

Abstract
Melatonin is produced in the dark by the pineal gland and is a key regulator of circadian and seasonal rhythms. A low melatonin level was reported in individuals with autism spectrum disorders (ASD), but the underlying cause of this deficit was unknown. The ASMT gene, encoding the last enzyme of melatonin synthesis, is located on the pseudo-autosomal region 1 of the sex chromosomes, deleted in several individuals with ASD. In this study, we sequenced all ASMT exons and promoters in individuals with ASD (n=250) and compared the allelic frequencies with controls (n=255). Non-conservative variations of ASMT were identified, including a splicing mutation present in two families with ASD, but not in controls. Two polymorphisms located in the promoter (rs4446909 and rs5989681) were more frequent in ASD compared to controls (P=0.0006) and were associated with a dramatic decrease in ASMT transcripts in blood cell lines (P=2×10−10). Biochemical analyses performed on blood platelets and/or cultured cells revealed a highly significant decrease in ASMT activity (P=2×10−12) and melatonin level (P=3×10−11) in individuals with ASD. These results indicate that a low melatonin level, caused by a primary deficit in ASMT activity, is a risk factor for ASD. They also support ASMT as a susceptibility gene for ASD and highlight the crucial role of melatonin in human cognition and behavior.
Keywords: autism, melatonin, circadian rhythm, sleep, HIOMT, ASMT
Introduction
Melatonin is a powerful antioxidant molecule, involved in the regulation of circadian and seasonal rhythms and immune function.1–4 It is released mainly by the pineal gland during the night and is produced by the conversion of serotonin to Nacetylserotonin by the rate-limiting enzyme AA-NAT (arylalkylamine N-acetyltransferase; EC 2.3.1.87), followed by the conversion of N-acetylserotonin to melatonin by ASMT (acetylserotonin methyltransferase; EC 2.1.1.4), also known as HIOMT (hydroxyindole O-methyltransferase).5 Melatonin secretion is highly heritable in humans,6 modulates neuronal plasticity7–9 and regulates circadian gene expression.10 It also plays a key role in communication behavior related to seasonal changes, such as song learning in birds.11 Abnormal melatonin concentrations can have a dramatic effect on human behavior, as shown in patients with Smith-Magenis syndrome, who have an inverted melatonin circadian rhythm and display autistic behavior.12 In autism spectrum disorders (ASD), low melatonin levels have been reported by three independent groups,13–15 but the underlying cause of this deficit and its relationship to susceptibility to ASD was unknown. ASD affect at least 6/1000 individuals and are characterized by impairments in communication skills and social interaction, as well as restricted, repetitive and stereotyped patterns of behavior.16–18 The genes responsible for ASD are largely unknown,19,20 but cytogenetic abnormalities are observed in at least 3–5% of the affected individuals.21 The PAR1 of the sex chromosomes, located at the tip of their short arms, has been found to be deleted in several individuals with ASD.22 Among the 12 PAR1 genes referenced, ASMT23 is an excellent candidate for susceptibility to ASD because it encodes the last enzyme in the melatonin biosynthesis pathway.24 In this study, using a combination of genetics and functional experiments, we report evidence showing that a low melatonin concentration caused by a primary deficit in ASMT activity is a risk factor for ASD.
Materials and Methods
Subjects
Families with ASD were recruited by the Paris Autism Research International Sibpair study at specialized clinical centers in seven countries (France, Sweden, Norway, Italy, Belgium, Austria, and the United States). Diagnosis was based on clinical evaluation by experienced clinicians, DSM-IV criteria, and the Autism Diagnostic Interview-Revised (ADI-R).25 In Sweden, the Diagnostic Interview for Social and Communication Disorders (DISCO-10)26 was used instead of the ADI-R in some cases. Patients with Asperger syndrome were evaluated with the Asperger Syndrome Diagnostic Interview.27 Patients diagnosed with medical disorders, such as fragile X syndrome or chromosomal anomalies, were excluded from the study.
For mutation screening, the study sample (n=250, 187 men and 63 women) was constituted of 250 independent families (71 subjects from multiplex families and 179 sporadic cases) and included 233 patients with autistic disorder and 11 with Asperger syndrome; six individuals narrowly missed the criteria for autistic disorder and were considered to have atypical autism (pervasive developmental disorder, PDD-NOS). There were 222 Caucasian, nine Black, three Asian, one Hispanic/Latin-American family, and 15 families of mixed ethnicity. For association studies, the ASD sample consisted of 278 patients of Caucasian origin (201 men and 77 women) from 72 multiplex families and 206 sporadic cases. There were 258 patients with autism, 14 with Asperger syndrome and six with atypical autism. The control sample (n=255) comprised 160 French and 95 Swedish individuals. An additional control group of 171 individuals from North Africa was screened for rare variants because one proband carrying the splice-site mutation (IVS5+2T>C) and one proband with the L326F mutation had parents originating from this region.
Blood and platelet biochemical analyses were performed in ASD probands (n=43; 14 female and 29 male patients, 14.8 ± 7 years old), their parents (n=34; 18 females and 16 males: 44 ± 9 years old) and in controls matched for sex and age (n=75; 30 females and 45 males; 27 ± 16 years old). The ASD patients were initially recruited for the analysis of serotonin levels and not chosen on the basis on their ASMT genotype. The controls were recruited at the Department of Orthopedics of the Lariboisière and Robert Debré hospitals in Paris. The control group for the B lymphoblastoid cell lines (BLCL) biochemical analyses comprised 14 French individuals also used in the association study and 19 healthy relatives of patients with Hirschsprung syndrome or mitochondrial diseases. The local research ethics boards reviewed and approved the study. Informed consent was obtained from probands (if possible), parents and controls.
