

Abstract
We discovered that many proteins located in the kinetochore outer domain, but not the inner core, are depleted from kinetochores and accumulate at spindle poles when ATP production is suppressed in PtK1 cells, and that microtubule depolymerization inhibits this process. These proteins include the microtubule motors CENP-E and cytoplasmic dynein, and proteins involved with the mitotic spindle checkpoint, Mad2, Bub1R, and the 3F3/2 phosphoantigen. Depletion of these components did not disrupt kinetochore outer domain structure or alter metaphase kinetochore microtubule number. Inhibition of dynein/dynactin activity by microinjection in prometaphase with purified p50 “dynamitin” protein or concentrated 70.1 anti-dynein antibody blocked outer domain protein transport to the spindle poles, prevented Mad2 depletion from kinetochores despite normal kinetochore microtubule numbers, reduced metaphase kinetochore tension by 40%, and induced a mitotic block at metaphase. Dynein/dynactin inhibition did not block chromosome congression to the spindle equator in prometaphase, or segregation to the poles in anaphase when the spindle checkpoint was inactivated by microinjection with Mad2 antibodies. Thus, a major function of dynein/dynactin in mitosis is in a kinetochore disassembly pathway that contributes to inactivation of the spindle checkpoint.
Keywords: mitosis; mitotic spindle checkpoint; Mad2; CENP-E; cytoplasmic dynein
Introduction
Mammalian kinetochores are functionally important for mitosis, as they attach chromosomes to spindle microtubule plus ends, contribute to force generation for chromosome movement, and play a major role in the mitotic spindle checkpoint (Rieder and Salmon, 1998; Maney et al., 1999; Shah and Cleveland, 2000). Kinetochores have an inner core of proteins embedded in the heterochromatin anchoring the kinetochore to the centromere, plus an outer domain containing binding sites for the plus ends of kinetochore microtubules and a fibrous corona (Rieder, 1982; McEwen et al., 1998; Rieder and Salmon, 1998; Maney et al., 1999). Protein components of the inner core include the CREST antigens CENP-A, CENP-B, and CENP-C (Maney et al., 1999). Outer domain proteins include the microtubule motor proteins CENP-E (a kinesin-related protein) and cytoplasmic dynein (and its associated protein complexes dynactin, Zw10, Rod, and CLIP-170), and the mitotic spindle checkpoint proteins Mad1, Mad2, Bub1, BubR1 (a yeast Mad3 homologue), Bub3, and the 3F3/2 phosphoantigen (Rieder and Salmon, 1998; Maney et al., 1999; Shah and Cleveland, 2000; Hoyt, 2001). Normally, as kinetochores interact with microtubules in mitosis and form kinetochore microtubules, the concentrations of many motor and spindle checkpoint proteins become reduced (Hoffman et al., 2001). This reduction appears to be important for preventing errors in chromosome alignment produced by individual kinetochores becoming attached to the ends of microtubules from opposite poles (merotelic attachment) (Cimini et al., 2001), as well as for inactivating the spindle checkpoint signal (Howell et al., 2000).
The mitotic spindle checkpoint prevents errors in chromosome segregation, thus preventing aneuploidy, tumor progression, and cell death (Wassmann and Benezra, 2001). Spindle checkpoint proteins were originally identified by yeast genetic screens and were shown to concentrate at unattached mammalian kinetochores with no or few kinetochore microtubules (Maney et al., 1999; Shah and Cleveland, 2000; Hoffman et al., 2001; Hoyt, 2001). In mammalian tissue cells, Mad2 is essential for spindle checkpoint activity (Chen et al., 1996; Li and Benezra, 1996; Gorbsky et al., 1998; Waters et al., 1998; Canman et al., 2000; Dobles et al., 2000) and exchanges rapidly at kinetochores with the cytoplasmic pool (half-life is ∼25 s at kinetochores) (Howell et al., 2000). This rapid exchange helps explain how a single unattached kinetochore may delay anaphase onset (Rieder et al., 1995), i.e., by rapidly catalyzing the formation of inhibitory Mad2–Cdc20 complexes and releasing them into the cytosol where they can prevent activation of the anaphase-promoting complex/cyclosome (APC/C)* (Kallio et al., 1998; Howell et al., 2000; Shah and Cleveland, 2000). BubR1 has also been shown to bind and inhibit Cdc20 activation of APC (Sudakin et al., 2001; Tang et al., 2001). Other spindle checkpoint proteins at unattached kinetochores are thought to catalyze Mad2 or BubR1 complex formation with Cdc20 (Shah and Cleveland, 2000; Hoyt, 2001; Skoufias et al., 2001; Sudakin et al., 2001).
The molecular mechanisms for checkpoint inactivation are not well understood, but checkpoint inactivation has been correlated with loss of kinetochore-bound Mad2 and dephosphorylation of the kinetochore 3F3/2 antigen (Gorbsky and Ricketts, 1993; Nicklas, 1997; Rieder and Salmon, 1998; Maney et al., 1999; Shah and Cleveland, 2000). Loss of kinetochore Mad2 depends mainly on kinetochore microtubule formation, whereas loss of 3F3/2 staining depends on tension generated by pulling forces at metaphase kinetochores (Nicklas et al., 1995; Waters et al., 1998). Mad2, cytoplasmic dynein, dynactin, CLIP-170, Rod, and Zw10 are unique in that they become substantially depleted at metaphase kinetochores (Williams et al., 1996; Starr et al., 1997, 1998; Maney et al., 1999; Scaërou et al., 1999; Howell et al., 2000; King et al., 2000; Hoffman et al., 2001), whereas Bub1, BubR1, Bub3, and the motor CENP-E are moderately depleted (Maney et al., 1999; Hoffman et al., 2001).
In our recent studies of Mad2 assembly dynamics in vivo (Howell et al., 2000), we developed a simple cellular ATP reduction assay and live cell imaging methods to demonstrate that Mad2 and its binding sites are transported from prometaphase kinetochores along spindle microtubules to the spindle poles. Upon ATP reduction, Mad2 disappeared from kinetochores and accumulated at spindle poles only in the presence of a spindle (i.e., no nocodazole treatment) and failed to accumulate when kinetochores lacked Mad2 as they do in metaphase (Howell et al., 2000). Here we use this ATP reduction assay to show that CENP-E, BubR1, cytoplasmic dynein, and the 3F3/2 antigen also undergo microtubule-dependent spindle pole accumulation in PtK1 cells. Cytoplasmic dynein, a minus-end–directed microtubule motor, has the correct polarity to produce depletion of kinetochore proteins and to transport them poleward. Inhibition of dynein/dynactin by microinjection of prometaphase cells with p50 “dynamitin” or the 70.1 anti-dynein antibody blocked protein redistribution from kinetochores to the poles, prevented anaphase onset, and resulted in accumulation of Mad2 at metaphase kinetochores with normal numbers of kinetochore microtubules. In contrast, dynein/dynactin inhibition did not prevent chromosome congression to the metaphase plate nor block anaphase chromosome segregation or cytokinesis when the spindle checkpoint was inactivated by microinjection of Mad2 antibodies in prometaphase.
Results
Redistribution of kinetochore proteins to the spindle poles occurs for outer domain but not inner core components
To test for kinetochore protein transport along spindle microtubules to the poles we used the ATP reduction assay developed by Howell et al. (2000). For outer domain proteins we tested Mad2, BubR1, CENP-E, and cytoplasmic dynein (Fig. 1, A and B) and the 3F3/2 antigen (Fig. 1, C and D). Consistent with previous studies (Kallio et al., 1998; Waters et al., 1998; Howell et al., 2000; Hoffman et al., 2001), prometaphase PtK1 cells treated with saline alone showed strong staining for Mad2, cytoplasmic dynein, and 3F3/2 at unattached and partially attached prometaphase kinetochores, whereas BubR1 and CENP-E localized to all kinetochores (Fig. 1, A and C). After a 30-min incubation with 5 mM sodium azide and 1 mM 2-deoxyglucose to reduce ATP to 5–10% of normal levels (see Materials and methods), Mad2, BubR1, CENP-E, and 3F3/2 fluorescence diminished at kinetochores and concentrated at spindle poles (Fig. 1, A–C). Cytoplasmic dynein fluorescence also increased at the spindle poles, but dim fluorescence was occasionally evident on a few kinetochores and along spindle microtubules (Fig. 1 A). After a 10-min washout period in saline, ATP recovered to 50–75% normal levels, and fluorescence returned at kinetochores and diminished at the spindle poles for all these outer domain proteins (Fig. 1, A and C). Notably, Mad2 and cytoplasmic dynein fluorescence returned only to unattached or partially attached kinetochores and were not evident on fully attached, metaphase-aligned kinetochores after inhibitor washout (Fig. 1 A). Unlike Mad2 and cytoplasmic dynein, the kinetochore components CENP-E and BubR1 were evident on all kinetochores after inhibitor washout, consistent with previous findings (Fig. 1 A; Hoffman et al., 2001). After washout, most kinetochores in 3F3/2 stained cells appeared labeled, perhaps because tension is not fully regained during the brief washout period (Fig. 1 C). Quantification of kinetochore and pole fluorescence for these experiments is presented in Table I. Microtubule depolymerization with nocodazole prevented redistribution of outer domain proteins from kinetochores to the poles (Fig. 1, B and D; Table II), showing that this process depends on spindle microtubules. Quantification of kinetochore fluorescence in nocodazole cells treated ± ATP inhibitors revealed similar values between controls and inhibitor treatments, with exception of the 3F3/2 antigen which was reduced 50% after azide/deoxyglucose treatment (Table II), an effect likely due to dephosphorylation of the 3F3/2 epitope. Unlike the outer domain proteins tested, we found no loss in kinetochore staining or increase in concentration at the spindle poles for the inner domain CREST antigens after 30-min treatment of cells with ATP inhibitors (Fig. 1 E; Table I).
Figure 1.
