

Abstract
Fluid accumulation into the subretinal space and the development of macular edema is a common condition in age-related macular degeneration, diabetic retinopathy, and following ocular surgery, or injury. Vascular endothelial growth factor (VEGF) and other cytokines have been implicated in the disruption of retinal pigment epithelium (RPE) barrier function and a reduction in the regulated removal of subretinal fluid; however, the cellular and molecular events linking these agents to the disruption of barrier function have not been established. In the current study, cultures of ARPE-19 and primary porcine retinal pigment epithelium (RPE) cells were utilized to investigate the effects of the VEGF-induced modifications to the barrier properties of the RPE. The barrier function was determined by transepithelial resistance (TER) measurements and morphology of the RPE monolayers. In both ARPE-19 and primary porcine RPE cells the administration of VEGF produced a significant drop in TER, and this response was only observed following apical administration. Maximum reduction in TER was reached 5 hours post VEGF administration. These responses were concentration-dependent with an EC50 of 502 pg/mL in ARPE-19 cells and 251 pg/mL in primary porcine cells. In both ARPE-19 and primary RPE cells, the response to VEGF was blocked by pretreatment with the relatively selective VEGF-R2 antagonists, SU5416 or ZM323881, or the protein tyrosine kinase inhibitor, genistein. Administration of the relatively selective VEGF-R2 agonist, VEGF-E, also reduced TER in a concentration-dependent manner (EC50 of 474 pg/mL), while VEGF-R1 agonist, placental growth factor (PlGF), did not significantly alter the TER. Immunolocalization studies demonstrated that confluent monolayers exhibited continuous cell-to-cell ZO-1 protein contacts and apical localization of the VEGF-R2 receptors. These data provide evidence that the VEGF-induced breakdown of RPE barrier function is mediated by the activation of apically-oriented VEGF-R2 receptors. Thus, VEGF-mediated increases in RPE permeability are initiated by a rise in intraocular levels of VEGF.
Keywords: RPE, resistance, VEGF, tight junction, macular edema, polarity, receptor
Introduction
Macular edema is a major cause of decreased visual acuity. It consists of accumulated fluid in the subretinal space within the macula, and it is often characterized by radially-oriented cysts (Tranos et al. 2004). Retinal edema can originate from a large variety of ocular insults, such as eye injuries, intraocular surgery, venous occlusive disease, diabetic retinopathy, posterior segment inflammatory disease, or age-related macular degeneration (AMD) (Tranos et al. 2004). A variety of treatment approaches have been attempted with varying success: topical and systemic steroids, non-steroidal anti-inflammatory agents, and laser photocoagulation treatment (Tranos et al. 2004). In recent clinical trials, anti-vascular endothelial growth factor (anti-VEGF) medications (e.g., pegaptanib sodium, bevacizumab, ranibizumab) have been shown to reduce the macular thickness in patients with macular edema and wet AMD, and an increase in visual acuity was reported to follow (Gragoudas et al. 2004; Heier et al. 2006; Spaide et al. 2006). These studies have provided evidence that the removal of VEGF from the intraocular environment can reduce macular edema, and supplement previous observations that intraocular VEGF plays a central role in the development of macular edema (Antcliff and Marshall 1999; Barber and Antonetti 2003). However, the cellular mechanisms by which VEGF induces the disruption of RPE barrier function, impairing the efficient removal of subretinal fluid are not completely understood.
Vascular endothelial growth factor was originally recognized as an endothelial angiogenic and vasopermeability factor (Neufeld et al. 1999), and more recently has also been shown to induce functional changes in the retinal pigment epithelium (RPE) (Hartnett et al. 2003; Zech et al. 1998). Cellular events regulated by VEGF are mediated by two receptor tyrosine kinases, VEGF-R1 and VEGF-R2 (Zachary 2001). The most common human isoform of VEGF is VEGF-A165; however, other VEGF proteins have been identified VEGF-B, VEGF-C, VEGF-D from human, VEGF-E from the Orf virus, and VEGF-F from snake venom. In contrast to VEGF-A, the other VEGF proteins show selectivity for one or the other VEGF-receptor (Yamazaki and Morita 2006).
Functionally intact tight-junctions are required for the efficient and regulated removal of fluid from the subretinal space and the barrier function of healthy RPE (Marmor 1999). It is thought that VEGF signaling modulates the paracellular permeability by altering the protein component of the junctional complexes (Schneeberger and Lynch 2004). Therefore, in order to begin to understand how VEGF contributes to the regulation of RPE barrier function, it is essential to understand how individual VEGF receptors effect RPE physiology. In vascular endothelial cells, there is growing evidence that increases in permeability and proliferation are mediated by VEGF-R2 activation, and within the choroid, that these actions can be opposed by VEGF-R1 stimulation (Autiero et al. 2003; Gille et al. 2001; Nozaki et al. 2006; Shen et al. 1999). However, a recent study of the RPE concluded that VEGF-R1 receptor activation is responsible for the VEGF-induced increase in epithelial permeability (Miyamoto et al. 2007). Our studies were designed to clarify how VEGF modulates RPE barrier properties. Results from these studies demonstrate that activation of VEGF-R2 receptors on the apical surface of the RPE are responsible for the VEGF-induced increase in epithelial permeability, and this response is blocked by the protein tyrosine kinase inhibitor, genistein. These results cast a new light on the regulation of RPE permeability and the way it can be treated pharmacologically.