Cell culture, DNA and RNA isolation
BLCL were established from EBV-transformed lymphocytes and grown at 37°C in RPMI-1640 medium (Life Technologies Inc., Grand Island, NY, USA), supplemented with 10% undialyzed fetal calf serum, 2 mM glutamine, 2.5 mM sodium pyruvate, 100 mg/ml streptomycin and 100 IU/ml penicillin, under standard conditions. DNA was extracted by the phenol/chloroform method, and RNA was isolated using the NucleoSpin RNA II kit (Macherey-Nagel, Duren, Germany). DNA/RNA concentrations were determined by measuring absorbance at 260 nm on a biophotometer (Eppendorf, Hamburg, Germany). Human pineal gland cDNAs were obtained from the Incyte cDNA library # LHS1565 (BioCat, Heidelberg, Germany).
Mutation screening and genotyping
Genotyping for the association study and mutation screening were performed by direct sequencing or TaqMan technology. PCR products were sequenced with the BigDye Terminator Cycle Sequencing Kit (V3.1, Applied Biosystems, Foster City, CA, USA). Samples were then subjected to electrophoresis, using an ABI PRISM genetic analyzer (Applied Biosystems). Absence of genotyping errors was controlled by sequencing the PCR product with the opposite primer in a subset of patients. For primers and PCR conditions, see Supplementary Table 1.
Association and statistical analyses
The linkage disequilibrium (LD) map for ASMT was calculated using pairwise LD (D′) between the 13 ASMT variations in 533 individuals (278 ASD and 255 controls). The LD calculation and the case-control study were performed with Haploview software.28 To detect population stratification bias, individuals with ASD and controls were screened for three single nucleotide polymorphisms (SNPs) (rs2289311, rs4782053, rs1921361), five ALU insertions (Ya5NBC27, Ya5NBC51, YaNBC102, YaNBC109, YbNBC65), and the mtDNA hypervariable region 1 (HVR1). In addition, all mothers from ASD patients were screened for the androgen receptor microsatellite. No significant genotype difference was observed for any of the markers tested (Supplementary Table 2). The transmission disequilibrium test (TDT) was performed using the family-based association test (FBAT)29 and haplotype-based association test (HBAT)30 using the empirical variance (‘-e’ option). For the TDT, only the four SNPs in promoter B were tested in 278 ASD families in which both parents had been genotyped. All SNPs were at Hardy-Weinberg equilibrium. We evaluated the distribution of the quantitative variables by the Kolmogorov-Smirnov test for Gaussian normality. Because the values for most of the samples were not normally distributed, we used the two-tailed non-parametric Mann-Whitney U-test to compare two groups and Spearman’s ρ test to evaluate the correlation between ASMT activity and melatonin concentration. We used SPSS version 13 for these tests.
RT-PCR and quantitative RT-PCR
Oligo(dT)-primed cDNA was prepared from 5 μg of BLCL RNA, using Superscript II (Invitrogen, Grand Island, NY, USA), according to the manufacturer’s instructions. The cDNAs were used directly in TaqMan assays, using the ABI PRISM 7500 Sequence Detection System (Applied Biosystems). Samples were run in duplicate or triplicate on 96-well optical PCR plates (ABgene, Surrey, UK). ASMT mRNA was quantified using commercially available assays. Two different assays were used, one covering the boundary between exon 1B and exon 2 (Hs00946625_ml), and the other covering the boundary between exon 8 and exon 9 (Hs00187839_ml). The two assays gave similar results and only the data obtained with Hs00946625_ml are presented. Relative values of expression were determined for each sample, using the standard curve method (ABI user’s manual), and these values were normalized to the threshold cycle (Ct) values of GAPDH, using the Hs99999905_ml assay. For ASMT and GAPDH, the thresholds were set at 0.2 and 0.25, respectively, within the linear region of the semi-logarithmic plot in all assays (data not shown). The consequence of the splice site mutation was investigated by sequencing the abnormal transcript after cloning the RT-PCR product.
Biochemical analyses
Blood samples were collected in the morning, between 0900 and 1100 h. The procedures for collecting and processing blood samples were designed to prevent release reactions. The anticoagulant was ACD-A (1 vol to 9 vol of whole blood). For the melatonin profile of family ASD 1, blood samples were collected every 2 h, from 1800 h to 1600 h the following day. Overnight, blood samples were collected in dimly lit conditions (<20 lux). Samples were drawn from an indwelling forearm catheter into Becton-Dickinson plastic tubes, centrifuged and frozen at −20°C. Platelets were counted with a Coulter ZBI electronic counter. ASMT enzymatic activities were determined, at least in duplicate, by radioenzymology,31 on the platelet pellet obtained by centrifugation of the platelet-rich plasma at 800 g for 20 min at room temperature and lysis with 100 hemolytic units of a purified SH-activated toxin (streptolysin O or alveolysin, generously provided by Prof. J. Alouf, Pasteur Institute, Paris). Melatonin content was measured in the resulting supernatant (i.e., platelet-poor plasma or plasma) by HPLC, with random controls by mass spectrometry.32
Sleep analysis
The three individuals from family ASD 1 were taking no medication that could interfere with melatonin secretion. Sleep analysis was performed by standard polysomnography (two EEG, two electro-oculograms, one submental electromyogram, two anterior tibialis muscle electromyograms and respiratory signals, Embla N7000, Flaga, Iceland), 1 week before blood was collected for the melatonin assay.