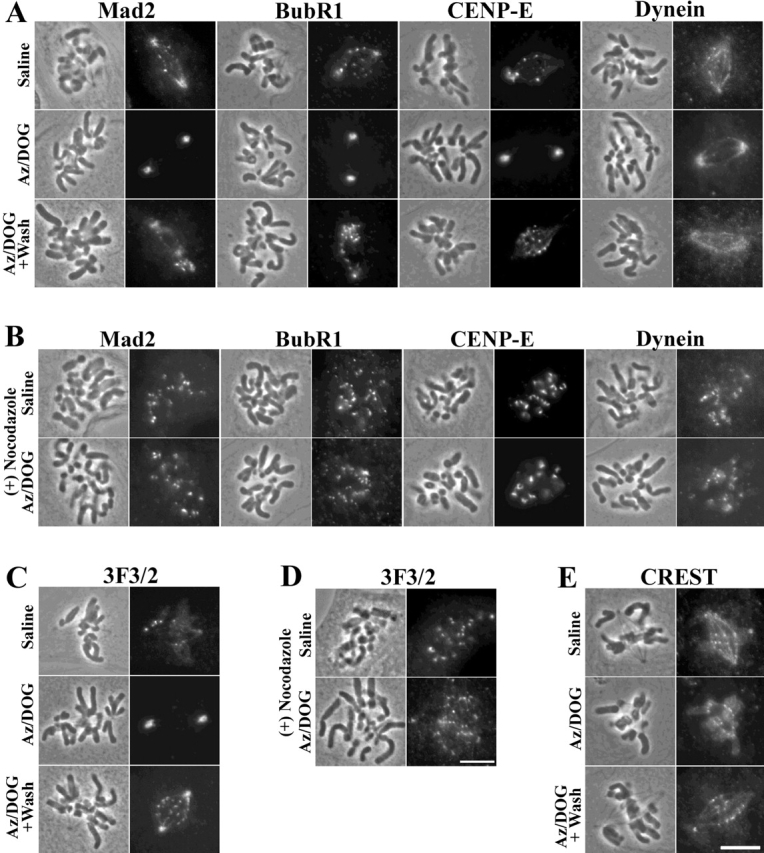
Outer domain kinetochore components localize to spindle poles after ATP reduction if the mitotic spindle is present. PtK1 cells were incubated for 30 min in either saline alone, saline supplemented with 5 mM Az/DOG, or saline + Az/DOG for 30 min followed by a 10-min rinse in saline (Az/DOG + wash). This assay was done both in the absence (A, C, and E) and presence (B and D) of 20 μM nocodazole. Mad2, BubR1, CENP-E, dynein (A), and 3F3/2 (C) fluorescence diminished at kinetochores and concentrated at spindle poles after inhibitor treatment in the absence of nocodazole. After washout of the inhibitors, Mad2, BubR1, CENP-E, and dynein fluorescence recovered at kinetochores and diminished at spindle poles, similar to the localization pattern seen with saline alone (A) (Az/DOG + Wash). 3F3/2 fluorescence reappeared on most kinetochores and was reduced at the spindle poles after inhibitor washout (C) (Az/DOG + wash). Incubation of cells with 20 μM nocodazole for 15 min before and during inhibitor treatment blocked this redistribution pattern for Mad2, BubR1, CENP-E, dynein, and 3F3/2 (B and D). In contrast to the outer domain kinetochore components, the inner domain CREST antigens remained localized to kinetochores after inhibitor treatment (E). For each cell, phase-contrast images are on the left and the corresponding fluorescent images are on the right. Bars, 10 μm.
Table I.
Integrated fluorescence of prometaphase kinetochores and spindle poles from cells treated ± ATP inhibitors and immunostained for various spindle checkpoint components and motor proteins
| Prometaphase kinetochores | Saline | Azide/DOG | Washout |
|---|---|---|---|
| Mad2 | |||
| n | 61 | 65 | 47 |
| mean ± SD | 4.3 ± 2.1 | 0.4 ± 0.8 | 5.3 ± 3.5 |
| ratio to control | 1 | 0.10 | 1.23 |
| BubR1 | |||
| n | 48 | 85 | 87 |
| mean ± SD | 19.9 ± 7.5 | 2.9 ± 2.9 | 22.1 ± 7.9 |
| ratio to control | 1 | 0.15 | 1.11 |
| CENP-E | |||
| n | 61 | 76 | 72 |
| mean ± SD | 10.7 ± 4.8 | 1.3 ± 2.3 | 8.1 ± 3.5 |
| ratio to control | 1 | 0.12 | 0.76 |
| Dynein | |||
| n | 105 | 72 | 113 |
| mean ± SD | 3.3 ± 2.7 | 0.8 ± 1.3 | 2.1 ± 1.2 |
| ratio to control | 1 | 0.24 | 0.64 |
| 3F3/2 | |||
| n | 51 | 51 | 43 |
| mean ± SD | 2.2 ± 0.8 | 0.3 ± 0.4 | 2.1 ± 0.6 |
| ratio to control | 1 | 0.12 | 0.93 |
| CREST | |||
| n | 47 | 34 | 51 |
| mean ± SD | 2.8 ± 0.7 | 2.9 ± 0.7 | 2.7 ± 0.6 |
| ratio to control | 1 | 1.04 | 0.96 |
| Spindle poles | |||
| Mad2 | 12 | 14 | 10 |
| n | 25.5 ± 11.7 | 147.8 ± 41.2 | 28.1 ± 15.8 |
| mean ± SD | 1 | 5.79 | 1.10 |
| ratio to control | |||
| BubR1 | 12 | 12 | 12 |
| n | 0.1 ± 0.04 | 0.3 ± 0.3 | 0.1 ± 0.06 |
| mean ± SD | 1 | 2.77 | 0.87 |
| ratio to control | |||
| CENP-E | 14 | 14 | 12 |
| n | 33.1 ± 37.7 | 141.8 ± 79.7 | 29.5 ± 30.1 |
| mean ± SD | 1 | 4.28 | 0.90 |
| ratio to control | |||
| Dynein | 16 | 12 | 10 |
| n | 43.8 ± 15.5 | 140.3 ± 45.8 | 26.8 ± 10.1 |
| mean ± SD | 1 | 3.20 | 0.61 |
| ratio to control | |||
| 3F3/2 | 10 | 10 | 12 |
| n | 39.9 ± 2.2 | 57.7 ± 10.4 | 36.0 ± 1.9 |
| mean ± SD | 1 | 1.45 | 0.90 |
| ratio to control | |||
| CREST | 12 | 12 | 10 |
| n | 23.3 ± 0.9 | 23.3 ± 1.0 | 22.7 ± 0.6 |
| mean ± SD | 1 | 1 | 0.97 |
| ratio to control |
To identify kinetichores, cells were double labeled with CREST antibodies for each treatment. Spindle poles were identified using phase contrast and/or fluorescence microscopy, and imaging conditions remained identical. With the exception of CREST, all other kinetochore components demonstrated significantly different mean fluorescence intensities at kinetochores and poles after azide/DOG treatment compared with saline and washout (P < 0.01). n = number of kinetochores and spindle poles measured per condition.
Table II.
Integrated fluorescence of kinetochores from nocodazole cells treated ± ATP inhibitors and immunostained for various spindle checkpoint components and motor proteins
| Kinetochores | Saline | Azide/DOG | Washout |
|---|---|---|---|
| Mad2 | |||
| n | 27 | 28 | 29 |
| mean ± SD | 4.6 ± 1.0 | 4.1 ± 0.8 | 4.4 ± 0.8 |
| ratio to control | 1 | 0.89 | 0.96 |
| BubR1 | |||
| n | 46 | 31 | 36 |
| mean ± SD | 9.2 ± 2.0 | 7.2 ± 2.9 | 9.0 ± 2.2 |
| ratio to control | 1 | 0.78 | 0.98 |
| CENP-E | |||
| n | 30 | 40 | 30 |
| mean ± SD | 21.6 ± 12.1 | 19.5 ± 7.4 | 23.1 ± 3.9 |
| ratio to control | 1 | 0.90 | 1.07 |
| Dynein | |||
| n | 40 | 26 | 24 |
| mean ± SD | 2.4 ± 1.1 | 2.5 ± 0.6 | 2.4 ± 0.5 |
| ratio to control | 1 | 1.04 | 1 |
| 3F3/2 | |||
| n | 46 | 30 | 44 |
| mean ± SD | 15.7 ± 5.1 | 7.4 ± 3.9 | 12.2 ± 3.1 |
| ratio to control | 1 | 0.47 | 0.78 |
Integrated kinetochore fluorescence was measured using identical imaging conditions. With the exception of BubR1 and 3F3/2, all other kinetochore components demonstrated similar mean fluorescence intensities after azide/DOG treatment when compared to saline and washout treatment (P > 0.01). n = number of kinetochores measured per condition. Mean ± SD = actual values/1,000.
ATP reduction does not disrupt kinetochore fibers or the kinetochore outer plate
ATP inhibitor treatment at similar concentrations has previously been shown to reduce spindle microtubule dynamics and induce astral microtubule growth, but have little effect on spindle morphology (Wadsworth and Salmon, 1988). To examine overall spindle structure during our ATP reduction assay, we obtained confocal images of immuofluorescently stained spindles in prometaphase PtK1 cells incubated with or without sodium azide (Az)/2-deoxyglucose (DOG) for 30 min. As seen in Fig. 2 A, prometaphase spindles remained bipolar and robust kinetochore fibers persisted after inhibitor treatment. Astral microtubule assembly was enhanced after inhibitor treatment as reported previously for BSC1 cells (Fig. 2 A; Wadsworth and Salmon, 1988). Cells progressed normally through mitosis after inhibitor washout (DeBrabender et al., 1981; unpublished data).
Figure 2.
