

Abstract
Aurora B is a mitotic protein kinase that phosphorylates histone H3, behaves as a chromosomal passenger protein, and functions in cytokinesis. We investigated a role for Aurora B with respect to human centromere protein A (CENP-A), a centromeric histone H3 homologue. Aurora B concentrates at centromeres in early G2, associates with histone H3 and centromeres at the times when histone H3 and CENP-A are phosphorylated, and phosphorylates histone H3 and CENP-A in vitro at a similar target serine residue. Dominant negative phosphorylation site mutants of CENP-A result in a delay at the terminal stage of cytokinesis (cell separation). The only molecular defects detected in analysis of 22 chromosomal, spindle, and regulatory proteins were disruptions in localization of inner centromere protein (INCENP), Aurora B, and a putative partner phosphatase, PP1γ1. Our data support a model where CENP-A phosphorylation is involved in regulating Aurora B, INCENP, and PP1γ1 targeting within the cell. These experiments identify an unexpected role for the kinetochore in regulation of cytokinesis.
Keywords: CENP-A; Aurora B; midbody; PP1γ1; cytokinesis
Introduction
The centromere is a specialized locus on chromosomes that mediates attachment of microtubules during mitosis via a large multiprotein complex called the kinetochore. Centromere protein A (CENP-A)* is an essential (Howman et al., 2000; Stoler et al., 1995) histone H3-like kinetochore protein incorporated specifically at active centromeres (Warburton et al., 1997), in direct contact with the DNA (Shelby et al., 1997). CENP-A has a similar histone fold domain to that of H3 and a divergent NH2-terminal tail (Sullivan et al., 1994). We showed recently that CENP-A is phosphorylated at serine 7 (Ser7) in early prophase after completion of H3 phosphorylation, and that CENP-A is subsequently dephosphorylated in early anaphase, before completion of histone H3 dephosphorylation (Zeitlin et al., 2001). The temporal differences between CENP-A and H3 phosphorylation and dephosphorylation suggest that although they share a similar phosphorylation site, they serve different functions.
The function of CENP-A phosphorylation is not known. Phosphorylation of other centromere proteins has been shown to control microtubule binding to chromosomes, spindle assembly checkpoint regulation, or degradation of sister chromatid cohesion components (Liao et al., 1994; Allshire, 1997; Waters et al., 1999). Mutational studies show that histone H3 phosphorylation at serine 10 (Ser10) is required for normal cell division in Tetrahymena (Wei et al., 1999). In G2 phase mammalian cells, histone H3 Ser10 phosphorylation begins at pericentromeric heterochromatin and extends throughout the chromosome arms in preparation for mitosis (Hendzel et al., 1997). H3 phosphorylation in G2 may be required to recruit proteins involved in remodeling and condensing chromatin (Schmiesing et al., 2000; Giet and Glover, 2001).
In Saccharomyces cerevisiae, Caenorhabditis elegans, and Drosophila melanogaster, histone H3 is phosphorylated by the respective Aurora kinase homologues Ipl1, AIR2, and Aurora B (Hsu et al., 2000; Giet and Glover, 2001); however, the equivalent Aurora kinase substrates have not been identified in mammalian cells. Recently, the inner centromere protein (INCENP) was demonstrated to be required for targeting Aurora B kinase to the centromere and central spindle. Aurora B follows a typical “passenger protein” pattern (Schumacher et al., 1998). The passenger protein hypothesis was originally conceived to account for the behavior of a group of proteins that transfer from the centromere region of metaphase chromosomes onto the central spindle microtubules during anaphase (for review see Earnshaw and Bernat, 1991). These “central spindle” microtubules are eventually bundled together along with the condensed contractile ring, resulting in formation of the midbody (for review see Straight and Field, 2000). Passenger proteins such as INCENP are detectable in the midbody until the two daughter cells complete separation (Cooke et al., 1987). The INCENP–Aurora B interaction is required for normal chromosome segregation and completion of cytokinesis in S. cerevisiae, X. laevis, C. elegans, and D. melanogaster (Kaitna et al., 2000; Adams et al., 2001a,b). Furthermore, the cytokinesis defects in cells lacking either Air-2 (the C. elegans Aurora B homologue) or ICP-1 (the C. elegans INCENP homologue) may be caused by disrupted localization of the midzone motor protein, ZEN-4/CeMKLP-1 (Kaitna et al., 2000). Air-2 is required for targeting CeMKLP-1 in C. elegans (Severson et al., 2000). However, Aurora B may (Giet and Glover, 2001) or may not (Adams et al., 2001b) be required for targeting the relevant homologue PAV-KLP/ZEN-4 in D. melanogaster. Based on these data, it appears that INCENP is required for Aurora B/Air-2 targeting, and that either or both of these proteins may be required for targeting of the MKLP-1 motor protein.
Another passenger protein, the C. elegans survivin homologue Bir-1, also appears to be required for Air-2 targeting (Skoufias et al., 2000; Speliotes et al., 2000; Uren et al., 2000). In the absence of Bir-1, Air-2 is undetectable on chromosomes and H3 phosphorylation is reduced or absent (Speliotes et al., 2000). Unfortunately, it was impossible to assess the interaction of Air-2 and Bir-1 in the central spindle, also called the spindle midzone, since the entire midzone was so severely disorganized in these embryos (Speliotes et al., 2000). Survivin and INCENP also appear to interact, since they have similar knockout phenotypes and localization, and interference with either one disrupts Aurora B/Air-2 localization (Kaitna et al., 2000). It has been proposed that these four proteins, survivin, INCENP, Aurora B, and MKLP-1 form a complex at the midzone that is required for cytokinesis (Adams et al., 2001a).
We have analyzed the function of CENP-A phosphorylation by expressing epitope-tagged mutant proteins in human cells. This approach has proven useful because the phosphorylation site mutants exert a dominant negative effect in the presence of wild-type, endogenous CENP-A. Because CENP-A is essential, knockout mice and deletion mutants in yeast have very low viability, characterized by arrest in mitosis, or extremely defective mitosis (Stoler et al., 1995; Howman et al., 2000). Expressing dominant mutations in the presence of wild-type allowed characterization of a role for CENP-A in events beyond those typically ascribed to histones. The mutant cells display delayed separation of daughter cells, joined by an elongated intercellular bridge with a prominent, long-lived Flemming body. We show that localization of the kinase Aurora B correlates with CENP-A and H3 phosphorylation in G2 and prophase, and Aurora B phosphorylates the NH2-terminal tails of both CENP-A and H3 in vitro. These data suggest that Aurora B is a mitotic H3/CENP-A kinase. In all cells examined, localization of survivin and MKLP-1 appear normal, even when Aurora B and INCENP localization are disrupted. Our work supports a role for the kinetochore chromatin protein CENP-A in the pathways that coordinate completion of cytokinesis in human cells.
Results
Human Aurora B is a mitotic histone H3/CENP-A kinase
In S. cerevisiae, C. elegans, and D. melanogaster, histone H3 is phosphorylated by the respective Aurora kinase homologues Ipl1, AIR2, and Aurora B (Hsu et al., 2000; Giet and Glover, 2001); however, the equivalent Aurora kinase substrates have not been identified in mammalian cells. CENP-A is 62% identical to histone H3, with almost no homology in the NH2 terminus (Sullivan et al., 1994). Human CENP-A is phosphorylated during mitosis at a site, serine 7, found within the only region of amino acid sequence similarity in the CENP-A and histone H3 NH2 termini (Zeitlin et al., 2001). We reasoned that human Aurora B, originally identified as Aurora, and Ipl-like midbody kinase (Aim-1; Terada et al., 1998) might also function as a mitotic CENP-A kinase.
Using phosphohistone H3 antibodies to stage cells from early to late G2 and into M phase, we were able to track the progression of Aurora B localization as cells entered mitosis (Fig. 1). Aurora B is first detectable in the nuclei of cells before the onset of histone H3 phosphorylation, where it is associated with pericentric regions surrounding many but not all centromeres (Fig. 1 a). Aurora B becomes concentrated in large pericentric foci (Fig. 1 b) before and during histone H3 phosphorylation initiation in pericentric chromatin. As histone H3 phosphorylation spreads throughout the chromatin during late G2, Aurora B is more uniformly distributed throughout the nucleus (Fig. 1 c). During prophase, when chromosome condensation is extensive, and CENP-A phosphorylation is detectable, Aurora B is detected primarily at centromeres, between paired sister kinetochores (Fig. 1 d, inset). These observations show, for the first time, a concordant program of Aurora B localization and histone phosphorylation events that spans G2 and prophase in human cells, complementing previous demonstrations that Drosophila and Xenopus Aurora B homologues localize to centromeres during mitosis (Adams et al., 2000). These data suggest that the control of intracellular localization is an element of Aurora B regulation.
Figure 1.