Results
Rare ASMT variations in ASD and ADHD
We investigated whether variations in ASMT were associated with ASD and ADHD by directly sequencing all ASMT exons and the two promoters, A and B, in 250 affected individuals. Several ASMT variants were identified (Figure 1), including a splice-site mutation (IVS5+2T>C), four non-synonymous variations (N17K, K81E, G306A, L326F) and two synonymous variations (N167N, Q205Q). Two of these variations, N17K (rs17149149) and L326F, were observed in the general population. N17K was found in one family with ASD from China (ASD 3) and is present in the SNP database at a frequency of 0.4–0.7% in the Han Chinese population. L326F was found in two ASD families (ASD 6 and ASD 7) and in 3 out of 426 controls (two from Sweden and one from North-Africa). The splice-site mutation (IVS5+2T>C) was present in two ASD families (ASD 1 and ASD 2) (Figure 1a), but not in controls (n=426). Using BLCL RNA from the ASD 1 proband, we detected abnormal ASMT transcripts, encoding a putative truncated ASMT protein, lacking the methyl-transferase domain (Figure 1a). Biochemical analysis of the IVS5+2T>C and L326F variations indicated that these mutations were associated with very low levels of ASMT activity and melatonin (Figure 2a, 2b). We could not analyze the functional consequences of the remaining mutations due to a lack of blood samples or cell lines. Interestingly, the nine individuals with ASD carrying rare mutations were also hyperactive and several had sleep problems (see clinical description of the patients in Supplementary Table 3).
Figure 1.
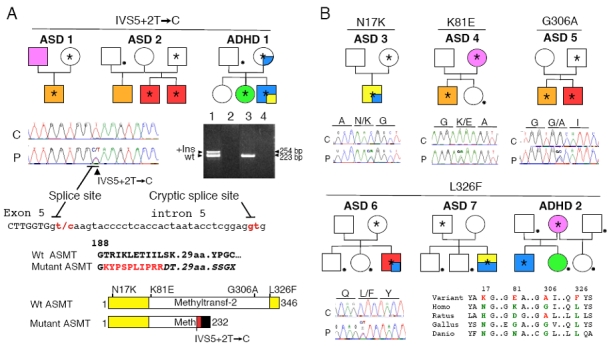
Non-synonymous ASMT variations in autism spectrum disorders (ASD) families. (a) Pedigree structure of the families carrying the splice–site mutation IVS5+2T>C; reverse transcriptase-polymerase chain reaction (RT-PCR) amplifying exons 4 to 6 of the ASMT cDNA from B lymphoblastoid cell lines of the ASD 1 proband carrying the splice– site mutation IVS5+2T > C (lane 1 +RT, lane 2 −RT) and a control (lane 3 +RT, lane 4 −RT). The insertion (+Ins) of 31 bp in the ASMT transcript originates from a cryptic donor splice–site downstream from exon 5. This insertion should lead to the additional sequence indicated in red and to a frame shift (characters in italics), causing premature truncation of the protein, lacking the methyl-transferase domain. Wt, wild-type. (b) Pedigree structure of the families carrying rare non-synonymous ASMT variations and conservation of the variant amino-acid in different species. Color codes in the pedigrees: autism with mild (orange) or severe (red) mental retardation, Asperger syndrome or high-functioning autism (yellow), attention-deficit/hyperactivity disorder ADHD (light blue) and depression (pink). The proband ASD 3 fulfilled diagnostic criteria for both high functioning autism and ADHD. The proband ASD 7 fulfilled diagnostic criteria for both Asperger syndrome and ADHD. The asterisk and the dot indicate the presence of the mutation and the absence of a DNA sample for analysis, respectively.
Figure 2.
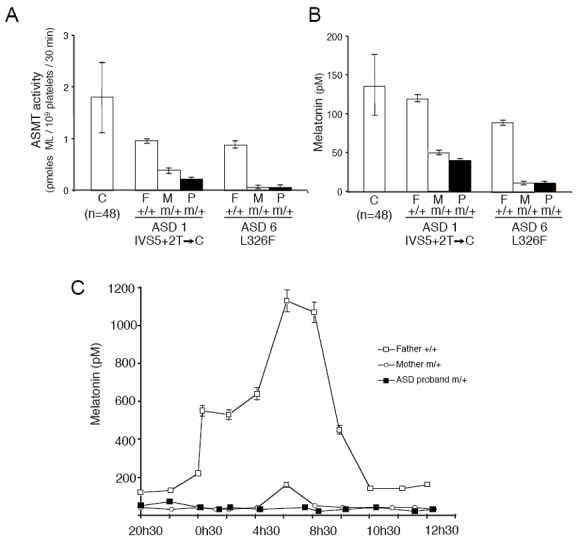
Impact of the ASMT mutations on enzyme activity and melatonin concentration. (a) ASMT activity, measured in platelets of the members of families autism spectrum disorder (ASD) 1 and ASD 6 carrying the splice–site IVS5+2T>C and the L326F ASMT mutations, respectively. (b) Blood melatonin concentration in the same individuals. (c) Nocturnal melatonin profile of family ASD 1. The proband (male, 24 years old) and his mother (53 years old) are heterozygous (m/+) for the splice–site mutation IVS5+2T>C. The proband’s father (55 years old) has no ASMT mutation (+/+). Error bars represent s.d. C: controls; F: father; M: mother; P: proband.
