

Abstract
The assembly of gap junctions (GJs) is a process coordinated by growth factors, kinases, and other signaling molecules. GJ assembly can be enhanced via the elevation of cAMP and subsequent stimulation of connexon trafficking to the plasma membrane. To study the positive regulation of GJ assembly, fibroblasts derived from connexin (Cx)43 knockout (KO) and wild-type (WT) mice were transfected with WT Cx43 (WTCx43) or mutant Cx43. GJ assembly between untransfected WT fibroblasts or stably transfected WTCx43/KO fibroblasts was increased two- to fivefold by 8Br-cAMP, and this increase could be blocked by inhibition of cAMP-dependent protein kinase (PKA) or truncation of the Cx43 COOH terminus (CT). Although serine 364 (S364) of the Cx43 CT was determined to be a major site of phosphorylation, the molar ratio of Cx43 phosphorylation was not increased by 8Br-cAMP. Importantly, GJ assembly between either S364ECx43/KO or S364ECx43/WT fibroblasts was stimulated by 8Br-cAMP, but that between S364ACx43/KO or S364PCx43/KO fibroblasts was not stimulated, indicating that phosphorylation or a negative charge at S364 is required for enhancement of GJ assembly by cAMP. Furthermore, GJ assembly between S364ACx43/WT fibroblasts could be stimulated by 8Br-cAMP, but could not be between S364PCx43/WT fibroblasts. Thus, S364PCx43 interferes with enhanced GJ assembly when coexpressed with WTCx43.
Keywords: connexin43; cAMP; protein kinase A; gap junction; site-directed mutagenesis
Introduction
Gap junction (GJ)*proteins (termed connexins [Cxs]), comprise a family of vertebrate transmembrane proteins that assemble intracellularly to form oligomeric channels (Musil and Goodenough, 1993). The GJ hemichannels, or connexons, are transported to the plasma membrane where they dock with connexons in the membranes of adjacent cells and aggregate to mediate intercellular transfer of various ions, signaling molecules, and metabolites (Simon and Goodenough, 1998). The essential role of GJ communication in the coordination of physiological processes within various tissues has become increasingly apparent, with defects in the different Cx proteins now linked to a wide variety of pathological conditions (Krutovskikh and Yamasaki, 2000).
The GJ protein Cx43 is one of the more ubiquitous of the GJ proteins. Its importance is underscored by the abnormal heart development, perinatal mortality, female sterility, altered bone development, and cataracts observed in Cx43 knockout (KO) mice (Lo, 1999). Its unique characteristics are further emphasized by studies of “knock-in” mice, in which replacement of Cx43 with Cx32 or Cx40 results in mice with germ-cell deficiencies and mild cardiac defects (Plum et al., 2000). Considerable research has focused on the gating or regulation of Cx43 in established GJs; much less attention has focused on the regulation of the assembly of Cx43-containing GJs. Yet, the relatively short half-life of Cx43, measuring only 2–3 h (Laird et al., 1991; Musil and Goodenough, 1991; Lampe, 1994), implies that assembly of Cx43-containing GJs must be a continual process in even fully differentiated tissues (Beardslee et al., 1998). The formation of new GJs likely plays an important role during development and differentiation (De Sousa et al., 1993; Zhang and Thorgeirsson, 1994), cell division (Lampe et al., 1998), wound repair (Larson et al., 1997), glandular secretion (Charollais et al., 2000), and parturition (Hendrix et al., 1995). The transient formation of GJs between blood cells and other cell types is also recognized, although the reasons for such formation remain speculative (Saez et al., 2000).
Cx43-mediated communication (Darrow et al., 1995), the size and number of GJs (Atkinson et al., 1995), and the assembly of new GJs (Paulson et al., 2000) can all be enhanced by agents that elevate intracellular cAMP. Such enhancement has been shown to result from increased synthesis (Mehta et al., 1992) and/or the increased trafficking of connexons to the plasma membrane (Burghardt et al., 1995; Holm et al., 1999; Paulson et al., 2000). We have previously shown that cAMP-enhanced GJ assembly is mediated by cAMP-dependent protein kinase (PKA), and that the activation of PKA is required prior to or during GJ assembly (Paulson et al., 2000). There are reports that microinjected PKA catalytic subunit affects GJ communication within minutes (Godwin et al., 1993; Britz-Cunningham et al., 1995), consistent with an effect on channel gating or open time. However, as we show here, the assembly of new GJs can also take place within minutes. Moreover, direct phosphorylation of Cx43 by PKA has not been documented, and activation of PKA does not result in a change in single channel conductance of Cx43 expressed in SKHep cells (Kwak et al., 1995).