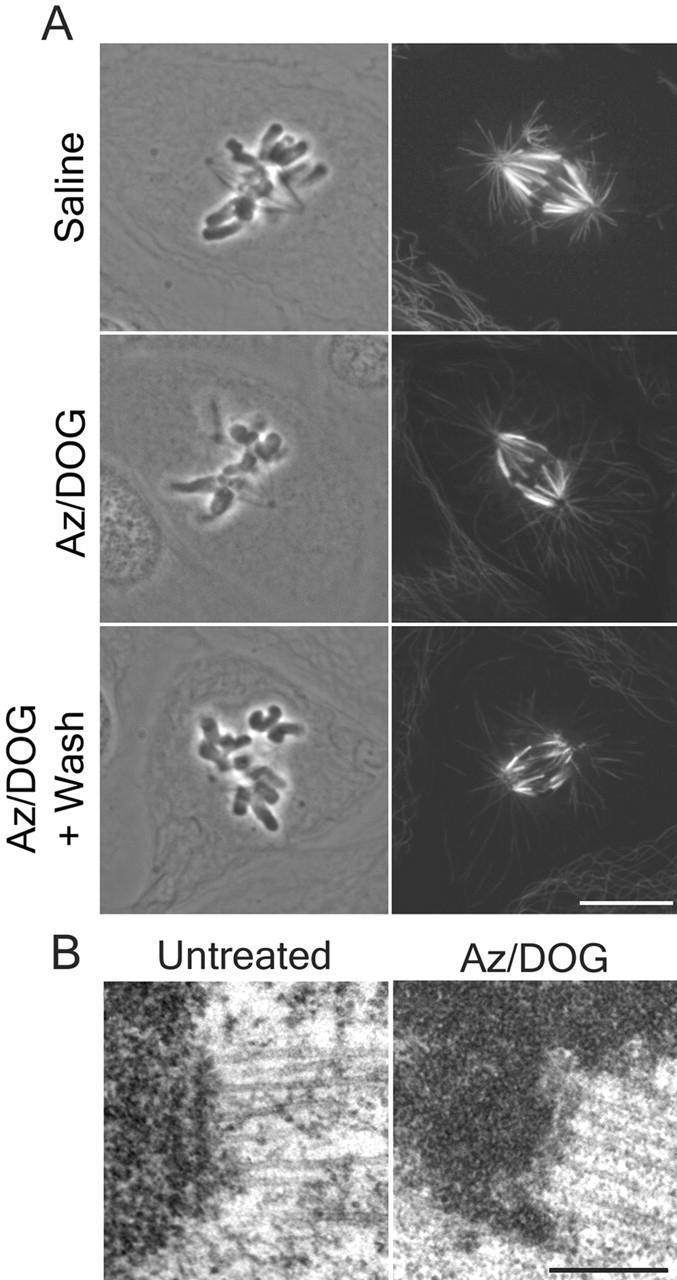
ATP reduction does not disrupt kinetochore fibers, kinetochore outer plate structure, or microtubule attachment. (A) Prometaphase PtK1 cells were processed for tubulin immunofluorescence after treatment with saline alone, saline plus ATP inhibitors, or saline + ATP inhibitors followed by a 10-min rinse. Single plane images were taken by confocal microscopy. (B) Electron micrographs of kinetochores from metaphase-aligned chromosomes from an untreated PtK1 cell and a cell treated with Az/DOG for 30 min. Bars: (A) 10 μm; (B) 0.2 μm.
Electron microscopy of metaphase-aligned chromosomes in cells fixed 30 min after ATP inhibitor treatment showed several important structural features of kinetochores (Fig. 2 B). First, significant reduction of the outer domain checkpoint and motor proteins tested did not disrupt the kinetochore outer plate or kinetochore microtubule plus-end anchorage within the outer plate (Fig. 2 B). Second, coronal filaments appeared reduced after inhibitor treatment (Fig 2 B). Third, kinetochores in inhibitor-treated cells had similar numbers of kinetochore microtubules (24.8 ± 4.8, n = 18) compared with untreated kinetochores (24.3 ± 4.9, n = 62) (McEwen et al., 1997). Thus, loss of most of the outer domain proteins tested did not alter the integrity of the outer plate or maintenance of kinetochore microtubule attachment.
Inhibition of dynein/dynactin blocks protein redistribution from kinetochores to spindle poles
We found 3F3/2 phosphorylation (Gorbsky and Ricketts, 1993; Fig. 1, C and D) and the motor activity responsible for the microtubule-dependent redistribution of outer domain proteins (Fig. 1, A and C) were not inhibited by the 30-min treatment with Az/DOG that reduced ATP to 5–10% of normal levels (see Materials and methods). Paschal and Vallee (1987) showed dynein has good motility at low ATP concentrations (10 μM) in in vitro motility assays, i.e., at ∼0.3–0.4% of the normal 2–3-mM cellular ATP concentration reported for tissue culture cells (Ikehara et al., 1984). Therefore, it seemed likely that dynein activity could be retained in our ATP reduction assays.
To examine dynein/dynactin function in the microtubule-dependent protein redistribution from kinetochores to the poles, we repeated our ATP inhibitor assay in prometaphase cells microinjected with high concentrations of the dynactin component, p50 dynamitin (Echeverri et al., 1996). High p50 levels have been shown to disrupt the dynactin complex (Echeverri et al., 1996; Whittman and Hyman, 1999; Merdes et al., 2000), prevent cytoplasmic dynein/dynactin localization to kinetochores (Echeverri et al., 1996), inhibit cytoplasmic dynein–dependent translocation of membrane vesicles in interphase cells (Burkhardt et al., 1997), and block poleward transport and accumulation at spindle poles of two non-kinetochore proteins, NuMA (Merdes et al., 2000) and TPX2 (Whittman et al., 2000). Dynein normally concentrates 55-fold at kinetochores in nocodazole-treated cells, compared with unattached kinetochores in untreated cells (Hoffman et al., 2001), but p50 injection depleted most if not all dynein from these kinetochores by 30 min (Fig. 3 A; Table III). Importantly, we found p50 injection into mid-prometaphase cells, 15 min before addition of Az/DOG, blocked redistribution from kinetochores to the spindle poles of all three outer domain proteins tested, i.e., Mad2, BubR1, and CENP-E, without substantially disrupting the spindle or spindle poles (Fig. 3 B).
Figure 3.
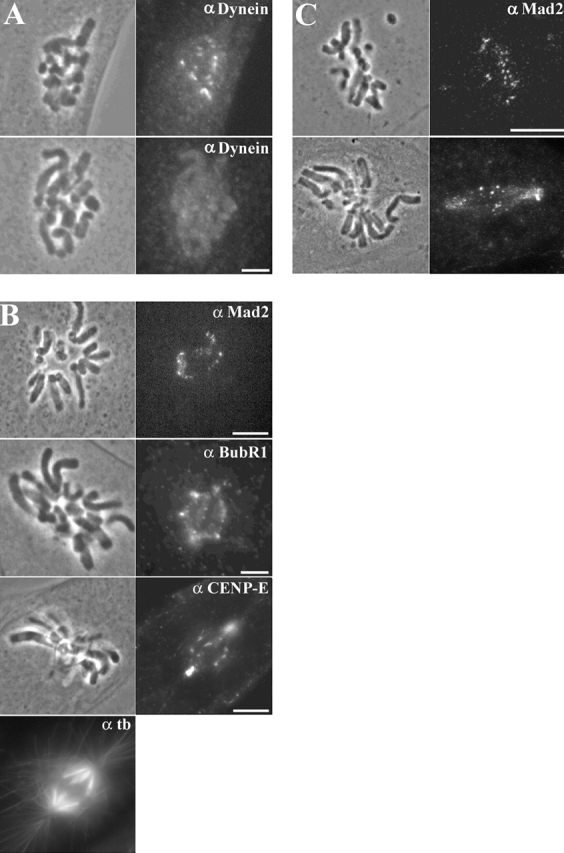
p50 injection prevents dynein localization to kinetochores (A) and blocks poleward transport of kinetochore outer domain components (B and C) in cells treated with Az/DOG. (A) Nocodazole-treated PtK1 cells were uninjected (top) or injected with p50 (bottom), incubated for 30 min, and then processed for dynein immunofluorescence. (B) p50 injected PtK1 cells incubated in Az/DOG for 30 min and processed for Mad2 (top), BubR1 (middle), and CENP-E + tubulin (bottom) immunofluorescence. (C) 70.1 mAb–injected PtK1 cells incubated in Az/DOG for 30 min and processed for Mad2 (top) and CENP-E (bottom) immunofluorescence. Bars, 5 μm.
Table III. Integrated fluorescence for dynein at kinetochores in nocodazole-treated noninjected and p50-injected cells.
| (+) 20 μM nocodazole (30 min) |
||
|---|---|---|
| Noninjected | p50 injected | |
| Dynein | ||
| n | 35 | 32 |
| mean ± SD | 3824 ± 1611 | 210 ± 559 |
| ratio to control | 1 | 0.055 |
Cells were incubated for 30 min in 20 μM nocodazole to enhance visualization of dynein at kinetochores (Hoffman et al., 2001). n = number of kinetochores.
Another approach commonly used to inhibit dynein function is microinjection of cells with concentrated antibodies to dynein intermediate light chain 70.1 (Heald et al., 1996; Gaglio et al., 1997; Faulkner et al., 2000; Merdes et al., 2000; Waterman-Storer et al., 2000). Therefore, we microinjected prometaphase cells with 70.1 anti-dynein antibody, waited 15 min before treating them with the ATP inhibitors for 30 min, and then processed the cells for Mad2 immunofluorescence. As expected from the p50 studies, Mad2 protein remained at kinetochores and did not accumulate at the spindle poles in 70.1 mAb–injected cells (Fig. 3 C). The same effect was seen for CENP-E and BubR1 (Fig. 3 C; unpublished data). These p50 and anti-70.1 studies suggest that dynein/dynactin produces poleward transport along microtubules of kinetochore outer domain proteins like Mad2, BubR1, and CENP-E.
Inhibition of dynein/dynactin function induces a Mad2-dependent mitotic arrest and blocks Mad2 depletion from attached kinetochores
To examine the effect of dynein/dynactin inhibition on mitotic progression in PtK1 cells, prometaphase cells containing an established bipolar spindle were microinjected with either p50 protein or 70.1 antibody and imaged by phase-contrast microscopy. Chromosome congression and oscillations continued in dynein-inhibited cells, yet cells arrested in metaphase, i.e., p50 or 70.1 antibody– injected cells never entered anaphase during the time of observation (up to 3.5 h after metaphase arrest; n = 12) (Fig. 4 A; Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200105093/DC1).
Figure 4.