ASMT polymorphisms in ASD and ADHD
We investigated whether frequent polymorphisms of the ASMT gene were associated with ASD by studying one insertion/deletion located in promoter A and 12 SNPs with a minor allele frequency greater than 5%, capturing most of the haplotype diversity (Figure 3a). ASD patients (n=278) differed significantly from controls (n=255) in terms of the allelic frequency of two SNPs — rs4446909 (P=0.006) and rs5989681 (P=0.007), located in promoter B (Figure 3b, Table 1 and Supplementary Figure 1). The H1 GGGC haplotype in promoter B was more frequent in ASD (P=0.002) than in controls, whereas the haplotype H3 ACGC was more frequent in controls than in ASD (P=0.005). We carried out a TDT using FBAT and HBAT on 278 families (Supplementary Table 4), and observed an overtransmission of haplotype H1 GGGC to probands (additive model P = 0.05; dominant model P = 0.02) and overtransmission of alleles G and C of the SNPs P1BC (dominant model P = 0.02) and rs6644635 (dominant model P = 0.04), respectively.
Figure 3.
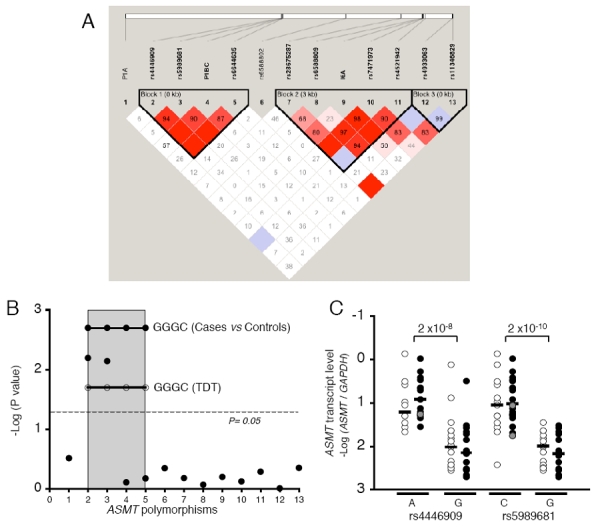
Association studies and transcript analyses of the ASMT gene. (a) Haplotype block structure of the ASMT gene. The relative physical position of each single nucleotide polymorphism (SNP) is given in the upper diagram, and the pairwise LD (D0) between all SNPs is given below each SNP combination. (b) Plot of the case–control P-values (−log10) for all variations studied within ASMT. 1: E1A; 2: rs4446909; 3: rs5989681; 4: P1BC; 5:rs6644635; 6: rs6588802; 7: rs28675287; 8: rs6588809; 9: I6A; 10: rs7471973; 11: rs5431942; 12: rs4933063; 13: rs11346829. SNPs located in promoter B are included in the shaded box. P-values for the risk haplotype GGGC are indicated as straight lines with close (cases vs controls) or open circles (transmission disequilibrium test (TDT)). (c) Quantification of ASMT transcripts relative to rs4446909 and rs5989681 genotypes (A represents the individuals with an A/A or A/G genotype; C represents the individuals with C/C or C/G genotype; G represents the individuals homozygous G/G). Black and white circles indicate individuals with autism spectrum disorders (ASD) and controls, respectively. The gray symbols indicate individuals homozygous A/A and C/C for rs4446909 and rs5989681, respectively. Real-time reverse transcriptase (RT)-PCR was performed with B lymphoblastoid cell lines from 38 ASD probands and 29 controls. Horizontal bars indicate medians. No statistical difference was observed between ASD and controls (Mann–Whitney U-test).
Table 1.
Frequencies of the polymorphisms located in ASMT promoter B in ASD patients and controls
| ASD (n= 278) | Controls (n= 255) | |
|---|---|---|
| SNPs | ||
| rs4446909 | ||
| f(G) | 0.77 | 0.70 |
| P value (Pc valuea) | 0.006 (0.10) | |
| OR [95% CI]b | 1.5 [1.1–2] | |
| rs5989681 | ||
| f(G) | 0.73 | 0.65 |
| P value (Pc value) | 0.007 (0.12) | |
| OR [95% CI] | 1.4 [1.1–2] | |
| P1BC | ||
| f(G) | 0.90 | 0.90 |
| P value | 0.78 | |
| rs6644635 | ||
| f(C) | 0.65 | 0.63 |
| P value | 0.66 | |
| Haplotypes3 | ||
| H1 GGGC | 0.36 | 0.27 |
| P value (Pc value) | 0.002 (0.04) | |
| H2 GGGT | 0.26 | 0.28 |
| P value | 0.54 | |
| H3 ACGC | 0.21 | 0.29 |
| P value (Pc value) | 0.005 (0.08) | |
| H4 GGAT | 0.097 | 0.09 |
| P value | 0.78 | |
| H5 GCGC | 0.055 | 0.057 |
| P value | 0.88 | |
Abbreviations: ASD, autism spectrum disorders; CI, confidence interval; OR, odds ratio; SNP, single nucleotide polymorphism.
