

Abstract
Despite the critical roles of the amyloid precursor protein (APP) in Alzheimer's disease pathogenesis, its physiological function remains poorly established. Our previous studies implicated a structural and functional activity of the APP family of proteins in the developing neuromuscular junction (NMJ). Here we performed comprehensive analyses of neurotransmission in mature neuromuscular synapse of APP deficient mice. We found that APP deletion led to reduced paired-pulse facilitation and increased depression of synaptic transmission with repetitive stimulation. Readily releasable pool size and total releasable vesicles were not affected, but probability of release was significantly increased. Strikingly, the amount of asynchronous release, a measure sensitive to presynaptic calcium concentration, was dramatically increased, and pharmacological studies revealed that it was attributed to aberrant activation of N- and L-type Ca2+ channels. We propose that APP modulates synaptic transmission at the NMJ by ensuring proper Ca2+ channel function.
Keywords: Amyloid precursor protein, synaptic transmission, neuromuscular junction, calcium channels, asynchronous release
Introduction
The defining pathological hallmark of Alzheimer's disease (AD) is the deposition of β amyloid plaques, of which the principal components are 40 and 42 amino acid β-amyloid peptides (Aβ) derived from proteolytic processing of the amyloid precursor protein (APP). Genetic studies establish a pivotal role of APP in AD pathogenesis, as both point mutations and gene duplications of APP are causal for a subset of early onset familial AD ((Rovelet-Lecrux et al., 2006) and reviewed in (Hardy and Selkoe, 2002)). Besides the amyloid pathology, synaptic dysfunction is widely recognized to play a critical role in dementia, and profound changes in the cholinergic system invariably accompany AD pathogenesis (Edeline, 1999; Kilgard, 2003; McKinney et al., 1983). The mechanisms linking APP with synaptic transmission and cholinergic activity are, however, poorly established.
APP is a conserved type I membrane protein. It is highly expressed in central and peripheral nervous systems, and can be detected in both pre- and postsynaptic compartments (Akaaboune et al., 2000; Schubert et al., 1991; Shigematsu et al., 1992). Full-length APP is processed by at least three proteases termed α-, β- and γ-secretases; the combination of β- and γ-secretase processing leads to the liberation of β amyloid peptides (Reviewed by (Zheng, 2006)). APP undergoes anterograde and retrograde transport; APP processing has been shown to occur en route (Zheng, 2006). Whereas the focus of AD research has been on β amyloid peptides, there is intense interest in understanding the basic biology of APP, as alterations in Aβ biogenesis simultaneously affect other APP processing products, which may exert complex effects on cellular function and may also contribute to AD pathogenesis.
Our previous analyses of APP deficient mice revealed reductions in grip strength, locomotor activity, long-term potentiation, and spatial learning in these animals (Dawson et al., 1999; Seabrook et al., 1999; Zheng et al., 1995). Studies of mice doubly deficient in APP and its homolog APP like protein 2 (APLP2) established an essential role of APP family proteins in the structure and function of the developing neuromuscular synapse (Wang et al., 2005). The mammalian NMJ is a highly specialized synaptic structure in which the presynaptic terminals of motor neuron axons are closely apposed to postsynaptic acetylcholine receptor (AChR)-rich endplates in the muscle fibers (reviewed in (Sanes and Lichtman, 1999)). In newborn mice, each postsynaptic endplate is innervated by multiple axons. The developing NMJ undergoes postnatal maturation such that by postnatal day 14 (P14), postsynaptic muscle fibers are singly innervated, which makes the mature neuromuscular synapse an ideal system to study cholinergic neurotransmission.
In the present study, we carried out extensive analyses of the properties of synaptic transmission at the mature NMJ of APP null mice, and report here that the APP null NMJ failed to show paired-pulse facilitation and displayed greater synaptic depression accompanied by enhanced asynchronous release during repetitive stimulation. We provide evidence that this is mediated by the aberrant activation of L- and N-type Ca2+ channels.
Experimental Procedures
Reagents
ω-conotoxin GVIa and ω-agatoxin IVa were purchased from Bachem, EGTA, tetra (acetoxymethyl ester) (EGTA-AM) was from Calbiochem. EGTA-AM and nifedipine stock solutions were prepared in dimethylsulfoxide (DMSO) and diluted in physiological saline before the experiment at final concentrations of 0.1% and 0.05%, respectively. The same concentration of DMSO was included in vehicle controls. FM1−43 and α-bungarotoxin (α-BTX) were purchased from Molecular Probes. Other reagents were from Sigma-Aldrich unless otherwise noted.
Animals
The APP knockout mice were described previously and were generated by intercrossing the APP heterozygous males and females (Zheng et al., 1995). These mice have been backcrossed to C57Bl/6J background for at least 10 generations. All efforts were made to minimize the number of animals used and their suffering throughout the experiments. Experiments were performed in accordance with the Baylor College of Medicine Institutional Animal Care and Use Committee and with national regulations and policies.
Electrophysiology
Ex vivo electrophysiological recording: P18-P22 mice were anesthetized with isoflurane (Abbott Laboratories). Diaphragm preparations of WT and APP−/− mice, with the phrenic nerve supply intact, were isolated and gently pinned flat in a sylgard-coated recording chamber. After the diaphragms had equilibrated for about an hour, the nerve was taken up into a suction electrode and the muscle fibers along the main intra-muscular branches nerve were impaled with 3 M KCl – filled glass micropipettes (20 − 35 MΩ). Intracellular sharp-electrode recording was performed to record mEPPs and evoked endplate potentials (EPPs) in normal Ringer's solution containing 2.3 μM μ-Conotoxin GIIIB (Bachem) to selectively block muscle voltage-gated sodium channels and therefore, muscle contraction. The saline was of the following composition (in mM): NaCl 116, KCl 4.5, MgSO4 1, NaHCO3, 23, NaH2PO4 1, Dextrose 11, CaCl2 2. Preparations were continuously perfused with oxygenated (95% O2 and 5% CO2) saline at a rate of 2 ml/min at 28°C ± 0.5°C. Initial resting membrane potentials were between −70 to −85 mV and, after an initial fall, were stable during the experiment and greater than −60 mV. Electrophysiological measurements were not recorded if the resting potential decreased by >15% of its original value. The stimulation pulses (0.1 msec) were applied via an AMPI Master-8 pulse generator and an AMPI Iso-Flex stimulus isolator (A.M.P. Instruments LTD). Potentials were amplified via a MultiClamp 700B amplifier (Axon Instruments), digitized at 10 KHz and recorded to a computer using pClamp 9 software (Axon Instruments). Offline data analysis was performed using Clamfit 9, (Axon Instruments), MiniAnalysis (Synaptosoft) and OriginPro 7.5 (OriginLab).