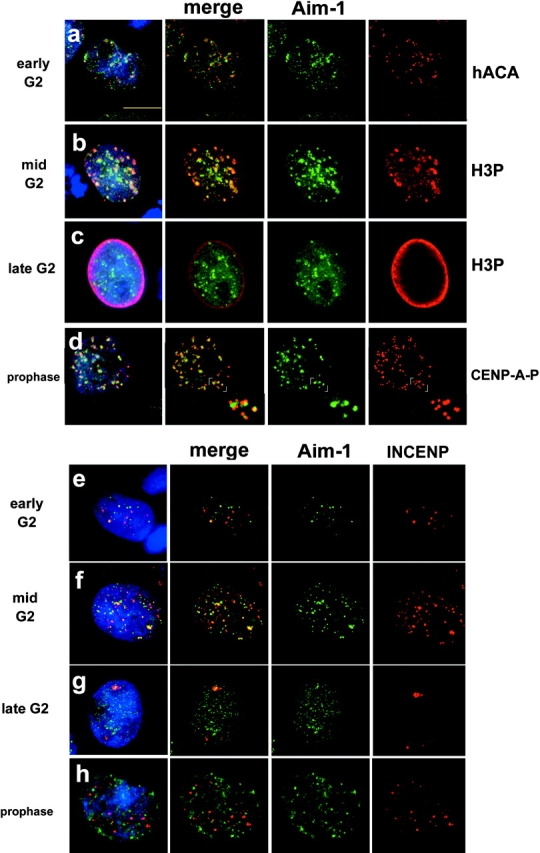
Aurora B and INCENP distribution in G2 and prophase. Aurora B kinase associates with centromeres during G2 and M. Immunofluorescence with anti–Aim-1 (Aim-1/Aurora B, green) and DAPI (DNA, blue). (a) Early G2 cells with hACA to detect centromeres (red), demonstrating that Aurora B associates with centromeres well before mitosis. (b) Mid G2 cells show clear colocalization of Aurora B and H3P-positive pericentric heterochromatin (red). (c) Late G2 cells with general distribution of Aurora B throughout the nuclei, at a time when H3P signal is present throughout the chromatin (red). (d) Prophase cells (with visible chromosome condensation judged by DAPI staining) with anti–CENP-A-Ser7P (red), showing that Aurora B associates with centromeres when CENP-A phosphorylation begins. Insets show that anti–CENP-A-Ser7P stains kinetochores, whereas Aurora B is detected in the inner centromere domain. (e) Early G2 cells show more Aurora B signals (green) than INCENP signals (red), with partial colocalization. (f) Mid G2 cells show more Aurora B signals, with an increase in INCENP signals and partial colocalization. (g) Late G2 cells with general distribution of Aurora B, and less INCENP signal in very few, larger foci. (h) Prophase cells with Aurora B distributed as in panel d, but INCENP does not colocalize with Aurora B concentrations at centromeres (yellow color would indicate overlap). Bar, 10 μm.
It has been shown in other systems that Aurora B localization to centromeres requires INCENP; however, the localization of INCENP during G2 has not been reported. We found that although Aurora B is present in numerous foci in early G2, INCENP is only present at a fraction of these sites (Fig. 1 e). Throughout G2, INCENP is present at sites that do not colocalize with Aurora B (Fig. 1 f), and in late G2 when Aurora B is detectable throughout the chromatin, INCENP is only present in a small number of much larger foci (Fig. 1 g). INCENP is again detectable in prophase, as expected (Fig. 1 h), although we did not observe INCENP distributed along chromosome arms as has been described by others (Earnshaw and Cooke, 1991).
To determine whether Aurora B possesses a CENP-A kinase activity, we asked whether immunoprecipitated Aurora B can utilize the NH2-terminal tail of CENP-A as a substrate. Aurora B was immunoprecipitated from nocodazole-arrested HeLa cells and incubated with CENP-A and H3 NH2 termini fused to glutathione S-transferase (GST) in an in vitro kinase reaction. Aurora B phosphorylated histone H3-GST, as expected from studies in other species (Hsu et al., 2000; Adams et al., 2001b; Giet and Glover, 2001; Murnion et al., 2001). CENP-A–GST was also used as a substrate with a somewhat lower efficiency (69% of the phosphate incorporated by H3-GST), while Aurora B did not phosphorylate GST alone (Fig. 2 a). This CENP-A kinase activity is highly concentrated by the immunoprecipitation procedure, since whole cell extracts do not have detectable kinase activity against these substrates, and therefore the immunoprecipitated activity is unlikely to represent a contaminating mitotic kinase (Fig. 2 b). To determine whether Ser7 of CENP-A is a substrate residue for Aurora B phosphorylation we performed two experiments. CENP-A–GST constructs were prepared in which Ser7 was mutated to alanine or glutamic acid. Phosphorylation of these proteins by Aurora B was reduced by 50%, demonstrating that Ser7 is a kinase substrate (Fig. 2 c). However, Aurora B is able to phosphorylate one or more of the other eight Ser residues in the NH2 terminus of CENP-A in vitro. In a second experiment, GST fusion proteins phosphorylated by Aurora B were probed by Western blot using an antibody specific for CENP-A phosphorylated at Ser7, demonstrating formation of the Ser7 phosphoepitope (Fig. 2 d). Together, the dynamic subcellular localization of Aurora B and in vitro kinase activity strongly suggest that Aurora B functions both as a CENP-A Ser7 kinase in mitosis as well as an H3 kinase in G2.
Figure 2.

Aurora B phosphorylates the NH 2 termini of histone H3 and CENP-A in vitro. Arrows indicate the migration of CENP-A–GST. (a) Immunoprecipitated Aurora B phosphorylates both H3 and CENP-A NH2 termini expressed as GST fusions. Top, representative Coomassie stain of SDS-PAGE. Lower bands are GST alone or fused to the NH2 terminus of either H3 or CENP-A; upper bands are antibody light chain, either anti–Aim-1 or control isotype-matched mouse IgG1. Also shown are mock reactions where GST fusions were mixed with the protein G sepharose beads used in immunoprecipitation. Bottom, autoradiogram of the same gel. Aurora B clearly phosphorylates the H3 and CENP-A NH2 termini. No signal is seen in either of the negative controls when the Aurora B antibody is not present. (b) Aurora B immunoprecipitation significantly concentrates the Aurora B kinase activity. Whole extract mixed with GST fusions does not result in phosphorylation of CENP-A or H3 NH2 termini (extract), and the Aurora B antibody alone contains no kinase activity (antibody). Whole extract alone allowed to autophosphorylate does not reveal CENP-A or H3 bands (auto). (c) Ser7 is a target Aurora B phosphorylation site in the CENP-A NH2 terminus. Aurora B immunoprecipitations were mixed with the CENP-A NH2-terminal GST fusion (WT), or site-directed mutants of this construct (S7A and S7E). CENP-A phosphorylation is reduced 50% by mutation of Ser7; however, it is not abolished. Immunoprecipitation with an irrelevant antibody (mouse IgG1) is negative. (d) Mutation of CENP-A Ser7 abolishes reactivity with the anti–CENP-A-Ser7P antibody. GST fusion proteins were incubated with immunoprecipitated Aurora B in the presence of cold ATP before Western blotting. Top, ponceau stain; bottom, Western blot. Only wild-type CENP-A reacts positively with the Ser7P antibody. There is no cross-reaction with GST or the NH2 terminus of H3, and mutation of Ser7 to alanine (S7A) or glutamate (S7E; unpublished data) abolishes reactivity.
Mutational analysis of CENP-A Ser7
To investigate the functional role of CENP-A phosphorylation, we constructed stable HeLa-TTA cell lines that inducibly express HA epitope–tagged CENP-A. The first, C4, expressing wild-type epitope–tagged CENP-A, was generated and characterized previously (Shelby et al., 1997). We also used site-directed mutagenesis to generate two point mutations in the CENP-A NH2 terminus. Conversion of Ser7 to alanine (S7A) preserves the size of the side chain, while removing the phosphorylation substrate hydroxyl group, and conversion of Ser7 to glutamate (S7E) introduces a stable negative charge at that position.
Several individual colonies were selected for each mutation based on immunofluorescence screening with anti-HA to detect the epitope-tagged proteins (see Materials and methods). Colonies that exhibited uniform expression within the population were further examined by Western blot with a human anticentromere antiserum which recognizes both the tagged and endogenous proteins (Fig. 3 a). For further study, two independent cell lines were chosen for each mutant which exhibited protein expression levels comparable to endogenous CENP-A. All experiments were performed using cells induced for 24 h.
Figure 3.

Expression of CENP-A Ser7 mutants in HeLa cells. (a) Western blot with sequential detection using two antibodies: 12CA5 to detect the epitope tag (HA), followed by hACA to detect endogenous CENP-A. C4 cells are epitope-tagged wild-type; S7A-8 and S7E-4 are representative mutant cell lines. Cells were collected either uninduced (”un”) or induced for 24 h by removal of tetracycline (”in”). (b) CENP-A mutant cell lines have normal kinetochore and spindle structure. Interphase (top) and mitotic cells (second row) with DNA (DAPI, blue), epitope tag (anti-HA, green), and centromeres (hACA, red). Bottom two rows, anti–CENP-C (red) and anti–CENP-E (red), respectively, with DAPI (DNA, blue) and antitubulin (green). (c) CENP-A mutant cell lines have normal cell-cycle profiles. DNA was detected with propidium iodide and analyzed by flow cytometry (representative profile shown as inset). TTA is the parental cell line with no epitope tag, C4 is wild-type epitope–tagged CENP-A, and S7A and S7E are each averaged values for two independent cell lines (S7A-8, S7A-18, S7E-4, and S7E-12). All cell lines appear to be similar within the standard deviation of five independent experiments (error bars). Bar, 10 μm.