Pc value: Significance levels corrected for multiple comparisons using a step-down permutation procedure (comprising 100,000 permutations).
Odds ratio: major allele vs minor allele.
Haplotype using rs4446909, rs5989681, P1BC and rs6644635.
Results with significance < 0.05 are indicated in bold.
We then explored the relationship between these frequent variations of the ASMT gene and ASMT expression. Promoter A activity is restricted to the retina,24 whereas promoter B is active in BLCL and in the pineal gland. In view of the results of quantitative RT-PCR, ASMT transcript level was found to be significantly associated with the two SNPs linked to ASD — rs4446909 and rs5989681 (Figure 3c). Interestingly, the G alleles of both SNPs were more frequent in ASD patients and were associated with a decrease in ASMT transcript levels by a factor of 4 to 20, respectively (rs4446909 P=2×10−8 and rs5989681 P=2×10−10).
Serotonin, ASMT activity and melatonin concentration in ASD
We then measured the serotonin concentration, the ASMT activity and melatonin concentration in blood platelets from 43 ASD patients, 34 parents of ASD patients and 48 control individuals. Consistent with previous studies,33 the serotonin level was significantly higher in individuals with ASD (P=2×10−11) and their parents (P=10−8) than in controls (Figure 4a). In contrast, the ASMT activity levels were significantly lower in individuals with ASD (P=2×10−12) and their parents (P=10−5) than in controls (Figure 4b). This deficit in ASMT activity was accompanied by a lower plasma melatonin concentration in patients with ASD (P=3×10−11) and their parents (P=9×10−5) than in controls (Figure 4c). Platelet ASMT activity and plasma melatonin levels were not correlated in controls (Figure 4d), whereas they were strongly correlated in patients with ASD (ρ=0.83; P=10−6). Thus, the decreased ASMT activity in ASD patients acted as a limiting factor for the production of melatonin by the pineal gland. We investigated this ASMT deficiency further by analyzing BLCLs from 53 individuals with ASD (for 15 of whom blood samples had already been tested) and 33 new independent controls (Figure 4e). We found that ASD patients had lower levels of ASMT activity than controls (P=7×10−8), as shown previously with platelets. These results obtained with cultured cells replicate our previous finding and exclude possible effects of environmental factors or regulation acting at a higher physiological level. We found no significant correlation between the severity of the deficit and clinical phenotype (IQ, language level or ADI-R scores in the three major domains of impairment: Reciprocal Social Interaction, Communication, and Repetitive Behaviors and Stereotyped Patterns) (data not shown).
Figure 4.
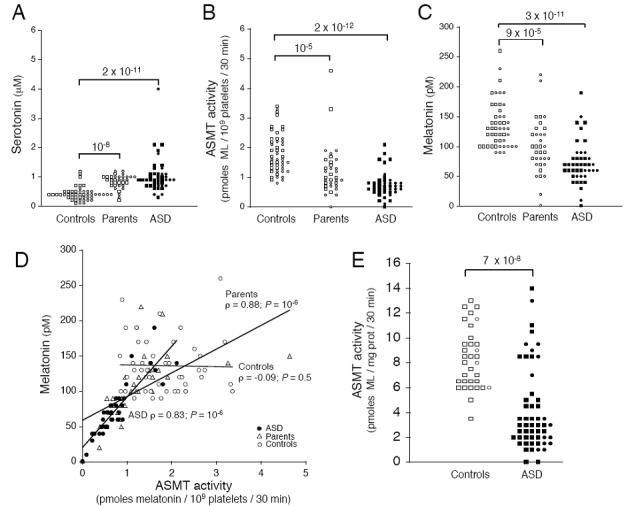
ASMT activity and melatonin concentration in patients, parents and controls. (a) Serotonin levels, measured in the platelets of 43 ASD patients (mean±s.d.; 1±0.6 μM), 34 parents (0.8±0.2 μM) and 48 controls (0.4±0.2 μM). (b) ASMT activity, measured in the platelets of 43 ASD patients (0.73±0.43 pmol/109 platelets/30 min), 34 parents (1.20±0.85 pmol/109 platelets/30 min) and 48 controls (1.81±0.68 pmol/109 platelets/30 min). (c) Plasma melatonin concentration in 43 autism spectrum disorders (ASD) patients (73±36 pmol), 34 parents (99±46 pmol) and 48 controls (136±39 pmol). (d) Blood melatonin concentration expressed with respect to ASMT activity in platelets. ASMT activity and melatonin concentration were not correlated in controls (white circles). In contrast, in ASD patients (black circles), and their parents (white triangles), a significant correlation was observed. Spearman’s rho test was used to evaluate the correlation between ASMT activity and melatonin concentration, (e) ASMT activity in B lymphoblastoid cell lines from 53 ASD patients (3.9±3.4) and 33 controls (8.3±2.4). Circles and squares indicate female and male subjects, respectively. Statistical significance was assessed with the Mann–Whitney U-test.