A large number of known and potential phosphorylation sites, protein kinase binding sites, and a ZO-1 binding site all reside on the COOH-terminal domain of Cx43 (Giepmans et al., 2000; Lampe and Lau, 2000; Toyofuku et al., 2000). A variety of COOH terminus (CT) sites and domains have also been identified that play a role in Cx43 channel gating, e.g., in a reduced pH environment (Ek-Vitorin et al., 1996). Importantly, when truncated Cx43 is expressed in mammalian cells, growth control and the response to PDGF are defective (Moorby and Gherardi, 1999). Therefore, it appears that although the Cx43 CT is not required for formation of functional GJs, the CT is critical for GJ regulation.
Several studies have indicated that serine 364 (S364) is an important site on the COOH-terminal domain of Cx43. S364 resides in a tandem RXSSR protein kinase C (PKC)-specific repeat found from R362–R374. An S-to-proline conversion at 364 (S364P) was first identified in vivo as a mutation in a subset of patients with visceral atrial heterotaxia, and GJs formed from S364PCx43 in vitro displayed an altered response to PKC- or PKA-related signaling cascades (Britz-Cunningham et al., 1995). In spite of the fact that Cx43 mutations have not been identified in other populations of visceral atrial heterotaxia patients (Debrus et al., 1997), S364PCx43 affects left-right patterning in the early Xenopus embryo when misexpressed dorsally, resulting in a significant increase in heterotaxia (Levin and Mercola, 1998). The S364P mutation also significantly alters the pH sensitivity of Cx43 gating in the Xenopus oocyte expression system (Ek-Vitorin et al., 1996). As a result of these findings, we investigate here whether mutation of this site also affects the regulation of GJ assembly.
Although PKA plays an important role in the regulation of Cx43 GJ assembly in cAMP-treated cells in vitro, it is yet unclear how such regulation is effected. To better understand this relationship, we have examined GJ assembly in several cell types transfected with COOH-terminal mutants of Cx43. These studies show that the upregulation of GJ assembly requires an intact COOH-terminal domain, and suggest a need for an appropriate conformation around position 364, potentially to facilitate Cx43CT interactions with other proteins that regulate GJ assembly. Furthermore, a negative charge at this site, such as would occur upon phosphorylation, appears to be a prerequisite for cAMP stimulation of GJ assembly.
Results
Enhancement of GJ assembly between mouse fibroblasts by activation of PKA
Established WT fibroblasts expressing endogenous WTCx43 typically display a two- to fourfold enhancement of GJ communication after exposure to cell permeant 8Br-cAMP for 3–4 h (e.g., clone 10-8 average recipient cells for control: 18.5 ± 0.31, n = 10; 8Br-cAMP: 37.0 ± 2.69, n = 10). A complement to the WT fibroblast, the KO fibroblast, was used for the majority of these studies, as it likely provides a similar physiological milieu suitable for the reintroduction and study of Cx43 (Fig. 1). Lucifer yellow dye transfer is absent between untransfected KO fibroblasts, and whole-cell conductance values are as low, if not lower, than those of other null cells (Kwak et al., 1995; Martyn et al., 1997).
Figure 1.