Upon induction, CENP-A Ser7 mutant proteins are expressed and target to centromeres efficiently in these cell lines (Fig. 3 b, top two rows). Thus, mutation of CENP-A Ser7 does not interfere with incorporation of the protein at centromeric loci in this system, nor does it result in significant mistargeting into chromosome arms. Localization of inner (CENP-C) and outer (CENP-E) kinetochore proteins was normal as was overall spindle structure (Fig. 3 b, bottom two rows) (see supplementary data on NuMA, CENP-F, Mad2, Bub1, and BubR1). Both mutations yield normal cell cycle profiles as analyzed by flow cytometry under these conditions (Fig. 3 c). Further, CENP-A mutant cells arrested normally after treatment with nocodazole and monastrol, demonstrating that the spindle checkpoint is intact in these cells (unpublished data). Therefore, under the short term induction conditions used in these experiments, we can conclude that CENP-A Ser7 mutations do not grossly interfere with kinetochore formation, spindle assembly, or cell cycle progression.
Delayed separation of CENP-A mutant cells
We directly examined the execution of mitosis in mutant cell lines by time-lapse phase-contrast microscopy (representative images are shown in Fig. 4, a–c). One phase image was taken every 2 min overnight on a heated stage. HeLa cells characteristically round up when they enter mitosis, allowing approximate staging of cell division. The time from the onset to completion of cell rounding was similar for all cell lines. Although this analysis cannot rule out subtle differences in mitotic progression, cells in all lines appeared to progress into anaphase B (oval-shape, with visibly separated chromatin masses) and telophase with similar kinetics. However, the kinetics of the final stage of cytokinesis, scored from the first appearance of an intercellular bridge to the disappearance of the Flemming body, were quite different in CENP-A Ser7 mutant cells (Fig. 4, a–c). Wild-type or C4 cells complete this process within 30–40 min (Fig. 4 d). S7E cells exhibit a dramatic delay, requiring ∼150 min on average, whereas S7A cells took an average of ∼70 min (Fig. 4 d). The midbody structure is very dynamic during this process, oscillating in length before finally breaking (see videos available at http://www.jcb.org/cgi/content/full/jcb.200108125/DC1).
Figure 4.
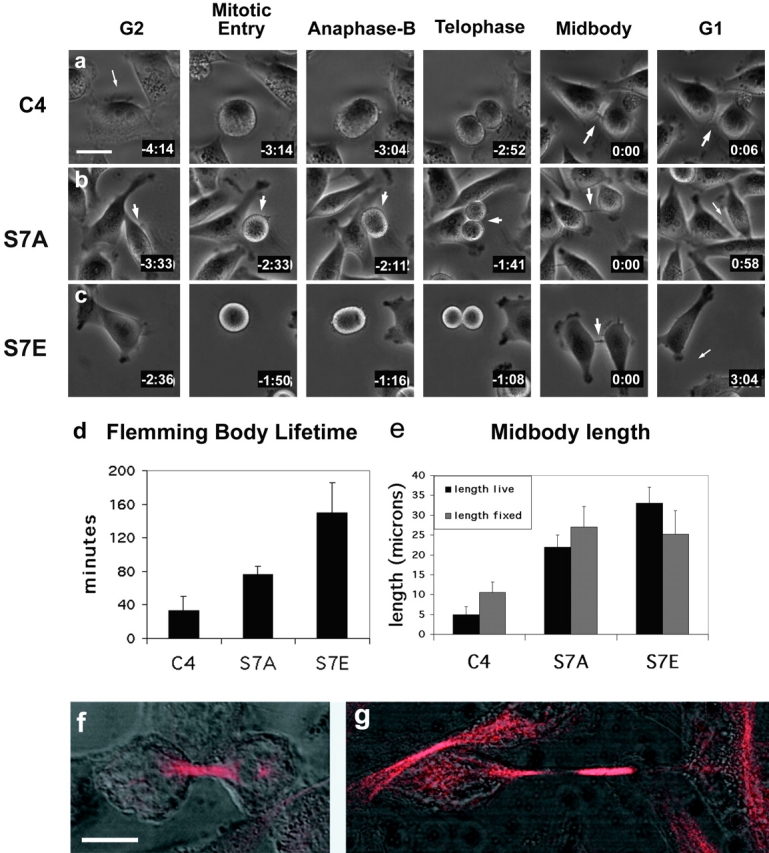
Live cell analysis: Flemming body lifetime and midbody length. (a–c) Cell division was visualized by live cell phase contrast microscopy at a magnification of 20×. Representative cells are shown for each stage of cell division (TTA cells closely resembled C4; unpublished data). For videos, see online supplemental material available at http://www.jcb.org/cgi/content/full/jcb.200108125/DC1. Cells were followed from interphase through initial rounding up, cell division, and reflattening until they ultimately separated. Midbodies were easily detectable in S7A and S7E cells but were smaller, and sometimes not visible, in C4. Arrows indicate cells of interest, midbodies, and newly separated cells. Times shown are hours:minutes, with the time of Flemming body appearance set to zero. (d) CENP-A mutant cell lines exhibit significantly longer midbody lifetimes than wild-type cells filmed under identical conditions of substrate attachment and cell seeding density. Images were taken once every 2 min. Midbody lifetimes were measured beginning when a visible Flemming body became visible between the two daughter cells, and ending when the midbody split, at which time the Flemming body was engulfed by one of the daughter cells and cell separation was considered complete. Standard error is shown for each cell line (bars). (e) Midbody lengths are shown in μm (± standard error) from videos of live cells (black) compared with lengths from fixed cells stained with antitubulin (gray). For live analysis, the length of the intercellular bridge was measured at each time point of 2 min, from when it first became visible to when the two cells completed separation and the Flemming body was engulfed by one of the daughter cells. These values were then averaged for each cell over time, and averaged again across the total number of cells filmed in this way (n = 7, 12, and 7 for C4, S7A, and S7E, respectively). Midbodies were observed to oscillate in a stretching motion before breaking at their maximum length. For fixed analysis, asynchronous cells were stained with antitubulin and a minimum of 150 cells were counted for each cell line. Midbody lengths for both mutants are significantly different from wild-type (P = 0) based on Chi-squared analysis. (f and g) Midbody morphology. The brightly labeled tubulin bundle (red) does not exactly match the length of the intercellular bridge (DIC). (f) In early cytokinesis, the tubulin bundle is longer than the intercellular bridge, defined as the distance between the two cell bodies. (g) In late cytokinesis, the tubulin bundle is shorter than the intercellular bridge, thus leaving an unstained gap between the end of the tubulin bundle and the edge of the cell body. Bars, 10 μm.
Measurements of midbody lengths from live films reveal that S7A midbodies are 22 μm on average, and the average length of S7E midbodies is 33 μm, whereas the average length for cells expressing wild-type epitope–tagged CENP-A is 5 μm (Fig. 4 e). Midbody lengths were also measured in fixed cells using α tubulin immunofluorescence in combination with DIC imaging. The trend of this analysis was similar to that obtained in live cells (Fig. 4 e). The brightly labeled tubulin bundle at the center of the intercellular bridge does not exactly span the distance between the two cell bodies. Early in cytokinesis, the tubulin bundle is longer than the bridge (Fig. 4 f), but in the mutants, as the cells pull apart, the tubulin bundle does not span the entire length of the bridge, leaving a prominent unstained gap between the edge of the tubulin bundle and the edge of the cell body (Fig. 4 g). These differences in lifetime, length, and morphology of the intercellular bridge point to a role for Ser7 of CENP-A in coordinating completion of cytokinesis.