Melatonin cycle and sleep pattern in family ASD 1
Finally, we investigated circadian melatonin synthesis in vivo and sleep patterns in family ASD 1, in which the unaffected mother and the son with ASD carr the splice-site mutation. Neither of the individuals carrying the ASMT mutation — the unaffected mother and the son with ASD — showed the normal increase in melatonin during the night (Figure 2c). They displayed modest sleep abnormalities, with poor sleep efficiency (proband 70% and mother 82%; controls > 85%) and a moderately high arousal index (22 and 17/h; controls <10/h), but had normal proportions of rapid eye movement sleep (26% and 24%; control range 15%–30%) and a normal amount of slow wave sleep (61 and 104 min; control range: 60–120 min).
Discussion
Abnormal melatonin concentration was observed in individuals with ASD by three independent groups using different methodological approaches.13–15 Our results confirm that low plasma melatonin concentration (half the mean of the control values) is a frequent trait in ASD patients, as observed in 65% of the patients tested, a proportion very similar (63%) to that previously reported by Tordjman et al.15 We show for the first time that abnormal melatonin levels are also present in the unaffected parents of ASD patients, suggesting a genetic origin. Indeed, the melatonin deficit observed in the patients was associated with low ASMT activity, suggesting that variations in the ASMT gene could be the cause of this deficit. This hypothesis was supported by the identification of genetic variations, which probably contribute to the enzymatic deficit by decreasing transcript levels, or altering the sequence of the ASMT protein. However, other unidentified genetic or epigenetic factors are contributing to the ASMT deficit since non-conservative mutations were observed only in a limited number of patients and the genetic association with the polymorphisms located in the ASMT promoter does not solely contribute to the enzymatic deficiency. Furthermore, we found unaffected relatives and controls with ASMT mutations and/or low melatonin concentration in the blood. Therefore, low ASMT activity cannot be considered as a direct cause of ASD, but as a susceptibility factor for this condition (less than 100 pmol melatonin/109 platelets/30 min; odds ratio: 77; 95% confidence interval: 19 <OR< 320).
Individuals with ASD frequently show irregularities in the circadian sleep–wake cycle34–36 and some show a free-running pattern, which is suppressed by melatonin treatment.37 In view of these clinical observations, it was postulated that one alteration at the origin of autism may occur as the child is entering into the day–night cycle.38 In agreement with this hypothesis, the deficit in melatonin may cause abnormal sleep–wake cycle in affected individuals. Additionally, since melatonin influences synaptic plasticity,5,7–9,11,39,40 a deficit in this molecule may also weaken neuronal networks, thereby increasing the effect of other pathological processes, such as abnormal synaptogenesis.41–14
Taken together, these findings indicate that a subgroup of individuals with ASD and low melatonin levels could benefit from the use of melatonin as a therapeutic compound. Melatonin treatment seems to help patients with ASD to fall asleep and to sleep through the night,37,45–48 but it remains unknown if melatonin could have a more beneficial effect if given before 3 years of age. Further studies are required to determine the role of the melatonin deficit in the affected individuals, and more generally of circadian and seasonal rhythms, in the susceptibility to neuropsychiatric disorders.
Supplementary Material
Acknowledgments
We thank the patients and their families for participating in this study. We also thank the DNA and cell bank of INSERM U679 (IFR des Neurosciences, Hôpital Pitié-Salpêtrière); the Centre d’Investigations Cliniques, Hôpital Robert Debré; C. Bouchier and S. Duthoy for the use of sequencing facilities at the Génopole Pasteur; A. Hchikat, L. Margarit, and G. Rouffet for technical assistance; and Luis Barietos, Jean-Pierre Hardelin, Ken McElreavey, Lluis Quintana-Murci and David Skuse for reading the manuscript and making helpful comments. This work was supported by the Pasteur Institute, INSERM, Assistance Publique-Hôpitaux de Paris, Fondation France Télécom, Cure Autism Now, Fondation de France, Fondation biomédicale de la Mairie de Paris, Fondation pour la Recherche Médicale, Fondation NRJ and the Swedish Medical Research Council.
