

Abstract
Phenoloxidase-catalyzed reactions are crucial to the survival of insects after a pathogen or parasite infection. In Manduca sexta, active phenoloxidase is generated from its precursor by a prophenoloxidase activating proteinase (PAP) in the presence of noncatalytic serine proteinase homologs (SPHs). The PAP and SPHs, located at the ends of a branched proteinase cascade, also require limited proteolysis to become functional. While the processing enzyme of M. sexta proPAP-2 and proPAP-3 is known, we are now investigating the proteolytic activation of proSPH-1 and proSPH-2. Here we report the development of a series of Bac-to-Bac plasmid vectors for co-expression, secretion, and affinity purification of proSPH-1 and proSPH-2 from insect cells infected by one baculovirus. The purified proteins were characterized and used as substrates in a search for their activating enzymes in plasma of the larvae injected with microorganisms. Proteolytic processing occurred after the proSPHs had been incubated with hydroxyapatite or gel filtration column fractions. The cleaved proteins were active as a cofactor for prophenoloxidase activation by PAP, and coexistence of SPH-1 and SPH-2 is essential for manifesting the auxiliary effect.
Keywords: clip domain, insect immunity, phenoloxidase, melanization, hemolymph protein
1. Introduction
With over 430 members listed in MEROPS (http://merops.sanger.ac.uk), chymotrypsin-like serine proteinases constitute the largest family of all peptidases. They participate in vital physiological processes including digestion, development and defense responses (Hedstrom, 2002). At the amino terminus of many arthropod serine proteinases, there is one or two clip domains connected to the carboxyl-terminal catalytic domain through a linker region and a disulfide bond (Jiang and Kanost, 2000). While their proteinase domains all contain a His-Asp-Ser catalytic triad, nonsynonymous substitutions lead to the loss of these key residues and enzymatic activity. Indeed, insect genomes encode serine proteinase homologs (SPHs) that are anticipated to resemble their ancestral enzymes in overall folding (Ross et al., 2003; Zou et al., 2006 and 2007) and possess functions other than peptide bond hydrolysis. For instance, clip-domain SPHs are essential for generating active phenoloxidase (PO) in some insects (Kwon et al., 2000; Yu et al., 2003).
Insect POs catalyze the formation of quinones and other reactive intermediates for melanin synthesis, microbe killing, cuticle sclerotization, and wound healing (Cerenius and Söderhäll, 2004; Nappi and Christensen, 2005; Zhao et al., 2007). They are produced as inactive zymogens and proteolytically activated by proPO activating proteinase (PAP, also known as PPAE or PPAF). PAPs have been isolated from several insects and characterized biochemically (Satoh et al., 1999; Jiang et al., 1998, 2003a and 2003b; Lee et al., 1998). In Manduca sexta, these enzymes cleave proPO at the correct bond but yield little PO activity. Only when a high Mr complex of SPH-1 and SPH-2 is present at the same time, does PAP generate active PO (Wang and Jiang, 2004; Gupta et al., 2005). In Holotrichia diomphalia, proPO are activated by PPAFI (a serine proteinase) and PPAFII (an SPH) (Lee et al., 1998). In Drosophila melanogaster, MP1 cuts proMP2 and MP2 activates proPO (Tang et al., 2006). In Anopheles gambiae, Clip-B3, -B4, -B8, -B14, and -B17 play a role in the proPO activation pathway (Volz et al., 2005; Paskewitz et al., 2006). Clip-A8 (an SPH) is required for parasite melanization whereas Clip-A2, -A5 and -A7 (SPHs) function synergistically to block this process (Volz et al., 2006).
While components of the proPO activation system (e.g. pathogen recognition proteins, serine proteinases, SPHs, serpins, and proPOs) have been elucidated in several insects (Kanost et al., 2004; Christophides et al., 2004; Iwanaga and Lee, 2005), limited information is available about the proteolytic activation of proSPHs: H. diomphalia PPAFIII is the only serine proteinase shown to cleave proPPAFII (Kim et al., 2002). In order to explore the proSPH activation branch of M sexta proPO activation system, we produced proSPH-1 and proSPH-2 in a baculovirus-insect cell expression system. The purified proteins, after being cleaved by column fractions of the induced larval plasma, manifested an auxiliary effect in the proPO activation reaction. Coexistence of SPH-1 and SPH-2 is essential for the cofactor activity.
2. Methods and materials
2.1. Modification of pFastBacDual
The plasmid pFastBacDual (Invitrogen Life Technology), which allows co-expression of two genes, was modified for efficient secretion and affinity purification of the recombinant proteins (Fig. 1). As described previously for pMHF (i.e. pMFH6) (Lu and Jiang, 2007), pMFBDpH was constructed by inserting a synthetic DNA fragment (#1) into the BamHI-EcoRI sites of pFastBacDual. After in-frame insertion of a foreign DNA to the EcoRI site of pMFBDpH, the encoded polypeptide is expected to be synthesized under the control of polyhedrin promoter and secreted into the medium using the honeybee melittin signal peptide.