Materials and Methods
Tissue culture
The ARPE-19/HPV-16 cell line was obtained from The American Type Culture Collections (passage 30 at the time of purchase; Manassas, VA). The cells were cultured in 75 cm2 tissue flasks according to the vendor’s instructions. Cells from passages 31-50 were used in experimentation. To establish primary porcine RPE cultures, porcine eyes were obtained from a local abattoir (Burbages, Ravenel, SC). The eyes were bisected; the anterior segment and the retina were removed. The eyecups were then rinsed with calcium- and magnesium-free balanced salt solution and incubated with 0.05% trypsin-EDTA (GIBCO BRL, Grand Island, NY) for 1 h at 37°C. The incubation buffer with the released cells was removed and ten-times the volume of DMEM/F12/HAM (Sigma Chemical Co., St. Louis, MO) with 10% fetal bovine serum (FBS; GIBCO), 1.2 g/L sodium bicarbonate (Fisher Scientific, Fair Lawn, NJ), and 10 mL/L L-glutamine-penicillin G-streptomycin (2 mM-100 U/mL-0.1 mg/mL; Sigma) was added. The primary cells were not passaged. Both ARPE-19 and primary RPE cells were plated on permeable-membrane inserts (Costar Clear Transwell, 0.4 μm pore; Costar, Cambridge, MA) containing DMEM/F12/HAM with 10% FBS, 1.2 g/L sodium bicarbonate, and 10 mL/L L-glutamine-penicillin-streptomycin in a humidified incubator and kept at 37°C in 5% CO2. After the cells attached, the medium was modified to contain only 1% FBS and changed every 2 to 3 days. Transepithelial resistance of cell monolayers was monitored with an epithelial volt-ohmmeter (WPI, Sarasota, FL) equipped with an STX2 electrode (WPI). Resistance measurements for individual wells were determined from four independent measurements, and corrected for the inherent transwell resistance. For each condition, at least three independent experiments were performed. Values are expressed as means ± SE and compared by the Student t test. A P value of less than 0.05 was considered statistically significant.
Cell treatments
Vascular endothelial growth factor (human VEGF-A165, Sigma), PlGF (PlGF-1; Fitzgerald Industries International, Inc., Concord, MA), or VEGF-E (Fitzgerald) was administered to the apical or basal sides in concentrations ranging from 15 pg/mL to 500 ng/mL (diluted with phosphate-buffered saline [PBS]). Baseline transepithelial resistance was measured at -60 min and immediately prior to agonist administration (t = 0). Following agonist administration, resistance was measured at 2 hours, unless otherwise noted. To evaluate the actions of VEGF-R2 antagonists, SU5416 (Calbiochem; San Diego, CA), ZM323881 (Tocris Bioscience, Ellisville, MO) or the tyrosine kinase inhibitor, genistein (Sigma), cells were treated with the agents (SU5416, 5 μM; ZM323881, 10 nM; and genistein, 100 μM) or vehicle control (DMSO; Sigma) one hour prior to agonist administration. The utilized DMSO concentrations in the vehicle are not expected to change the TER (Konari et al. 1995). Dose-response curves were analyzed with Prism 4.02 (GraphPad Software Inc., San Diego, CA).
Immunohistochemistry
Cell monolayers were washed 3-times in PBS, fixed in 2% paraformaldehyde (Mallinckrodt Baker, Inc.) in PBS for 15 min, and again washed 3-times in PBS at room temperature (RT). Cells were then permeabilized by exposure to 0.2% Triton X-100 (VWR, West Chester, PA) in PBS for 15 min, washed in PBS (3 times), and incubated with primary antibodies (mouse anti-ZO-1, diluted 1:100; Zymed Laboratory, Inc., San Francisco, CA; and rabbit anti-VEGF-R2, diluted 1:100; Chemicon, Temecula, CA) for 1 hour at 4°C. Cells were then washed 3-times in PBS and incubated with secondary antibodies (fluorescein-conjugated goat anti-mouse, diluted 1:50; and rhodamine-conjugated goat anti-rabbit, diluted 1:50; Chemicon, Temecula, CA) for 30 min at room temperature. The inserts with attached monolayers were mounted on slides and examined using a Leica DM LFSA confocal microscope (Leica Microsystems, Wetzlar, Germany). The confocal microscope allowed obtaining a stack of 100 images on the Z-axis to visualize the whole volume of the stains. Image transformations to show cross sections of the volumes, remove autofluorescence, and calculate color distribution profiles were performed by the Leica microscope software (LCS 2.6, Leica). The intensity profile of optical confocal sectioning of three fluorochromes along the z-axis was determined for 10 selected regions of interest. Averages for the profile maxima from these regions were used to obtain the localization data for the individual stains.
Measurement of barrier function
Paracellular permeability of ARPE-19 cell monolayers was determined by measuring the apical to basolateral movement of inulin over a time-course of 100 minutes. Lyophilized carboxyl-14C-inulin (50 μCi, 1.6 mCi/g, Moravek Biochemicals, Brea, CA) was suspended in 2 mL of PBS at a temperature of 60°C. 20 μL aliquots, containing 0.5 μCi (31.25 μg) were diluted with 100-times as much cold inulin (MP Biochemicals, Solon, OH) in 340 μL tissue culture media (DMEM/F12/HAM with 1% FBS) and added to the apical chambers of transwell filters five hours after treatment with either apical VEGF (10 ng/mL) or vehicle. The five-hour delay between the VEGF and inulin administrations ensured that the inulin flux was measured in steady state, as TER changed little between 5 and 24 hours. Aliquots of 100 μL were removed from the basolateral chamber at the time of inulin administration, and every 20 minutes afterwards up to 100 min and replaced by equal amounts of inulin-free media. The samples were placed into individual wells of 24-well microplates, mixed with 900 μL Microscint-20 scintillation fluid (Packard, Meriden, CT), and counted in a scintillation chamber (TopCount, Packard). Values are expressed as the mean of six independent wells ± SE and compared by the Student t test. Inulin flux was determined from linear regression analysis of the quantity of inulin present in the basolateral chamber versus elapsed time using Prism 4.02.
Results
Initial experiments were carried out using the human ARPE-19 cell line. Upon confluence, TER began to increase, reaching a maximum TER of 45 ± 4 Ωcm2 within 10 to 14 days (Fig. 1A). These TER values are similar to those previously reported for this cell line in confluent culture (Dunn et al. 1996; Geisen et al. 2006). There was no significant difference between the TER levels developed in different passages of cultured cells (31-50) and the resistance levels were not influenced by the presence or absence of serum in the culture media. Confluent cultures of ARPE-19 cells were routinely maintained for three weeks; however, selected RPE cultures were evaluated for up to 8 weeks without significant reduction in their TER levels.
Figure 1.