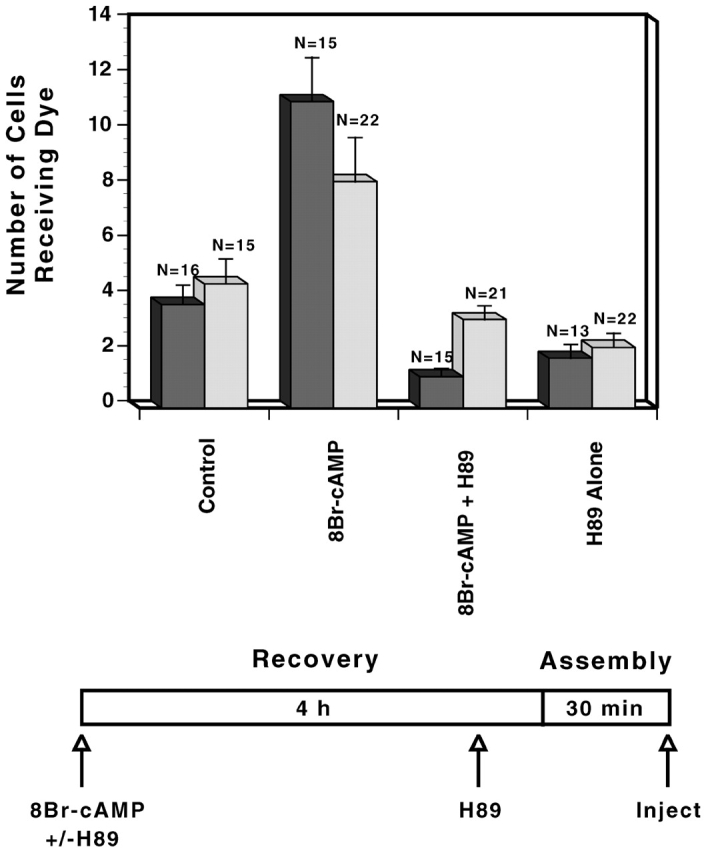
Stimulation of GJ assembly after the elevation of cAMP. Two separate, representative experiments (dark vs. light bars) comparing GJ assembly between untreated WTCx43/KO fibroblasts (clone 22C-3) and cells treated with 8Br-cAMP, with both 8Br-cAMP and H89, or with H89 alone. (Dark bars) When present, H89 was added at recovery. (Light bars) When present, H89 was added 15 min before reaggregation.
To evaluate the cAMP responsiveness of GJ assembly, cells were treated with the cell-permeant cAMP analogue, 8Br-cAMP, and an inhibitor of PKA, N-(2-[{p-bromocinnamyl} amino]ethyl)-5-isoquinolinesulfonamide dihydrochloride (H89), was used to establish whether cAMP-enhanced assembly was dependent on PKA (Paulson et al., 2000). To induce the assembly of new GJs, cells were trypsin dissociated, recovered for a period of time sufficient to ensure removal of GJ remnants from the cell surface, and finally reaggregated as a confluent monolayer for 7–60 min depending on the experiment (Materials and methods). A two- to threefold increase in GJ assembly, as measured by transfer of microinjected dye, was seen following treatment of WTCx43/KO fibroblasts with 8Br-cAMP (350 μM) for both recovery and reaggregation (assembly) (Fig.1). This increase was blocked when H89 (30 μM) was added either at the beginning of recovery or 15 min before reaggregation. Nearly identical results were achieved using WT fibroblasts (e.g., 30 min assembly, clone 10-7 control: 13.0 ± 1.5, n = 10; 8Br-cAMP [4.5 h]: 24.8 ± 1.8, n = 10; 8Br-cAMP + H89 [4.5 h]: 13.8 ± 2.2, n = 10; H89 alone [4.5 h]: 10.8 ± 1.4, n = 10).
Synthesis of Cx43 is not required for enhanced GJ assembly
To determine whether elevated cAMP stimulated the synthesis of Cx43, Western immunoblotting and densitometry were used to monitor Cx43 levels in cell homogenates from assembly experiments. 8Br-cAMP treatment during the assembly assay elevated total Cx43 in WT fibroblasts by 26% on average (n = 6; 4.5-h treatment time) (Fig. 2 A, inset). In contrast, expression levels were not significantly elevated by 8Br-cAMP in WTCx43- or mutant Cx43-transfected KO fibroblasts after correcting for loading differences (unpublished data; see Fig. 5 B).
Figure 2.