Fig. 1. Development of a series of Bac-to-Bac plasmid vectors for protein expression in baculovirus-infected insect cells.

(A) Cloning scheme (upper left) and pFastBacDual features (lower left). DNA and protein sequences of fragments 1, 2, 3, H1 and H2, as well as the flanking restriction sites are listed on the right. These five fragments were inserted into the same or compatible sites of the recipient plasmids (upper left) as described in Methods and materials. (B, H, I) Sequences of the multiple cloning regions of pFastBacDual (Invitrogen Life Technology), pHF (i.e. pFH6) (Ji et al., 2003) and pMHF (i.e. pMFH6) (Lu and Jiang, 2007); (C–G) Plasmid vectors constructed in this work for co-expressing two proteins under the control of late promoters (pH and p10) in one baculovirus. For proteins or domains that lack a signal peptide, efficient secretion is expected from the honeybee melittin signal peptide (underlined) fused with the protein(s) of interest. The signal peptidase cuts the fusion protein as the position marked “||”. In-frame insertion of the coding sequence(s) allows a hexahistidine tag attached to the carboxyl terminus of expressed protein(s) for affinity purification. The primer binding regions, restriction enzyme recognition sites, polyadenylation signals and stop codons (*) are indicated.
The plasmids pMMFBD and pMFBDp10 were constructed as follows: 1) amplification of the signal peptide-coding region in pMHF (i.e. pMFH6) using vector-specific primers j023 (5′-TTCCGGATTATTCATACC, + strand) and j044 (5′-CCATGGATCGATCCCGGGCATAGAT GTAAGAAATG, - strand). Primer j044 includes NcoI, ClaI and SmaI restriction sites fused with the reverse complement sequence encoding Ile-Ser-Tyr-Ile-Tyr-Ala, the end of the signal peptide; 2) T/A cloning and sequence verification of the PCR product; 3) Insertion of the BamHI-NcoI fragment (#2) into pMFBDpH and pFastBacDual digested with BbsI and NcoI – in this case BbsI cleavage left an overhang compatible with the BamHI site. The resulting plasmids (pMMFBD and pMFBDp10) (Fig. 1) allows co-expression of two polypeptides under the control of polyhedrin and p10 promoters. In-frame insertion of two different coding regions to the EcoRI (pH side) and ClaI (p10 side) sites of pMMFBD allows secretion of the corresponding proteins into the culture medium.
Following sequence verification, pMMFBD and pMFBDp10 were improved by incorporating a synthetic DNA fragment (H2) to the KpnI site: primers j045 (5′-CGGGCCCATCACCATCACC ATCACTAAGTAC, + strand) and j046 (3′-CATGGCCCGGGTAGTGGTAGTGGTAGTGATT -5′, − strand) were phosphorylated, annealed, and non-directionally inserted to KpnI-digested and dephosphorylated vectors. After sequence analysis, plasmids containing a single copy of the fragment in the correct orientation (designated pMMHFBDp10 and pMHFBDp10, one of the two KpnI sites destroyed and one ApaI site added in the p10 side) (Fig. 1) were kept. In-frame insertion at the ApaI site is required for fusing the hexahistidine tag to the carboxyl-terminus of an expressed protein.
The multiple cloning sites downstream of the polyhedrin promoter in pMHFBDp10 and pFastBacDual were modified by inserting the BamHI-HindIII fragment (#3) of pMHF (i.e. pMFH6) to the same sites to generate pMHMHFBD and pMHFBDpH (Fig. 1). In-frame insertion of one coding region to the EcoRI and XhoI sites and another to the ClaI and ApaI sites of pMHMHFBD allows co-expression, secretion, and affinity purification of two proteins.
2.2. Construction of recombinant baculoviruses for proSPH-1 and proSPH-2 expression
M. sexta SPH-1 cDNA was amplified using j410 (5′-CTGAATTCAGTCCGAAGATCT, + strand) and j411 (5′-GTCCTCGAGTTCGTAAACCGT, − strand). The PCR product was cloned into pGem-T (Promega) and verified by DNA sequencing. From the resulting plasmid, a 1.2 kb EcoRI-XhoI fragment was retrieved by partial digestion (because of an internal XhoI site in the cDNA) and inserted into the same sites in pMHMHFBD to generate SPH-1/pMHMHFBD. M. sexta SPH-2 cDNA was amplified using j412 (5′-GCTCATCGATCCACTATCGAC, + strand) and j413 (5′-CTGGGCCCCGTAAGTGGAGCT, − strand). Following T/A cloning and sequenceverification, the ClaI-ApaI fragment was subcloned into the same sites in pMHMHFBD and SPH-1/pMHMHFBD to yield SPH-2/pMHMHFBD and SPH-1&2/pMHMHFBD, respectively. In this cloning step, the parental plasmids were isolated from E. coli GM2163 (F−, dam−, dcm−, hsdR−, chloramphenicolr) so that the ClaI site was not methylated.