Resistance and morphology of RPE cell monolayers. A, Increase in transepithelial resistance of ARPE-19 cell monolayers following cell plating on transwell inserts. The dashed line represents the average resistance (45 ± 4 Ωcm2) for confluent monolayers between weeks 2 and 5. Cultures were tested for up to 8 weeks; however, routine experiments were performed following three weeks in culture. B, Increase in transepithelial resistance of porcine primary RPE cell monolayers following cell plating on transwell inserts. The dashed line represents the average resistance (76 ± 9 Ωcm2) achieved for confluent monolayers between weeks 5 and 11. Values are means ±SE. Immunohistochemistry of confluent ARPE-19 cells (C) and porcine primary RPE cells (D) with mouse anti-ZO-1 as primary, and FITC-conjugated anti-mouse as secondary antibodies. The magnification was ×200.
Primary cultures of porcine RPE cells were also established. Because passaged primary cells changed their phenotypic appearances (loss of pigmentation, loss of uniform morphology), only unpassaged primary porcine RPE cells were used. Due to a slower growth rate, these cells took 5 to 6 weeks to reach confluence and a maximum TER value of 76 ± 9 Ωcm2 (Fig. 1B). These cultures maintained their TER values for up to three months with no visible change of cell pigmentation or morphology.
To determine if confluent RPE monolayers exhibited morphological evidence of tight junctions necessary for RPE barrier function, the immuno-localization of ZO-1 was assessed by fluorescent microscopy. Although both ARPE-19 and primary porcine cell monolayers exhibited continuous cell-to-cell ZO-1 protein contacts, the primary porcine cells were more uniform in size and shape when compared to the ARPE-19 cell line (Fig. 1C and D). In addition, there were sporadic binuclear ARPE-19 cells (data not shown).
VEGF-induced changes in transepithelial resistance
To investigate the cellular site and mechanism that mediates VEGF-induced changes in RPE permeability, TER across monolayers of ARPE-19 and primary porcine cells was monitored. Previous studies have shown that TER is proportional to permeability in both ARPE-19 cells and primary RPE cell cultures (Dunn et al. 1996; Jin et al. 2002). Administration of 10 ng/mL VEGF to the apical surface of the RPE monolayers, produced a significant drop in TER across both ARPE-19 and primary porcine cells. However, administration of VEGF to the basal surface of either cell type did not significantly alter TER (Fig. 2A and B). These apical responses were time-dependent with maximum reduction in resistance of 46 ± 2% in ARPE-19 cells and 31.4 ± 4% in primary porcine monolayers measured at 5 hours. In cultures where the incubation with VEGF was continued for 24 hours, no recovery of TER was observed (Fig. 3A and B).
Figure 2.

Polarized response of RPE cell monolayer to VEGF-A165. In both ARPE-19 cells (A) and porcine primary RPE cells (B), the treatment with VEGF-A165 (10 ng/mL) to the apical side significantly decreased TER, while the treatment of the basal side did not significantly alter resistance measurements. Values are means ±SE of individual measurements normalized to the average TER at t = 0. (* P <0.05, ** P <0.01).
Figure 3.

Time course of TER changes in response to VEGF-A165. The apical side of confluent monolayers were treated with 10 ng/mL of VEGF-A165 for 30 minutes to 24 hours. Addition of 10 ng/mL VEGF-A165 ARPE-19 cells reduced TER to approximately 50% of its original value within 5 hours (A). The addition of VEGF-A-165 to primary porcine RPE cells reduced TER by 70% within 5 hours (B). Values are means ±SE of individual measurements normalized to the average TER values at t = 0. (* P <0.05, ** P <0.01).
The apical VEGF-induced reduction in TER was paralleled by a decrease in the barrier function. Barrier function was assessed by measuring the apical to basolateral movement of carboxyl-14C-inulin in 10 ng/mL apical VEGF-treated versus vehicle-treated ARPE-19 cell monolayers. The inulin flux was 780 μg/hour after VEGF-treatment, while in controls without VEGF it was only 580 μg/hour, which meant a 36% reduction in barrier function 5 hours after VEGF administration.
As shown in Figure 4, the apical VEGF-induced reduction in TER was concentration-dependent. In ARPE-19 (Fig. 4A) and primary porcine cells (Fig. 4B), the EC50 values were 502 and 251 pg/mL, respectively. The Hill coefficients for these curves were not significantly different from 1.0.
Figure 4.
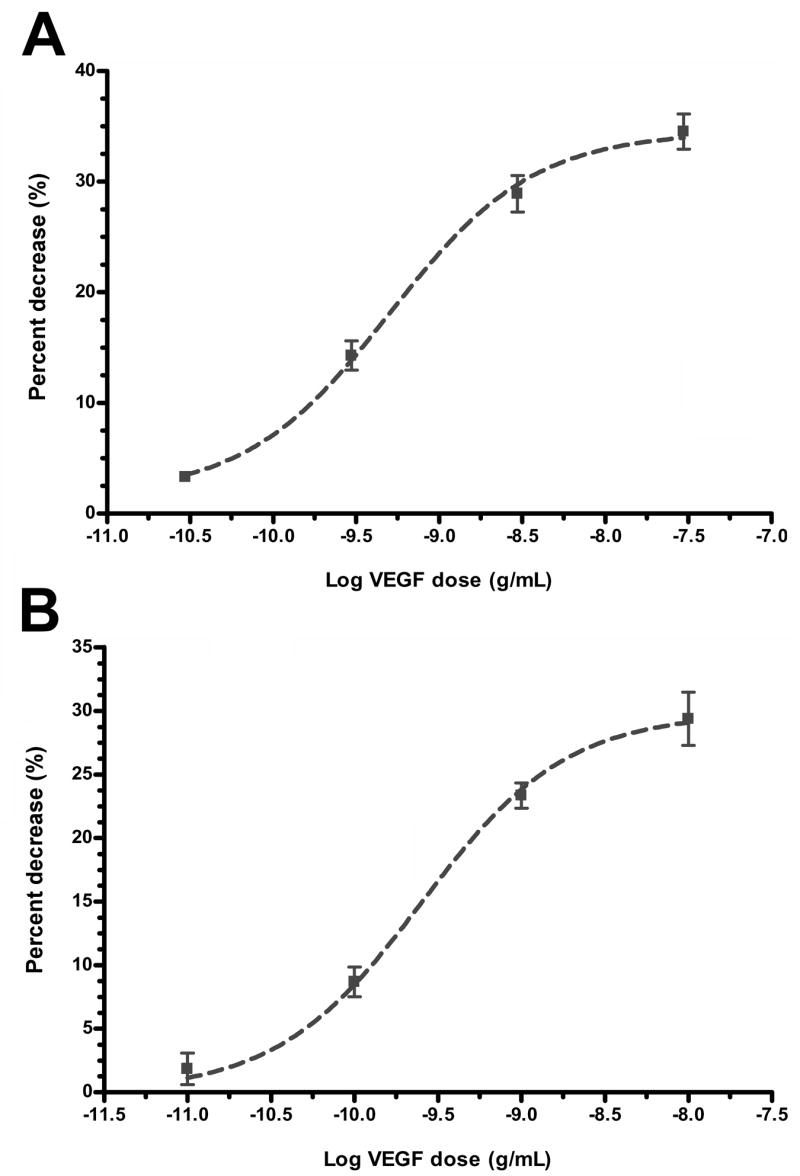
Concentration-dependent reduction in TER induced by VEGF-A165. In both ARPE-19 cells (A) and porcine primary RPE cells (B), the percent decrease in resistance was concentration dependent with an EC50 of 502 pg/mL (LogEC50 = -9.30 ±0.09) for ARPE-19 cells and 251 pg/mL (LogEC50 = -9.60 ±0.05) for porcine primary RPE cells. The Hill coefficients were not significantly different from 1.0. Values are means ±SE of individual measurements normalized to the average TER values at t = 0.