Mislocalization of Aurora B and INCENP during anaphase in CENP-A mutant cells
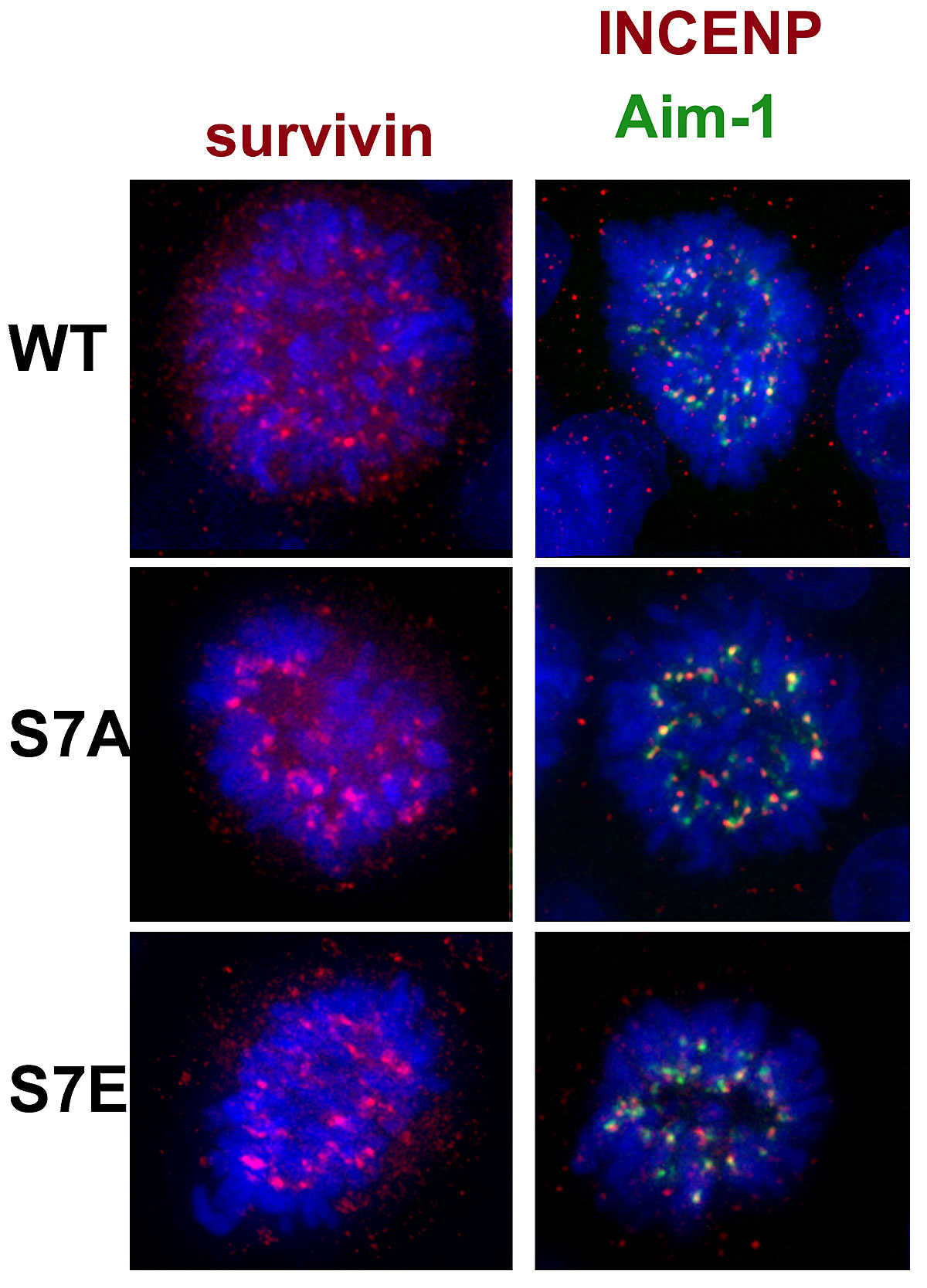
Aurora B itself has recently been shown to exist in a complex with INCENP and survivin, in addition to having a known functional role in cytokinesis (Tatsuka et al., 1998; Terada et al., 1998; Adams et al., 2000). We observed a qualitative difference in Aurora B and INCENP localization during anaphase. Aurora B and INCENP normally transfer completely to the midzone at anaphase (Fig. 5 a). In CENP-A–S7A cells, both proteins have normal anaphase midzone localization (Fig. 5 b). In contrast, in CENP-A–S7E cells, a detectable portion of both Aurora B and INCENP is bound to anaphase chromosomes, while a portion of both proteins are localized to the midzone (Fig. 5 c). Thus, the S7E mutation interferes with Aurora B and INCENP dynamics at the stage when passenger proteins transfer to the midzone. This is not a general defect in anaphase midzone assembly, since MKLP-1 (Fig. 5 d) and CENP-E (Fig. 5 e) are localized normally during this stage. The passenger proteins ultimately adopt a normal localization in the midbody at the end of telophase (survivin, Fig. 5 f; INCENP, Fig. 5 g; Aurora B, Fig. 6). Together, CENP-A Ser7 mutations appear to alter the dynamics of Aurora B and INCENP partitioning between chromosomes and the midzone during anaphase, but do not interfere with the localization of passenger proteins at midbodies during cytokinesis.
Figure 5.
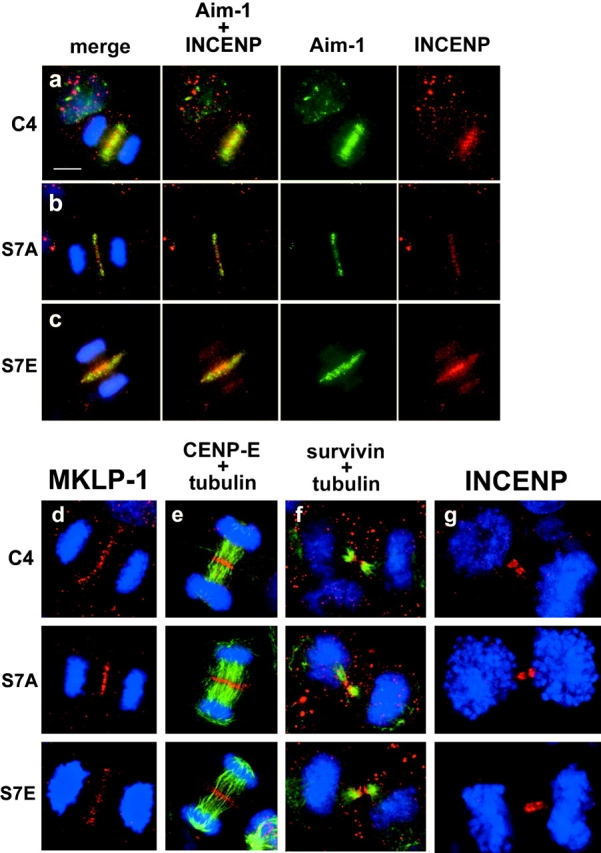
Passenger and motor protein localization in CENP-A mutant cell lines. (a–c) Immunofluorescence to detect DNA (blue), Aurora B (green), and INCENP (red). In both wild-type (C4) and S7A cells, Aurora B and INCENP are localized only at the midzone during anaphase, while in S7E cells Aurora B and INCENP are also detected on chromosomes (bottom row, third panel). (d–i) DNA is shown in blue (DAPI) in all panels. d, MKLP-1 (red); e, CENP-E (red, with tubulin in green) at the midzone. f, survivin (red, with tubulin in green); g, INCENP (red) at the midbody.
Figure 6.

PP1γ1 is mislocalized in CENP-A mutant cell lines. (a–c) Protein phosphatase 1γ1 localizes to midbodies in wild-type but is delocalized in S7A and S7E cells. Midbodies are shown with Aurora B in red and PP1γ1 in green. (a) PP1γ1 is detectable in the midbodies of wild-type S7A (b) cells (colocalization appears yellow in “merge”), but is not detected in midbodies of S7E cells (arrow) (c). PP1γ1 is delocalized throughout S7A cells, but is undetectable in telophase nuclei of wild-type cells. Insets show detail of the midbody. Bar, 10 μm.
Mistargeting of a PP1γ1 in CENP-A Ser7 cells
Protein phosphorylation reflects the steady-state balance of two opposing enzyme activities: a kinase and a phosphatase. We selected protein phosphatase 1γ1 (PP1γ1) for analysis in CENP-A Ser7 mutant cells for four reasons. First, the yeast PP1 homologue Glc7 acts as a phosphatase that functions in Ipl1-dependent phosphorylation pathways in S. cerevisiae (Francisco et al., 1994). Second, PP1 itself has been shown to dephosphorylate histone H3 (Chadee et al., 1995; Murnion et al., 2001). Third, PP1γ1 localizes to the mitotic spindle in mitosis (Andreassen et al., 1998). Finally, antisense interference with PP1γ1 expression causes human cells to delay and fail in late-stage cytokinesis, similar to the phenotypic effects of Aurora B overexpression and CENP-A Ser7 mutations reported here (Cheng et al., 2000).
Immunofluorescence with an isoform-specific antibody demonstrates that PP1γ1 is primarily localized at the midbody during cytokinesis in HeLa Tta (and C4 wild-type cells; unpublished data), with no detectable signal elsewhere in the cell (PP1γ1 in green, Aurora B in red, overlap in yellow; Fig. 6 a). In CENP-A S7A, PP1γ1 is inefficiently targeted to the midbody, so that a large portion of the signal is distributed throughout the cells (Fig. 6 b). In S7E cells, PP1γ1 fails to concentrate at the midbody at all (Fig. 6 c). We conclude that mutation of CENP-A Ser7 disrupts the trafficking of PP1γ1 during cytokinesis. Together, these experiments identify a remarkably specific role of the CENP-A NH2 terminus in modulating the cellular localization of three proteins required for cytokinesis: Aurora B, INCENP, and PP1γ1. Based on the known involvement of these proteins in cytokinesis, we infer that the delay in cytokinesis produced by CENP-A Ser7 mutations results from misregulation of this pathway through aberrant enzyme localization within the cell.