In vivo transposition of the expression cassette, selection of bacterial colonies carrying the recombinant bacmids, and isolation of bacmid DNA were performed according to the manufacturer’s protocols (Invitrogen Life Technologies). The initial viral stocks (V0) for proSPH-1 and proSPH-2 production were separately obtained by transfecting Spodoptera frugiperda Sf21 cells with a bacmid DNA-CellFECTIN mixture, and their titers were improved through serial infections. The V6 viral stocks, containing the highest levels of the baculoviruses, were stored at −70°C for further experiments. Expression conditions were optimized as described previously (Ji et al., 2003).
2.3. Analysis of proSPH-1 and proSPH-2 expressed individually and together
Sf21 cells in 5.0 ml insect serum-free medium (Invitrogen Life Technologies) (7.6×105 cells/ml, T-25 flask) were infected with 0.5 ml V3 viral stock at 27°C for 72 h. The recombinant proteins were captured from 0.5 ml of V4 culture medium using 50 μl of Ni-NTA beads (Qiagen). After washing with 150 μl of buffer A (50 mM sodium phosphate, pH 8.0) containing 0.5 M NaCl for three times, bound proteins were eluted with 50 μl of 0.3 M imidazole in the washing buffer three times and combined for electrophoretic analysis on native or denaturing gels. The proteins were visualized by Coomassie blue staining or immunoblotting using SPH-1 or SPH-2 antibodies.
2.4. Large-scale expression and purification of recombinant proSPH-1 and proSPH-2
Sf21 cells (at 2.4×106 cells/ml) in 1.4 L of insect serum-free medium (Invitrogen Life Technologies) were separately infected with the baculovirus stocks at a multiplicity of infection of 10 and grown at 27°C for 96 h with gentle agitation (100 rpm). After the cells were removed by centrifugation at 5,000g for 10 min, the pH of the conditioned medium was adjusted to 8.0 using 1.0 M Tris base. The cell debris and fine particles were spun down by centrifugation at 10,000g, and the supernatant was diluted with 20 mM Tris-HCl, pH 8.0 (buffer A) to a final volume of 4.2 L. The solution was applied to a Q-Sepharose FF column (20 ml bed volume) at a flow rate of 5.0 ml/min and, following a washing step with 100 ml buffer A, bound proteins were eluted from the column with a linear gradient of 0–1.0 M NaCl in 240 ml of buffer A at a flow rate of 1.0 ml/min. The fractions containing proSPH-1 or proSPH-2 (as seen by immunoblotting) were combined and loaded onto a 10 ml Ni2+-NTA agarose column. After washing with 50 ml of 50 mM sodium phosphate, pH 8.0 (buffer B), the bound proteins were eluted with a gradient of 0–0.3 M imidazole in 90 ml of buffer B. Fractions containing proSPH-1 or proSPH-2 were combined, dialyzed against 20 mM Tris-HCl (pH 7.6) and stored at −80°C.
2.5. Insect rearing, bacterial challenge, and hemolymph collection
M. sexta eggs were purchased from Carolina Biological Supply and the larvae were reared on an artificial diet (Dunn and Drake, 1983). Day 2, 5th instar larvae were injected with a mixture of formaldehyde-killed E. coli (3×107 cells), M. luteus (30 μg) and curdlan (30 μg) in 50 μl of H2O. Hemolymph was directly collected into saturated (NH4)2SO4 from cut prolegs of the larvae 24 h after the immune challenge. The final saturation of (NH4)2SO4 was adjusted to 50% in order toprevent spontaneous melanization, and the protein suspension was stored in −80°C.
2.6. Fractionation of M. sexta induced hemolymph on hydroxyapatite and gel filtration columns
The thawed hemolymph suspension (40 ml) was centrifuged at 22,000g for 20 min, and the pellet was dissolved with 60 ml of buffer C (5 mM sodium phosphate, pH 6.5, 0.5 M NaCl) supplemented with 0.002% 1-phenyl-2-thiourea. After centrifugation, the 20–50% (NH4)2SO4 fraction was collected as described previously (Jiang et al., 2003a). This fraction was dissolved in 60 ml of buffer C and dialyzed against the same buffer (1.2 L) overnight. Following centrifugation at 22,000g for 20 min, the protein sample was loaded onto a hydroxyapatite column (2.5 cm i.d. × 8 cm) equilibrated with buffer C. After washing with 150 ml of the same buffer, bound proteins were eluted with a linear gradient of 0–125 mM sodium phosphate, pH 6.5 in 200 ml, 0.5 M NaCl. Fractions (4.0 ml/tube) were collected for the proSPH processing assays. Active fractions were pooled and precipitated with 50% saturated (NH4)2SO4. After centrifugation, the pellet was dissolved in 3.0 ml of buffer D (50 mM Tris-HCl, 0.5 M NaCl, pH 7.5) and applied to Sephacryl S-100 column (2.5 cm i.d. × 100 cm) equilibrated with the same buffer. The fractions were collected at 6 ml/tube for the activity assays.
