperature (RT) or on ice for 30 s for filipin stainings, and blocked in 2% FCS and 0.2% gelatin in PBS. The cells were incubated in the presence of primary antibody for 1 h at RT, washed three times for 5 min with PBS, incubated with the appropriate secondary antibodies, washed again, and mounted. For filipin labeling, cells were incubated for 20 min at RT with filipin diluted in PBS (0.5 mg/ml), washed two times for 5 min in PBS, and mounted. Fluorescent images were obtained using Axiophot (ZEISS) or a confocal microscope (TCS confocal system; Leica). For immunocytochemistry on brain sections, mice were anesthetized and fixed by vascular perfusion with 4% paraformaldehyde in 0.1 M sodium phosphate, pH 7.2. Brains were dissected and infiltrated with 20% sucrose in phosphate buffer at 4°C before cryostat sectioning. Frozen sections (10–20 μm) were mounted, permeabilized for 10 min with methanol (−20°C), and labeled with primary antibody (overnight at RT) and the respective secondary antibody (4 h at RT). For filipin labeling, sections were incubated in quenching solution (1.5 mg/ml glycine, 1% BSA, 0.02% saponin–PBS) at RT for 30 min. This was followed by incubating sections with filipin (0.005 mg/ml) for 10 min in the dark while shaking. Cells that showed colocalization of PLP and LAMP-1 were counted from 50 random fields from four different sagittal sections of wild-type or transgenic mice. 119 cells were counted from the sections of the transgenic animal, and 8 cells were counted from the sections of the wild-type animal.
Acknowledgments
We wish to thank M. Kiebler (Max Planck Institute for Developmental Biology) for his generous support throughout the study. Most of the work was performed at the Max Planck Institute for Developmental Biology. We thank G. Wittig, M. Schiffmann, and M. Serada for technical assistance and I. Bünzli-Ehret for preparation of oligodendrocytes. We are also indebted to J. Gruenberg (University of Geneva, Geneva, Switzerland) for anti-LBPA antibodies, B. Hoflack (Institut de Biologie de Lille, Lille, France) for anti-M6PR antibodies, C. Linington (Max Planck Institute for Neurobiology, Martinsried, Germany) for anti-PLP and MOG antibodies, and P. Keller (Max Planck Institute for Cell Biology and Genetics, Dresden, Germany) for GPI–YFP cDNA.
This work was supported by Fortüne (University of Tübingen) and by SFB446.
Footnotes
Abbreviations used in this paper: CNS, central nervous system; EEA1, early endosomal antigen 1; GFP, green fluorescence protein; GPI, glucosylphosphatidylinositol; LAMP-1, lysosome-associated membrane protein 1; LBPA, lysobisphosphatidic acid; MOG, myelin oligodendrocyte glycoprotein; M6PR, mannose-6-phosphate receptor; mβCd, methyl-β-cyclodextrin; NPC, -C1, -A, or -B, Niemann-Pick type C, C1, A, or B; PLP, proteolipid protein, PMD, Pelizaeus-Merzbacher disease, RT, room temperature; SFV, Semliki Forest virus; YFP, yellow fluorescence protein.