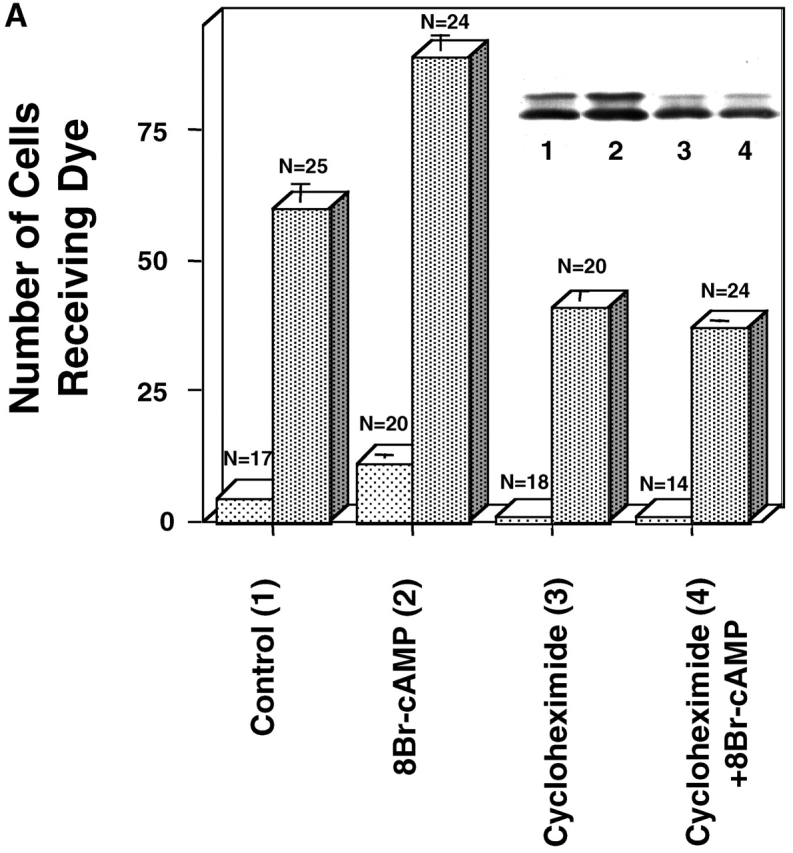

The effects of cycloheximide on GJ assembly. Representative experiments comparing GJ assembly between WT fibroblasts (A) (clone 10-3) untreated (1) or treated with 8Br-cAMP (2), cycloheximide (3), or both cycloheximide and 8Br-cAMP (4). Cells were recovered for 4 h 23 min in suspension and reaggregated for 7 min (light bars) or recovered for 3.5 h in suspension and reaggregated for 1 h (dark bars). (Inset) Western immunoblot of WT fibroblasts after 1-h assembly (dark bars). (B) Immunolocalization of Cx43 using Cx43IF1 mouse monoclonal antibody (1:10), detected by goat anti–mouse Alexafluor 568 (1:500). WT fibroblasts were recovered for 3.5 h in suspension and reaggregated for 1 h before fixation. When present, 8Br-cAMP and/or cycloheximide were added at the beginning of recovery. (A) control; (B) 8Br-cAMP; (C) cycloheximide; (D) cycloheximide + 8Br-cAMP.
Figure 5.



GJ assembly between S364PCx43, S364ECx43 and S364ACx43/KO fibroblasts. (A) Example of Lucifer yellow dye transfer between S364PCx43 and S364ECx43/KO fibroblasts assembled overnight into confluent monolayers (*, injected cell). (B) Representative immunoblot of S364PCx43, S364ECx43, or S364ACx43/KO homogenates after GJ assembly and treatment without (1, 3, 5) or with (2, 4, 6) 8Br-cAMP for the entire assay. To detect S364PCx43 and S364ECx43 (arrowhead), membranes were labeled with a Cx43 antibody (1:8,000; Sigma-Aldrich) and detected with peroxi- dase-conjugated secondary antibody (1:10,000). A cross-reactive product, which was not consistently detected, is indicated below NP S364ECx43 (small arrowhead). (C) Compilation of representative GJ assembly experiments from S364PCx43, S364ECx43, and S364ACx43/KO fibroblasts. Cells were recovered for 4 h, reaggregated for 30 min, and, where indicated, treated with 8Br-cAMP for the entire 4.5 h of the assembly experiment. N values are above the error bars. *0.05 < P < 0.1; **0.01 < P < 0.02.