2.7. Processing of proSPH-1 and proSPH-2 by the column fractions
Column fractions (3 μl) were individually incubated on ice with the purified proSPH-1 (0.140 mg/ml, 2 μl), proSPH-2 (0.120 mg/ml, 2 μl), or both (2 μl + 2 μl) in a total volume of 40 μl, 20 mM Tris-HCl, pH 7.6. After 60 min incubation, the reaction mixtures were treated with 10 μl, 5×SDS sample buffer at 95°C for 5 min, separated by 10% SDS polyacrylamide gel electrophoresis, transferred to a nitrocellulose membrane and reacted with 1:5000 diluted SPH-1 or SPH-2 antiserum. The antibody-antigen complex was recognized by goat-anti-rabbit IgG conjugated to horseradish peroxidase (Bio-Rad), and chemiluminescence emitted from the hydrolysis of SuperSignal West Pico Chemiluminescent substrate (Pierce) was detected by X-ray film exposure and development.
2.8. ProPO activation and PO activity assay
PO activity was determined on a microplate reader according to Jiang et al (2003a). Briefly, M. sexta proPO (1 μl, 40 μg/ml) and a column fraction (3 μl) were incubated on ice with proSPH-1 (2 μl), proSPH-2 (2 μl), or both (2 μl + 2 μl) in a total volume of 40 μl Tris-HCl, pH 7.6. Dopamine (150 μl, 2.0 mM) was added to the reaction mixtures 60 min later and PO activity was measured immediately.
3. Results and discussion
3.1. Construction of a series of expression vectors
In order to facilitate synthesis, secretion and purification of recombinant proteins in the baculovirus-insect cell expression system, we have developed a series of Bac-to-Bac plasmids based on pFastBacDual. With expanded multiple cloning regions (Fig. 1), one or more of these vectors can be selected with ease to meet special requirements of the protein production. For instance, co-expression of two proteins using a single recombinant virus is expected to enhance cell infection and reduce inclusion body formation caused by the lack of its partner. Introduction of the honeybee melittin signal peptide may increase the expression and secretion of proteins or their constituents which do not contain a secretion peptide. As demonstrated previously (Jarvis et al., 1993), this leader peptide can also be used for producing proteins with an inefficient signal peptide. The classical secretion pathway allows the correctly folded and properly modified proteins to enter an oxidized environment, away from the majority of cellular proteins. Moreover, the fusion of carboxyl-terminal hexahistidine tag facilitates the capture of recombinant proteins from a large volume of conditioned medium by affinity chromatography.
3.2. Purification and characterization of proSPH-1 and proSPH-2 from the insect cells
We have constructed three plasmids (SPH-1/pMHMHFBD, SPH-2/pMHMHFBD and SPH-1&2/pMHMHFBD) and generated respective baculoviruses. Sf21 infected with these viruses produced proSPH-1, proSPH-2 and both proSPHs. The secreted proteins were separately isolated from the culture media by nickel affinity chromatography (Fig. 2). The purified proteins migrated to about the 60 and 50 kDa positions on the SDS-polyacrylamide gel. Immunoblot analysis showed SPH-2 antibodies also weakly reacted with proSPH-1. On the native gel, proSPH-2 ran as two diffused bands slower than the proSPH-1 band did. The co-expressed proteins had the same migration patterns as the individual ones, suggesting there was no strong association between proSPH-1 and proSPH-2 under the experimental conditions. Since proSPH-1 level was unstable during large-scale co-expression tests (data not shown) and there was no apparent association between the SPH precursors, we chose to produce the two proteins separately and carry out the activation assay using one or both precursors.
Fig. 2. Expression analysis of proSPH-1 and proSPH-2 in baculovirus-infected insect cells.