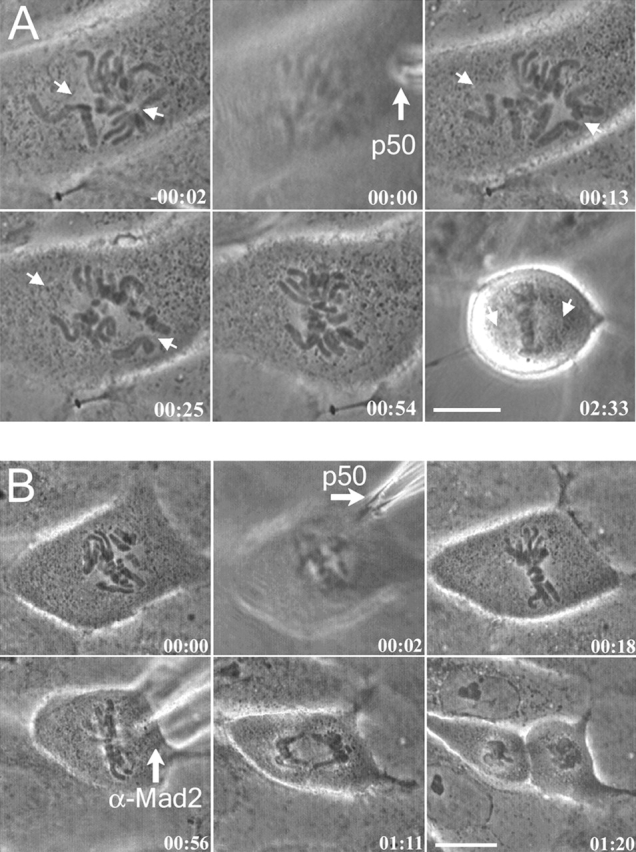
Dynein/dynactin inhibition induces a Mad2-dependent mitotic arrest in PtK1 cells, but does not block chromosomes congression to the spindle equator or anaphase chromosome segregation and cytokinesis after the checkpoint is inactivated by microinjection of Mad2 antibody. (A) Time-lapse of early prometaphase cell microinjected with p50 (time, 00:00) shows chromosome congression and metaphase arrest. (B) Time-lapse of a prometaphase PtK1 microinjected with p50 (time, 00:02) to induce a mitotic block, and then further injected with Mad2 antibodies to inactivate the checkpoint (00:56), inducing anaphase chromosome segregation and cytokinesis. Time, hrs:min. (C) Graph of anaphase A kinetochore to pole movements for four chromosomes measured (K-P, ♦, ⋄) and anaphase B spindle pole elongation (P-P, ▪) for cell in (B). Not all measured chromosomes are plotted for clarity. Bar, 10 μm.
Normally, PtK1 cells enter anaphase 22 min after the last chromosome begins congression to the metaphase plate (Rieder et al., 1995) and within 11 ± 2 min after the last kinetochore becomes depleted of Mad2 (Howell et al., 2000). Therefore, we asked whether the metaphase block induced by inhibition of dynein/dynactin function depends on Mad2 activity. Prometaphase cells were microinjected with p50 protein (Fig. 4 B) or 70.1 antibodies (unpublished data), allowed to arrest in metaphase for at least 45 min, and subsequently microinjected with function-blocking Mad2 antibody (Fig. 4 B). Previous studies have shown that microinjection of prometaphase PtK1 cells with this Mad2 antibody results in precocious anaphase onset (Canman et al., 2000). Consistent with Canman et al. (2000), we found control cells injected with Mad2 antibodies alone entered precociously into anaphase ∼14.5 min after microinjection (n = 8). Microinjection of anti-Mad2 antibodies into p50 or 70.1 mAb–blocked cells resulted in a release from the metaphase arrest and entry into anaphase ∼16 min after Mad2 antibody injection (n = 7) (Fig. 4 B; Video 2, available at http://www.jcb.org/cgi/ content/full/jcb.200105093/DC1). Therefore, Mad2 activity is necessary for the observed mitotic block in dynein-inhibited cells.
Next, we tested whether dynein/dynactin inhibition prevents the normal depletion of Mad2 from kinetochores of metaphase-aligned chromosomes (Waters et al., 1998; Fig. 5 A). Prometaphase cells were microinjected with p50 or 70.1 antibodies, allowed to arrest in metaphase, and then processed for Mad2 and tubulin immunofluorescence. Mad2 was seen at almost all kinetochores on metaphase-aligned chromosomes in either p50 (Fig. 5 A) or 70.1 mAb–injected cells (unpublished data). Quantitative analysis of Mad2 fluorescence showed that kinetochores on aligned chromosomes in the p50 microinjected cells had 25 times more Mad2 than kinetochores on aligned chromosomes in uninjected cells (Table IV). This amount of Mad2 represents ∼25% of the Mad2 that binds to unattached prometaphase kinetochores.
Figure 5.
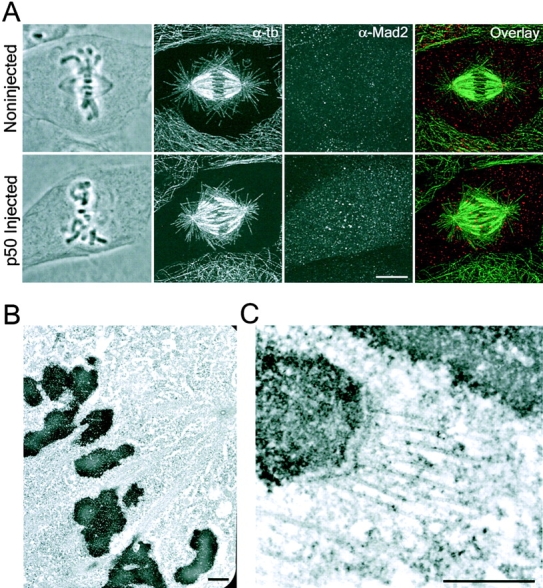
Dynein/dynactin inhibition prevents Mad2 depletion from attached kinetochores with normal numbers of metaphase kinetochore microtubules. (A) Phase contrast and fluorescence images of metaphase PtK1 cells were either uninjected (top) or injected with p50 in early prometaphase (bottom). Cells were fixed 60 min after p50 microinjection and processed for tubulin (α-tb, green) and Mad2 (α-Mad2, red). Note, the gluteraldhyde fixation used to preserve microtubules results in particulate Mad2 throughout the cytoplasm. Low magnification (B) and high magnification (C) electron micrographs of metaphase kinetochore fibers in cells microinjected with p50 in early prometaphase. Counts of kinetochore microtubule numbers at metaphase kinetochores in p50 injected cells (25.5 ± 6.1) were nearly identical to those in uninjected cells (24.3 ± 4.9). Bars: (A) 5 μm; (B) 1 μm; (C) 0.5 μm.
Table IV. Integrated fluorescence of Mad2 at aligned versus unaligned kinetochores in noninjected and p50-injected cells.
| Noninjected | Noninjected | p50 injected | |
|---|---|---|---|
| Aligned | Unaligned | Aligned | |
| Mad2 | |||
| n | 36 | 62 | 59 |
| mean ± SD | 104 ± 227 | 9298 ± 8554 | 2441 ± 1534 |
| ratio to control | 1 | 89 | 24 |
p50-injected cells were processed for Mad2 immunofluorescence after a 45-min block in metaphase. n = number of kinetochores.
One way that dynein/dynactin inhibition could prevent depletion of Mad2 from kinetochores is by blocking kinetochore microtubule formation (Waters et al., 1998). However, kinetochores on metaphase-aligned chromosomes in p50-injected cells exhibited robust fluorescent kinetochore fibers in tubulin immunofluorescence images (Fig. 5 A) and in thin-section electron micrographs (Fig. 5, B and C). Microtubule attachment to metaphase kinetochores in the p50-microinjected cells appeared normal (Fig. 5 C), and counts of kinetochore microtubule numbers (25.5 ± 6.1, n = 47) were nearly identical to those in control cells (24.3 ± 4.9, n = 62), as determined by electron microscopy. In addition, microinjection of p50 into metaphase cells pretreated with 10 μM taxol to stabilize kinetochore microtubules (McEwen et al., 1997; Waters et al., 1998) induced Mad2 accumulation at almost all kinetochores (unpublished data), although very few kinetochores in uninjected cells exhibit Mad2 (Waters et al., 1998). Collectively, these results demonstrate the inhibition of dynein/dynactin in prometaphase prevents depletion of Mad2 from kinetochores on metaphase-aligned chromosomes without preventing normal kinetochore microtubule formation.
Live-cell analysis of Mad2 at kinetochores and loss of poleward transport following dynein/dynactin inhibition
Recently, we found by live cell analysis that Mad2 binding sites are moved along spindle fibers to the poles (Howell et al., 2000). This transport explains why Mad2 disappears from spindle fibers and poles after Mad2 is depleted at kinetochores during kinetochore microtubule formation and chromosome alignment (Howell et al., 2000). If cytoplasmic dynein produces the transport of Mad2 and/or Mad2 binding sites to the poles along spindle microtubules as predicted from the ATP reduction assay, then we expected inhibition of dynein/dynactin activity should also induce loss of Mad2 localization to the spindle fibers and spindle poles.
To visualize Mad2 dynamics in dynein-inhibited cells, we microinjected late prometaphase PtK1 cells containing 1–3 unaligned chromosomes with fluorescent Mad2, and then immediately imaged the cell with fluorescence microscopy. As seen in Fig. 6 and demonstrated in our previous studies (Howell et al., 2000), fluorescent Mad2 localizes strongly to partially attached kinetochores, to the proximal spindle pole, and along the spindle in between the labeled kinetochore and pole. Fluorescent Mad2 is not detectable on fully attached, metaphase-aligned chromosomes. In contrast to previous observations of Mad2 dynamics (Howell et al., 2000), we found Mad2 fluorescence at the spindle poles and fibers proximal to unattached and partially attached kinetochores disappeared quickly (10–15 min after p50 injection) after p50 microinjection, despite the fact that those kinetochores nearby retained Mad2 fluorescence for >1 h (Fig. 6). Mad2 fluorescence also reappeared gradually on kinetochores of some metaphase-aligned chromosomes (Fig. 6). Fluorescent Mad2 remained at kinetochores in p50-injected cells as chromosomes congressed to the spindle equator, further supporting dynein/dynactin activity in Mad2 depletion from kinetochores.
Figure 6.

Fluorescent Mad2 transport and localization to spindle poles disappears in vivo upon inhibition of dynein/dynactin. Time-lapse of a late prometaphase PtK1 cell microinjected with fluorescence Mad2 protein before (time, −00:10:25) and after further injection with p50 (time, 00:00:00). Fluorescent Mad2 localized to unattached or partially attached kinetochores, the spindle fibers in between these kinetochores and the pole, and the proximal spindle pole (arrowheads). No Mad2 fluorescence was evident on the kinetochores of metaphase-aligned chromosomes (top). Following microinjection with p50, fluorescent Mad2 disappeared at the spindle pole but remained bright on the unattached or partially attached kinetochores. Fluorescent Mad2 also accumulated over time on kinetochores of metaphase-aligned chromosomes (arrows). Bar, 5 μm.