VEGF-receptor subtype
The Hill coefficients calculated from the concentration response curves provide evidence that VEGF-induced changes in TER across the RPE monolayers are mediated by a single class of non-interacting VEGF receptors. To begin to investigate the receptor subtype activated by VEGF on the apical surface of the RPE cells, TER responses to the VEGF-R1 receptor agonist, PlGF, and the VEGF-R2 receptor agonist, VEGF-E, were determined in both ARPE-19 cells (Fig. 5A) and in porcine primary RPE cells (Fig. 5B). Apical administration of 5 ng/mL PlGF did not significantly change TER in either culture. However, 5 ng/mL apical VEGF-E significantly reduced TER by 36 ± 2% in ARPE-19 cells and 37 ± 2% in porcine primary RPE cells. Administration of either agonist to the basal surface did not significantly alter TER.
Figure 5.

Responses of RPE monolayers following VEGF-E and PlGF administration. In both ARPE-19 cells (A) and porcine primary RPE cells (B), apically administered VEGF-E (5 ng/mL) significantly reduced TER; however, apically-administration of PlGF (5 ng/mL) did not significantly alter resistance. The administration of either agonist to the basolateral side did not significantly alter TER. C, Concentration-response curves of the ARPE-19 cells to apical VEGF-E and PlGF and basal VEGF-E. The apical administration of VEGF-E induced a concentration dependent decrease in TER. The EC50 to this response was 474 pg/mL (LogEC50 = -9.32 ±0.04). The Hill coefficient was not significantly different from 1.0. Apically-applied PlGF or basolaterally-applied VEGF-E were not effective in significantly altering TER. Values are means ±SE normalized to the average resistance at t = 0. (** P <0.01)
As shown in Figure 5C, the reduction in TER following administration of VEGF-E to the apical surface of ARPE-19 cells was concentration-dependent with an EC50 value of 474 pg/mL. However, increasing VEGF-E concentrations from 5 – 500 ng/mL on the basal surface did not significantly alter TER. Similarly, increasing the apical concentration of PIGF from 5 – 500 ng/mL did not produce any significant change in TER.
To confirm that the VEGF-induced changes in TER measured in this study were mediated by the VEGF-R2 receptor and begin to investigate the down-stream signaling mechanism, monolayers were pretreated with two VEGF-R2 antagonists, SU5416 or ZM323881, or the pan-tyrosine kinase inhibitor, genistein. SU5416 is a relatively selective VEGF-R2 inhibitor, but it was also shown to inhibit VEGF-R1 with an IC50 of 90 nM (Glass et al. 2006). Therefore, we used ZM323881, which is three orders of magnitude more selective for VEGF-R2 and is not known to inhibit VEGF-R1. One hour pretreatment with 5 μM SU5416 or 10 nM ZM323881 blocked the VEGF-E-induced decrease in resistance in both ARPE-19 (Fig. 6A and C) and porcine primary RPE cells (Fig. 6B and D). The addition of SU5416, ZM323881, or vehicle alone, did not significantly alter TER. Pretreatment of ARPE-19 cells with genistein (100 μM) or vehicle for 1 hour also abolished the resistance drop induced by VEGF (Fig. 7). In contrast, apical VEGF alone resulted in a 30 ± 2% drop in the resistance within 2 hours of treatment.
Figure 6.

Inhibition of VEGF-E165 response by VEGF-R2 antagonists. Pretreatment of ARPE-19 (A) or primary porcine RPE (B) cells with with 5 μM SU5416 blocked the response to VEGF-E. In monolayers pretreated with vehicle, VEGF-E induced a significant drop in TER within 120 minutes after treatment. Pretreatment of ARPE-19 cells (C) or primary porcine RPE cells (D) with the more selective VEGF-R2 antagonist, ZM323881, also blocked the response to VEGF-E. Again, vehicle pretreatment did not significantly alter VEGF-E-induced reduction in TER. Values are means ±SE of individual measurements normalized to the average TER values at t = 0. (* P <0.05, ** P <0.01).
Figure 7.