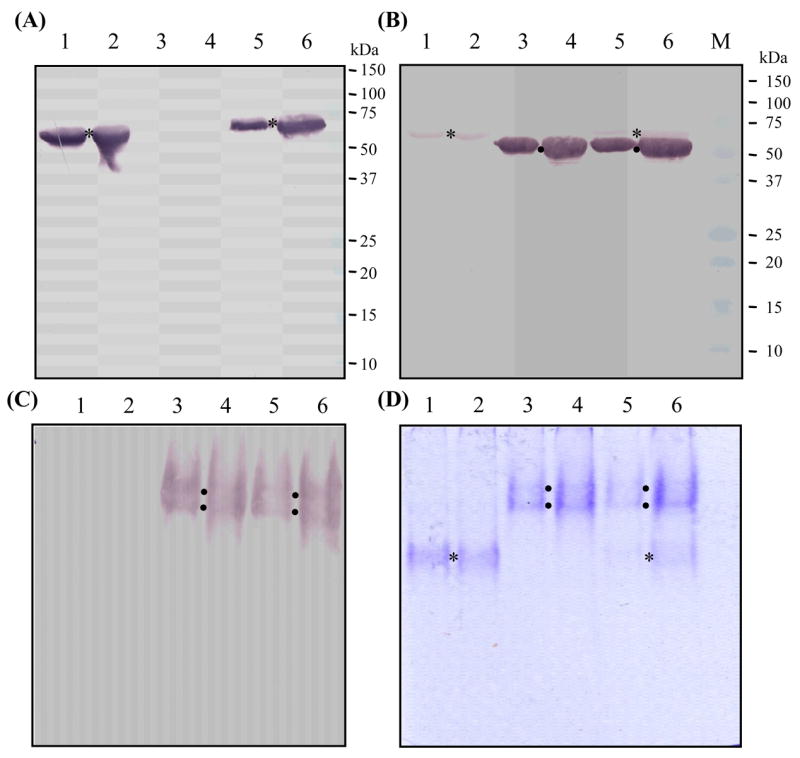
Affinity-purified proSPH-1, proSPH-2 and co-expressed proSPHs were separated by 10% SDS-PAGE followed by immunoblotting using SPH-1 (A) or SPH-2 (B) antibodies. The same protein samples were resolved on 7.5% native polyacrylamide gels and detected using SPH-2 antibodies (C) or Coomassie blue staining (D). Lanes 1 and 2, proSPH-1 only; lanes 3 and 4, proSPH-2 only; lanes 5 and 6, both proSPHs. SPH-2 antibodies recognized proSPH-2 and also weakly reacted with denatured proSPH-1, whereas SPH-1 antibodies did not recognize native proSPH-1 (data not shown). The positions of proSPH-2 (●) were revealed by its antibodies, whereas those of proSPH-1 (*) on the native gels were deduced from results shown in the other panels. The positions and sizes of the protein markers are indicated on the right of the denaturing gels.
3.3. Large-scale expression of proSPH-1 and proSPH-2 in insect cells
As shown in Fig. 3 and Fig. 4, the recombinant proteins were soluble and secreted into the cell culture medium. After removing the Sf21 cells, we captured the proSPHs by ion exchange chromatography and eluted them from the Q-Sepharose column in a small volume. The proSPH fractions were pooled and loaded onto Ni-NTA agarose columns. A linear gradient of imidazole was applied to elute the proteins. Silver staining and immunoblot analysis following SDS-polyacrylamide gel electrophoresis indicated that the affinity purified proteins were essentially pure and intact. ProSPH-1 ran as a 60 kDa band whereas proSPH-2 had an apparent Mr of 52 kDa. Heterogeneity in glycosylation may be responsible for the band broadening. While a streak was observed on the native gel behind the diffused bands of proSPHs, a majority of the proteins seem to exist in a low association state and migrated deep into the gel. Consistent with that, most of the purified proSPH-1 or proSPH-2 mobilized on the HPLC size exclusion column as a single peak. While proSPH-1 eluted at 9.40 min, corresponding to a calculated Mr of 78 kDa, proSPH-2 eluted at 9.73 min, corresponding to 62 kDa. These results indicated that the proSPHs may exist as monomers.
Fig. 3. Isolation of M. sexta proSPH-1 from the baculovirus-infected insect cells.

(A) 10% SDS-PAGE and silver staining. (B) Immunoblot analysis using SPH-1 first antibody and goat-anti-rabbit IgG conjugated to alkaline phosphatase. (C) 7.5% native PAGE and silver staining. (D) Analysis of purified proSPH-1 on an HPLC size exclusion column. 1, conditioned cell culture medium; 2, proteins eluted from the Q-Sepharose column; 3, affinity-purified protein from the Ni-NTA agarose column; M, molecular weight markers. The positions and sizes of the marker proteins are indicated on the right.
Fig. 4. Isolation of M. sexta proSPH-2 from the baculovirus-infected insect cells.

(A) 10% SDS-PAGE and silver staining. (B) Immunoblot analysis using SPH-2 first antibody and goat-anti-rabbit IgG conjugated to alkaline phosphatase. (C) 7.5% native PAGE and silver staining. (D) Analysis of purified proSPH-2 on an HPLC size exclusion column. 1, conditioned cell culture medium; 2, proteins eluted from the Q-Sepharose column; 3, affinity-purified protein from the Ni-NTA agarose column; M, molecular weight markers. The positions and sizes of the marker proteins are indicated on the right.
3.4. Detection of a proSPH-1 and proSPH-2 processing activity in the induced plasma
Using the purified proSPHs as substrates, we attempted to isolate their activating enzyme from the induced M. sexta hemolymph. After (NH4)2SO4 fractionation, hydroxyapatite and gel filtration chromatography, we found that certain fractions slightly cleaved proSPH-1 and proSPH-2 (Fig. 5, A and B). The processing of proSPH-1 was incomplete, and we only detected its 38 kDa proteinase-like domain using SPH-1 antibodies. These column fractions contained PAP-1, hemolymph proteinase 1 (HP1) and small amounts of other HPs (data not shown). Unfortunately, we have not yet been able to sort out which one of these enzymes is responsible for proSPH-1 cleavage. The same column fractions also cleaved over 50% of proSPH-2, yielding a 37 kDa heavy chain and a 16 kDa light chain. While the latter was identical in migration rate to the amino-terminal fragment of SPH-1 from M. sexta hemolymph (Wang and Jiang, 2004), the 37 kDa band is slightly larger than the carboxyl-terminal fragment of natural SPH-1 (36 kDa). This small difference may be caused by the carboxyl-terminal histidine tag in the recombinant protein.