To test whether Mad2 is exchanging with cytoplasmic pools at normal rates in p50-blocked cells, we measured fluorescence recovery after photobleaching (FRAP) as described by Howell et al. (2000). The mean half-life of Mad2 at kinetochores in p50-injected cells (33 ± 16 s, n = 8 kinetochores) was similar to that measured in uninjected cells (28 ± 15 s, n =19) (Howell et al., 2000). These results show that Mad2 has a normal turnover rate at kinetochores in the absence of kinetochore dynein/dynactin or dynein/dynactin-driven poleward transport of kinetochore proteins.
Kinetochore tension on metaphase chromosomes is reduced 40% by dynein/dynactin inhibition
Cytoplasmic dynein has been proposed to have a major role in generation of poleward forces for chromosome movement (Maney et al., 1999; Sharp et al., 2000; Banks and Heald, 2001). However, we found chromosomes were able to congress and align near the spindle equator (Fig. 4), and kinetochores accumulated normal numbers of kinetochore microtubules in prometaphase cells microinjected with p50 protein (Fig. 5). To obtain an estimate of the average strength of the pulling forces at metaphase kinetochores in dynein/dynactin-inhibited cells, we measured the average stretch of the centromere between sister kinetochores. Cells were microinjected with p50 and allowed to arrest at metaphase. Control cells and microinjected cells were processed for Mad2 and tubulin immunofluorescence and viewed using confocal microscopy. We found the average interkinetochore distance measured between the ends of sister kinetochore fibers in p50 injected cells (1.9 ± 0.4 μm, n = 61) was significantly shorter than for noninjected metaphase cells (2.6 ± 0.6 μm, n = 44; P < 0.01). We recently determined the length of an unstretched centromere by measuring the distance between late prophase centromeres in PtK1 cells (0.9 ± 0.1 μm, n = 48) (Hoffman et al., 2001). From these numbers, the centromeric stretch in dynein-inhibited cells is ∼ 60% that of control cells, (i.e., [1.9 − 0.9]/[2.6 − 0.9] = 0.59).
Anaphase A velocity is not substantially reduced by the dynein/dynactin inhibition that activates the spindle checkpoint
In our experiments, control Ptk1 cells induced prematurely into anaphase with anti-Mad2 antibodies during mid-prometaphase (n = 6 cells) showed anaphase A kinetochore-to-pole movements at rates of ∼1.3 ± 0.3 μm/min (18 chromosomes), and anaphase B spindle pole separation at rates of ∼1.0 ± 0.1 μm/min. Similarly, in all healthy, double-injected, dynein-inhibited cells like the one shown in Fig. 4 B (four cells injected with p50 and three injected with 70.1 antibody), anaphase chromosome segregation and subsequent cytokinesis between separated chromosomes occurred after anaphase onset with timing similar to controls. For the cell seen in Fig. 4 B, we found anaphase A kinetochore-to-pole movement occurred at ∼0.9 um/min (6 chromosomes measured), whereas anaphase B spindle pole separation occurred at ∼1.1 um/min (Fig. 4 C), similar to rates seen in control cells (see above). Overall, we were able to obtain reliable measurements of anaphase A and B from three out of seven dynein-inhibited cells in which visibility of the spindle poles was not inhibited by the cell-rounding that occurred during the metaphase block. The average values (n = 3 cells, 13 chromosomes) for dynein-inhibited cells were 0.9 ± 0.2 μm/min (P < 0.01) for anaphase A, and 1.2 ± 0.2 μm/min (P > 0.01) for anaphase B pole-to-pole elongation. Thus, dynein/dynactin inhibition appears to significantly reduce anaphase A velocities by ∼33%, and anaphase B movements remain similar to controls.
Discussion
The dynamic assembly of motor and checkpoint proteins at kinetochores depends on dynein/dynactin interactions with spindle microtubules
Our results indicate that outer domain kinetochore motors CENP-E and cytoplasmic dynein and the checkpoint-related proteins Mad2, BubR1, and the 3F3/2 antigen, are in a dynamic assembly at kinetochores with the rate of association from cytoplasmic pools balanced by either the rate of dissociation driven by dynein/dynactin interactions with spindle microtubules, or by direct dissociation into the cytoplasm (Fig. 7). Our ATP reduction assay should be useful for identifying other kinetochore proteins which undergo dynamic assembly at kinetochores, which proteins are more stable (e.g., like the inner core CREST antigens), and which outer plate proteins are responsible for attachment to the plus ends of kinetochore microtubules (Fig. 2 B).
Figure 7.
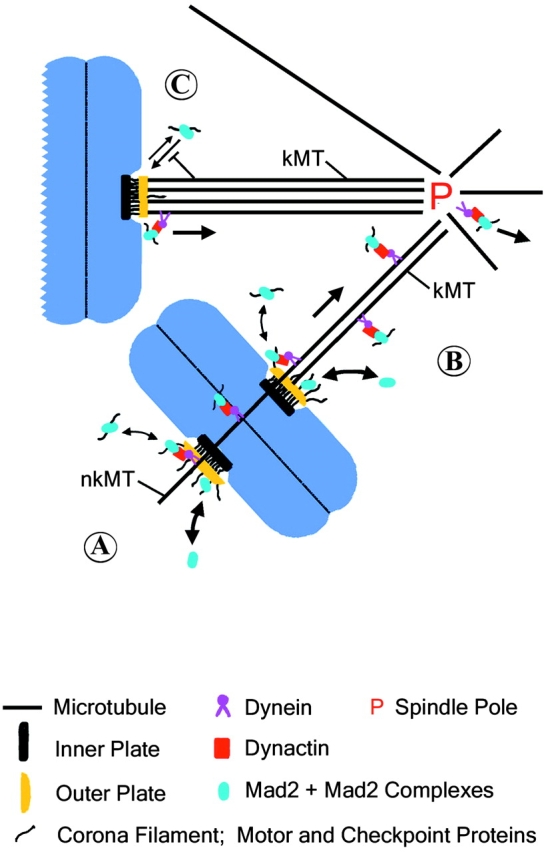
Model of dynein/dynactin-driven poleward transport of kinetochore proteins along spindle microtubules, and the role of this transport in inactivation of the spindle checkpoint activity at kinetochores. (A) Mad2 + Mad2 complexes (blue oval) and other motor and checkpoint proteins (corona filament) assemble from cytoplasmic pools onto unattached kinetochores where the checkpoint proteins catalyze formation of Mad2-Cdc20 inhibitory complexes. (B) Dissociation of Mad2 and other outer domain components occurs either by direct exchange with cytoplasmic pools or through dynein/dynactin-interactions with non-kinetochore (nkMT) or kinetochore (kMT) microtubules. Motor and checkpoint protein complexes are transported poleward by dynein/dynactin where they dissociate into the cytoplasm. (C) Full kinetochore microtubule occupancy on metaphase-aligned chromosomes prevents association of outer domain components, thereby blocking formation of Mad2–Cdc20 inhibitory complexes and allowing for spindle checkpoint inactivation.
At normal ATP concentrations, our data predict that dynein/dynactin continuously remove outer domain motor and spindle checkpoint proteins from kinetochores and transport them poleward, as we previously proposed for the Mad2 binding site from our live-cell fluorescent Mad2 imaging (Howell et al., 2000). In support of this prediction, Drosophila embryos with mutations in cytoplasmic dynein have recently been shown to accumulate dynein, p50 dynamitin and ZW10 at metaphase kinetochores, implying that their depletion from metaphase kinetochores depends on dynein motor function (Wojcik et al., 2001).
It is important to recognize that the poleward transport we infer from our ATP inhibitor assay or by direct observations of fluorescent Mad2 in living cells occurs poleward from unattached, partially attached, and fully attached kinetochores. This indicates that dynein/dynactin-dependent transport of proteins from kinetochores occurs by kinetochore lateral interactions with non-kinetochore microtubules (Fig. 7 A) as well as by lateral and end-on interactions with kinetochore microtubules (Fig. 7, B and C). We found dynein/dynactin-dependent poleward transport of Mad2 particles occurs at velocities of 2–3 μm/min (Howell et al., 2000). Interestingly, similar velocities have been reported for dynein/dynactin-dependent poleward transport of NuMA (Merdes et al., 2000).
Our results show that after washout of the ATP inhibitors, the outer domain kinetochore proteins tested, including cytoplasmic dynein, reaccumulate rapidly (within 10 min) to normal levels at kinetochores (Table I), indicating that the association of these complexes to kinetochores is prevented by the ATP reduction. Because these proteins do not decrease from kinetochores upon ATP depletion in nocodazole-treated cells (with exception of 3F3/2) (Table II) or in dynein/dynactin-inhibited cells, interactions between microtubules and dynein/dynactin are required for protein depletion by ATP inhibition. The conclusion from our ATP inhibitor assay that dynein/dynactin can transport various kinetochore outer domain components along microtubules to the poles suggests that dynein/dynactin is likely involved in removing a protein responsible for regulating or contributing attachment of other outer domain proteins, perhaps, for example, a protein localized to the base of the corona filament (Fig. 7 B), a structure which likely contains binding sites for CENP-E, dynein/dynactin, and various checkpoint proteins (Howell et al., 2000; Maney et al., 1999). Interestingly, ZW10 and Rod are required for accumulation of dynein/dynactin at unattached or partially attached kinetochores (Starr et al., 1998; Chan et al., 2000). Whether ZW10 or Rod are essential for dynein/dynactin-dependent transport of kinetochore proteins poleward along microtubules is an important unanswered question (Chan et al., 2000).
Kinetochore microtubule formation may normally function to block the association pathways (Fig. 7 C) such that constitutive dissociation pathways, including direct dissociation into the cytosol and dynein/dynactin-driven depletion from kinetochores, results in protein depletion from metaphase kinetochores. So far, we only know the approximate relative contributions of these two dissociation pathways for the Mad2 binding site at kinetochores. Inhibition of dynein/dynactin-dependent transport prevents the complete depletion of Mad2 from metaphase kinetochores (Tables III and IV) that are capable of accumulating normal numbers of kinetochore microtubules (Fig. 5), but a 75% reduction does occur indicating the presence of dissociation pathways independent of dynein/dynactin.