Inhibition of VEGF-A165 response by Genistein. Pretreatment of ARPE-19 cells with genistein (100 μM) for 1 hour blocked the reduction in TER induced by VEGF-A165. The addition of Genistein alone or vehicle did not significantly alter TER. Values are means ±SE normalized to the average resistance at t = 0. (** P <0.01)
VEGF-R2 is localized to the apical side of RPE cells
Only apical administration of VEGF agonists significantly altered the TER across RPE monolayers. These data support the idea that the VEGF-R2 receptor expression is limited to the apical membrane in these cells. Figure 8A shows a flatmount confocal image of immunohistochemically stained porcine primary RPE cell monolayer for ZO-1 (green) and VEGF-R2 (red) proteins. Cell nuclei were stained with DRAQ5 (blue). In these cells, ZO-1 protein again was shown to form continuous cell-to-cell contact, while VEGF-R2 protein was exhibited as a punctate staining pattern across the monolayer. To investigate the cellular localization of these proteins at various depths, 100 confocal images were obtained and the signal profiles were determined (Fig. 8B). This profile does not distinguish between membrane, cytosol or nuclear location, but rather the relative apical to basal distribution along the z-axis for ZO-1 and VEGF-R2 antibodies, and the nucleus. As expected from RPE cell anatomy, the ZO-1 antibody stained close to the apical surface of the cells. The VEGF-R2 antibody stained even closer to the apical surface, while the nuclear stain was observed within the central regions along the Z-axis. The average (n = 10) peak intensities for VEGF-R2 and ZO-1 in porcine RPE cells were 2.84 ± 0.12 and 2.97 ± 0.10 μm, respectively, from the top of the image stack.
Figure 8.
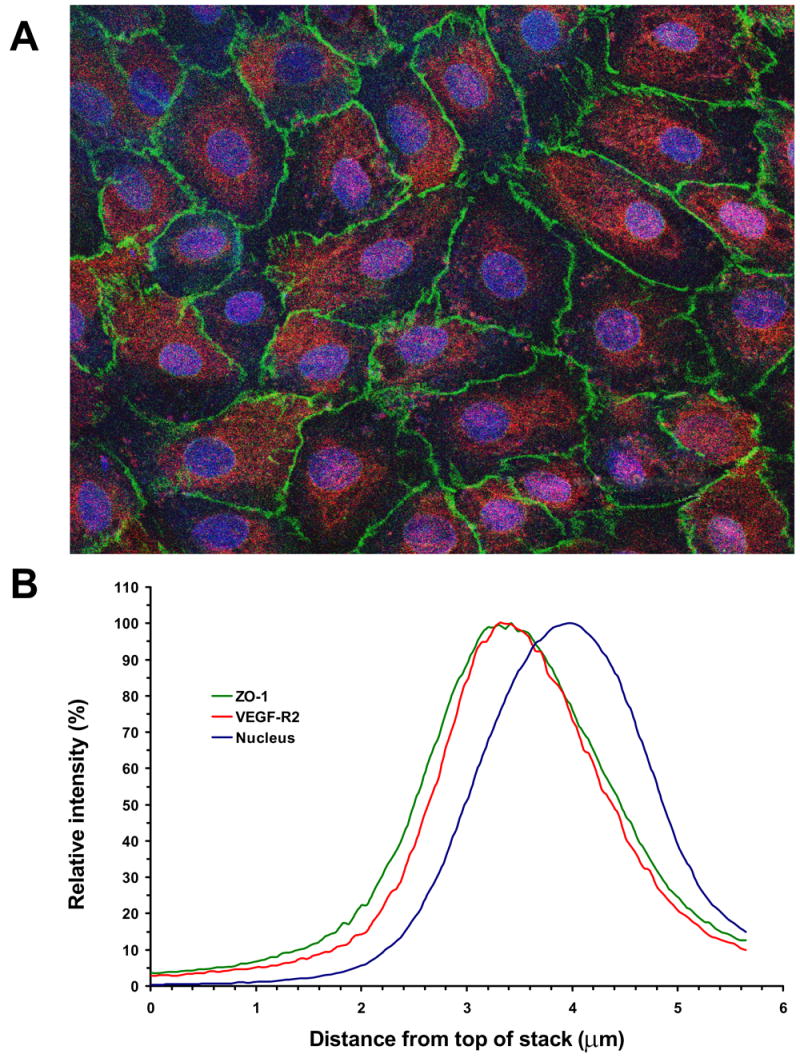
Apical localization of VEGF-R2 in confluent RPE monolayers. A, Immunohistochemical staining of the porcine primary RPE cell monolayer with mouse anti-ZO-1 and rabbit anti-VEGF-R2 as primary antibodies, and FITC-conjugated goat anti-mouse (green) and rhodamine-conjugated goat anti-rabbit (red) as secondary antibodies, respectively. The cell nuclei were visualized with DRAQ5 (blue). The image was obtained as a Z-stack on the confocal microscope. The VEGF-R2 staining was only observed in the apical regions and was absent at depths below the nuclei. The magnification was ×400. B, Color profile in the Z-stack images. The figure shows the profile for a representative region, containing a single RPE cell. Optical sectioning revealed that single color profiles represent the localization of the individual stains in the depth of the image. Analysis of color profiles revealed the close co-localization of VEGF-R2 receptor and ZO-1 protein in the apical region of the RPE cell.
Discussion
When an eye is injured or inflamed, the paracellular permeability of the RPE cells can increase resulting in the breakdown of the outer blood retinal barrier, and the regulated removal of subretinal fluid is impaired. Previous in vitro studies have provided evidence that cytokines, such as HGF or VEGF may mediate this response (Caldwell et al. 2005; Jin et al. 2002; Pe’er et al. 1995). Moreover, recent clinical studies showed that anti-VEGF medications can reduce macular fluid accumulation (Gragoudas et al. 2004; Heier et al. 2006; Spaide et al. 2006).