To further test whether increased synthesis of Cx43 was required for cAMP-enhanced assembly, the protein synthesis inhibitor, cycloheximide, was added either at the beginning of recovery or just prior to reaggregation (assembly) of the WT or WTCx43/KO fibroblasts. GJ assembly was measured by the degree of dye coupling, and the amount of Cx43 protein was evaluated by Western immunoblotting. When cycloheximide was added to WTCx43/KO at the beginning of a 30-min reaggregation, GJ assembly was inhibited by 50%. These lower levels of assembly were still doubled by 8Br-cAMP and Cx43 levels remained unchanged (clone 22C-3, control: 6.5 ± 0.8, n = 17; 8Br-cAMP: 14.1 ± 1.3, n = 15; cycloheximide: 3 ± 0.4, n = 16; 8Br-cAMP + cycloheximide 6.5 ± 0.5, n = 17). However, when cycloheximide was introduced at the beginning of recovery with either the WT or WTCx43/KO fibroblasts, the inhibition of control and enhanced levels of GJ assembly was nearly complete in spite of the detection of significant levels of Cx43 (Fig. 2 A, inset; unpublished data). Although significant levels of coupling developed by 7–15 min of assembly, immunolabeling failed to show discernible GJ punctae at interfaces in either controls or 8Br-cAMP–treated samples (unpublished data). Interestingly, when reaggregation was extended, to allow for a greater degree of GJ assembly, samples treated with cycloheximide for the entire assay developed detectable punctae at interfaces and relatively high levels of coupling (Fig. 2, A and B), indicating that GJ assembly was slowed but not blocked by cycloheximide. In spite of this, 8Br-cAMP stimulation of GJ assembly detected by either dye transfer or immunolabel was completely blocked by a 4.5-h treatment of cycloheximide with the longer assembly (Fig. 2, A and B). Thus, the enhancement of GJ assembly by cAMP is not dependent on new synthesis of Cx43, but may be dependent on the synthesis of another protein, possibly one that possesses a relatively short half-life. In addition, the level of unstimulated GJ assembly appears also to be affected by a cycloheximide-sensitive protein.
Gap junction assembly between M257Cx43 transfectants
A primary objective of this study was to determine whether specific domains of the Cx43 CT are required for the positive regulation of GJ assembly by intracellular cAMP. As a first step in addressing this objective, M257Cx43 cDNA was transfected into the KO fibroblasts to determine whether removal of most of the predicted Cx43 phosphorylation and regulatory sites would interfere with either control or enhanced levels of GJ assembly. Of two transfections, only 31% (29/93) of the stably transfected M257Cx43/KO fibroblast clones were found to minimally transfer Lucifer yellow dye, compared with 72% (36/50) of the WT Cx43/KO fibroblast clones. Importantly, and in contrast to the WTCx43/KO fibroblasts, treatment of weakly coupled M257Cx43/KO fibroblasts with 8Br-cAMP, as either assembling or established monolayers, had no effect on dye transfer (e.g., clone TR22B, control: 1.82 ± 0.87, n = 11; 8Br-cAMP: 1.55 ± 0.62, n = 11). Dye transfer disappeared between the M257Cx43/KO fibroblast subclones with increased passage in spite of continued expression of significant levels of protein. Immunolabel of M257Cx43/KO fibroblasts confirmed that significant levels of truncated Cx43 resided within the cell cytoplasm, with very little localizing to interfaces (unpublished data).
Because M257Cx43 did not form significant GJs in the fibroblast, N2A cells that had been previously transfected with WTCx43 or M257Cx43 were also examined. Prior to transfection, N2A cells have a very low level of electrical coupling measured by double whole-cell patch clamp that is reportedly the result of Cx45 expression (Veenstra et al., 1992). We detected no Lucifer yellow dye coupling between parental N2A cells (0.18 ± 0.4; n = 10), and 8Br-cAMP treatment (4.5 h) failed to stimulate coupling (0 ± 0; n = 10). Confluent monolayers of WTCx43/N2A and M257Cx43/N2A, cells were found to be equally well dye coupled, and treatment of established WTCx43/N2A monolayers with 8Br-cAMP for 4.5 h increased coupling three- to fourfold, whereas coupling between the M257Cx43/N2As remained at control levels (Fig. 3 A). When the same cells were dissociated, recovered, and reaggregated in the presence or absence of 8Br-cAMP, GJ assembly between the WTCx43/N2As was significantly increased, but that between the M257Cx43/N2As remained unchanged (Fig. 3 B). The M257Cx43/N2A cells also assembled GJs more slowly than the WTCx43/N2A cells did, in spite of communicating equally well in established monolayers (Fig. 3, compare A and B). Thus, as was found using the less well-coupled M257Cx43/KO fibroblasts, these data show that the Cx43 CT affects the kinetics of basal GJ assembly and that the CT is required for enhanced GJ assembly.
Figure 3.