Fig. 5. Processing of the proSPHs by column fractions of the M. sexta hemolymph from larvae injected with bacteria and its relationship with proPO activation. (A, B).

Processing of proSPH-1 and proSPH-2 by the hydroxyapatite (HT#18) and Sephacryl S-100 (GF#52) column fractions. The reaction mixtures were separated by 10% SDS-PAGE and detected using SPH-1 (A) or SPH-2 (B) antibodies. (C) ProPO and HT#18/GF#52 were incubated with proSPH-1, proSPH-2, or both for 60 min. The PO activities in the reactions and controls were determined. (D) To test if processed SPH-1 and SPH-2 are both required for enhancing in proPO activation, we incubated proPO and GF#52 with proSPH-2 (4) or proSPH-1 (5) for 60 min. PO activities in the control group (1, proSPH-1; 2, proSPH-2; 3, proSPH-1 and proSPH-2, all incubated with proPO and GF#52) and treatment group (4, proSPH-1; 5, proSPH-2 added just before the activity assay) were measured. The activities are plotted in the bar graphs (C, D) as mean ± SEM (n = 3).
3.5. Proteolytically processed SPH-1 and SPH-2 precursors as a cofactor for proPO activation
After the hydroxyapatite column fraction and proPO were incubated with one proSPH, we did not detect much PO activity (Fig. 5C) even though proSPH-1 was cleaved by the fractions. Similar results were obtained when a Sephacryl S-100 column fraction was used. However, after both proSPH-1 and proSPH-2 were incubated with the column fraction, we observed a large increase in proPO activation. While this increase likely resulted from cleaved SPH-1 and SPH-2, we could not rule out the possibility that one SPH and the other proSPH (e.g. SPH-1 and proSPH-2) have the auxiliary effect. Therefore, we examined if the simultaneous presence of cleaved SPH-1 and SPH-2 is required for this cofactor activity. The addition of proSPH-2 to cleaved SPH-1 and PO, or adding proSPH-1 to processed SPH-2 and PO, only generated a low level of PO activity (Fig. 5D). The processed SPH-1 and SPH-2 functioned synergistically to enhance proPO activation by PAP-1 in the column fractions. This result extended our previous findings that proPO, PAP and SPHs which loosely associated with each other (Wang and Jiang, 2004; Gupta et al., 2005). However, we did not detect any increase in proSPH cleavage by the column fractions in the presence of proPO (data not published). Association of proPO and SPHs (even in the presence of APMSF-treated PAP-1) did not lead to proPO activation (Jiang et al., 2003b). Proteolytic activation of M. sexta proPO requires an active PAP and SPHs at the same time (Gupta et al., 2005).
In comparison with the complex of SPH-1 and SPH-2 isolated from the hemolymph, we noticed that the cofactor activity generated by proSPH-1 and proSPH-2 cleavage activation was much lower than that of the same amounts of SPHs isolated from the hemolymph (Fig. 6). We examined the electrophoretic migration behaviors of processed proSPH-1 and proSPH-2 and found that they had a high mobility on the native polyacrylamide gels. In contrast, the plasma SPH complex had a high Mr and migrated slightly into the stacking gel. Since only specific cleavage occurred in these samples (Fig. 5), the smears were caused by association of SPH, proSPH and other reaction components (rather than protein degradation which could also lead to a lower cofactor activity). We speculate that the low auxiliary effect of SPH-1 and SPH-2 generated in vitro was related to the failure to form higher Mr complexes. Several possible reasons may account for the assembly problem: 1) proteolytic cleavage of proSPH-2 and especially proSPH-1 was incomplete (Fig. 5); 2) SPH-1 isolated from the hemolymph existed as 31 and 36 kDa bands on a reducing SDS gel (Wang and Jiang, 2004); 3) mass fingerprint analysis and cDNA cloning revealed heterogeneity of SPH-1 from the hemolymph of M. sexta (Jiang and Yu, unpublished results); 4) certain plasma factors may contribute to proper cleavage of proSPHs and formation of a high Mr SPH complex.
Fig. 6. Comparison of PO cofactor activity from processed recombinant proSPH-1 and proSPH-2 with that from the hemolymph SPH-1/SPH-2.

(A) PO activity assay. ProSPH-1 (1 μl, 120 ng/μl), proSPH-2 (1 μl, 120 ng/μl), GF#52 (1 μl), and proPO (1 μl, 40 ng/μl) were incubated on ice for 40 min. Or, GF#52 (1 μl), proPO (1 μl, 40 ng/μl), and hemolymph SPH-1/SPH-2 (3 μl, 35 ng/μl) were incubated under the identical condition, along with the controls. After the incubation, 150 μl of dopamine (2 mM) was added for the PO activity measurement. Mobility of processed proSPH-1 (B) and of proSPH-2 (C) on 7.5 % native PAGE detected by proSPH-1 and proSPH-2 antisera, respectively.