Discussion
We have identified a functional relationship between Aurora B kinase and the centromeric histone CENP-A and defined a role for kinetochore chromatin in coordinating cytokinesis in human cells. The dynamic cellular localization of Aurora B and its enzymatic activity in vitro make it a likely mitotic kinase for CENP-A as well as histone H3 in human cells. Two amino acid substitutions at CENP-A Ser7, S7A, and S7E act as dominant negative mutants to delay cytokinesis in HeLa cells. In mutant cells, during the terminal event of what appears to be an otherwise normal mitosis, daughter cell separation is significantly delayed, with cells linked by a prominent midbody. We characterized the localization of 22 proteins in mutant cells representing chromosomal, kinetochore, spindle, passenger, and regulatory proteins. Only three proteins showed defects in localization: Aurora B, INCENP, and PP1γ1. All of these proteins have been shown previously to function in cytokinesis (Mackay et al., 1998; Tatsuka et al., 1998; Cheng et al., 2000). The severity of the cytokinesis delay phenotypes in the two mutants is correlated with the severity of Aurora B, INCENP, and PP1γ1 localization defects. We suggest that the cytokinesis delay in CENP-A Ser7 mutant cells is caused by defects in the localization of the Aurora B, INCENP, and PP1γ1.
Previous work has shown that Aurora B and its homologues function as histone H3 kinases in S. cerevisiae, C. elegans, Drosophila, and Xenopus (Hsu et al., 2000; Giet and Glover, 2001), although other serine kinases such as NimA have been proposed to function as histone H3 kinases in mitosis as well (De Souza et al., 2000). In this paper, we have presented data that suggest Aurora B functions as both a histone H3 and CENP-A mitotic kinase in human cells. The NH2-terminal tails of CENP-A homologues exhibit a high degree of evolutionary divergence. While Ser- or Thr-containing motifs similar to that in human CENP-A are found in Drosophila and S. cerevisiae (unpublished data), CENP-A residue Ser7 is not conserved in mouse. However, our data indicate that Ser7 is not the only Aurora B substrate in the CENP-A NH2 terminus. It is plausible that one of these other substrate residues may serve a similar function in other organisms. For example, functionally equivalent phosphorylation sites on annexin I (Haigler et al., 1992) and phosphorylases (Hwang and Fletterick, 1986) have arisen through convergent evolution. Alternatively, a functionally equivalent site could be present on a different kinetochore component in other species, similar to evolutionary division of labor between Rap1 and TRF1 at yeast and human telomeres (Li et al., 2000). Indeed, the high degree of evolutionary divergence of kinetochore proteins CENP-A and CENP-C suggest significant structural plasticity in kinetochore proteins.
Histones are clearly not the only substrates for Aurora B. Human Aurora B is concentrated in the spindle midzone, contractile ring, and midbody during telophase and cytokinesis, and appears to have a functional role in formation of these structures (Tatsuka et al., 1998; Terada et al., 1998; Murata-Hori et al., 2000). Indeed, myosin light-chain kinase, present in the cleavage furrow, is phosphorylated by Aurora B (Murata-Hori et al., 2000). Thus, subcellular localization is likely to be a critical determinant of Aurora B substrate specificity at different stages of G2 and M.
Association with targeting subunits is another mechanism for regulating protein kinase substrate specificity (for review see Johnson et al., 1996). It has been reported previously that Aurora B targeting to chromosomes depends on INCENP (Kaitna et al., 2000; Adams et al., 2000, 2001b) and survivin/BIR-1 (Speliotes et al., 2000) and that INCENP initially localizes along chromosomes, eventually becoming concentrated at centromeric regions in prometaphase (Cooke et al., 1987). Our results show Aurora B clearly localized to pericentric regions in early G2, while INCENP is present in foci that only partially colocalize with Aurora B. We were unable to detect survivin at centromeres during G2 (unpublished data). We conclude that Aurora B localizes to centromeres in G2 in human cells before INCENP or survivin. Thus, if Aurora B utilizes targeting subunits to direct its substrate-specific localization, it is likely that regulation of Aurora B targeting before prometaphase is distinct from that in the latter half of mitosis.
Studies of INCENP trafficking during mitosis have led to the idea that centromeric localization early in mitosis is required for a ‘priming step’ that renders the passenger protein complex competent to transfer to the spindle midzone later in mitosis (Ainsztein et al., 1998). Thus, we propose that CENP-A chromatin functions in part to mediate the assembly or priming of Aurora B and INCENP in the passenger protein complex, thus activating the complex for its late mitotic functions. Such a kinetochore-dependent assembly process has been proposed to function in the spindle assembly checkpoint, where a complex containing Mad2 and p55cdc20 is thought to be assembled by unattached kinetochores, forming active APC inhibitor which then functions elsewhere in the cell (Waters et al., 1999).
There are two general ways CENP-A could be involved in a kinetochore-dependent assembly process. CENP-A could play an indirect role by providing a permissive chromatin environment for the productive interaction of Aurora B and INCENP. Alternatively, CENP-A could play a direct role by binding to Aurora B, INCENP, or survivin. Such a direct interaction would necessarily be transient, since by prometaphase most Aurora B and INCENP are present in the inner centromere domain which is visibly distinct from the CENP-A-containing kinetochore chromatin. Together, these considerations lead to a model in which CENP-A chromatin mediates assembly of a “primed” Aurora B and INCENP complex. We suggest that while CENP-A remains stably bound to kinetochore chromatin, its phosphorylation cycle contributes to the function of the passenger protein complex, which acts at distal sites in the cell.
During anaphase, Aurora B, INCENP, and survivin normally relocate together to the spindle midzone, where all three of these proteins are required for successful cytokinesis (Mackay et al., 1998; Schumacher et al., 1998; Tatsuka et al., 1998; Adams et al., 2000; Chen et al., 2000; Kaitna et al., 2000; Giet and Glover, 2001; Speliotes et al., 2000). We have shown here that in anaphase in CENP-A S7E cells, some Aurora B and INCENP molecules are uncoupled from survivin. A portion of Aurora B and INCENP is bound to anaphase chromatin, at a point when survivin is detected only in the spindle midzone. In fact, the anaphase chromosome association of Aurora B and INCENP is quite similar to the behavior of an INCENP mutant, INCENP1–405, which fails to bind to central spindle microtubules (Mackay et al., 1998). If our model is correct, a portion of the Aurora B and INCENP in CENP-A S7E cells is not primed, and therefore does not bind to the midzone in anaphase, but instead binds general chromatin. Consistent with this finding, we occasionally observed persistent histone H3 phosphorylation in anaphase CENP-A S7E cells (unpublished data). The fact that only a portion of Aurora B and INCENP are affected may be related to the fact that CENP-A S7E is coassembled with wild-type CENP-A nucleosomes at kinetochores in these cells.
At the final step of cytokinesis, PP1γ1 is absent from the midbody of CENP-A S7E cells. It has been shown by others that PP1 and Aurora B/Ipl1 act opposite each other on the substrates histone H3 (Hsu et al., 2000; Murnion et al., 2001) and the yeast kinetochore protein, Ndc10 (Francisco and Chan, 1994; Francisco et al., 1994). Our data are consistent with a model where PP1γ1 and Aurora B act opposite each other to regulate the phosphorylation state of CENP-A in human cells.
Does the phosphorylation state of CENP-A directly influence the enzyme localization processes we have identified? CENP-A S7A and S7E could simply act as structural mutants in the NH2 terminus. Alternatively, our data are consistent with the proposal that the localization of Aurora B, INCENP, and PP1γ1 in the latter half of mitosis requires dephosphorylation of CENP-A Ser7.
What could cause a midbody delay?
The localization pattern of all passenger proteins, including Aurora B, ends with concentration at the midbody, after which time these proteins are no longer detectable. Cytokinesis may not be required for degradation of these proteins, as shown for CENP-E (Brown et al., 1994), but could degradation of these proteins be required for cytokinesis? It was shown recently that Aurora B can be degraded in vitro and in vivo by one form of the anaphase promoting complex, a ubiquitin protein ligase called APC–Cdh (Sorensen et al., 2000). The APC–Cdh complex is active in late mitosis and G1 (Fang et al., 1998), and in yeast the APC must remain active in G1 to prevent premature accumulation of mitotic cyclins (Amon et al., 1994). It was proposed previously that mitotic proteins are degraded or thrown away at the end of mitosis to prevent their interference with the following G1 (Mullins and McIntosh, 1982). A mechanism and location for this process have not yet been found. The protease caspase-9 has been detected in midbodies and associates with survivin, supporting the idea that proteolytic activities require special regulation to prevent apoptosis (O'Connor et al., 2000). Based on these observations, we arrive at a model where proteolytic activities could be required to degrade specific targets at the midbody before daughter cells can separate, and if this process is not regulated correctly, cells may undergo apoptosis. Overexpression of either Aurora B or PP1γ1 is associated with polyploidy and multinucleate cells, both indicators of failed cell division (Sogawa et al., 1994, 1995, 1997; Yamada et al., 1994; Tatsuka et al., 1998), supporting the idea that Aurora B levels must be regulated to allow for normal cytokinesis. By this reasoning, one possible explanation of the long midbody lifetime in CENP-A mutant cells is delayed degradation of Aurora B. Alternatively, Aurora B and PP1γ1 are themselves each required for cytokinesis (Terada et al., 1998; Cheng et al., 2000). It is plausible that defects in either Aurora B or PP1γ1 alone would be sufficient to account for the midbody delay phenotype induced by CENP-A Ser7 mutations.