References
- Brown, D.A., and E. London. 2000. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 275:17221–17224. [DOI] [PubMed] [Google Scholar]
- Brown, D.A., and J.K. Rose. 1992. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical surface. Cell. 68:533–544. [DOI] [PubMed] [Google Scholar]
- Brown, M.S., and J.L. Goldstein. 1986. A receptor-mediated pathway for cholesterol homeostasis. Science. 232:34–47. [DOI] [PubMed] [Google Scholar]
- Brotherus, J., and O. Renkonen. 1977. Subcellular distribution of lipids in cultured BHK cells: evidence for the enrichment of lysobisphosphatic acid and neutral lipids in lysosomes. J. Lipid Res. 18:191–202. [PubMed] [Google Scholar]
- Gagescu, R., N. Demaurex, R.G. Parton, W. Hunzinger, L.A. Huber, and J. Gruenberg. 2000. The recycling endosome of Madin-Darby canine kidney cells is a mildly acidic compartment rich in raft compartments. Mol. Biol. Cell. 11:2775–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbern, J., F. Cambi, M. Shy, and J. Kamholz. 1999. The molecular pathogenesis of Pelizaeus-Merzbacher disease. Arch. Neurol. 56:1210–1214. [DOI] [PubMed] [Google Scholar]
- Gow, A., and R.A. Lazzarini. 1996. A cellular mechanism governing the severity of Pelizaeus-Merzbacher disease. Nat. Genet. 13:422–428. [DOI] [PubMed] [Google Scholar]
- Gow, A., V.L. Friedrich, Jr., and R.A. Lazzarini. 1994. Intracellular transport and sorting of the oligodendrocyte transmembrane proteolipid protein. J. Neurosci. Res. 37:563–573. [DOI] [PubMed] [Google Scholar]
- Gow, A., C.M. Southwood, and R.A. Lazzarini. 1998. Disrupted proteolipid protein trafficking results in oligodendrocyte apoptosis in an animal model of Pelizaeus-Merzbacher disease. J. Cell Biol. 140:925–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodes, M.E., and S.R. Dlouhy. 1996. The proteolipid protein gene: double, double, … and trouble. Am. J. Hum. Genet. 59:12–15. [PMC free article] [PubMed] [Google Scholar]
- Hornick, C.A., D.Y. Hui, and J.G. DeLamatre. 1997. A role for retrosomes in intracellular cholesterol transport from endosomes to the plasma membrane. Am. J. Physiol. 273:C1075–C1081. [DOI] [PubMed] [Google Scholar]
- Inoue, Y., T. Kagawa, Y. Matsumura, K. Ikenaka, and K. Mikoshiba. 1996. Cell death of oligodendrocytes or demyelination induced by overexpression of proteolipid protein depending on expressed gene dosage. Neurosci. Res. 25:161–172. [DOI] [PubMed] [Google Scholar]
- Jung, M., E. Krämer, M. Grzenkowski, K. Tang, W. Blakemore, A. Aguzzi, K. Khazaie, K. Chlichlia, G. von Blakenfeld, H. Kettenmann, and J. Trotter. 1995. Lines of murine oligodendroglial precursor cells immortalized by an activated neu tyrosine kinase show distinct degrees of interaction with axons in vitro and in vivo. Eur. J. Neurosci. 7:1245–1265. [DOI] [PubMed] [Google Scholar]
- Kagawa, T., K. Ikenaka, Y. Inoue, S. Kuriyama, T. Tsujii, J. Nakao, K. Nakajima, J. Aruga, H. Okano, and K. Mikoshiba. 1994. Glial cell degeneration and hypomyelination caused by overexpression of myelin proteolipid protein gene. Neuron. 13:427–442. [DOI] [PubMed] [Google Scholar]
- Kalway, S.A., R. Smith, and G.J. Kidd. 1997. Myelin proteolipid protein expressed in COS-1 cells is targeted to actin-associated surfaces. J. Neurosci. Res. 48:201–211. [PubMed] [Google Scholar]
- Keller, P., and K. Simons. 1998. Cholesterol is required for surface transport of influenza virus hemagglutinin. J. Cell Biol. 140:1357–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller, P., D. Toomre, E. Diaz, J. White, and K. Simons. 2001. Multicolour imaging of post-Golgi sorting and trafficking in live cells. Nat. Cell Biol. 3:140–149. [DOI] [PubMed] [Google Scholar]