In addition to the dynein/dynactin-dependent pathway of disassembly, outer domain proteins may also have individual dissociation rates from their binding sites into the cytosol (Fig. 7). Mad2 clearly does, as its turnover at Mad2 binding sites has a half-life of 20–30 s, even in the presence of dynein/dynactin inhibition as measured by FRAP analysis. Similar data is needed to identify the other motor and checkpoint proteins that dynamically exchange with cytoplasmic pools.
How dynein/dynactin may contribute to spindle checkpoint inactivation
Consistent with others, we found inhibition of dynein/dynactin induces a strong mitotic block (Echeverri et al., 1996; Gaglio et al., 1997; Faulkner et al., 2000; Merdes et al., 2000; Wojcik et al., 2001). Inhibition of dynein/dynactin before mitosis and spindle formation can profoundly disrupt bipolar spindle organization and prevent chromosome congression to a metaphase plate (Vaisberg et al., 1993; Echeverri et al., 1996). In contrast, our studies show that inhibition of dynein/dynactin by p50 microinjection after bipolar spindle formation in prometaphase cells slows but does not block chromosome congression and alignment at the metaphase plate (Fig. 4), and spindle bipolarity is not substantially disrupted during the 45–60-min metaphase block (Fig. 5); however, occasionally, spindles broadened and poles became more displaced during extended metaphase arrests (>1 h). Kinetochores on aligned chromosomes accumulated normal numbers of kinetochore microtubules (Fig. 5). Thus, unlike CENP-E, which appears to be required for recruitment and attachment of kinetochore microtubules (Schaar et al., 1997; Yao et al., 2000; McEwen et al., 2001), dynein/dynactin primarily contributes to checkpoint inactivation by another mechanism.
One way dynein/dynactin may contribute to checkpoint inactivation is by removing Mad2 binding sites from kinetochores by microtubule-dependent transport. This mechanism is supported by our ATP inhibition studies (Figs. 1 and 3), the persistence of substantial Mad2 at kinetochores of metaphase-aligned chromosomes (Table IV) with normal numbers of kinetochore microtubules in cells blocked in metaphase by dynein/dynactin inhibition (Fig. 5), and the transport and the inhabition of Mad2 transport from kinetochores to poles seen by live cell imaging (Fig. 6; Howell et al., 2000).
A second way dynein/dynactin may contribute to checkpoint inactivation is by removing ZW10 and Rod from kinetochores. In support of this hypothesis, depletion of ZW10 or Rod from kinetochores by antibody inhibition (Chan et al., 2000) or protein mutations (Basto et al., 2000) disrupts the spindle checkpoint. In addition, Drosophila embryos with mutant dynein become blocked in metaphase with elevated concentrations of ZW10 at their kinetochores (Wojcik et al., 2001).
Dynein/dynactin could also help inactivate the checkpoint by contributing to tension at kinetochores of aligned chromosomes. In our studies, inhibition of dynein/dynactin induced a 40% reduction in this tension either by reducing force production at kinetochores or by disrupting microtubule anchorage at the spindle poles (Gordon et al., 2001). CENP-E depletion from mammalian tissue cell kinetochores or microtubule stabilization with taxol is accompanied by 80–90% reduction of tension without preventing MaD2 depletion from kinetochores with nearly normal numbers of kinetochore microtubules (Yao et al., 2000; McEwen et al., 2001). As a result, it seems unlikely that the 40% reduction of tension upon dynein/dynactin inhibition contributes to maintenance of kinetochore Mad2. However, this reduction in tension could contribute to checkpoint activation by other mechanisms, such as those that regulate BubR1 inhibition of Cdc20 activation of the APC/C (Skoufias et al., 2001; Sudakin et al., 2001; Tang et al., 2001).
Dynein/dynactin may have a more critical function in spindle checkpoint inactivation than in chromosome movement
Typically in mammalian tissue cells, cytoplasmic dynein is barely, if at all, detectable at kinetochores in metaphase and anaphase (Hoffman et al., 2001). Here we found that inhibition of dynein/dynactin in prometaphase cells did not block pre-anaphase chromosome oscillations, chromosome congression to the spindle equator, or anaphase chromosome segregation when the spindle checkpoint was inactivated. Anaphase A velocities were reduced by ∼30%, similar to the 40% reduction in tension measured for kinetochores on chromosomes at the metaphase plate. In general, chromosome movements in mammalian cells appear less sensitive to dynein/dynactin inhibition (Echeverri et al., 1996; Gaglio et al., 1997; Faulkner et al., 2000; O'Connell and Wang, 2000), in comparison with recent findings for Drosophila meiosis and embryonic mitosis (Savoian et al., 2000; Sharp et al., 2000). The insensitivity of chromosome movement to dynein/dynactin inhibition and our ATP reduction results indicate that, for mammalian tissue cells, proteins other than cytoplasmic dynein and CENP-E are critical for the anchorage of kinetochore microtubule plus ends to the outer plate, and for dynamically coupling plus end polymerization/depolymerization to chromosome movement (Inoue and Salmon, 1995; Desai and Mitchison, 1997).
Materials and methods
ATP inhibitor assay
All chemicals were purchased from Sigma-Aldrich unless otherwise noted. PtK1 cells (American Type Culture Collection) were cultured as described previously (Howell et al., 2000). For the ATP inhibitor studies, cells were rinsed in saline (140 mM NaCl, 5 mM KCl, 0.6 mM MgSO4, 0.1 mM CaCl2, 1 mM Na2HPO4, 1 mM KH2PO4, pH 7.3) to remove tissue culture medium and placed in either saline g (saline with 4.5 g/liter d-glucose) or saline plus 5 mM Az and 1 mM DOG for 30 min at 37°C. Cells were then either processed for immunofluorescence (see below) or rinsed two times for 5 min each in saline g and then processed. For the microtubule depolymerization assays, 20 μM nocodazole was added to all solutions. Using a Trypan blue exclusion assay, we found no decrease in cell viability after ATP inhibitor treatment.
ATP levels were quantitatively measured using a luciferan/luciferase-based assay essentially as described previously (Kangas et al., 1984; Garewal et al., 1986). Two different methods, trichloric acetic acid (TCA) (Kangas et al., 1984) and boiling (Garewal et al., 1986), were used to release ATP from PtK1 cells with both yielding similar results. Each ATP assay was performed in duplicate or triplicate per assay condition and the overall experiment was repeated three times for each method described. An ATP determination kit containing substrate, enzyme, and buffer was purchased from Molecular Probes. In brief, PtK1 cells were grown in 6-well plates to cell density of 2 × 105 cells/mL. For the boiling method, cells were trypsinized, pelleted at 1,000 rpms, rinsed in HBSS (GIBCO BRL), repelleted, and then resuspended in either 1 mL of saline g or saline + ATP inhibitors for 30 min at 37°C. Cell counts were determined during this incubation period using a hemacytometer. Cells were then pelleted at 1,000 rpms for 4 min, resuspended in 100 μL deionized water ± ATP inhibitors, immediately boiled for 5 min, and frozen at −20°C until ATP measurements were made. For washout conditions, cells were incubated with ATP inhibitors for 30 min, pelleted and resuspended in saline g for 10 min at 37°C, counted, and then repelleted and boiled as described above. For TCA method, cells were grown to 2 × 105 cells/mL, rinsed with HBSS, and incubated in either saline g or saline ± inhibitors for 30 min. After the removal of saline ± inhibitors, 250 μL of 1% cold TCA was added, incubated on ice for 10 min, and then removed and frozen at –20°C. Washout conditions were performed as described above.
ATP measurements were made essentially as described in the ATP-determining kit protocol (Molecular Probes). In brief, a SLM-Aminco model 8100 Spectrofluorometer was used to measure light output at 560 nm, and ATP levels were quantitated by comparing light output from a test sample with that from a standard ATP solution. We found ATP to be 5–10% the level of saline g controls after a 30-min treatment with 5 mM Az and 1 mM DOG, and a recovery of 50–75% after a 10-min washout period. These measurements are similar to those reported previously for PtK1 cells (DeBrabender et al., 1981; Bershadsky and Gelfand, 1983; Spurck et al., 1986).
Immunofluorescence
For Mad2, BubR1, CENP-E, and cytoplasmic dynein staining, cells were rinsed briefly in PHEM (60 mM Pipes, 25 mM Hepes, pH 6.9, 10 mM EGTA, 4 mM MgSO4) and lysed in 0.5% Triton-X 100/PHEM for 5 min at 37°C. Cells were fixed in 4% formaldehyde/PHEM for 20 min at 37°C. For 3F3/2 staining, 100 nM Microcystin was added to the lysis buffer, and cells were fixed in 1% formaldehyde/PHEM as described above. After a brief rinse in PBS (0.14 M NaCl, 2.5 mM KCl, 10 mM Na2HPO4, 1.5 mM KH2PO4, pH 7.4), cells were blocked in 10% boiled donkey serum/PHEM at 25°C for 1 h. Primary antibody dilutions were performed in 10% boiled donkey serum/PHEM for 1 h at 25°C as follows: 1:100 anti-Mad2; 1:500 BubR1, a gift of Dr. Tim Yen (Fox Chase Cancer Center, Philadelphia, PA); 1:750 CENP-E, a gift of Dr. Gordan Chan (Fox Chase Cancer Center); 1:2,000 cytoplasmic dynein 70.1 intermediate chain (Sigma-Aldrich); 1:5,000 3F3/2, a gift of Dr. Gary Gorbsky (University of Oklahoma, Oklahoma City, OK); 1:750 CREST serum, a gift of Dr. Bill Brinkley (Baylor College of Medicine, Houston, TX), and 1:300 DM1A tubulin (Sigma-Aldrich). After four 5-min rinses in PBST (PBS supplemented with 0.05% Tween-20), cells were incubated with appropriate secondary antibodies diluted into 5% boiled donkey serum/PHEM at 25°C for 1 h. Secondary antibodies were conjugated to either Rhodamine Red X (Jackson Laboratories) or Alexa 488 (Molecular Probes), and dilutions were as follows: 1:100 donkey anti–rabbit (Mad2, BubR1, CENP-E); 1:200 donkey anti–mouse (cytoplasmic dynein, 3F3/2, tubulin), and 1:100 donkey anti–human (CREST). Cells were then rinsed four times for 5 min in PBST, and mounted as described previously (Howell et al., 2000). For immunofluorescent studies of Mad2 and tubulin, cells were lysed (see above), fixed for 20 min in PHEM containing 4% formaldehyde/0.5% grade gluteraldehyde, quenched with 1 mg/ml NaBH4 in PHEM three times for 5 min at 25°C, and stained for Mad2 and tubulin.