Materials and methods
Cell lines and culture conditions
HeLa TTA cells (Gossen and Bujard, 1992) and derivations thereof were cultured at 37°C in DME medium containing 10% fetal calf serum (GIBCO BRL) with 5% CO2 in the presence of 1 μg/ml tetracycline and geneticin (400 μg/ml; GIBCO BRL).
Construction, culture, and analysis of stably transfected mutant cell lines
Site-directed mutagenesis was performed using QuikChange (Stratagene) on the vector pUHD-CENP-A-HA (Shelby et al., 1997). HeLa TTA cell lines were constructed as described previously (Shelby et al., 1997) by cotransfection with mutated pUHD-CENP-A-HA and puromycin resistance plasmids (pBS-PAC) (de la Luna et al., 1988). Colonies were expanded and assayed by Western blot with antibody against the HA tag (mAb12CA5; a gift from Ian Wilson, Scripps Research Institute, La Jolla, CA) and immunofluorescence with anti-HA mAb 16B12 (Babco) to assess relative levels of expression and centromere targeting. Cell lines that exhibited ≥95% expressing cells with uniform expression as judged by immunofluorescence and protein levels comparable to wild-type CENP-A as judged by Western blot were selected for further analysis. To concentrate on primary defects, all experiments shown here were performed on low-passage cells induced for only 24 h. Upon chronic induction (>2 wk) or long-term passage without induction (>12 wk), CENP-A S7A cells accumulated cells with aberrant nuclear shapes, while CENP-A S7E cells exhibited reduced viability, likely due to low-level leakage of the Tet-regulated promoter. For this reason, cells expressing wild-type epitope–tagged CENP-A (C4 cells, previously called HeLa tTA-CAHA; Shelby et al., 1997) or parental TTA cells, which behave identically under the assay conditions employed here, were used as negative controls, rather than uninduced cells. Aliquots of low-passage cells were thawed from frozen stocks every 2 mo.
SDS-PAGE and Western Blotting
Conditions used were modified slightly from those described previously (Shelby et al., 1997). Briefly, samples were loaded onto 12.5% polyacrylamide gels containing SDS or 15% gels with no SDS. For Western Blot, samples were transferred onto PVDF membrane (Gelman Sciences) and blocked in 5% nonfat milk in TBST. All antibodies were used at a dilution of 1:1,000. Detection was performed using the SuperSignal West Pico Chemiluminescent detection kit (Pierce Chemical Co.).
Immunofluorescence
Immunofluorescence with histone H3 and CENP-A phosphorylation–specific antibodies were performed as described previously (Zeitlin et al., 2001). Tubulin staining was performed using mouse mAb Dm1a (Sigma-Aldrich) at a dilution of 1:1,000, on methanol-fixed cells (10 min, −20°C). Other antibodies were used at the following dilutions: anti-HA mAb 16B12 (1:1,000; Babco), anti–Aim-1 (1:100; Transduction Labs), anti-PP1γ1 (1:200; Boston Express), anti–CENP-E (1:500; a gift from Don Cleveland, Ludwig Institute for Cancer Research, UC San Diego, La Jolla, CA), anti–CENP-C (1:500; a gift from Peter Warburton, Mount Sinai School of Medicine, New York, NY), Ki67 (1:50; Upstate Biotechnology), anti–MKLP-1 (1:100; Santa Cruz Biotechnology, Inc.), antisurvivin (1:100; Novus Biologicals), anti-Mad2 (1:50; a gift from Jennifer Waters, University of North Carolina, Chapel Hill, NC), anti-Bub1 and BubR1 (1:1,000; given by Tim Yen, Institute for Cancer Research, Fox Chase Cancer Center, Philadelphia, PA), anti-NuMA (1:2,500; a gift from Duane Compton, Dartmouth Medical School, Hannover, NH), C4 antiactin (1:1,000; a gift from Velia Fowler, The Scripps Research Institute, La Jolla, CA), 1186 anti-INCENP (1:2,000; a gift from Bill Earnshaw, Wellcome Institute for Cell Biology, University of Edinburgh, Edinburgh, Scotland, UK); anti-GMP1 (1:100; Zymed Labs); anti-SUMO 2/3 (1:100; a gift from Hisato Saitoh, Graduate School of Medical Science, Kyushu University, Fukuoka, Japan), antilamin (1:500; a gift from Larry Gerace, The Scripps Research Institute, La Jolla, CA), MCP21 (1:100; Affiniti Research Products), and anti–CENP-F (1:100; a gift from Carlos Casiano, Loma Linda University School of Medicine, Loma Linda, CA). Secondary antibodies were used at a dilution of 1:100 (Jackson ImmunoResearch Laboratories).
Live cell microscopy
Cells were grown directly on glass coverslips and mounted after 1 d induction. Cells were maintained during imaging in DME plus 10 mM Hepes buffer in a Rose chamber on a heated stage at 37°C (Bionomic Controller BC-100; 20/20 Technologies, Inc.). Cells were imaged at 2 min intervals on a Nikon TE-300 using a 20× Nikon lens (NA = 0.50), a long-working distance condenser (NA = 0.52), and an ORCA II (Hamamatsu) cooled CCD camera coupled to an electronic shutter (Uniblitz). Metamorph 4.5 was used for computer-based image acquisition and analysis of live cell data (Universal Imaging Co.) Single images shown were prepared using Adobe Photoshop® 5.5. Data on midbody lengths, average times, and errors were analyzed with Adobe Photoshop® and Microsoft Excel.
Deconvolution microscopy
Images were collected at intervals of 0.2 or 0.3 μm in the Z axis on a DeltaVision wide field optical sectioning microscope system, based on an Olympus IX70 epifluorescence microscope (Applied Precision, Issaquah, WA). A 100x 1.35 NA neofluor oil immersion lens was used for all images. Images were processed using a constrained iterative deconvolution algorithm. All images shown are projections of 3-dimensional image stacks (Softworx analysis software, Applied Precision). Composite montages of projections were assembled using Adobe® Photoshop 5.5.
Flow cytometry
For flow cytometry, cells were collected by trypsinization and quenched in complete media. Cells were washed with PBS and fixed overnight in 70% ethanol at −20°C. Samples were then washed and resuspended with RNase (10 μg/ml) and propidium iodide (5 μg/ml) and filtered through mesh (Nitex 3–53/36; Sefar) before detection on a FACS Calibur I (Becton Dickinson). 20,000 cells were counted for each cell line, and these experiments were performed five times. Data were analyzed using CellQuest (Becton Dickinson).
GST fusion proteins
CENP-A and H3 NH2 termini were fused to GST using the vector pGEX-KG (Guan and Dixon, 1991). Plasmids were transformed into BL21 cells (Novagen) and grown to OD = 0.6–1.0 in LB with 100 μg/ml carbenicillin (Sigma-Aldrich) before induction with 200 μM IPTG for 1–2 h at 37°C. Cell pellets were resuspended in ice cold PBS containing 1 mM DTT and 1 mM EDTA plus 0.5 mg/ml of lysozyme and incubated on ice for 30 min. Cells were then incubated on ice for 30 min after addition of Triton X-100 to a final concentration of 1%, in the presence of 10 mM MgCl2 and 100 μg/ml DNase1 (Boehringer). Lysates were cleared of debris by centrifugation and preswollen glutathione agarose beads were added to the supernatant and incubated for a minimum of 1 h at 4°C with gentle rotation. Beads were collected by centrifugation, washed four times by resuspension and centrifugation in buffer containing PBS, 1 mM DTT, and 1% Triton X-100, and finally washed in kinase buffer 2–3 times (see below).