In paired–pulse experiments, EPPs were elicited by nerve stimulation with supramaximal double pulses with an interpulse interval range from 10 to 100 ms. Facilitation was evaluated by calculating the ratio P2/P1: where P2 is the EPP amplitude elicited by the second stimulus and P1 is the EPP amplitude elicited by the first stimulus. EPP ratio in train stimuli were calculated the same way.
Measurement of readily releasable pool (RRP) size: Quantal content (QC) was determined by the direct method and corrected for nonlinear summation, using the formula: QC=E/ (McLachlan and Martin, 1981), where E is the amplitude of the first EPP of the train and q is the mean mEPP amplitude during the train, E1 is the difference between the resting membrane potential and the reversal potential which was assumed to be 0 mV for the endplate response (Magleby and Stevens, 1972), and f is 0.8 for mouse muscle (McLachlan and Martin, 1981). Since change of train frequency didn't affect the estimated size of readily releasable pool (RRP) (Elmqvist and Quastel, 1965), 30 Hz train was used in the present study to avoid induction of asynchronous release in the NMJs of APP−/− mice. The QC of EPP was plotted against cumulative number of quanta. RRP was estimated by back-extrapolation from the linear portion of the curve to the x axis intercept. The x-intercept gives an estimate of RRP based upon the assumption of negligible mobilization into the RRP (Elmqvist and Quastel, 1965).
Analysis of asynchronous, total, evoked and delayed response: Total release for the 10 min 30 Hz train was estimated by integrating the EPPs and miniature-like responses during the stimulus train; asynchronous release was evaluated by counting the frequency of miniature-like response during the train. For the 30 pulses at 200 Hz experiment, evoked release was quantified as the integral of the response during the short train. Delayed release was examined by integrating the responses occurring during the 10 to 200 msec following the last EPP of the train. Baseline measured before the first pulse of the train was subtracted before integration.
FM1−43 labeling of synaptic vesicles
Diaphragms preparations were placed in 8 μM FM1−43 (Molecular Probes) in oxygenated Ringer's solution at 28°C. Muscle contraction was blocked with 2.3 μM μ-conotoxin GIIIB. FM1−43 labeling of WT and APP−/− NMJs was obtained by stimulating the phrenic nerve at 10 Hz for 10 min, a protocol that loads the exo-and endocytosis pool as well as the reserved pool with FM1−43 at mouse NMJ motor nerve terminals (Polo-Parada et al., 2001). After stimulation, muscles were rinsed with 10 min washes in oxygenated Ringer's solution and 2 min washes in Ringer's solution with 1 mM ADVASEP-7 (Biotium, Hayward, CA) over a 30 min period. After FM1−43 labeling, AChRs were labeled by incubation in 10 μg/ml Alexa Fluor 594-conjugated α-BTX (Molecular Probes) for 20 min. Following rinsing, endplates were visualized with a 40× water immersion objective on a Nikon Eclipse E600FN fluorescence microscope equipped with the appropriate filter cubes. Images were captured using a digital camera (Photometrics Cascade 512B, Roper Scientific) operated by Metamorph Acquisition and Device Control (Molecular Devices) and stored digitally. FM1−43 destaining by titanic nerve stimulation in WT and APP−/− NMJs were determined FM1−43 positive terminals were quantified off-line for pixel intensity using Metamorph image analysis tool (Molecular Devices).
Statistics
All data shown in the text and figures are mean ± SEM. (N=number of fibers/neurons). Possible statistical differences were analyzed with a paired or unpaired student's t test. Nonparametric Kolmogorov-Smirnov test (Urbano et al., 2003) was used when the values were expressed as percentages. p<0.05 was regarded as significant.
Results
Normal Miniature Endplate Potential but Altered Short-term Dynamics at APP null NMJ
Our previous electron microscopy studies documented that mice doubly deficient in APP and APLP2 exhibit significant reduction of vesicle numbers at presynaptic terminals, which is associated with lower frequency of miniature endplate potential (mEPP) at postnatal day 0 (P0) of developing NMJ. This reduction was not seen in P0 APP single knockout mice (Wang et al., 2005). In the present study, we examined whether the mEPP was normal in the mature NMJ of APP null mice. mEPP at P18−22 diaphragm nerve-muscle preparations of littermate wild-type (WT) and APP null (APP−/−) mice was recorded and, in agreement with that of the developing NMJ, we detected no differences in either mEPP amplitude or frequency in APP−/− samples as compared with WT controls (Figure 1, A-C). However, single EPP amplitude, 10−90% rise time and half width were significantly increased in APP−/− NMJ compared to WT controls (Table 1). To examine short-term dynamics in APP−/− NMJ, we first performed paired-pulse experiments, a protocol commonly used to determine presynaptic function, by applying two pulses at 10, 20, 50 and 100 millisecond intervals in WT and APP−/− NMJ (Fig. 1, D and E). At physiological calcium level (2 mM Ca2+) and 1 mM Mg2+, a significantly lower paired-pulse ratio (P2/P1) was observed in APP−/− mice at all intervals tested, with the largest effects occurring at the shortest interpulse interval. Similar paired-pulse defects were observed in NMJs of 1−2 month old APP−/− mice (N=3, data not shown), make the possibility that the defect is caused by a developmental delay less likely. We then carried out the paired-pulse experiments by varying external Ca2+ concentrations from 0.25 to 2 mM in the presence of 1 mM of constant Mg2+ at 10 msec interpulse intervals. The APP null junction showed facilitation similar to WT controls at low extracellular Ca2+ levels (<1 mM) (Figure 1F). In agreement with this result, we found similar increases of the EPP amplitude in WT and APP null NMJ in response to rising Ca2+ concentrations from 0.25 to 2 mM (Fig. 1G). However, while the EPP amplitude continued to increase with higher Ca2+ levels in WT controls, in APP−/− NMJ, the EPP amplitude reached to near maximal value at 2 mM Ca2+ (Figure 1G).