Comparison of cells expressing WTCx43 or M257Cx43. (A) A representative experiment in which established monolayers of WTCx43/N2A and M257Cx43/N2A cells were treated with or without 8Br-cAMP for 4.5 h and coupling measured by microinjection of Lucifer yellow dye. (B) A representative experiment comparing de novo GJ assembly between WTCx43/N2A and M257Cx43/N2A cells treated for the entire experiment with or without 8Br-cAMP. *0.001 < P < 0.01
Cx43 is phosphorylated at S364 in unstimulated cells
PKA regulation of GJ communication is reportedly altered when S364 of Cx43 is mutated to a proline (Britz-Cunningham et al., 1995). This information, in addition to a need for the CT of Cx43 in basal and enhanced assembly, and the indication that PKA is required for enhanced and possibly basal GJ assembly, led us to question whether specific sites on the CT were phosphorylated during GJ assembly. As previous studies have suggested that Cx43 is phosphorylated during GJ assembly to a P2 phospho-form (Musil and Goodenough, 1991), we first purified P2 from Cx43 in untreated confluent T51B cells for analysis. T51B cells were used because they express abundant Cx43 with a phosphorylation pattern resembling that of Cx43 in WT fibroblasts. After trypsin digestion and separation of phosphorylated from nonphosphorylated peptides, matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectometry (MS) indicated that one of the major fragments had a mass of 2,224. This matches the calculated mass of tryptic peptide V347-R366 plus 80 (the mass of a single phosphate). Consistent with this tentative identification, the nonphosphorylated fraction contained a fragment of 2,144, the predicted mass of unphosphorylated V347-R366. The nonphosphorylated fraction also contained several other fragments with masses consistent with Cx43 (e.g., Y265-K287). Finally, tandem MS analysis yielded secondary fragments that had masses consistent with those expected from phosphorylated and nonphosphorylated peptide V347-R366. V347-R366 contains no threonine or tyrosine residues and only two serine residues, S364 and S365, near the COOH-terminal end of the peptide. Their adjacency and position near the CT make positive identification of the specific phosphorylated residue(s) impractical using these techniques. We know from previous research that the P2 form of Cx43 is phosphorylated at multiple serine residues found on several different peptides after trypsin digestion (Berthoud et al., 1993; Saez et al., 1997; Lampe and Lau, 2000). As the peptide containing S364 and S365 isolated from T51B cells was prominent, it is likely that phosphorylation of this peptide is a major phosphorylation in P2. Data from many of the other peptides detected by MS will need to be verified in other studies.
In an attempt to ascertain which residue(s) of the S364–S365-containing peptide was phosphorylated, WTCx43/ or S364PCx43/KO fibroblasts were metabolically labeled with [32P]i, Cx43 was immunoprecipitated, and tryptic peptides were analyzed by HPLC. The WTCx43-expressing cells incorporated 3,400 cpm, and the S364PCx43 cells incorporated 1,200 cpm that could be recovered in tryptic fragments. HPLC separation of labeled Cx43 from WTCx43 cells revealed peaks of radioactivity eluting at 8, 21–22, and 24 min, whereas separation of labeled S364PCx43 revealed peaks at 8 and 20–21 min, consistent with WTCx43 cells, and several other smaller peaks (Fig. 4 A). Comparison of common peaks showed a 29-fold greater incorporation of 32P in the 8-min fraction from WTCx43 as related to the same fraction from S364PCx43, whereas the 20–21-min fractions contained much closer to equal levels of radioactivity. We have previously shown that when the sequence A367-R370 was phosphorylated at S368, it eluted at 8 min using this HPLC system (Lampe et al., 2000). We expect P363S(32P)SR to also elute in the 8-min fraction, as its charge is nearly identical and its mass only differs from phosphorylated A367-R370 by 26 Da.
Figure 4.