References
- 1.Axelrod J. The pineal gland: a neurochemical transducer. Science. 1974;184:1341–1348. doi: 10.1126/science.184.4144.1341. [DOI] [PubMed] [Google Scholar]
- 2.Brzezinski A. Melatonin in humans. N Engl J Med. 1997;336:186–195. doi: 10.1056/NEJM199701163360306. [DOI] [PubMed] [Google Scholar]
- 3.Arendt J. Melatonin, circadian rhythms, and sleep. N Engl J Med. 2000;343:1114–1116. doi: 10.1056/NEJM200010123431510. [DOI] [PubMed] [Google Scholar]
- 4.Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–1263. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- 5.Simonneaux V, Ribelayga C. Generation of the melatonin endocrine message in mammals: a review of the complex regulation of melatonin synthesis by norepinephrine, peptides, and other pineal transmitters. Pharmacol Rev. 2003;55:325–395. doi: 10.1124/pr.55.2.2. [DOI] [PubMed] [Google Scholar]
- 6.Hallam KT, Olver JS, Chambers V, Begg DP, McGrath C, Norman TR. The heritability of melatonin secretion and sensitivity to bright nocturnal light in twins. Psychoneuroendocrinology. 2006;31:867–875. doi: 10.1016/j.psyneuen.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 7.Liu C, Weaver DR, Jin X, Shearman LP, Pieschl RL, Gribkoff VK, et al. Molecular dissection of two distinct actions of melatonin on the suprachiasmatic circadian clock. Neuron. 1997;19:91–102. doi: 10.1016/s0896-6273(00)80350-5. [DOI] [PubMed] [Google Scholar]
- 8.Wan Q, Man HY, Liu F, Braunton J, Niznik HB, Pang SF, et al. Differential modulation of GABAA receptor function by Mel1a and Mel1b receptors. Nat Neurosci. 1999;2:401–403. doi: 10.1038/8062. [DOI] [PubMed] [Google Scholar]
- 9.El-Sherif Y, Tesoriero J, Hogan MV, Wieraszko A. Melatonin regulates neuronal plasticity in the hippocampus. J Neurosci Res. 2003;72:454–460. doi: 10.1002/jnr.10605. [DOI] [PubMed] [Google Scholar]
- 10.von Gall C, Garabette ML, Kell CA, Frenzel S, Dehghani F, Schumm-Draeger PM, et al. Rhythmic gene expression in pituitary depends on heterologous sensitization by the neurohormone melatonin. Nat Neurosci. 2002;5:234–238. doi: 10.1038/nn806. [DOI] [PubMed] [Google Scholar]
- 11.Jansen R, Metzdorf R, van der Roest M, Fusani L, ter Maat A, Gahr M. Melatonin affects the temporal organization of the song of the zebra finch. FASEB J. 2005;19:848–850. doi: 10.1096/fj.04-2874fje. [DOI] [PubMed] [Google Scholar]
- 12.Smith AC, Dykens E, Greenberg F. Behavioral phenotype of Smith-Magenis syndrome (del 17p11.2) Am J Med Genet. 1998;81:179–185. doi: 10.1002/(sici)1096-8628(19980328)81:2<179::aid-ajmg10>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 13.Nir I, Meir D, Zilber N, Knobler H, Hadjez J, Lerner Y. Brief report: circadian melatonin, thyroid-stimulating hormone, prolactin, and cortisol levels in serum of young adults with autism. J Autism Dev Disord. 1995;25:641–654. doi: 10.1007/BF02178193. [DOI] [PubMed] [Google Scholar]
- 14.Kulman G, Lissoni P, Rovelli F, Roselli MG, Brivio F, Sequeri P. Evidence of pineal endocrine hypofunction in autistic children. Neuroendocrinol Lett. 2000;21:31–34. [PubMed] [Google Scholar]
- 15.Tordjman S, Anderson GM, Pichard N, Charbuy H, Touitou Y. Nocturnal excretion of 6-sulphatoxymelatonin in children and adolescents with autistic disorder. Biol Psychiatry. 2005;57:134–138. doi: 10.1016/j.biopsych.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 16.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4. APA; Washington, DC: 1994. [Google Scholar]
- 17.Baird G, Simonoff E, Pickles A, Chandler S, Loucas T, Meldrum D, et al. Prevalence of disorders of the autism spectrum in a population cohort of children in South Thames: the Special Needs and Autism Project (SNAP) Lancet. 2006;368:210–215. doi: 10.1016/S0140-6736(06)69041-7. [DOI] [PubMed] [Google Scholar]
- 18.Chakrabarti S, Fombonne E. Pervasive developmental disorders in preschool children: confirmation of high prevalence. Am J Psychiatry. 2005;162:1133–1141. doi: 10.1176/appi.ajp.162.6.1133. [DOI] [PubMed] [Google Scholar]
- 19.Veenstra-VanderWeele J, Cook EH. Molecular genetics of autism spectrum disorder. Mol Psychiatry. 2004;9:819–832. doi: 10.1038/sj.mp.4001505. [DOI] [PubMed] [Google Scholar]
- 20.Persico AM, Bourgeron T. Searching for ways out of the autism maze: genetic, epigenetic and environmental clues. Trends Neurosci. 2006;29:349–358. doi: 10.1016/j.tins.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 21.Vorstman JA, Staal WG, van Daalen E, van Engeland H, Hochstenbach PF, Franke L. Identification of novel autism candidate regions through analysis of reported cytogenetic abnormalities associated with autism. Mol Psychiatry. 2006;11(1):18–28. doi: 10.1038/sj.mp.4001781. [DOI] [PubMed] [Google Scholar]
- 22.Thomas NS, Sharp AJ, Browne CE, Skuse D, Hardie C, Dennis NR. Xp deletions associated with autism in three females. Hum Genet. 1999;104:43–48. doi: 10.1007/s004390050908. [DOI] [PubMed] [Google Scholar]
- 23.Yi H, Donohue SJ, Klein DC, McBride OW. Localization of the hydroxyindole-O-methyltransferase gene to the pseudoautosomal region: implications for mapping of psychiatric disorders. Hum Mol Genet. 1993;2:127–131. doi: 10.1093/hmg/2.2.127. [DOI] [PubMed] [Google Scholar]