Figure1.

Parameters of synaptic transmission in P18−22 WT and APP−/− NMJs. A. Representative mEPP traces in NMJs of WT and APP−/− mice. Scale: 1 mV/200 msec. B. Mean mEPP amplitude and C. Mean mEPP frequency of WT (N=40) and APP−/− (N=41) mice. D. Representative traces show EPPs to pairs of pulses separated by 10, 20, 50 and 100 millisecond (msec) intervals at normal levels of extracellular Ca2+ (2 mM) and Mg2+ (1 mM). Scale: 10 mV/20 msec. E. Summary of paired-pulse ratio (P2/P1) in WT (N=22, 15 muscles) and APP−/− mice (N=25, 18 muscles) in response to different interpulse intervals. F. Average P2/P1 at 10 msec interpulse interval as a function of extracellular Ca2+ (0.25−2 mM) (N is 5−8 and 7−12 at each Ca2+ dose for WT and APP−/−, respectively). Insert shows sample recordings at 1 mM Ca2+. Scale: 2 mV/10 msec. G. Ca2+ dependence of EPPs at [Ca2+]o ranging from 0.25 to 10 mM in APP−/− and WT NMJs (N is 5 to 9 at each [Ca2+]o concentration for both genotypes). Error bars indicate SEM. *p<0.05; **p<0.005.
Table 1.
Summary of EPP Biophysical Properties
| WT |
APP−/− |
p-value | |||||
|---|---|---|---|---|---|---|---|
| Mean | SEM | N | Mean | SEM | N | ||
| Amplitude (mV) |
15.3 |
0.76 |
45 |
18.7 |
1.04 |
45 |
0.01* |
| 10−90% Rise time (ms) |
0.69 |
0.04 |
45 |
0.84 |
0.04 |
45 |
0.006* |
| Decay time (ms) |
5.13 |
0.01 |
45 |
5.33 |
0.01 |
45 |
0.2 |
| Half width (ms) | 2.69 | 0.09 | 45 | 3.13 | 0.13 | 45 | 0.006* |
Differences between WT and APP−/− samples reached statistical significance (student's t-test).
We next monitored the responses of APP null NMJs to 1 second stimulus train at four different frequencies (20, 50, 100 and 200 Hz) (Figure 2). WT NMJs displayed an initial facilitation followed by a general decrease at all frequencies tested (Fig. 2A, WT), depicted by the ratios of 10th to first (Fig. 2B) or last to first (Fig. 2C) of EPP amplitude. The depression was greater by the end of the 1 sec train as compared to the 10th stimulation, especially at higher frequencies (compare Fig. 2B with Fig. 2C, 100 and 200 Hz). Consistent with the paired-pulse data, the APP−/− NMJ failed to show initial facilitation (Fig. 2A, compare second (P2) to first (P1) response, APP−/−). With one exception (20 Hz, 10th/1st ratio), both the ratios of 10th/1st (Fig. 2B) and last/1st EPP amplitude (Fig. 2C) were significantly lower in APP null samples compared to WT controls under the same frequencies.
Figure 2.

Increased depression of synaptic transmission in APP−/− NMJs at high-frequency stimulation. A. Representative traces showing EPP depression elicited by 20, 50, 100 and 200 Hz nerve stimulation at WT or APP−/− endplates. Scale: 5 mV/100 msec. Enlarged trace under 200 Hz recording shows the amplitude values. B and C. Ratios of EPP at 10th to first (B) or the end of train to first (C) stimulus at 20, 50, 100 and 200 Hz. N=12−14 at each frequency for both genotypes. *p<0.05; **p<0.005.
Identical Readily Releasable and Total Releasable Vesicle Pools but Increased Probability of Release at APP−/− NMJ
The lack of paired-pulse facilitation and greater depression under high frequency repetitive stimulation seen in APP−/− NMJ could be attributed by higher initial probability of release (Pr) (Rozov et al., 2001; Zucker and Regehr, 2002) or smaller vesicle pool size, which includes readily releasable pool (RRP) and total releasable vesicle pool, leaving the nerve terminals unable to sustain the response during repetitive stimulation. To differentiate these possibilities, we employed various calculations to estimate the RRP, total vesicle pool size and Pr in WT and APP−/− NMJs. The quantal size was defined for each tested NMJ as the mean amplitude of miniature-like responses during the 30 Hz train, the values of which were similar in WT and APP−/− preparations (WT: 0.47 ± 0.04 mV; APP−/−: 0.48 ± 0.03 mV), suggesting that the greater depression during repetitive stimulation was not due to a decrease in the sensitivity of postsynaptic receptors to transmitters. Quantal content (QC) was determined by the ratio of EPP/miniature-like EPP and corrected for nonlinear summation (see Method section for detail). QC was 31 ± 4 for WT and 43 ± 4 for APP−/−; the difference was statistically significant (p<0.05). The method of Elmqvist and Quastel (Elmqvist and Quastel, 1965) was used to estimate the RRP size by plotting the QC of EPPs against cumulative number of quanta and back-extrapolated from the linear portion of the curve to the x-axis intercept (Figure 3A). The x-intercept gives an estimate of RRP based on the assumption of negligible mobilization into the RRP (Elmqvist and Quastel, 1965; Lu and Trussell, 2000). The RRP values calculated by this method did not yield significant differences between the control and APP null samples (Figure 3B). Having an estimate of RRP, Pr was calculated by dividing the QC of the first evoked EPP by the RRP size (Juttner et al., 2005), which represents the fraction of vesicles released during the first stimulation. APP−/− NMJs showed a significant increase in Pr (Figure 3C). We also examined the rate of recovery of the EPP following depletion of the RRP by a 200 stimulus train at 100 Hz. We detected no abnormality in the recovery kinetics of the EPP when two stimulus trains were delivered at 1 sec interval (82.1% recovery in WT versus 84.6% in APP−/−; N=9 for both genotypes).
Figure 3.