Aurora B immunoprecipitation and kinase assay
Aurora B was immunoprecipitated from HeLa cells using a commercial antibody (anti–Aim-1; Transduction Labs). Cells were lysed in Pagano buffer (Pagano et al., 1994) (0.1% Triton X-100, 5 mM EDTA, 50 mM Tris, 250 mM NaCl, 10 mM MgCl2, 1 mM DTT plus microcystin, and protease inhibitor cocktail; Sigma-Aldrich) on ice for 30 min and sonicated for 3–5 min at 4°C. 7.5 μg of antibody (anti–Aim-1 or IgG1) were added to the lysates, in addition to gammabind G sepharose beads (Amersham Pharmacia Biotech) for a minimum of 1 h at 4°C. Samples were spun down and washed 3–5 times with Pagano buffer and 2 times with kinase buffer. Kinase assays were performed in buffer containing 50 mM Tris, 10 mM MgCl2, 1 mM DTT, 100 mM NaCl, 10 mM glutathione, and 1 μM ATP-γ32P. Peptide or GST fusion beads were mixed with gammabind G-IP beads in kinase buffer (total volume 50 μl per reaction) and incubated for 30 min at 37°C before running on 12.5% SDS-PAGE and detection with autoradiography (Kodak X-OMAT film). Band intensities were quantitated from short exposures using the program ImagePro Plus (Media Cybernetics).
Online supplemental material
Videos of mitosis (see Fig. 4) and additional immunolocalization data are available as online supplements at http://www.jcb.org/cgi/content/full/jcb.200108125/DC1.
Supplemental Material
Acknowledgments
This paper is dedicated to the memory of our good friend and colleague Rich Shelby. We would like to thank CD Allis for helpful discussions. We would also like to thank Tarun Kapoor for donating a sample of Monastrol; Malcolm Wood, Clare Waterman-Storer, and Wendy Salmon for microscopy facilities and technical assistance; Joe Trotter and Alan Saluk for flow cytometry assistance; and Clare McGowan for technical advice and reagents.
S.G. Zeitlin was supported in part by a fellowship from the ARCS foundation. This work was funded by a grant (GM39068) to K.F. Sullivan from the National Institute for General Medical Sciences.
The online version of this article contains supplemental material.
Footnotes
Abbreviations used in this paper: CENP-A, centromere protein A; GST, glutathione S-transferase; INCENP, inner centromere protein.
References
- Adams, R.R., M. Carmena, and W.C. Earnshaw. 2001. a. Chromosomal passengers and the (aurora) ABCs of mitosis. Trends Cell Biol. 11:49–54. [DOI] [PubMed] [Google Scholar]
- Adams, R.R., H. Maiato, W.C. Earnshaw, and M. Carmena. 2001. b. Essential roles of Drosophila inner centromere protein (INCENP) and aurora B in histone H3 phosphorylation, metaphase chromosome alignment, kinetochore disjunction, and chromosome segregation. J Cell Biol. 153:865–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams, R.R., S.P. Wheatleya, A.M. Gouldsworthy, S.E. Kandels-Lewis, M. Carmena, C. Smythe, D.L. Gerloff, and W.C. Earnshaw. 2000. INCENP binds the Aurora-related kinase AIRK2 and is required to target it to chromosomes, the central spindle and cleavage furrow. Curr. Biol. 10:1075–1078. [DOI] [PubMed] [Google Scholar]
- Ainsztein, A.M., S.E. Kandels-Lewis, A.M. Mackay, and E.C. Earnshaw. 1998. INCENP centromere and spindle targeting: identification of essential conserved motifs and involvement of heterochromatin protein HP1. J. Cell Biol. 143:1763–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allshire, R.C. 1997. Centromeres, checkpoints and chromatid cohesion. Curr. Opin. Genet. Dev. 7:264–273. [DOI] [PubMed] [Google Scholar]
- Amon, A., S. Irniger, and K. Nasmyth. 1994. Closing the cell cycle circle in yeast: G2 cyclin proteolysis initiated at mitosis persists until the activation of G1 cyclins in the next cycle. Cell. 77:1037–1050. [DOI] [PubMed] [Google Scholar]
- Andreassen, P.R., F.B. Lacroix, E. Villa-Moruzzi, and R.L. Margolis. 1998. Differential subcellular localization of protein phosphatase-1 α, γ1, and Δ isoforms during both interphase and mitosis in mammalian cells. J. Cell Biol. 141:1207–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, K.D., R.M. Coulson, T.J. Yen, and D.W. Cleveland. 1994. Cyclin-like accumulation and loss of the putative kinetochore motor CENP-E results from coupling continuous synthesis with specific degradation at the end of mitosis. J. Cell Biol. 125:1303–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadee, D.N., W.R. Taylor, R.A. Hurta, C.D. Allis, J.A. Wright, and J.R. Davie. 1995. Increased phosphorylation of histone H1 in mouse fibroblasts transformed with oncogenes or constitutively active mitogen-activated protein kinase kinase. J. Biol. Chem. 270:20098–20105. [DOI] [PubMed] [Google Scholar]
- Chen, J., W. Wu, S.K. Tahir, P.E. Kroeger, S.H. Rosenberg, L.M. Cowsert, F. Bennett, S. Krajewski, M. Krajewska, K. Welsh, et al. 2000. Down-regulation of survivin by antisense oligonucleotides increases apoptosis, inhibits cytokinesis and anchorage-independent growth. Neoplasia. 2:235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, A., N.M. Dean, and R.E. Honkanen. 2000. Serine/threonine protein phosphatase type 1gamma1 is required for the completion of cytokinesis in human A549 lung carcinoma cells. J. Biol. Chem. 275:1846–1854. [DOI] [PubMed] [Google Scholar]
- Cooke, C.A., M.M. Heck, and W.C. Earnshaw. 1987. The inner centromere protein (INCENP) antigens: movement from inner centomere to midbody during mitosis. J. Cell Biol. 105:2053–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Luna, S., I. Soria, D. Pulido, J. Ortin, and A. Jimenez. 1988. Efficient transformation of mammalian cells with constructs containing a puromycin-resistance marker. Gene. 62:121–126. [DOI] [PubMed] [Google Scholar]
- De Souza, C.P., A.H. Osmani, L.P. Wu, J.L. Spotts, and S.A. Osmani. 2000. Mitotic histone H3 phosphorylation by the NIMA kinase in Auspergillus nidulans. Cell. 102:293–302. [DOI] [PubMed] [Google Scholar]
- Earnshaw, W.C., and R.L. Bernat. 1991. Chromosomal passengers: toward an integrated view of mitosis. Chromosoma. 100:139–146. [DOI] [PubMed] [Google Scholar]
- Earnshaw, W.C., and C.A. Cooke. 1991. Analysis of the distribution of the INCENPs throughout mitosis reveals the existence of a pathway of structural changes in the chromosomes during metaphase and early events in cleavage furrow formation. J. Cell Sci. 98:443–461. [DOI] [PubMed] [Google Scholar]
- Fang, G., H. Yu, and M.W. Kirschner. 1998. Direct binding of CDC20 protein family members activates the anaphase-promoting complex in mitosis and G1. Mol. Cell. 2:163–171. [DOI] [PubMed] [Google Scholar]
- Francisco, L., and C.S. Chan. 1994. Regulation of yeast chromosome segregation by Ipl1 protein kinase and type 1 protein phosphatase. Cell Mol. Biol. Res. 40:207–213. [PubMed] [Google Scholar]
- Francisco, L., W. Wang, and C.S. Chan. 1994. Type 1 protein phosphatase acts in opposition to IpL1 protein kinase in regulating yeast chromosome segregation. Mol. Cell. Biol. 14:4731–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giet, R., and D.M. Glover. 2001. Drosophila aurora B kinase is required for histone H3 phosphorylation and condensin recruitment during chromosome condensation and to organize the central spindle during cytokinesis. J. Cell Biol. 152:669–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossen, M., and H. Bujard. 1992. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA. 89:5547–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan, K.L., and J.E. Dixon. 1991. Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal. Biochem. 192:262–267. [DOI] [PubMed] [Google Scholar]
- Haigler, H.T., J.A. Mangili, Y. Gao, J. Jones, and N.D. Horseman. 1992. Identification and characterization of columbid annexin Icp37. Insights into the evolution of annexin I phosphorylation sites. J. Biol. Chem. 267:19123–19129. [PubMed] [Google Scholar]
- Hendzel, M.J., Y. Wei, M.A. Mancini, A. Van Hooser, T. Ranalli, B.R. Brinkley, D.P. Bazett-Jones, and C.D. Allis. 1997. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 106:348–360. [DOI] [PubMed] [Google Scholar]