3.6. Concluding remarks
In this work, we produced M. sexta proSPH-1 and proSPH-2 using an improved baculovirus-insect cell expression system. The purified proteins were functional as substrates in the search of their activating proteinases. We observed their proteolytic processing by column fractions, and the cleaved SPHs were partially assembled and active as a cofactor for proPO activation. We further demonstrated that the coexistence of processed SPH-1 and SPH-2 is required for the auxiliary effect. These results serve as a foundation for future mechanistic studies on proSPH activation.
Acknowledgments
This work was supported by National Institutes of Health Grants GM58634. Dr. Xiaoqiang Yu at University of Missouri-Kansas City kindly provided SPH-1 and SPH-2 cDNA clones and antisera for this research. We would like to thank Drs. Jack Dillwith and Andrew Mort for their helpful comments on the manuscript. This article was approved for publication by the Director of Oklahoma Agricultural Experimental Station and supported in part under project OKLO2450.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Christophides GK, Vlachou D, Kafatos FC. Comparative and functional genomics of the innate immune system in the malaria vector Anopheles gambiae. Immunol Rev. 2004;198:127–148. doi: 10.1111/j.0105-2896.2004.0127.x. [DOI] [PubMed] [Google Scholar]
- Cerenius L, Söderhäll K. The prophenoloxidase-activating system in invertebrates. Immunol Rev. 2004;198:116–126. doi: 10.1111/j.0105-2896.2004.00116.x. [DOI] [PubMed] [Google Scholar]
- Dunn P, Drake D. Fate of bacteria injected into naïve and immunized larvae of the tobacco hornworm, Manduca sexta. J Invert Pathol. 1983;41:77–85. [Google Scholar]
- Gupta S, Wang Y, Jiang H. Manduca sexta prophenoloxidase (proPO) activation requires proPO-activating proteinase (PAP) and serine proteinase homologs (SPHs) simultaneously. Insect Biochem Mol Biol. 2005;35:241–248. doi: 10.1016/j.ibmb.2004.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom L. Serine protease mechanism and specificity. Chem Rev. 2002;102:4501–4524. doi: 10.1021/cr000033x. [DOI] [PubMed] [Google Scholar]
- Iwanaga S, Lee BL. Recent advances in the innate immunity of invertebrate animals. J Biochem Mol Biol. 2005;38:128–150. doi: 10.5483/bmbrep.2005.38.2.128. [DOI] [PubMed] [Google Scholar]
- Jarvis DL, Summers MD, Garcia A, Jr, Bohlmeyer DA. Influence of different signal peptides and prosequences on expression and secretion of human tissue plasminogen activator in the baculovirus system. J Biol Chem. 1993;268:16754–16762. [PubMed] [Google Scholar]
- Ji C, Wang Y, Ross J, Jiang H. Expression and in vitro activation of Manduca sexta prophenoloxidase-activating proteinase-2 precursor (proPAP-2) from baculovirus-infected insect cells. Protein Exp Purif. 2003;29:235–243. doi: 10.1016/s1046-5928(03)00020-2. [DOI] [PubMed] [Google Scholar]
- Jiang H, Kanost MR. The clip-domain family of serine proteinases in arthropods. Insect Biochem Mol Biol. 2000;30:95–105. doi: 10.1016/s0965-1748(99)00113-7. [DOI] [PubMed] [Google Scholar]
- Jiang H, Wang Y, Kanost MR. Pro-phenoloxidase activating proteinase from an insect, Manduca sexta: a bacteria-inducible protein similar to Drosophila easter. Proc Natl Acad Sci USA. 1998;95:12220–12225. doi: 10.1073/pnas.95.21.12220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Wang Y, Yu XQ, Kanost MR. Prophenoloxidase-activating proteinase-2 from hemolymph of Manduca sexta: a bacteria-inducible serine proteinase containing two clip domains. J Biol Chem. 2003a;278:3552–3561. doi: 10.1074/jbc.M205743200. [DOI] [PubMed] [Google Scholar]
- Jiang H, Wang Y, Yu XQ, Zhu Y, Kanost MR. Prophenoloxidase-activating proteinase-3 from Manduca sexta hemolymph: a clip-domain serine proteinase regulated by serpin-1J and serine proteinase homologs. Insect Biochem Mol Biol. 2003b;33:1049–1060. doi: 10.1016/s0965-1748(03)00123-1. [DOI] [PubMed] [Google Scholar]
- Kanost MR, Jiang H, Yu XQ. Innate immune responses of a lepidopteran insect, Manduca sexta. Immunol Rev. 2004;198:97–105. doi: 10.1111/j.0105-2896.2004.0121.x. [DOI] [PubMed] [Google Scholar]