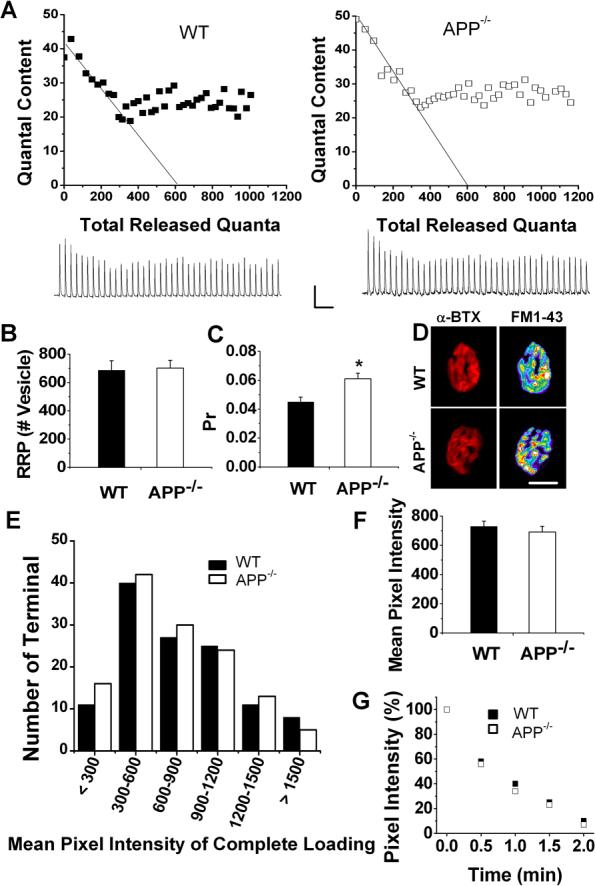
APP deficiency affects probability of release (Pr) but not readily-releasable pool (RRP) or total releasable pool size. A. Quantal content in a 40 pulse-stimulus-train at 30 Hz plotted against the number of cumulative quanta. Interrupted lines were extrapolated from the data points obtained during the initial rapid decline phase. The x-intercepts gave maximum number of quanta releasable without replenishment, i.e. RRP. Traces underneath are recordings from above WT and APP−/− NMJs, respectively. Scale: 5mV/100 msec. B and C. Summary results of RRP (B) and Pr (C) of WT (N=17, 12 mice) and APP−/− (N=20, 13 mice) NMJs. Pr was calculated as initial QC/RRP, *p<0.05. D. Representative AChR-positive endplates marked by Alexa Fluor 594-conjugated α-BTX (red) and their corresponding presynaptic vesicle pool size visualized by FM1−43 loading (pseudo color), respectively. Scale: 20 μm. E. Distributions of mean pixel intensity of FM1−43 loaded endplates in WT and APP−/− NMJs. F. Total pool size revealed by measuring mean pixel intensity of FM1−43 loaded endplates in WT (N=122) and APP−/− (N=130) mice. G. Representative time courses of FM1−43 destaining in APP−/− and WT NMJs. Error bars indicate SEM.
We next measured the total vesicle pool size in WT and APP null NMJ by FM1−43 dye incorporation, which is internalized into vesicles during cycles of exo-endocytosis (Betz and Bewick, 1992). Total vesicle pool size was quantified by measuring the mean pixel intensity of FM1−43 loaded by 10 Hz-10 min stimulation — a protocol that results in loading of total releasable vesicles at mouse motor nerve terminals (Polo-Parada et al., 2001). Endplates were visualized by incubating, after FM1−43 loading, with Alexa Fluor 594-conjugated α-bungarotoxin (α-BTX) to label the postsynaptic acetylcholine receptors (Fig. 3D). No obvious differences in the overall morphology of the APP−/− NMJs were observed (Figure 3D). Distributions of mean pixel intensities of FM1−43 loaded endplates from WT and APP−/− mice were similar (Figure 3E), as were the total pixel intensities (Figure 3F), indicating that sizes of vesicle pool that could be loaded by the stimulation was similar for both genotypes. Similar RRP and endplate morphology are likely the reason for the comparable mEPP frequency between APP−/− and WT NMJ (Kuno et al., 1971; Priller et al., 2006; Reim et al., 2001). FM1−43 destaining by 200 Hz nerve stimulation was indistinguishable between the two genotypes (figure 4G).
Figure 4.

Reduced synchronous but increased asynchronous release in APP−/− endplates during repetitive stimulation. A. Representative traces from WT and littermate APP−/− NMJ in response to 10 min, 30 Hz stimulation (scale: 5 mV/30 ms). B. Average EPP amplitude during 10 min 30 Hz train normalized to the first response. Each point represents average EPP amplitude of 1 sec responses. C. Increased frequency of asynchronous release in APP−/− NMJs during the train. D. Bar graphs showing identical total release, which is the integral to 10 min, 30 Hz train, in both genotypes. (WT, N=7; APP−/−, N=9). E. Representative traces of WT and APP−/− NMJs in response to 30 stimuli applied at 200 Hz before (Ctrl) and after addition of 0.1 mM EGTA-AM (Scale: 5 mV/30 msec). F and G. Significantly increased delayed release (F), but similar evoked release (G) in APP−/− NMJs in the absence of EGTA-AM (N is 23 and 27 for WT and APP−/−, respectively). H. Blockade of evoked, delayed release and amplitude of first EPPs by EGTA-AM in WT (6 pairs, 6 muscles) and APP−/− NMJs (5 pairs, 5 muscles). Error bars indicate SEM. *p<0.05, **p<0.005.
Increased Asynchronous Release Accompanied Greater Depression in APP Null NMJ
It has been reported that in response to trains of action potentials, synchronous release eventually declines, whereas asynchronous release often progressively increases, an effect that is primarily caused by the buildup of intracellular Ca2+ during repetitive stimulation (Atluri and Regehr, 1998). The asynchronous release competes with synchronous release for the recovered quanta since they are derived from the same pool of readily releasable vesicles (Elmqvist and Quastel, 1965; Hagler and Goda, 2001; Lu and Trussell, 2000). As such, elevated asynchronous release can reduce future synchronous release (Otsu and Murphy, 2004). To test whether enhanced asynchronous release might be involved in the greater depression seen in APP−/− mice, we first measured asynchronous release by quantifying the frequency of mini-like responses during a 10 min train at 30 Hz (Figure 4). As expected, the evoked EPP in APP−/− NMJ was attenuated more rapidly by repeated stimuli (∼87% depression at last EPP in APP null compared to ∼60% in the controls) (Figure 4B). Interestingly, this greater degree of depression was accompanied by and positively correlated with increases in frequencies of asynchronous release with a peak rate of 54 ± 15/sec in APP null samples compared to 11 ± 5 /sec in WT controls (Figure 4C). Total release, estimated by integrating the responses during the 10 min-30 Hz stimulus train, was similar in both genotypes (Figure 4D). These results suggest that greater depression of synchronous release in APP−/− involves a shift to asynchronous release.