Primary porcine RPE cells and the ARPE-19 cell line were utilized to study the mechanisms responsible for the VEGF-mediated changes in RPE permeability. The ARPE-19 cell line expresses several RPE-specific markers (CRALBP and RPE65) and forms polarized monolayers; however, the TER generated by these cells is normal at 40-70% less than the primary RPE cells in culture (Dunn et al. 1996; Geisen et al. 2006; Hartnett et al. 2003; Jin et al. 2002). In the current study, both primary cells and the ARPE-19 cell line produced monolayers that formed continuous tight-junctioned complexes between cells and generated TER that was consistent with previous studies (Dunn et al. 1996; Geisen et al. 2006; Miyamoto et al. 2007). Although the TER values obtained in ARPE-19 cells were lower than those in primary porcine cells, the administration of VEGF to either cell type resulted in a robust (30-50%) drop in the TER within 5 hours of treatment. These responses are consistent with previous studies on HGF or VEGF-induced reduction in TER in primary cultures of bovine RPE cells (Hartnett et al. 2003; Jin et al. 2002). The results, however, contradict those of Ghassemifar and colleagues (Ghassemifar et al. 2006), where they found that VEGF slightly (~10%) increases the TER of ARPE-19 cells and tightens the RPE junctions. The route of VEGF administration is not reported in that paper, and it is possible that they may have administered VEGF basolaterally. In our hands, the basolateral route of administration did not significantly change TER. However, slight increases within the experimental error were seen with administration of vehicle, PlGF, and basolateral VEGF or VEGF-E.
VEGF-induced reductions in TER were only measurable following apical administration. This apical selectivity for VEGF-induced reductions in TER is additionally supported by morphological data demonstrating that the expression of VEGF-R2 receptors is limited to the apical surface region of cultured RPE monolayers, similar to that observed with the ZO-1 protein. Although apical VEGF secretion from the RPE has been identified, under most conditions basolateral VEGF secretion from the RPE cells predominates (Blaauwgeers et al. 1999; Maminishkis et al. 2006). The basolateral secretion of VEGF is approximately 10-times greater than the apical secretion, and has been shown to stimulate VEGF-R2 in the adjacent choriocapillaris endothelium in a paracrine manner (Blaauwgeers et al. 1999; Marneros et al. 2005). Hence, it is unlikely that VEGF derived from the RPE initiates the breakdown of the outer blood retinal barrier in vivo. However under conditions where the RPE permeability is compromised, RPE-derived VEGF may accelerate or prolong increased permeability changes in the RPE. In light of these results, increase in RPE permeability appears to be initiated by VEGF secreted from other cells in the neural retina (Antcliff and Marshall 1999) or a shift toward apical VEGF secretion from the RPE (Kannan et al. 2006; Slomiany and Rosenzweig 2004). Additional studies are needed to determine the factors which regulate the direction and the degree of VEGF secretion from the RPE in vivo. Finally, these studies provide evidence why intraocular administration of anti-VEGF medication is effective in reducing macular edema.
Molecular and pharmacological studies have demonstrated the existence of two VEGF receptor subtypes: VEGF-R1 and VEGF-R2 (Zachary 2001). In the vascular endothelium, VEGF increases paracellular permeability by the activation of VEGF-R2 receptors (Autiero et al. 2003; Gille et al. 2001; Shen et al. 1999). However, a recent study by Miyamoto and colleagues concluded that the VEGF-R1 receptor subtype mediated VEGF-induced changes in the barrier properties of ARPE-19 cell monolayers (Miyamoto et al. 2007). Like Miyamoto and colleagues, we identified VEGF-A165 as an effective cytokine in regulating RPE barrier function. However, we found that the VEGF-R2 agonist, VEGF-E, but not the VEGF-R1 agonist increased RPE permeability. In our hands, the VEGF-R1 agonist did not induce a permeability change when used in the same or in higher concentrations than they utilized. In addition, the VEGF-R2 agonist-induced increases in RPE permeability were abolished by pretreatment with the relatively selective SU5416, and the three orders of magnitude more selective ZM323881, both VEGF-R2 antagonists. There are many differences between the two papers that may explain the contradictory results. The utilized cell line and culture conditions were different, as well as the source of the agonist ligands. Also the route of VEGF administration is not clear in their paper (Miyamoto et al. 2007). These seemingly minor changes may make a considerable difference, because the activity of VEGF receptors can be complicated by a series of additional factors including agonist-induced crosstalk between VEGF-R1 and VEGF-R2, hetero-dimerization between individual receptor subunits, and hetero-dimerization of the agonists themselves (Autiero et al. 2003); moreover, additional receptors may enhance VEGF receptor signaling (Neuropilin-1) (Becker et al. 2005). Our data provide convincing evidence that the RPE permeability change measured in this study resulted from activation of the VEGF-R2 receptor subtype. This conclusion is reinforced by studies showing that VEGF-R1 signaling reduced laser-induced CNV through an inhibitory action on VEGF-R2 phosphorylation (Nozaki et al. 2006); moreover, experiments in ARPE-19 cells indicated that VEGF-R1 activation induced the expression of pigment epithelium-derived factor (Ohno-Matsui et al. 2003), a potent anti-angiogenic agent that blocks VEGF-R2 signaling in endothelial cells (Cai et al. 2006). Thus, these papers also contradict the hypothesis of Miyamoto and coworkers that VEGF-R1 activity promotes RPE permeability.
Genistein is a known protein tyrosine kinase inhibitor that has been found to disrupt multiple signaling pathways such as NF-κB, AKT and MAP kinases (Davis 1995; Li and Sarkar 2002). Pretreatment with genistein blocked VEGF-induced increases in RPE permeability. Although these results can not identify a specific signaling pathway linking VEGF-R2 receptors to changes in permeability, they do support the idea that the use of cell-signaling pathway inhibitors will be effective in RPE permeability and macular edema.
In summary, in both human ARPE-19 cells and primary porcine RPE cells we have found that only apical administration of VEGF produced a rapid, significant and concentration dependent drop in TER. Studies of antagonists are relatively selective and agonists provided clear evidence that VEGF-R2 receptors were responsible for the mediation of the VEGF effect, and that these receptors were localized to the apical surface. These results support the idea that RPE permeability increases are initiated by a rise in intraocular levels of VEGF, and favoring the intraocular route for anti-VEGF treatment of macular edema.