Quantitation of fluorescence intensities was performed exactly as described in detail by Hoffman et al. (2001). Computer generated 9 × 9 and 13 × 13 pixel circles (for kinetochores) and 18 × 18 and 24 × 24 pixel circles (spindle poles) were used to measure kinetochore, spindle pole, and background fluorescence (Hoffman et al., 2001). Each coverslip was costained for CREST to allow precise identification of kinetochores. Spindle poles were identified by fluorescence and/or phase-contrast microscopy. Images were collected using identical imaging settings.
Microinjection
PtK1 cells were grown on coverslips, mounted into modified Rose chambers, and microinjected as described previously (Howell et al., 2000). Stage temperature was maintained at 36–38°C using a Sage air curtain incubator. Immediately after injection, growth media was replaced with dye-free F12 medium, and time-lapse images were obtained as described by Canman et al. (2000). Cells were microinjected with either 10 mg/ml purified p50 (Whittman and Hyman, 1999) or concentrated 70.1 anti-dynein antibody (∼7–8 mg/ml) (Waterman-Storer et al., 2000) in injection buffer (10 mM Na2HPO4, pH 7.4, 100 mM KCl, 1 mM MgCl2), and time-lapse images were taken every 20 s using a MetaView image acquisition system (Universal Imaging Corp.) For the Mad2 antibody microinjection studies, prometaphase cells were first injected with either p50 or concentrated anti-70.1 dynein antibody, allowed to block in metaphase for 45–60 min, and finally microinjected with affinity-purified, anti-Mad2 antibody as previously described (Waters et al., 1998; Canman et al., 2000).
For studies of fluorescent Mad2 localization in dynein-inhibited cells, prometaphase cells were microinjected with Alexa 488-Mad2 protein (Fig. 6; Howell et al., 2000) and subsequently injected ∼5 min later with p50. Fluorescent and phase-contrast images were collected as described below.
Microscopy and laser photobleaching
Immunofluorescence and live cell digital images were acquired using wide-field eip-fluorescence and phase microscopy using procedures and instrumentation described by Howell et al. (2000). Confocal images of fixed cells were obtained with an Orca-ER CCD camera (Hamamatsu Photonics) on a Nikon TE300 inverted microscope equipped with a spinning disk confocal module (PerkinElmer Wallac) and scanner (Yokagawa), and acquired using a 100× 1.4 NA Plan Apochromat phase objective, a 100mW Argon-Krypton laser, and a Sutter filter wheel for selecting and shuttering of 488-, 568-, or 647-nm wavelengths. All images were acquired at 0.2-μm steps using MetaMorph software controlling the Nikon TE300 focus motor. Laser photobleaching and FRAP analysis were as described by Howell et al. (2000).
Electron microscopy
After cell treatment and fixation (see above), coverslips were flat embedded in Epon and serial 80-nm-thick sections were cut, stained, and imaged at 5,000× as previously described (McEwen et al., 1997; Rieder and Cassels, 1999). Microtubule counts for individual kinetochores were determined in duplicate counting trials as described by McEwen et al. (1997). Counting variation was 1.3% for unextracted cells, and 0.4% for extracted cells. Statistical computations were preformed using Microsoft Excel.
Measurements of interkinetochore distances and chromosome movements
Distances between sister kinetochores were measured in control and p50 blocked metaphase cells, as described by Hoffman et al. (2001). Anaphase chromosome movements and spindle elongation were measured from time-lapse images of cells obtained from the microinjection microscope system (see above) using MetaMorph image analysis software (Universal Imaging Corp.). Distance measurements were obtained between the leading edges of separating sister chromosomes, the leading edges of chromosomes and their poles, and the separation of the poles. The leading edges of separating chromosomes were easy to detect in the phase-contrast images, but the position of the poles was more difficult, particularly when poles were disrupted by dynein/dynactin inhibition. In phase-contrast, the spindle excludes dark cytoplasmic organelles. The spindle pole was usually identified as the polar end of the less dense region occupied by the spindle at the focal point of most chromosome movements.
Online supplemental material
Video 1 is a QuickTime movie of a prometaphase PtK1 cell microinjected with 10 mg/ml p50 dynamitin. Images were collected every 1–2 min. Time, hr:min:sec. Video 2 is a QuickTime movie of a prometaphase PtK1 cell microinjected with 10 mg/ml p50 dynamitin that arrested in metaphase and was subsequently injected with Mad2 antibodies. Images were collected every 1–2 min. Time, hr:min:sec. Videos are available at http://www.jcb.org/cgi/content/full/jcb.200105093/DC1.
Supplemental Material
Acknowledgments
We would like to thank Tarun Kapoor (Rockefeller University, New York, NY) and Viviana Cantillana (University of North Carolina, Chapel Hill, NC) for providing p50 protein, and Rita M. Bernard and Grisel Cassels for electron microscopy assistance. Thanks to Ben Moree and Dr. Ashotush Tripathy for assistance measuring ATP levels, Drs. Tom Hays, Lisa Cameron, Katie Shannon, and Leana Topper for their critical readings of this manuscript, and Jeff Molk and Dr. Daniela Cimini for helpful discussions.
This work is supported by an American Cancer Society Postdoctoral Fellowship to B.J. Howell (00-147-01-CCG), National Institutes of Health grant to E.D. Salmon (GM-24364), National Science Foundation grant to B.F. McEwen (MCB0110821), and National Institutes of Health grant to C.L. Rieder (GMS-R01-40198).
The online version of this article contains supplemental material.
B.F. McEwen and J.C. Cantman contributed equally to this work.
Footnotes
Abbreviations used in this paper: APC/C, anaphase-promoting complex/cyclosome; DOG, 2-deoxyglucose; Az, sodium azide; TCA, trichloric acetic acid.
References
- Banks, J.D., and R. Heald. 2001. Chromosome movement: Dynein-out at the kinetochore. Curr. Biol. 11: R128–R131. [DOI] [PubMed] [Google Scholar]
- Basto, R., R. Gomes, and R.E. Karess. 2000. Rough Deal and Zw10 are required for the metaphase checkpoint in Drosophila. Nat. Cell Biol. 2:939–943. [DOI] [PubMed] [Google Scholar]
- Bershadsky, A.D., and V.I. Gelfand. 1983. Role of ATP in the regulation and stability of cytoskeletal structures. Cell Biol. Int. Rep. 5:173–187. [DOI] [PubMed] [Google Scholar]
- Burkhardt, J.K., C.J. Echeverri, T. Nilsson, and R.B. Vallee. 1997. Overexpression of Dynamitin (p50) subunit of the dynactin complex disrupts dynein-dependent maintenance of membrane organelle distribution. J. Cell Biol. 139:469–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canman, J.C., D.B. Hoffman, and E.D. Salmon. 2000. The role of pre-and post-anaphase microtubules in the cytokinesis phase of the cell cycle. Curr. Biol. 10:611–614. [DOI] [PubMed] [Google Scholar]
- Chan, G.K.T., S.A. Jablonski, D.A. Starr, M.L. Goldberg, and T.J. Yen. 2000. Human Zw10 and ROD are mitotic checkpoint proteins that bind to kinetochores. Nat. Cell Biol. 2:944–947. [DOI] [PubMed] [Google Scholar]
- Chen, R-H., J.C. Waters, E.D. Salmon, and A.W. Murray. 1996. Association of spindle assembly checkpoint component XMAD2 with unattached kinetochores. Science. 274:242–246. [DOI] [PubMed] [Google Scholar]
- Cimini, D., B. Howell, P. Maddox, A. Khodjakov, F. Degrassi, and E.D. Salmon. 2001. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J. Cell Biol. 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBrabender, M., G. Guens, R. Nuydens, R. Willebrords, and J. DeMey. 1981. Microtubule assembly in living cells after release from nocodazole block: The effects of metabolic inhibitors, taxol and pH. Cell Biol. Int. Rep. 5:913–920. [DOI] [PubMed] [Google Scholar]
- Desai, A., and T.J. Mitchison. 1997. Microtubule polymerization dynamics. Ann. Rev. Cell Dev. Biol. 13:83–117 [DOI] [PubMed] [Google Scholar]
- Dobles, M., V. Liberal, M.L. Scott, R. Benezra, and P.K. Sorger. 2000. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell. 101:635–645. [DOI] [PubMed] [Google Scholar]
- Echeverri, C.J., B.M. Paschal, K.T. Vaughan, and R.B. Vallee. 1996. Molecular characterization of the 50-kD subunit of dynactin reveals function for the complex in chromosome alignment and spindle organization during mitosis. J. Cell Biol. 132:617–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner, N.E., D.L. Dujardin, C.-Y. Tai, K.T. Vaughan, C.B. O'Connell, Y.-L. Wang, and R.B. Vallee. 2000. A role for the lissencephaly gene LIS1 in mitosis and cytoplasmic dynein function. Nat. Cell Biol. 2:784–791. [DOI] [PubMed] [Google Scholar]
- Gaglio, T., M.A. Dionne, and D.A. Compton. 1997. Mitotic spindle poles are organized by structural and motor proteins in addition to centrosomes. J. Cell Biol. 138:1055–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garewal, H.S., F.R. Ahmann, R.B. Schifman, and A. Celniker. 1986. ATP assay: ability to distinguish cytostatic from cytocidal anticancer drug effects. J. Natl. Cancer Inst. 77:1039–1045. [PubMed] [Google Scholar]
- Gorbsky, G.J., and W.A. Ricketts. 1993. Differential expression of a phosphoepitope at the kinetochores of moving chromosomes. J. Cell Biol. 122:1311–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbsky, G.J., R.-H., Chen, and A.W. Murray. 1998. Microinjection of antibody to Mad2 protein into mammalian cells in mitosis induces premature anaphase. J. Cell Biol. 141:1193–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, M.B., L. Howard, and D.A. Compton. 2001. Chromosome movement in mitosis requires microtubule anchorage at spindle poles. J. Cell Biol. 152:425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heald, R., R. Tournebize, T. Blank, R. Sandaltzopoulos, P. Beker, A. Hyman, and E. Karsenti. 1996. Self-organization of microtubules into bipolar spindles around artificial chromosomes in Xenopus egg extracts. Nature. 382:420–425. [DOI] [PubMed] [Google Scholar]