Asynchronous release induced by high-frequency stimulation consists of two types: release during the stimulus train when synchronous release still operates but is out competed by asynchronous release; and release after the stimulus train, i.e., delayed asynchronous release which is induced by residual Ca2+ after the stimulus train ends (Barrett and Stevens, 1972; Hefft and Jonas, 2005; Maximov and Sudhof, 2005). Having detected a profound increase in asynchronous release in APP null NMJs during prolonged stimulation, we next used a high-frequency short-train protocol consisting of 30 pulses at 200 Hz to further evaluate asynchronous release by comparing the delayed asynchronous response between two genotypes (Figure 4, E-H). Similar to that of prolonged stimulation, delayed asynchronous release was significantly higher in APP−/− NMJ as compared to WT controls under the short-train condition (Figure 4F), while evoked release, obtained by integrating the response during the stimulus train that includes synchronous, asynchronous release and temporal summation, was almost identical in both genotypes (Figure 4G). Since residual Ca2+ buildup has been implicated in asynchronous release, we therefore examined the effects of EGTA-AM, a membrane-permeable Ca2+ chelator with a slow binding rate for Ca2+, on evoked and delayed release to see if the increased asynchronous release in APP−/− terminals during repetitive stimulation was due to Ca2+ accumulation. After 20−30 min incubation with 100 μM EGTA-AM, reduction of evoked release, which was the integral of the response during 30-pulse stimulus train, was significantly higher in APP−/− NMJs than WT controls (Figure 4H). Similar to the total evoked release during the train, the amplitude of the first EPPs was significantly eliminated by EGTA-AM in APP−/− NMJs compared to WT controls (Figure 4H). Our data suggested a significantly more asynchronous component not only after the train, but also during the stimulus train in the absence of APP since asynchronous release is much more sensitive to EGTA-AM than synchronous release (Hefft and Jonas, 2005). Consistent with the notion that prolonged Ca2+ transient underlies asynchronous release, EGTA-AM similarly reduced delayed asynchronous release in both genotypes after the train (Figure 4H).
Aberrant Ca2+ Channel Activities at APP−/− NMJs
In mature NMJs, the P/Q-type voltage-dependent Ca2+ channel (VDCC) is predominantly responsible for the control of ACh release. Aberrant activation of N- or L-type calcium channels has been observed when P/Q type VDCC or the adhesion protein NCAM are disrupted (Pagani et al., 2006; Polo-Parada et al., 2001; Urbano et al., 2001; Urbano et al., 2003). Of note, the N-type Ca2+ channel is located farther away from the Ca2+ sensor as compared with the P/Q-type channel (Wu et al., 1999), which increase the probability of Ca2+ being bound by a calcium buffer like EGTA-AM, and would contribute to a higher extent of asynchronous release and greater synaptic depression at the presynaptic terminals (Hefft and Jonas, 2005; Pagani et al., 2006; Zakharenko et al., 1999). We thus reasoned that a disturbed calcium channel regulation may lead to aberrant Ca2+ entry and contribute to the markedly increased asynchronous release and EGTA-AM sensitivity in APP deficient animals. To test the idea, we compared effects of specific Ca2+ channel blockers, including the N-type blocker ω-conotoxin GVIA, the L-type blocker nifedipine, and the P/Q blocker ω-agatoxin-IVA, on evoked and delayed release in APP−/− and WT controls by measuring the integral of the response before and after bath application of the blockers (Figure 5, A-C). Consistent with the notion that P/Q-type VDCC is the predominant channel mediating synaptic transmission in mature NMJ, incubation with 1 μM N- or 20 μM L-type calcium channel blockers ω-Conotoxin GVIA or nifedipine, respectively, did not lead to appreciable effects in WT controls (Figure 5, D and E). However, these treatments resulted in significantly higher blockage of evoked (Figure 5D) and delayed response (Figure 5E) in APP−/− NMJ, suggesting the ectopic activation of N- and L-type calcium channels. Accordingly, the P/Q channel blocker ω-Agatoxin-IVA was significantly less effective on evoked response in APP−/− NMJs compared to WT control (Figure 5, C and D), implicating existence of a P/Q channel blocker-insensitive component in the evoked release in the absence of APP. Kinetics of N- and L-type Ca2+ channel mediated EPPs in APP−/− NMJs were obtained by analyzing the first EPP of the train in the presence of P/Q channel blocker. The halfwidth, 10−90% rise time and decay time was 1.93±0.35, 0.88±0.25 and 1.03±0.19, respectively. ω-Conotoxin GVIA also resulted in a significantly higher reduction in the amplitude of first EPPs in APP−/− NMJs when compared to the WT controls (Figure 5, F). These results suggest that although P/Q-type Ca2+ channels were still the predominant Ca2+ channel mediating synaptic transmission at APP−/− NMJs, N- and L-type Ca2+ channels were coupled to neurotransmitter release and are at least partially responsible for the increased asynchronous release at APP−/− NMJs. We found that the sum of individual blockers' effects on evoked transmitter release exceeded 100% in APP null preparations (Figure 5D). This is probably due to the nonlinear relationship between Ca2+ current and transmitter release (Mintz et al., 1995; Takahashi and Momiyama, 1993).
Figure 5.

Effect of VDCC blockers on evoked and delayed release in APP−/− and WT NMJs. A-C. Representative traces showing evoked and delayed release triggered by 30 pulse-stimulation at 200 Hz in the absence of blockers (Ctrl), with 1 μM ω-CgTx-GVIA (A, N-blocker), 20 μM nifedipine (B, L-blocker) or 100 nM ω-Agatoxin-IVA (C-D, P/Q-blocker) Scale: 5 mV/50 msec. D - E. Bar diagrams illustrate the blockade exerted by ω-CgTx-GVIA (WT, N=5; APP−/−, N=11), nifedipine (WT, N=5; APP−/−, N=6) or ω-Agatoxin-IVA (WT, N=5; APP−/−, N=8) on evoked, delayed release and 1st EPP amplitude in APP−/− and WT controls. Error bars indicate SEM. *p<0.05.