- Howman, E.V., K.J. Fowler, A.J. Newson, S. Redward, A.C. MacDonald, P. Kalitsis, and K.H. Choo. 2000. Early disruption of centromeric chromatin organization in centromere protein A (Cenpa) null mice. Proc. Natl. Acad. Sci. USA. 97:1148–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, J.Y., Z.W. Sun, X. Li, M. Reuben, K. Tatchell, D.K. Bishop, J.M. Grushcow, C.J. Brame, J.A. Caldwell, D.F. Hunt, R. Lin, M.M. Smith, and C.D. Allis. 2000. Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell. 102:279–291. [DOI] [PubMed] [Google Scholar]
- Hwang, P.K., and R.J. Fletterick. 1986. Convergent and divergent evolution of regulatory sites in eukaryotic phosphorylases. Nature. 324:80–84. [DOI] [PubMed] [Google Scholar]
- Johnson, L.N., M.E. Noble, and D.J. Owen. 1996. Active and inactive protein kinases: structural basis for regulation. Cell. 85:149–158. [DOI] [PubMed] [Google Scholar]
- Kaitna, S., M. Mendoza, V. Jantsch-Plunger, and M. Glotzer. 2000. Incenp and an aurora-like kinase form a complex essential for chromosome segregation and efficient completion of cytokinesis. Curr. Biol. 10:1172–1181. [DOI] [PubMed] [Google Scholar]
- Li, B., S. Oestreich, and T. de Lange. 2000. Identification of human Rap1: implications for telomere evolution. Cell. 101:471–483. [DOI] [PubMed] [Google Scholar]
- Liao, H., G. Li, and T.J. Yen. 1994. Mitotic regulation of microtubule cross-linking activity of CENP-E kinetochore protein. Science. 265:394–398. [DOI] [PubMed] [Google Scholar]
- Mackay, A.M., A.M. Ainsztein, D.M. Eckley, and W.C. Earnshaw. 1998. A dominant mutant of inner centromere protein (INCENP), a chromosomal protein, disrupts prometaphase congression and cytokinesis. J. Cell Biol. 140:991–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins, J.M., and J.R. McIntosh. 1982. Isolation and initial characterization of the mammalian midbody. J. Cell Biol. 94:654–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata-Hori, M., K. Fumoto, Y. Fukuta, T. Iwasaki, A. Kikuchi, M. Tatsuka, and H. Hosoya. 2000. Myosin II regulatory light chain as a novel substrate for AIM-1, an aurora/Ipl1p-related kinase from rat. J. Biochem. 128:903–907. [DOI] [PubMed] [Google Scholar]
- Murnion, M.E., R.R. Adams, D.M. Callister, C.D. Allis, W.C. Earnshaw, and J.R. Swedlow. 2001. Chromatin-associated protein phosphatase 1 regulates aurora-B and histone H3 phosphorylation. J. Biol. Chem. 276:26656–26665. [DOI] [PubMed] [Google Scholar]
- O'Connor, D.S., D. Grossman, J. Plescia, F. Li, H. Zhang, A. Villa, S. Tognin, P.C. Marchisio, and D.C. Altieri. 2000. Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc. Natl. Acad. Sci. USA. 97:13103–13107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano, M., A.M. Theodoras, S.W. Tam, and G.F. Draetta. 1994. Cyclin D1-mediated inhibition of repair and replicative DNA synthesis in human fibroblasts. Genes Dev. 8:1627–1639. [DOI] [PubMed] [Google Scholar]
- Schmiesing, J.A., H.C. Gregson, S. Zhou, and K. Yokomori. 2000. A human condensin complex contianing hCAP-C-hCAP-E and CNAP1, a homolog of Xenopus XCAP-D2, colocalizes with phosphorylated histone H3 during the early stage of mitotic chromosome condensation. Mol. Cell. Biol. 20:6996–7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher, J.M., A. Golden, and P.J. Donovan. 1998. AIR-2: An Aurora/Ipl1-related protein kinase associated with chromosomes and midbody microtubules is required for polar body extrusion and cytokinesis in Caenorhabditis elegans embryos. J. Cell Biol. 143:1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severson, A.F., D.R. Hamill, J.C. Carter, J. Schumacher, and B. Bowerman. 2000. The aurora-related kinase AIR-2 recruits ZEN-4/CeMKLP1 to the mitotic spindle at metaphase and is required for cytokinesis. Curr. Biol. 10:1162–1171. [DOI] [PubMed] [Google Scholar]
- Shelby, R.D., O. Vafa, and K.F. Sullivan. 1997. Assembly of CENP-A into centromeric chromatin requires a cooperative array of nucleosomal DNA contact sites. J. Cell Biol. 136:501–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoufias, D.A., C. Mollinari, F.B. Lacroix, and R.L. Margolis. 2000. Human survivin is a kinetochore-associated passenger protein. J. Cell Biol. 151:1575–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogawa, K., T. Yamada, Y. Funamoto, K. Kohno, H. Nishikawa, F. Kishida, F. Hamazaki, N. Yamashita, and K. Matsumoto. 1994. Selective increase in expression of isoform PP1 gamma 1 of type-1 protein phosphatase in chondrosarcoma cells. Res. Commun. Mol. Pathol. Pharmacol. 86:375–378. [PubMed] [Google Scholar]
- Sogawa, K., T. Yamada, S. Oka, K. Kawasaki, S. Mori, H. Tanaka, H. Norimatsu, Y. Cai, H. Kuwabara, H. Shima, et al. 1995. Enhanced expression of catalytic subunit isoform PP1 γ1 phosphatase type 1 associated with malignancy of osteogenic tumor. Cancer Lett. 89:1–6. [DOI] [PubMed] [Google Scholar]
- Sogawa, K., T. Masaki, A. Miyauchi, A. Sugita, K. Kito, N. Ueda, K. Miyamoto, K. Okazaki, K. Okutani, and K. Matsumoto. 1997. Enhanced expression of PP1 γ1, a catalytic subunit isoform of protein phosphatase type 1, in invasive ductal carcinoma of the breast. Cancer Lett. 112:263–268. [DOI] [PubMed] [Google Scholar]
- Sorensen, C.S., C. Lukas, E.R. Kramer, J.M. Peters, J. Bartek, and J. Lukas. 2000. Nonperiodic activity of the human anaphase-promoting complex-Cdh1 ubiquitin ligase results in continuous DNA synthesis uncoupled from mitosis. Mol. Cell. Biol. 20:7613–7623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speliotes, E.K., A. Uren, D. Vaux, and H.R. Horvitz. 2000. The survivin-like C. elegans BIR-1 protein acts with the Aurora-like kinase AIR-2 to affect chromosomes and the spindle midzone. Mol. Cell. 6:211–223. [DOI] [PubMed] [Google Scholar]
- Stoler, S., K.C. Keith, K.E. Curnick, and M. Fitzgerald-Hayes. 1995. A mutation in CSE4, an essential gene encoding a novel chromatin- associated protein in yeast, causes chromosome nondisjunction and cell cycle arrest at mitosis. Genes Dev. 9:573–586. [DOI] [PubMed] [Google Scholar]
- Straight, A.F., and C.M. Field. 2000. Microtubules, membranes and cytokinesis. Curr. Biol. 10:R760–R770. [DOI] [PubMed] [Google Scholar]
- Sullivan, K.F., M. Hechenberger, and K. Masri. 1994. Human CENP-A contains a histone H3–related histone fold domain that is required for targeting to the centromere. J. Cell Biol. 127:581–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsuka, M., H. Katayama, T. Ota, T. Tanaka, S. Odashima, F. Suzuki, and Y. Terada. 1998. Multinuclearity and increased ploidy caused by overexpression of the aurora- and Ipl1-like midbody-associated protein mitotic kinase in human cancer cells. Cancer Res. 58:4811–4816. [PubMed] [Google Scholar]
- Terada, Y., M. Tatsuka, F. Suzuki, Y. Yasuda, S. Fujita, and M. Otsu. 1998. AIM-1: a mammalian midbody-associated protein required for cytokinesis. EMBO J. 17:667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uren, A.G., L. Wong, M. Pakusch, K.J. Fowler, F.J. Burrows, D.L. Vaux, and K.H. Choo. 2000. Survivin and the inner centromere protein INCENP show similar cell-cycle localization and gene knockout phenotype. Curr. Biol. 10:1319–1328. [DOI] [PubMed] [Google Scholar]
- Warburton, P.E., C.A. Cooke, S. Bourassa, O. Vafa, B.A. Sullivan, G. Stetten, G. Gimelli, D. Warburton, C. Tyler-Smith, K.F. Sullivan, et al. 1997. Immunolocalization of CENP-A suggests a distinct nucleosome structure at the inner kinetochore plate of active centromeres. Curr. Biol. 7:901–904. [DOI] [PubMed] [Google Scholar]
- Waters, J.C., R.H. Chen, A.W. Murray, G.J. Gorbsky, E.D. Salmon, and R.B. Nicklas. 1999. Mad2 binding by phosphorylated kinetochores links error detection and checkpoint action in mitosis. Curr. Biol. 9:649–652. [DOI] [PubMed] [Google Scholar]
- Wei, Y., L. Yu, J. Bowen, M.A. Gorovsky, and C.D. Allis. 1999. Phosphorylation of histone H3 is required for proper chromosome condensation and segregation. Cell. 97:99–109. [DOI] [PubMed] [Google Scholar]
- Yamada, T., K. Sogawa, T. Masaki, Y. Funamoto, K. Kohno, S. Oka, H. Norimatsu, and K. Matsumoto. 1994. Enhanced expression of catalytic subunit isoform PP1 γ 1 of protein phosphatase type 1 in malignant fibrous histiocytoma. Res. Commun. Mol. Pathol. Pharmacol. 86:125–128. [PubMed] [Google Scholar]
- Zeitlin, S.G., C.M. Barber, C.D. Allis, and K.F. Sullivan. 2001. Differential regulation of CENP-A and histone H3 phosphorylation in G2/M. J. Cell Sci. 114:653–661. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}