- Kim, T., K. Fiedler, D.L. Madison, W.H. Krueger, and S.E. Pfeiffer. 1995. Cloning and characterization of MVP17: a developmentally regulated myelin protein in oligodendrocytes. J. Neurosci. Res. 42:413–422. [DOI] [PubMed] [Google Scholar]
- Klugmann, M., M.H. Schwab, A. Puhlhofer, A. Schneider, F. Zimmermann, I.R. Griffiths, and K.-A. Nave. 1997. Assembly of CNS myelin in the absence of proteolipid protein. Neuron. 18:59–70. [DOI] [PubMed] [Google Scholar]
- Kobayashi, T., E. Stang, K.S. Fang, P. den Moerloose, R.G. Parton, and J. Gruenberg. 1998. A lipid associated with the antiphosphospholipid syndrome regulates endosome structure and function. Nature. 392:193–197. [DOI] [PubMed] [Google Scholar]
- Kobayashi, T., M.-H. Beuchat, M. Lindsay, S. Frias, R.D. Palmiter, H. Skuraba, R.G. Parton, and J. Gruenberg. 1999. Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nat. Cell Biol. 1:113–118. [DOI] [PubMed] [Google Scholar]
- Kobayashi, T., A. Yamji-Hasegawa, and E. Kiyokawa. 2001. Lipid domains in the endocytic pathway. Semin. Cell Dev. Biol. 12:173–182. [DOI] [PubMed] [Google Scholar]
- Krämer, E.-M., T. Koch, A. Niehaus, and J. Trotter. 1997. Oligodendrocytes direct glucosylphosphatidylinositol-anchored proteins to the myelin sheath in glycosphingolipid-rich complexes. J. Biol. Chem. 272:8937–8945. [DOI] [PubMed] [Google Scholar]
- Krämer, E.-M., A. Schardt, and K.-A. Nave. 2001. Membrane traffic in myelinating oligodendrocytes. Microsc. Res. Tech. 52:656–671. [DOI] [PubMed] [Google Scholar]
- Liljeström, P., and H. Garoff. 1991. A new generation of animal cell expression vectors based on the Semliki forest virus replication. Biotechnology (NY). 9:1356–1361. [DOI] [PubMed] [Google Scholar]
- Linington, C., and T.V. Waehneldt. 1990. Conservation of the carboxyl terminal epitope of myelin proteolipid protein in the tetrapods and lobe-finned fish. J. Neurochem. 54:1354–1359. [DOI] [PubMed] [Google Scholar]
- Liscum, L. 2000. Niemann-Pick type C mutations cause lipid traffic jam. Traffic. 1:218–225. [DOI] [PubMed] [Google Scholar]
- Lusa, S., T.S. Blom, E.-L. Eskelinen, E. Kuismanen, J.-E. Mansson, K. Simons, and E. Ikonen. 2001. Depletion of rafts in late endocytic membrane is controlled by NPC1-dependent recycling of cholesterol to the plasma membrane. J. Cell Sci. 114:1893–1900. [DOI] [PubMed] [Google Scholar]
- Macklin, W.B., B.D. Trapp, T. Kagawa, and K. Ikenaka. 1995. Hypomyelination in transgenic mice with multiple copies of the myelin proteolipid protein gene. J. Neurochem. 64:S86. [Google Scholar]
- Martin, O.C., and R.E. Pagano. 1994. Internalization and sorting of a fluorescent analog of glucosylceramide to the Golgi apparatus of human skin fibroblasts: utilization of endocytic and nonendocytic transport mechanism. J. Cell Biol. 125:769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee, S., X. Zha, I. Tabas, and F.R. Maxfield. 1998. Cholesterol distribution in living cells: fluorescence imaging using dehydroergosterol as a fluorescent cholesterol analog. Biophys. J. 75:1915–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols, A.J., A.K. Kenworthy, R.S. Polishchuk, R. Lodge, T.H. Roberts, K. Hirschberg, R.D. Phair, and J. Lippincott-Schwartz. 2001. Rapid recycling of lipid raft markers between the cell surface and Golgi complex. J. Cell Biol. 153:529–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterhout, D.J., A. Wolven, R.M. Wolf, M.D. Resh, and M.V. Chao. 1999. Morphological differentiation of oligodendrocytes requires activation of fyn tyrosine kinase. J. Cell Biol. 145:1209–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentchev, P.G., M.T. Vanier, K. Suzuki, and M. Patterson. 1995. Niemann-Pick disease type C: a cellular cholesterol lipidosis. The Metabolic and Molecular Bases of Inherited Disease. C.R. Scriver, W.S. Beudet and D. Sly, editors. McGraw-Hill, New York. 2625–2639.