Discussion
Altered Synaptic Transmission at NMJ in the Absence of APP
The aim of this study was to examine the physiological role of APP in synaptic transmission, in particular, cholinergic transmission, taking advantage of the mature NMJ system. Our analysis of neuromuscular synapses showed that although mEPP frequency and amplitude occurred normally, lack of APP significantly reduced the paired-pulse ratio under physiological Ca2+ levels and caused greater depression during repetitive stimulation, which is likely the cause for the reduced forelimb grip strength observed in the animals (Zheng et al., 1995). Similar paired-pulse abnormality at physiological, but not lower, Ca2+ concentrations was also seen in the NMJs of mice deficient in the cell adhesion protein NCAM (Polo-Parada et al., 2001). In line with these findings, we observed a significantly increased Pr, whereas the RRP and total releasable vesicle pool sizes remained unchanged. The increased Pr is also consistent with elevated EPP amplitude in APP−/− mice at physiological Ca2+ levels (Table 1), implicating a possible alteration of calcium-dependent release. Increases in Pr and changes in EPP kinetics due to activation of Ca2+ dependent K+ channels were not examined in the present study, and cannot be excluded as a potential contributor (Raffaelli et al., 2004). Ca2+ dependence of the paired-pulse ratio and near-saturating EPP amplitude at 2 mM Ca2+ suggest that the Pr might be close to maximum levels at APP−/− junctions at in vivo Ca2+ levels. The possibility of Pr compensation at physiological Ca2+ was also discussed in mice lacking NCAM or P/Q-type Ca2+ channels (Polo-Parada et al., 2001; Rafuse et al., 2000; Urbano et al., 2003). In addition to reduced paired-pulse ratio and greater depression during stimulus train, we observed a significantly increased asynchronous release in APP−/− NMJs, while the total release during repetitive stimulation was similar between the two genotypes. This is consistent with unchanged numbers of releasable vesicles and the notion that synchronous and asynchronous release share a common pool of releasable vesicles (Hagler and Goda, 2001; Otsu and Murphy, 2004).
The increased Pr, unchanged RRP and mEPP frequency obtained in our study are different from a recent report documenting that unchanged Pr but increased RRP and mEPSC frequency result from an increased number of synapses in APP−/− hippocampal autaptic cultures (Priller et al., 2006). This could be attributed to differences in the experimental systems and/or region-specific regulation of APP-mediated pathways. In particular, Priller et al. (2006) used hippocampal autaptic cultures in which one axon forms many synapses with its own somatodendrites, while the mature neuromuscular synapses we studied are singly innervated. Furthermore, we attribute much of the APP null phenotypes to deregulated Ca2+ channel subtype activity. Therefore, although APP may indeed play important roles in diverse neuronal cell types, they may be mediated though distinct pathways.
Mechanisms of Increased Asynchronous Release in APP−/− NMJ
A novel finding of the present study is the increased asynchronous release during repetitive stimulation in APP−/− NMJs. Asynchronous release is typically regarded as a result of Ca2+ accumulation, and N-type Ca2+ channels in the presynaptic terminals have been implicated in regulating the extent of asynchronous versus synchronous release (Hefft and Jonas, 2005; Pagani et al., 2006; Zakharenko et al., 1999). Of relevance to the present study, P/Q-type Ca2+ channels are the predominant VDCCs involved in mediating synaptic transmission at the mature mammalian NMJ. N- and L-type calcium channels become active only under certain non-physiological conditions such as genetic or pharmacological manipulations of VDCCs or diseases (Pagani et al., 2006; Polo-Parada et al., 2001; Urbano et al., 2001; Urbano et al., 2003). Since administration of N- and L-type Ca2+ channel blockers was more effective on inhibiting the evoked and delayed release in APP−/− NMJs as compared to the wild-type controls, our results indicate that loss of APP leads to ectopic activation of N- and L-type Ca2+ channels, which contribute, at least in part, to increased asynchronous release and greater depression. The increased 10−90% rise time in APP null NMJs (Table 1) is consistent with enhanced asynchronous release and L-type Ca2+ channel-dependent neurotransmission (Hefft and Jonas, 2005; Maximov and Sudhof, 2005; Urbano et al., 2001). Alterations of high affinity calcium sensors (Hui et al., 2005; Nishiki and Augustine, 2004; Yoshihara and Littleton, 2002) and/or mitochondrial Ca2+ uptake (David and Barrett, 2003) as possible contributors in the aberrant asynchronous release in APP null NMJ were not examined in the current study and cannot be excluded.
Western blotting and immunohistochemical staining of nerve-muscle preparations for the VDCC proteins using various antibodies met with low signals and high background, preventing quantitative measurement of the protein levels or possible alterations as a function of APP dosage. Although the exact mechanism that underlies APP-induced changes in L- and N-type Ca2+ channel activity is unclear, it is worth noting that the maturation of NMJ during postnatal development involves the gradual reduction of N-type Ca2+ channel activity and its functional replacement by P/Q type channels upon maturation (Nudler et al., 2003). It is conceivable that APP may play a role in the switching process. The present results uncover a potent activity of APP in mediating the balance between synchronous and asynchronous release, and we provide functional evidence for the regulation of Ca2+ channel subtype activities by APP in cholinergic synaptic transmission.
Synaptic dysfunction is an important contributor to AD dementia and deregulation of calcium homeostasis has been implicated in AD pathogenesis (reviewed in (LaFerla, 2002)). Although AD is unlikely a disease of APP loss-of-function, mis-regulation of APP-mediated pathways may contribute to AD pathogenesis. It is interesting that presenilins, another class of molecules intimately linked to AD, have also been documented to mediate calcium homeostasis (Leissring et al., 2000; Yoo et al., 2000), and were recently shown to display properties of a calcium leak channel (Tu et al., 2006). Although it is likely that multiple distinct pathways are involved in functional regulation of calcium by APP and presenilins, deregulation of any pathway may lead to the disturbance of calcium homeostasis, which may contribute to synaptic impairment and AD pathogenesis.
Acknowledgements
We wish to thank N. Aithmitti and X. Chen for expert technical assistance, D. J. Shim and other members of the Zheng laboratory for discussion. We are grateful to Drs. J. Dani and C. Rosenmund for guidance during the course of the study. This work was supported by grants from NIH (NS40039 and AG20670) and Alzheimer's Association (ZEN-03-4757 and IIRG-06-25779). BW is a trainee of the NIH training grant T32 AG000183.