- Kim MS, Baek MJ, Lee MH, Park JW, Lee SY, Söderhäll K, Lee BL. A new easter-type serine protease cleaves a masquerade-like protein during prophenoloxidase activation in Holotrichia diomphalia larvae. J Biol Chem. 2002;277:39999–40004. doi: 10.1074/jbc.M205508200. [DOI] [PubMed] [Google Scholar]
- Kwon TH, Kim MS, Choi HW, Joo CH, Cho MY, Lee BL. A masquerade-like serine proteinase homologue is necessary for phenoloxidase activity in the coleopteran insect, Holotrichia diomphalia larvae. Eur J Biochem. 2000;267:6188–6196. doi: 10.1046/j.1432-1327.2000.01695.x. [DOI] [PubMed] [Google Scholar]
- Lee SY, Kwon TH, Hyun JH, Choi JS, Kawabata SI, Iwanaga S, Lee BL. In vitro activation of pro-phenol-oxidase by two kinds of pro-phenol-oxidase-activating factors isolated from hemolymph of coleopteran, Holotrichia diomphalia larvae. Eur J Biochem. 1998;254:50–57. doi: 10.1046/j.1432-1327.1998.2540050.x. [DOI] [PubMed] [Google Scholar]
- Lu Z, Jiang H. Regulation of phenoloxidase activity by high and low molecular weight inhibitors from the larval hemolymph of Manduca sexta. Insect Biochem Mol Biol. 2007;37:478–485. doi: 10.1016/j.ibmb.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nappi AJ, Christensen BM. Melanogenesis and associated cytotoxic reactions: applications to insect innate immunity. Insect Biochem Mol Biol. 2005;35:443–459. doi: 10.1016/j.ibmb.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Paskewitz SM, Andreev O, Shi L. Gene silencing of serine proteases affects melanization of Sephadex beads in Anopheles gambiae. Insect Biochem Mol Biol. 2006;36:701–711. doi: 10.1016/j.ibmb.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Ross J, Jiang H, Kanost MR, Wang Y. Serine proteases and their homologs in the Drosophila melanogaster genome: an initial analysis of sequence conservation and phylogenetic relationships. Gene. 2003;304:117–131. doi: 10.1016/s0378-1119(02)01187-3. [DOI] [PubMed] [Google Scholar]
- Satoh D, Horii A, Ochiai M, Ashida M. Prophenoloxidase-activating enzyme of the silkworm, Bombyx mori: purification, characterization, and cDNA cloning. J Biol Chem. 1999;274:7441–7453. doi: 10.1074/jbc.274.11.7441. [DOI] [PubMed] [Google Scholar]
- Tang H, Kambris Z, Lemaitre B, Hashimoto C. Two proteases defining a melanization cascade in the immune system of Drosophila. J Biol Chem. 2006;281:28097–28104. doi: 10.1074/jbc.M601642200. [DOI] [PubMed] [Google Scholar]
- Volz J, Osta MA, Kafatos FC, Muller HM. The roles of two clip domain serine proteases in innate immune responses of the malaria vector Anopheles gambiae. J Biol Chem. 2005;280:40161–40168. doi: 10.1074/jbc.M506191200. [DOI] [PubMed] [Google Scholar]
- Volz J, Muller HM, Zdanowicz A, Kafatos FC, Osta MA. A genetic module regulates the melanization response of Anopheles to Plasmodium. Cell Microbiol. 2006;8:1392–1405. doi: 10.1111/j.1462-5822.2006.00718.x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Jiang H. Prophenoloxidase (proPO) activation in Manduca sexta: an analysis of molecular interactions among proPO, proPO-activating proteiase-3, and a cofactor. Insect Biochem Mol Biol. 2004;34:731–742. doi: 10.1016/j.ibmb.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Yu XQ, Jiang H, Wang Y, Kanost MR. Nonproteolytic serine protease homologs are involved in prophenoloxidase activation in the tobacco hornworm, Manduca sexta. Insect Biochem Mol Biol. 2003;33:197–208. doi: 10.1016/s0965-1748(02)00191-1. [DOI] [PubMed] [Google Scholar]
- Zhao P, Jiajing Li Yang Wang, Jiang H. Broad-spectrum antimicrobial activity of the reactive compounds generated in vitro by Manduca sexta phenoloxidase. Insect Biochem Mol Biol. 2007;37:952–959. doi: 10.1016/j.ibmb.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Z, Lopez D, Kanost MR, Evans JD, Jiang H. Comparative analysis of serine protease-related genes in the honey bee genome: possible involvement in embryonic development and innate immunity. Insect Mol Biol. 2006;15:603–614. doi: 10.1111/j.1365-2583.2006.00684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Z, Evans J, Lu Z, Zhao P, Williams M, Sumathipala N, Hetru C, Hultmark D, Jiang H. Comparative genome analysis of the Tribolium immune system. Genome Biol. 2007 doi: 10.1186/gb-2007-8-8-r177. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]