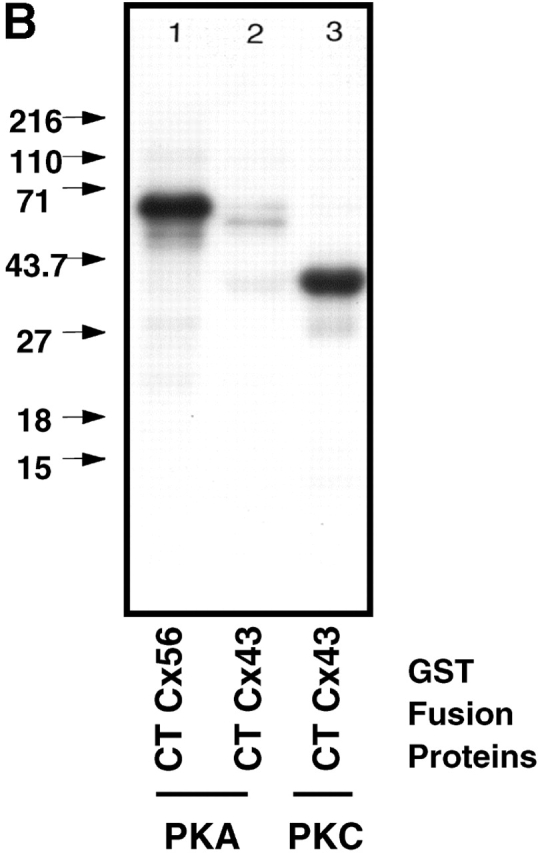
S364 of Cx43 is phosphorylated in unstimulated WT Cx43/KO fibroblasts. (A) HPLC elution profile for 32P-labeled Cx43 peptides isolated from cells expressing WTCx43 (•) or S364PCx43 (□). The elution position of certain peptides is indicated. Amino acids 346–382 of Cx43 and tryptic cleavage sites are depicted above the profile (arrows). Asterisks denote sites that may only be partially cleaved by trypsin. (B) Autoradiogram depicting in vitro phosphorylation of Cx GST fusion proteins. (Lane 1) Purified GST CTCx56 phosphorylated by PKA. (Lane 2) Purified GST CTCx43 phosphorylated by of PKA. (Lane 3) Purified GST CTCx43 phosphorylated by PKC. (C) Phosphorylation of Cx43 after treatment with 8Br-cAMP. Autoradiogram showing results of two separate experiments in which confluent monolayers of WT fibroblasts (10-3) were loaded with radiolabeled [32P]i in the presence or absence of 8Br-cAMP (Materials and methods). (1 and 3) Untreated. (2 and 4) Treated with 8Br-cAMP.
MALDI-TOF MS indicated that the radiolabeled fractions that eluted at 20–21 min (Fig. 4 A) contained peptides (derived from GST–Cx43CT) with masses of 1355 and 2778, consistent with the mass expected for A371-I382 and M320-K345 (see Materials and methods). Because the 20–21-min fractions from either WTCx43 or S364PCx43 cells held significant radioactivity, the S364P mutation did not appear to affect phosphorylation in these two peptides. However, WTCx43-expressing cells showed a third peak eluting at 24 min that was not found with S364PCx43 cells (Fig. 4 A). MALDI-TOF MS showed that the radiolabeled WTCx43 peak fraction at 24 min contained tryptic fragments with masses of 2,144 and 1,717, consistent with V347-R366 and V347-R362, which are distinguished by reduced cleavage of the R-P bond. V347-R366 contains phosphorylatable serine residues at 364 and 365. Only limited radioactivity was recovered in this same fraction from the S364PCx43 cells, indicating that the same peptide was not phosphorylated when S364 was mutated. Likewise, in a different set of experiments, phosphorylation and processing of S364A- and S364E-expressing cells resulted in only background levels of radioactivity in the HPLC fractions associated with V347-R366, whereas WTCx43-expressing cells showed 98 cpm (unpublished data). These data support the conclusion that the presence of S364 is necessary for phosphorylation to occur on this fragment, and that S365 is not a viable alternative phosphorylation site in cells when S364 is mutated. However, these results do not explain why S364PCx43 yet displays a shift in migration of Cx43 in SDS-PAGE in untreated cells (Fig 5 B). The most likely explanation is that phosphorylation within the A371-I382 or M320-K345 fragment, or compensatory phosphorylation represented by the radioactivity eluting at 11, 14, and 17 min in the S364PCx43 run, is sufficient to shift Cx43 to a slower migrating form.
Treatment with 8Br-cAMP does not increase the phosphorylation of Cx43
MS is an excellent method for detecting phosphorylation, but it is not a good method for quantifying such phosphorylation, and therefore could not be used to study relative changes in phosphorylation during enhanced assembly or the kinases involved. Thus, a Cx43 glutathione sulphotransferase (GST)–CT fusion protein was used to first test for phosphorylation by PKA in vitro. As can be seen in Fig. 4 B, the Cx43 fusion protein was efficiently phosphorylated by purified PKC, but not by PKA. In contrast, the CT of Cx56 was highly phosphorylated by PKA, indicating that the preparation was active.