Abbreviations
- AChR
acetylcholine receptor
- AD
Alzheimer's disease
- APP
amyloid precursor protein
- α-BTX
α-bungarotoxin
- Aβ
β-amyloid peptide
- DMSO
dimethylsulfoxide
- EPP
endplate potential
- mEPP
miniature endplate potential
- NMJ
neuromuscular junction
- Pr
probability of release
- QC
quantal content
- RRP
readily releasable pool
- EGTA-AM, EGTA
tetra(acetoxymethyl ester)
- VDCC
voltage-dependent Ca2+ channel
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akaaboune M, Allinquant B, Farza H, Roy K, Magoul R, Fiszman M, Festoff BW, Hantai D. Developmental regulation of amyloid precursor protein at the neuromuscular junction in mouse skeletal muscle. Mol Cell Neurosci. 2000;15:355–367. doi: 10.1006/mcne.2000.0834. [DOI] [PubMed] [Google Scholar]
- Atluri PP, Regehr WG. Delayed release of neurotransmitter from cerebellar granule cells. J Neurosci. 1998;18:8214–8227. doi: 10.1523/JNEUROSCI.18-20-08214.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett EF, Stevens CF. The kinetics of transmitter release at the frog neuromuscular junction. J Physiol. 1972;227:691–708. doi: 10.1113/jphysiol.1972.sp010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz WJ, Bewick GS. Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science. 1992;255:200–203. doi: 10.1126/science.1553547. [DOI] [PubMed] [Google Scholar]
- David G, Barrett EF. Mitochondrial Ca2+ uptake prevents desynchronization of quantal release and minimizes depletion during repetitive stimulation of mouse motor nerve terminals. J Physiol. 2003;548:425–438. doi: 10.1113/jphysiol.2002.035196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson GR, Seabrook GR, Zheng H, Smith DW, Graham S, O'Dowd G, Bowery BJ, Boyce S, Trumbauer ME, Chen HY, et al. Age-related cognitive deficits, impaired long-term potentiation and reduction in synaptic marker density in mice lacking the beta-amyloid precursor protein. Neuroscience. 1999;90:1–13. doi: 10.1016/s0306-4522(98)00410-2. [DOI] [PubMed] [Google Scholar]
- Edeline JM. Learning-induced physiological plasticity in the thalamo-cortical sensory systems: a critical evaluation of receptive field plasticity, map changes and their potential mechanisms. Prog Neurobiol. 1999;57:165–224. doi: 10.1016/s0301-0082(98)00042-2. [DOI] [PubMed] [Google Scholar]
- Elmqvist D, Quastel DM. A quantitative study of end-plate potentials in isolated human muscle. J Physiol. 1965;178:505–529. doi: 10.1113/jphysiol.1965.sp007639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagler DJ, Jr., Goda Y. Properties of synchronous and asynchronous release during pulse train depression in cultured hippocampal neurons. J Neurophysiol. 2001;85:2324–2334. doi: 10.1152/jn.2001.85.6.2324. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hefft S, Jonas P. Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat Neurosci. 2005;8:1319–1328. doi: 10.1038/nn1542. [DOI] [PubMed] [Google Scholar]
- Hui E, Bai J, Wang P, Sugimori M, Llinas RR, Chapman ER. Three distinct kinetic groupings of the synaptotagmin family: candidate sensors for rapid and delayed exocytosis. Proc Natl Acad Sci U S A. 2005;102:5210–5214. doi: 10.1073/pnas.0500941102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juttner R, More MI, Das D, Babich A, Meier J, Henning M, Erdmann B, Mu Ller EC, Otto A, Grantyn R, Rathjen FG. Impaired synapse function during postnatal development in the absence of CALEB, an EGF-like protein processed by neuronal activity. Neuron. 2005;46:233–245. doi: 10.1016/j.neuron.2005.02.027. [DOI] [PubMed] [Google Scholar]
- Kilgard M. Cholinergic modulation of skill learning and plasticity. Neuron. 2003;38:678–680. doi: 10.1016/s0896-6273(03)00327-1. [DOI] [PubMed] [Google Scholar]
- Kuno M, Turkanis SA, Weakly JN. Correlation between nerve terminal size and transmitter release at the neuromuscular junction of the frog. J Physiol. 1971;213:545–556. doi: 10.1113/jphysiol.1971.sp009399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat Rev Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Akbari Y, Fanger CM, Cahalan MD, Mattson MP, LaFerla FM. Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J Cell Biol. 2000;149:793–798. doi: 10.1083/jcb.149.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Trussell LO. Inhibitory transmission mediated by asynchronous transmitter release. Neuron. 2000;26:683–694. doi: 10.1016/s0896-6273(00)81204-0. [DOI] [PubMed] [Google Scholar]
- Magleby KL, Stevens CF. A quantitative description of end-plate currents. J Physiol. 1972;223:173–197. doi: 10.1113/jphysiol.1972.sp009840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Sudhof TC. Autonomous function of synaptotagmin 1 in triggering synchronous release independent of asynchronous release. Neuron. 2005;48:547–554. doi: 10.1016/j.neuron.2005.09.006. [DOI] [PubMed] [Google Scholar]
- McKinney M, Coyle JT, Hedreen JC. Topographic analysis of the innervation of the rat neocortex and hippocampus by the basal forebrain cholinergic system. J Comp Neurol. 1983;217:103–121. doi: 10.1002/cne.902170109. [DOI] [PubMed] [Google Scholar]
- McLachlan EM, Martin AR. Non-linear summation of end-plate potentials in the frog and mouse. J Physiol. 1981;311:307–324. doi: 10.1113/jphysiol.1981.sp013586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz IM, Sabatini BL, Regehr WG. Calcium control of transmitter release at a cerebellar synapse. Neuron. 1995;15:675–688. doi: 10.1016/0896-6273(95)90155-8. [DOI] [PubMed] [Google Scholar]
- Nishiki T, Augustine GJ. Synaptotagmin I synchronizes transmitter release in mouse hippocampal neurons. J Neurosci. 2004;24:6127–6132. doi: 10.1523/JNEUROSCI.1563-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nudler S, Piriz J, Urbano FJ, Rosato-Siri MD, Renteria ES, Uchitel OD. Ca2+ channels and synaptic transmission at the adult, neonatal, and P/Q-type deficient neuromuscular junction. Ann N Y Acad Sci. 2003;998:11–17. doi: 10.1196/annals.1254.003. [DOI] [PubMed] [Google Scholar]