- Hoffman, D.B., C.G. Pearson, T.J. Yen, B.J. Howell, and E.D. Salmon. 2001. Microtubule dependent changes in the assembly of microtubule motor proteins and mitotic spindle checkpoint proteins at PtK1 kinetochores. Mol. Biol. Cell. 12:1995–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell, B.J., D.B. Hoffman, G. Fang, A.W. Murray, and E.D. Salmon. 2000. Visualization of Mad2 dynamics at kinetochores, along spindle fibers, and at spindle poles in living cells. J. Cell Biol. 150:1233–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt, M.A. 2001. A new view of the spindle checkpoint. J. Cell Biol. 154:909–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikehara, T., H. Yamaguchi, K. Hosokawa, T. Sakai, and H. Miyamoto. 1984. Rb+ influx in response to changes in energy generation effect of the regulation of the ATP content of HeLa cells. J. Cell Physiol. 119:273–282. [DOI] [PubMed] [Google Scholar]
- Inoue, S., and E.D. Salmon. 1995. Force generation by microtubule assembly/disassembly in mitosis and related movements. Mol. Biol. Cell. 6:1619–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallio, M., J. Weinstein, J.R. Daum, D.J. Burke, and G.J. Gorbsky. 1998. Mammalian p55CDC mediates the association of the spindle checkpoint protein Mad2 with the cyclosome/anaphase promoting complex and is involved in the regulation of anaphase onset and late mitotic events. J. Cell Biol. 141:1393–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kangas, L., M. Grönroos, and A.-L. Nieminen. 1984. Bioluminescence of cellular ATP: a new method for evaluating cytotoxic agents in vitro. Med. Biol. 62:338–343. [PubMed] [Google Scholar]
- King, J.M., T.S. Hays, and R.B. Nicklas. 2000. Dynein is a transient kinetochore component whose binding is regulated by microtubule attachment, not tension. J. Cell Biol. 151:739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y., and R. Benezra. 1996. Identification of a human mitotic checkpoint gene: hsMAD2. Science. 274:246–248. [DOI] [PubMed] [Google Scholar]
- Maney, T., L.M. Ginkel, A.W. Hunter, and L. Wordeman. 1999. The kinetochore of higher eukaryotes: A molecular view. Int. Rev. Cytol. 194:67–131. [DOI] [PubMed] [Google Scholar]
- McEwen, B.F., A.B. Heagle, G.O. Cassels, K.F. Buttle, and C.L. Rieder. 1997. Kinetochore fiber maturation in PtK1 cells and its implications for the mechanisms of chromosome congression and anaphase onset. J. Cell Biol. 137:1567–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen, B.F., C.-E. Hsieh, A.L. Mattheyses, and C.L. Rieder. 1998. A new look at kinetochore structure in vertebrate somatic cells using high-pressure freezing and freeze substitution. Chromosoma. 107: 366–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen, B.F., G.K.T. Chan, B. Zubrowski, M.S. Savoian, M.T. Sauer, T.J. Yen. 2001. CENP-E is essential for reliable bioriented spindle attachment, but chromosome alignment can be achieved via redundant mechanisms in mammalian cells. Mol. Biol. Cell. 12:2776–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merdes, A., R. Heald, K. Samejima, W.C. Earnshaw, and D.W. Clevelanc. 2000. Formation of spindle poles by dynein/dynactin-dependent transport of NuMA. J. Cell Biol. 149:851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicklas, R.B. 1997. How cells get the right chromosomes. Science. 274:632–637. [DOI] [PubMed] [Google Scholar]
- Nicklas, R.B., S.C. Ward, and G.J. Gorbsky. 1995. Kinetochore chemistry is sensitive to tension and may link mitotic forces to a cell cycle checkpoint. J. Cell Biol. 130:929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell, C.B., and Y. Wang. 2000. Mammalian spindle orientation and position respond to changes in cell shape in a dynein-dependent fashion. Mol. Biol. Cell. 11:1765–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschal, B.M., and R.B. Vallee. 1987. Retrograde transport by the microtubule associated protein MAP 1C. Nature. 330:181–183. [DOI] [PubMed] [Google Scholar]
- Rieder, C.L. 1982. The formation, structure, and composition of the mammalian kinetochore and kinetochore fiber. Intl. Rev. Cytol. 79:1–58. [DOI] [PubMed] [Google Scholar]
- Rieder, C.L., and E.D. Salmon. 1998. The vertebrate cell kinetochore and its roles during mitosis. Trends Cell Biol. 8:310–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder, C.L. and G. Cassels. 1999. Correlative light and electron microscopy of mitotic cells in monolayer cultures. Meth. Cell Biol. 61:297–315. [DOI] [PubMed] [Google Scholar]
- Rieder, C.L., R.W. Cole, A. Khodjakov, and G. Sluder. 1995. The checkpoint delaying anaphase in response to chromosome monorientation is mediated by an inhibitory signal produced by unattached kinetochores. J. Cell Biol. 130:941–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savoian, M.S., M.L. Goldberg, and C.L. Rieder. 2000. The rate of poleward chromosome motion is attenuated in Drosophila zw10 and rod mutants. Nat. Cell Biol. 2:948–952. [DOI] [PubMed] [Google Scholar]
- Scaërou, F., I. Aguilera, R. Saunders, N. Kane, L. Blottière, and R. Karess. 1999. The rough deal protein is a new kinetochore component for accurate chromosome segregation in Drosophila. J. Cell Sci. 112:3757–3768. [DOI] [PubMed] [Google Scholar]
- Schaar, B.T., G.K.T. Chan, P. Maddox, E.D. Salmon, and T.J. Yen. 1997. CENP-E function at kinetochores is essential of chromosome alignment. J. Cell Biol. 139:1373–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah, J.V., and D.W. Cleveland. 2000. Waiting for anaphase Mad2 and the spindle assembly checkpoint. Cell. 103:997–1000 [DOI] [PubMed] [Google Scholar]
- Sharp, D.J., G.C. Rogers, and J.M. Scholey. 2000. Cytoplasmic dynein is required for poleward chromosome movement during mitosis in Drosophila embryos. Nat. Cell Biol. 2:922–930. [DOI] [PubMed] [Google Scholar]
- Skoufias, D.A., P.R. Andreassen, F.B. Lacroix, L. Wilson, and R.L Margolis. 2001. Mammalian mad2 and bub1/bubR1 recognize distinct spindle-attachment and kinetochore-tension checkpoints. Proc. Natl. Acad. Sci. 98:4492–4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurck, T.P., J.D. Pickett-Heaps, and M.W. Klymkowsky. 1986. I. Effects of dinitrophenol/deoxyglucose on the live spindle. Protoplasma. 131:47–59. [Google Scholar]
- Starr, D.A., B.C. Williams, Z. Li, B. Eternad-Moghadam, R.K. Dawe, and M.L. Goldberg. 1997. Conservation of the centromere/kinetochore protein ZW10. J. Cell Biol. 138:1289–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr, D.A., B.C. Williams, T.S. Hays, and M.L. Goldberg. 1998. ZW10 helps recruit dynactin and dynein to the kinetochore. J. Cell Biol. 142:762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudakin, V., G.K.T. Chan, and T.J. Yen. 2001. Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BubR1, Bub3, Cdc20, and Mad2. J. Cell Biol. 154:925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, Z.R., B. Bharadwaj, B. Li, and H. Yu. 2001. Mad2-independent inhibition of APCCdc20 by the mitotic checkpoint protein BubR1. Dev. Cell. 1:227–237. [DOI] [PubMed] [Google Scholar]
- Vaisberg, E.A., M.P. Koonce, and J.R. McIntosh. 1993. Cytoplasmic dynein plays a role in mammalian mitotic spindle formation. J. Cell Biol. 123:849–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadsworth, P., and E.D. Salmon. 1988. Spindle microtubule dynamics: modulation by metabolic inhibitors. Cell Motil. Cytoskel. 11:97–105. [DOI] [PubMed] [Google Scholar]
- Wassmann, K., and R. Benezra. 2001. Mitotic checkpoints: from yeast to cancer. Curr. Opin. Gen. Devel. 11:83–90. [DOI] [PubMed] [Google Scholar]
- Waterman-Storer, C., D.Y. Duey, K.L. Weber, J. Keech, R.E. Cheney, E.D. Salmon, and W.M. Bement. 2000. Microtubules remodel actomyosin networks in Xenopus egg extracts via two mechanisms of F-actin transport. J. Cell Biol. 150:361–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters, J.C., R.-H. Chen, A.W. Murray, and E.D. Salmon. 1998. Localization of Mad2 to kinetochores depends on microtubule attachment, not tension. J. Cell Biol. 141:1181–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, B.C., M. Gatti, and M. Golberg. 1996. Bipolar spindle attachments affect redistributions of ZW10, a Drosophila centromere/kinetochore component required for accurate chromosome segregation. J. Cell Biol. 134:1127–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittman, T., and T. Hyman. 1999. Recombinant p50/Dynamitin as a tool to examine the role of dynactin in intracellular processes. Meth. Cell Biol. 61:137–143. [DOI] [PubMed] [Google Scholar]
- Whittman, T., M. Wilm, E. Karsenti, and I. Vernos. 2000. TPX2, a novel Xenopus MAP involved in spindle pole organization. J. Cell Biol. 149:1405–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik, E., R. Basto, F. Scaerou, R. Karess, and T. Hays. 2001. Kinetochore dynein: its dynamics and role in the metaphase checkpoint. Nat. Cell Biol. 3:1001–1007. [DOI] [PubMed] [Google Scholar]
- Yao, X., A. Abrieu, Y. Zheng, K.F. Sullivan, and D.W. Cleveland. 2000. CENP-E forms a link between attachment of spindle microtubules to kinetochores and the mitotic checkpoint. Nat. Cell Biol. 2:484–491. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.