- 24.Rodriguez IR, Mazuruk K, Schoen TJ, Chader GJ. Structural analysis of the human hydroxyindole-O-methyltransferase gene. Presence of two distinct promoters. J Biol Chem. 1994;269:31969–31977. [PubMed] [Google Scholar]
- 25.Lord C, Rutter M, Le Couteur A. Autism diagnostic interviewrevised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord. 1994;24:659–685. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- 26.Wing L, Leekam SR, Libby SJ, Gould J, Larcombe M. The diagnostic interview for social and communication disorders: background, inter-rater reliability and clinical use. J Child Psychol Psychiatry. 2002;43:307–325. doi: 10.1111/1469-7610.00023. [DOI] [PubMed] [Google Scholar]
- 27.Gillberg C, Rastam M, Wentz E. The Asperger Syndrome (and high functioning autism) Diagnostic Interview (ASDI): a preliminary study of a new structured clinical interview. Autism. 2001;5:57–66. doi: 10.1177/1362361301005001006. [DOI] [PubMed] [Google Scholar]
- 28.Barrett JC, Fry B, Mailer J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 29.Horvath S, Xu X, Laird NM. The family based association test method: strategies for studying general genotype-phenotype associations. Eur J Hum Genet. 2001;9:301–306. doi: 10.1038/sj.ejhg.5200625. [DOI] [PubMed] [Google Scholar]
- 30.Horvath S, Xu X, Lake SL, Silverman EK, Weiss ST, Laird NM. Family-based tests for associating haplotypes with general phenotype data: application to asthma genetics. Genet Epidemiol. 2004;26:61–69. doi: 10.1002/gepi.10295. [DOI] [PubMed] [Google Scholar]
- 31.Axelrod J, Wurtman RJ, Snyder SH. Control of hydroxyindole O-methyltransferase activity in the rat pineal gland by environmental lighting. J Biol Chem. 1965;240:949–954. [PubMed] [Google Scholar]
- 32.Finocchiaro LM, Nahmod VE, Launay JM. Melatonin biosynthesis and metabolism in peripheral blood mononuclear leucocytes. Biochem J. 1991;280:727–731. doi: 10.1042/bj2800727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leboyer M, Philippe A, Bouvard M, Guilloud-Bataille M, Bondoux D, Tabuteau F, et al. Whole blood serotonin and plasma betaendorphin in autistic probands and their first-degree relatives. Biol Psychiatry. 1999;45:158–163. doi: 10.1016/s0006-3223(97)00532-5. [DOI] [PubMed] [Google Scholar]
- 34.Limoges E, Mottron L, Bolduc C, Berthiaume C, Godbout R. Atypical sleep architecture and the autism phenotype. Brain. 2005;128(Part 5):1049–1061. doi: 10.1093/brain/awh425. [DOI] [PubMed] [Google Scholar]
- 35.Wiggs L, Stores G. Sleep patterns and sleep disorders in children with autistic spectrum disorders: insights using parent report and actigraphy. Dev Med Child Neurol. 2004;46:372–380. doi: 10.1017/s0012162204000611. [DOI] [PubMed] [Google Scholar]
- 36.Richdale AL, Prior MR. The sleep/wake rhythm in children with autism. Eur Child Adolesc Psychiatry. 1995;4:175–186. doi: 10.1007/BF01980456. [DOI] [PubMed] [Google Scholar]
- 37.Hayashi E. Effect of melatonin on sleep-wake rhythm: the sleep diary of an autistic male. Psychiatry Clin Neurosci. 2000;54:383–384. doi: 10.1046/j.1440-1819.2000.00725.x. [DOI] [PubMed] [Google Scholar]
- 38.Segawa M. Epochs of development of the sleep-wake cycle reflect the modulation of the higher cortical function particular for each epoch. Sleep Biol Rhythms. 2006;4:4–15. [Google Scholar]
- 39.Wang LM, Suthana NA, Chaudhury D, Weaver DR, Colwell CS. Melatonin inhibits hippocampal long-term potentiation. Eur J Neurosci. 2005;22:2231–2237. doi: 10.1111/j.1460-9568.2005.04408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Larson J, Jessen RE, Uz T, Arslan AD, Kurtuncu M, Imbesi M, et al. Impaired hippocampal long-term potentiation in melatonin MT(2) receptor-deficient mice. Neurosci Lett. 2006;393:23–26. doi: 10.1016/j.neulet.2005.09.040. [DOI] [PubMed] [Google Scholar]
- 41.Belmonte MK, Bourgeron T. Fragile X syndrome and autism at the intersection of genetic and neural networks. Nat Neurosci. 2006;9:1221–1225. doi: 10.1038/nn1765. [DOI] [PubMed] [Google Scholar]
- 42.Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2006;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, Liu XQ, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007;39:319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garstang J, Wallis M. Randomized controlled trial of melatonin for children with autistic spectrum disorders and sleep problems. Child Care Health Dev. 2006;32:585–589. doi: 10.1111/j.1365-2214.2006.00616.x. [DOI] [PubMed] [Google Scholar]
- 46.Giannotti F, Cortesi F, Cerquiglini A, Bernabei P. An open-label study of controlled-release melatonin in treatment of sleep disorders in children with autism. J Autism Dev Disord. 2006;36:741–752. doi: 10.1007/s10803-006-0116-z. [DOI] [PubMed] [Google Scholar]
- 47.Tjon Pian Gi CV, Broeren JP, Starreveld JS, Versteegh FG. Melatonin for treatment of sleeping disorders in children with attention deficit/hyperactivity disorder: a preliminary open label study. Eur J Pediatr. 2003;162:554–555. doi: 10.1007/s00431-003-1207-x. [DOI] [PubMed] [Google Scholar]
- 48.Malow BA. Sleep disorders, epilepsy, and autism. Ment Retard Dev Disabil Res Rev. 2004;10:122–125. doi: 10.1002/mrdd.20023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.