- Otsu Y, Murphy TH. Optical postsynaptic measurement of vesicle release rates for hippocampal synapses undergoing asynchronous release during train stimulation. J Neurosci. 2004;24:9076–9086. doi: 10.1523/JNEUROSCI.2060-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagani MR, Reisin RC, Uchitel OD. Calcium signaling pathways mediating synaptic potentiation triggered by amyotrophic lateral sclerosis IgG in motor nerve terminals. J Neurosci. 2006;26:2661–2672. doi: 10.1523/JNEUROSCI.4394-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo-Parada L, Bose CM, Landmesser LT. Alterations in transmission, vesicle dynamics, and transmitter release machinery at NCAM-deficient neuromuscular junctions. Neuron. 2001;32:815–828. doi: 10.1016/s0896-6273(01)00521-9. [DOI] [PubMed] [Google Scholar]
- Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J. Synapse formation and function is modulated by the amyloid precursor protein. J Neurosci. 2006;26:7212–7221. doi: 10.1523/JNEUROSCI.1450-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffaelli G, Saviane C, Mohajerani MH, Pedarzani P, Cherubini E. BK potassium channels control transmitter release at CA3-CA3 synapses in the rat hippocampus. J Physiol. 2004;557:147–157. doi: 10.1113/jphysiol.2004.062661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafuse VF, Polo-Parada L, Landmesser LT. Structural and functional alterations of neuromuscular junctions in NCAM-deficient mice. J Neurosci. 2000;20:6529–6539. doi: 10.1523/JNEUROSCI.20-17-06529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reim K, Mansour M, Varoqueaux F, McMahon HT, Sudhof TC, Brose N, Rosenmund C. Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell. 2001;104:71–81. doi: 10.1016/s0092-8674(01)00192-1. [DOI] [PubMed] [Google Scholar]
- Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriere A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38:24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- Rozov A, Burnashev N, Sakmann B, Neher E. Transmitter release modulation by intracellular Ca2+ buffers in facilitating and depressing nerve terminals of pyramidal cells in layer 2/3 of the rat neocortex indicates a target cell-specific difference in presynaptic calcium dynamics. J Physiol. 2001;531:807–826. doi: 10.1111/j.1469-7793.2001.0807h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Development of the vertebrate neuromuscular junction. Annu Rev Neurosci. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- Schubert W, Prior R, Weidemann A, Dircksen H, Multhaup G, Masters CL, Beyreuther K. Localization of Alzheimer beta A4 amyloid precursor protein at central and peripheral synaptic sites. Brain Res. 1991;563:184–194. doi: 10.1016/0006-8993(91)91532-6. [DOI] [PubMed] [Google Scholar]
- Seabrook GR, Smith DW, Bowery BJ, Easter A, Reynolds T, Fitzjohn SM, Morton RA, Zheng H, Dawson GR, Sirinathsinghji DJ, et al. Mechanisms contributing to the deficits in hippocampal synaptic plasticity in mice lacking amyloid precursor protein. Neuropharmacology. 1999;38:349–359. doi: 10.1016/s0028-3908(98)00204-4. [DOI] [PubMed] [Google Scholar]
- Shigematsu K, McGeer PL, McGeer EG. Localization of amyloid precursor protein in selective postsynaptic densities of rat cortical neurons. Brain Res. 1992;592:353–357. doi: 10.1016/0006-8993(92)91697-d. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Momiyama A. Different types of calcium channels mediate central synaptic transmission. Nature. 1993;366:156–158. doi: 10.1038/366156a0. [DOI] [PubMed] [Google Scholar]
- Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell. 2006;126:981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbano FJ, Depetris RS, Uchitel OD. Coupling of L-type calcium channels to neurotransmitter release at mouse motor nerve terminals. Pflugers Arch. 2001;441:824–831. doi: 10.1007/s004240000489. [DOI] [PubMed] [Google Scholar]
- Urbano FJ, Piedras-Renteria ES, Jun K, Shin HS, Uchitel OD, Tsien RW. Altered properties of quantal neurotransmitter release at endplates of mice lacking P/Q-type Ca2+ channels. Proc Natl Acad Sci U S A. 2003;100:3491–3496. doi: 10.1073/pnas.0437991100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Yang G, Mosier DR, Chang P, Zaidi T, Gong YD, Zhao NM, Dominguez B, Lee KF, Gan WB, Zheng H. Defective neuromuscular synapses in mice lacking amyloid precursor protein (APP) and APP-Like protein 2. J Neurosci. 2005;25:1219–1225. doi: 10.1523/JNEUROSCI.4660-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Westenbroek RE, Borst JG, Catterall WA, Sakmann B. Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx-type synapses. J Neurosci. 1999;19:726–736. doi: 10.1523/JNEUROSCI.19-02-00726.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo AS, Cheng I, Chung S, Grenfell TZ, Lee H, Pack-Chung E, Handler M, Shen J, Xia W, Tesco G, et al. Presenilin-mediated modulation of capacitative calcium entry. Neuron. 2000;27:561–572. doi: 10.1016/s0896-6273(00)00066-0. [DOI] [PubMed] [Google Scholar]
- Yoshihara M, Littleton JT. Synaptotagmin I functions as a calcium sensor to synchronize neurotransmitter release. Neuron. 2002;36:897–908. doi: 10.1016/s0896-6273(02)01065-6. [DOI] [PubMed] [Google Scholar]
- Zakharenko S, Chang S, O'Donoghue M, Popov SV. Neurotransmitter secretion along growing nerve processes: comparison with synaptic vesicle exocytosis. J Cell Biol. 1999;144:507–518. doi: 10.1083/jcb.144.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, Smith DW, Heavens RP, Dawson GR, Boyce S, Conner MW, et al. beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell. 1995;81:525–531. doi: 10.1016/0092-8674(95)90073-x. [DOI] [PubMed] [Google Scholar]
- Zheng H, Koo EH. The amyloid precursor protein: Beyond amyloid. Molecular Neurodegeneration. 2006;1:6. doi: 10.1186/1750-1326-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]