Acknowledgments
I want to thank Mr. Phil Yates and Ms. Kioina Meyers for technical assistance; and Dr. David E. Potter for helpful discussions. Supported by NIH/NEI EY014793, NIH/NEI EY009741, MUSC/URC P-28918, and an unrestricted grant to MUSC from Research to Prevent Blindness, New York, NY. The Leica DM LFSA confocal microscope was obtained through a supplement to NIH/NEI EY-13520.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Antcliff RJ, Marshall J. The pathogenesis of edema in diabetic maculopathy. Seminars in ophthalmology. 1999;14(4):223–232. doi: 10.3109/08820539909069541. [DOI] [PubMed] [Google Scholar]
- Autiero M, Waltenberger J, Communi D, Kranz A, Moons L, Lambrechts D, Kroll J, Plaisance S, De Mol M, Bono F, Kliche S, Fellbrich G, Ballmer-Hofer K, Maglione D, Mayr-Beyrle U, Dewerchin M, Dombrowski S, Stanimirovic D, Van Hummelen P, Dehio C, Hicklin DJ, Persico G, Herbert JM, Shibuya M, Collen D, Conway EM, Carmeliet P. Role of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat Med. 2003;9(7):936–943. doi: 10.1038/nm884. [DOI] [PubMed] [Google Scholar]
- Barber AJ, Antonetti DA. Mapping the blood vessels with paracellular permeability in the retinas of diabetic rats. Invest Ophthalmol Vis Sci. 2003;44(12):5410–5416. doi: 10.1167/iovs.03-0244. [DOI] [PubMed] [Google Scholar]
- Becker PM, Waltenberger J, Yachechko R, Mirzapoiazova T, Sham JS, Lee CG, Elias JA, Verin AD. Neuropilin-1 regulates vascular endothelial growth factor-mediated endothelial permeability. Circulation research. 2005;96(12):1257–1265. doi: 10.1161/01.RES.0000171756.13554.49. [DOI] [PubMed] [Google Scholar]
- Blaauwgeers HG, Holtkamp GM, Rutten H, Witmer AN, Koolwijk P, Partanen TA, Alitalo K, Kroon ME, Kijlstra A, van Hinsbergh VW, Schlingemann RO. Polarized vascular endothelial growth factor secretion by human retinal pigment epithelium and localization of vascular endothelial growth factor receptors on the inner choriocapillaris. Evidence for a trophic paracrine relation. Am J Pathol. 1999;155(2):421–428. doi: 10.1016/S0002-9440(10)65138-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Jiang WG, Grant MB, Boulton M. Pigment epithelium-derived factor inhibits angiogenesis via regulated intracellular proteolysis of vascular endothelial growth factor receptor 1. J Biol Chem. 2006;281(6):3604–3613. doi: 10.1074/jbc.M507401200. [DOI] [PubMed] [Google Scholar]
- Caldwell RB, Bartoli M, Behzadian MA, El-Remessy AE, Al-Shabrawey M, Platt DH, Liou GI, Caldwell RW. Vascular endothelial growth factor and diabetic retinopathy: role of oxidative stress. Curr Drug Targets. 2005;6(4):511–524. doi: 10.2174/1389450054021981. [DOI] [PubMed] [Google Scholar]
- Davis RJ. Transcriptional regulation by MAP kinases. Mol Reprod Dev. 1995;42(4):459–467. doi: 10.1002/mrd.1080420414. [DOI] [PubMed] [Google Scholar]
- Dunn KC, Aotaki-Keen AE, Putkey FR, Hjelmeland LM. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp Eye Res. 1996;62(2):155–169. doi: 10.1006/exer.1996.0020. [DOI] [PubMed] [Google Scholar]
- Geisen P, McColm JR, King BM, Hartnett ME. Characterization of barrier properties and inducible VEGF expression of several types of retinal pigment epithelium in medium-term culture. Current eye research. 2006;31(9):739–748. doi: 10.1080/02713680600837408. [DOI] [PubMed] [Google Scholar]
- Ghassemifar R, Lai CM, Rakoczy PE. VEGF differentially regulates transcription and translation of ZO-1alpha+ and ZO-1alpha- and mediates trans-epithelial resistance in cultured endothelial and epithelial cells. Cell and tissue research. 2006;323(1):117–125. doi: 10.1007/s00441-005-0046-7. [DOI] [PubMed] [Google Scholar]
- Gille H, Kowalski J, Li B, LeCouter J, Moffat B, Zioncheck TF, Pelletier N, Ferrara N. Analysis of biological effects and signaling properties of Flt-1 (VEGFR-1) and KDR (VEGFR-2). A reassessment using novel receptor-specific vascular endothelial growth factor mutants. J Biol Chem. 2001;276(5):3222–3230. doi: 10.1074/jbc.M002016200. [DOI] [PubMed] [Google Scholar]
- Glass CA, Harper SJ, Bates DO. The anti-angiogenic VEGF isoform VEGF165b transiently increases hydraulic conductivity, probably through VEGF receptor 1 in vivo. The Journal of physiology. 2006;572(Pt 1):243–257. doi: 10.1113/jphysiol.2005.103127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gragoudas ES, Adamis AP, Cunningham ET, Jr, Feinsod M, Guyer DR. Pegaptanib for neovascular age-related macular degeneration. N Engl J Med. 2004;351(27):2805–2816. doi: 10.1056/NEJMoa042760. [DOI] [PubMed] [Google Scholar]
- Hartnett ME, Lappas A, Darland D, McColm JR, Lovejoy S, D’Amore PA. Retinal pigment epithelium and endothelial cell interaction causes retinal pigment epithelial barrier dysfunction via a soluble VEGF-dependent mechanism. Exp Eye Res. 2003;77(5):593–599. doi: 10.1016/s0014-4835(03)00189-1. [DOI] [PubMed] [Google Scholar]
- Heier JS, Antoszyk AN, Pavan PR, Leff SR, Rosenfeld PJ, Ciulla TA, Dreyer RF, Gentile RC, Sy JP, Hantsbarger G, Shams N. Ranibizumab for treatment of neovascular age-related macular degeneration: a phase I/II multicenter, controlled, multidose study. Ophthalmology. 2006;113(4):642, e641–644. doi: 10.1016/j.ophtha.2005.10.052. [DOI] [PubMed] [Google Scholar]