- Pereyra, P.M., E. Horvath, and P.E. Braun. 1988. Triton X-100 extractions of central nervous system myelin indicate a possible role for the minor myelin proteins in stability of lamellae. Neurochem. Res. 13:583–595. [DOI] [PubMed] [Google Scholar]
- Pfeiffer, S.E., A.E. Warrington, and R. Bansal. 1993. The oligodendrocyte and its many cellular processes. Trends Cell Biol. 3:191–197. [DOI] [PubMed] [Google Scholar]
- Puri, V., R. Watanabe, M. Dominguez, X. Sun, C.L. Wheatley, D.L. Marks, and R.E. Pagano. 1999. Cholesterol modulates membrane traffic along the endocytic pathway in sphingolipid-storage diseases. Nat. Cell Biol. 1:386–388. [DOI] [PubMed] [Google Scholar]
- Readhead, C., A. Schneider, I. Griffiths, and K.-A. Nave. 1994. Premature arrest of myelin formation in transgenic mice with increased proteolipid protein gene dosage. Neuron. 12:583–595. [DOI] [PubMed] [Google Scholar]
- Simons, K., and J. Gruenberg. 2000. Jamming the endosomal system: lipid rafts and lysosomal storage disease. Trends Cell Biol. 10:459–462. [DOI] [PubMed] [Google Scholar]
- Simons, K., and E. Ikonen. 1997. Functional rafts in cell membranes. Nature. 387:569–572. [DOI] [PubMed] [Google Scholar]
- Simons, M., E.-M. Krämer, C. Thiele, W. Stoffel, and J. Trotter. 2000. Assembly of myelin by association of proteolipid protein with cholesterol- and galactosylceramide-rich membrane domains. J. Cell Biol. 151:143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen, A., R. Lippe, S. Christoforidis, J.M. Gaullier, A. Brech, J. Callaghan, B.H. Toh, C. Murphy, M. Zerial, and H. Stenmark. 1998. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature. 394:494–498. [DOI] [PubMed] [Google Scholar]
- Sinoway, M.P., K. Kitagawa, S. Timsit, G.A. Hashim, and D.R. Colman. 1994. Proteolipid protein interactions in transfectants: implications for myelin assembly. J. Neurosci. Res. 37:551–562. [DOI] [PubMed] [Google Scholar]
- Sperber, B.R., E.A. Boyle-Walsh, M.J. Engleka, P. Gadue, A.C. Peterson, P.L. Stein, S.S. Scherer, and F.A. McMorris. 2001. A unique role for fyn in CNS myelination. J. Neurosci. 21:2039–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter, U., and G.J. Snipes. 1995. Biology and genetics of hereditary motor and sensory neuropathies. Annu. Rev. Neurosci. 18:45–75. [DOI] [PubMed] [Google Scholar]
- Umemori, H., S. Sato, T. Yagi, S. Alzawa, and T. Yamamoto. 1994. Initial events of myelination involve fyn tyrosine kinase signalling. Nature. 367:572–576. [DOI] [PubMed] [Google Scholar]
- van der Haar, M.E., H.W. Visser, H. de Vries, and D. Hoekstra. 1998. Transport of proteolipid protein to the plasma membrane does not depend on glycosphingolipid cotransport in oligodendrocyte cultures. J. Neurosci. Res. 51:371–381. [DOI] [PubMed] [Google Scholar]
- Viola, A. 2001. The amplification of TCR signaling by dynamic membrane microdomains. Trends Immunol. 22:322–327. [DOI] [PubMed] [Google Scholar]
- Weimbs, T., and W. Stoffel. 1992. Proteolipid protein (PLP) of CNS myelin: position of free, disulfide-bonded, and fatty acid thioester-linked cysteine residues and implication for the membrane topology of PLP. Biochemistry. 31:12289–12296. [DOI] [PubMed] [Google Scholar]