- Jin M, Barron E, He S, Ryan SJ, Hinton DR. Regulation of RPE intercellular junction integrity and function by hepatocyte growth factor. Invest Ophthalmol Vis Sci. 2002;43(8):2782–2790. [PubMed] [Google Scholar]
- Kannan R, Zhang N, Sreekumar PG, Spee CK, Rodriguez A, Barron E, Hinton DR. Stimulation of apical and basolateral VEGF-A and VEGF-C secretion by oxidative stress in polarized retinal pigment epithelial cells. Molecular vision. 2006;12:1649–1659. [PubMed] [Google Scholar]
- Konari K, Sawada N, Zhong Y, Isomura H, Nakagawa T, Mori M. Development of the blood-retinal barrier in vitro: formation of tight junctions as revealed by occludin and ZO-1 correlates with the barrier function of chick retinal pigment epithelial cells. Exp Eye Res. 1995;61(1):99–108. doi: 10.1016/s0014-4835(95)80063-8. [DOI] [PubMed] [Google Scholar]
- Li Y, Sarkar FH. Inhibition of nuclear factor kappaB activation in PC3 cells by genistein is mediated via Akt signaling pathway. Clin Cancer Res. 2002;8(7):2369–2377. [PubMed] [Google Scholar]
- Maminishkis A, Chen S, Jalickee S, Banzon T, Shi G, Wang FE, Ehalt T, Hammer JA, Miller SS. Confluent monolayers of cultured human fetal retinal pigment epithelium exhibit morphology and physiology of native tissue. Invest Ophthalmol Vis Sci. 2006;47(8):3612–3624. doi: 10.1167/iovs.05-1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmor MF. Mechanisms of fluid accumulation in retinal edema. Doc Ophthalmol. 1999;97(34):239–249. doi: 10.1023/a:1002192829817. [DOI] [PubMed] [Google Scholar]
- Marneros AG, Fan J, Yokoyama Y, Gerber HP, Ferrara N, Crouch RK, Olsen BR. Vascular endothelial growth factor expression in the retinal pigment epithelium is essential for choriocapillaris development and visual function. Am J Pathol. 2005;167(5):1451–1459. doi: 10.1016/S0002-9440(10)61231-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto N, de Kozak Y, Jeanny JC, Glotin A, Mascarelli F, Massin P, Benezra D, Behar-Cohen F. Placental growth factor-1 and epithelial haemato-retinal barrier breakdown: potential implication in the pathogenesis of diabetic retinopathy. Diabetologia. 2007;50(2):461–470. doi: 10.1007/s00125-006-0539-2. [DOI] [PubMed] [Google Scholar]
- Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. Faseb J. 1999;13(1):9–22. [PubMed] [Google Scholar]
- Nozaki M, Sakurai E, Raisler BJ, Baffi JZ, Witta J, Ogura Y, Brekken RA, Sage EH, Ambati BK, Ambati J. Loss of SPARC-mediated VEGFR-1 suppression after injury reveals a novel antiangiogenic activity of VEGF-A. J Clin Invest. 2006;116(2):422–429. doi: 10.1172/JCI26316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno-Matsui K, Yoshida T, Uetama T, Mochizuki M, Morita I. Vascular endothelial growth factor upregulates pigment epithelium-derived factor expression via VEGFR-1 in human retinal pigment epithelial cells. Biochem Biophys Res Commun. 2003;303(3):962–967. doi: 10.1016/s0006-291x(03)00446-7. [DOI] [PubMed] [Google Scholar]
- Pe’er J, Shweiki D, Itin A, Hemo I, Gnessin H, Keshet E. Hypoxia-induced expression of vascular endothelial growth factor by retinal cells is a common factor in neovascularizing ocular diseases. Lab Invest. 1995;72(6):638–645. [PubMed] [Google Scholar]
- Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol. 2004;286(6):C1213–1228. doi: 10.1152/ajpcell.00558.2003. [DOI] [PubMed] [Google Scholar]
- Shen BQ, Lee DY, Zioncheck TF. Vascular endothelial growth factor governs endothelial nitric-oxide synthase expression via a KDR/Flk-1 receptor and a protein kinase C signaling pathway. J Biol Chem. 1999;274(46):33057–33063. doi: 10.1074/jbc.274.46.33057. [DOI] [PubMed] [Google Scholar]
- Slomiany MG, Rosenzweig SA. IGF-1-induced VEGF and IGFBP-3 secretion correlates with increased HIF-1 alpha expression and activity in retinal pigment epithelial cell line D407. Invest Ophthalmol Vis Sci. 2004;45(8):2838–2847. doi: 10.1167/iovs.03-0565. [DOI] [PubMed] [Google Scholar]
- Spaide RF, Laud K, Fine HF, Klancnik JM, Jr, Meyerle CB, Yannuzzi LA, Sorenson J, Slakter J, Fisher YL, Cooney MJ. Intravitreal bevacizumab treatment of choroidal neovascularization secondary to age-related macular degeneration. Retina. 2006;26(4):383–390. doi: 10.1097/01.iae.0000238561.99283.0e. [DOI] [PubMed] [Google Scholar]
- Tranos PG, Wickremasinghe SS, Stangos NT, Topouzis F, Tsinopoulos I, Pavesio CE. Macular edema. Surv Ophthalmol. 2004;49(5):470–490. doi: 10.1016/j.survophthal.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Yamazaki Y, Morita T. Molecular and functional diversity of vascular endothelial growth factors. Molecular diversity. 2006;10(4):515–527. doi: 10.1007/s11030-006-9027-3. [DOI] [PubMed] [Google Scholar]
- Zachary I. Signaling mechanisms mediating vascular protective actions of vascular endothelial growth factor. Am J Physiol Cell Physiol. 2001;280(6):C1375–1386. doi: 10.1152/ajpcell.2001.280.6.C1375. [DOI] [PubMed] [Google Scholar]
- Zech JC, Pouvreau I, Cotinet A, Goureau O, Le Varlet B, de Kozak Y. Effect of cytokines and nitric oxide on tight junctions in cultured rat retinal pigment epithelium. Invest Ophthalmol Vis Sci. 1998;39(9):1600–1608. [PubMed] [Google Scholar]