Next, to test whether PKA might act indirectly to increase the phosphorylation of Cx43 (e.g., at S364), monolayers of WT fibroblasts were radiolabeled with [32P]i (1 mCi/ml), treated with 8Br-cAMP, and Cx43 was immunoprecipitated. A 24% average increase in radiolabeled Cx43 was detected in both trials after a 4-h treatment with 8Br-cAMP (300 μM) (Fig. 4 C). Treatment of parallel cultures with 8Br-cAMP resulted in a significant elevation of dye coupling (unpublished data). Because Cx43 levels in the same clone were similarly found to increase by an average of 26% after 8Br-cAMP treatment (as described above; Fig. 2 A, inset), the small increase in radiolabeled Cx43 detected in these two experiments is almost certainly due to an increase in immunoprecipitated Cx43 (phosphorylated and nonphosphorylated) rather than an increase in the molar ratio of phosphorylation. Furthermore, the proportion of P forms remained constant. Thus, an overall increase in phosphorylation of Cx43 during enhanced assembly is not likely.
Conversion of S364 to alanine or proline, but not to glutamic acid, inhibits enhanced assembly
Because S364 of Cx43 was significantly phosphorylated in resting cells, we next asked whether phosphorylation at S364 was a prerequisite for basal and/or enhanced levels of GJ assembly. To address this question, S364PCx43, S364ECx43, and S364ACx43 mutants were stably expressed in KO fibroblasts, and GJ assembly was examined between several subclones of each transfectant. All S364 mutant/KO transfectants communicated and expressed high levels of Cx protein (Fig. 5, A and B; unpublished data). Interestingly, GJ assembly was only enhanced between the S364ECx43/KO fibroblasts in response to 8Br-cAMP, and not between the S364PCx43 or the S364ACx43/KO fibroblasts (Fig. 5 C). Because the negative charge of the acidic glutamic acid (E) may mimic a phosphate group, these results suggest that phosphorylation of S364 is a prerequisite for enhanced but not basal levels of GJ assembly.
Coexpression of S364PCx43 with native Cx43 in WT fibroblasts inhibits enhanced GJ assembly
Because homomeric S364PCx43 and S364ACx43 GJs in KO fibroblasts were not positively regulated by 8Br-cAMP, we questioned whether these mutants, coexpressed with WTCx43, would affect the regulation of GJ assembly. Thus, WT fibroblasts were transiently transfected with S364PCx43, S364ECx43, S364ACx43, or pIRES vector alone, and assembly was studied. Transfection with each of these mutants increased both GJ communication after assembly (Fig. 6 A) and Cx43 levels on westerns (Fig. 6 B) by at least twofold, indicating that the mutants were strongly expressed and were forming functional GJs. In keeping with the experiments utilizing stably transfected KO fibroblasts, the S364PCx43-transfected WT fibroblasts did not display enhanced assembly, whereas control pIRES-transfected and S364ECx43-transfected WT fibroblasts did (Fig. 6, A and C). In contrast to the results with S364ACx43/KO fibroblasts, enhanced assembly was observed between the S364ACx43/WT fibroblasts (Fig. 6 C). However, levels of assembly due to endogenous Cx43 in S364ACx43/WT fibroblasts were not as significantly augmented by S364ACx43 as they were by the other two mutants (Fig. 6 C), in spite of similar levels of expression by Western immunoblotting (unpublished data). Note that transfection of WTCx43 into the WT fibroblasts doubled both basal and enhanced GJ assembly (Fig 6 C).
Figure 6.



GJ assembly following the expression of vector, S364PCx43, S364ECx43, S364ACx43, or WTCx43 in WT fibroblasts. (A) Comparison of GJ assembly between WT fibroblasts (10-3) transfected with either S364PCx43 or S364ECx43 and treated with or without 8Br-cAMP for the entire experiment (*, injected cell). (B) Example of immunoblots of cell homogenates following two different assembly experiments using either WT fibroblasts transiently transfected with M257Cx43 or S364PCx43. Cells were transfected 48 h prior to experimentation with either vector alone (pIRES; 1, 2, 5, and 6) or mutant construct (3, 4, 7, and 8). Cells were dissociated and recovered for 4 h 15 or 23 min, reaggregated for 15 (M257Cx43) or 7 (S364PCx43) min, and treated either without (1 and 3) or with (2 and 4) 8Br-cAMP for the entire GJ assembly experiment. To detect endogenous WTCx43 and transfected M257Cx43 (arrowhead, first panel) membranes were labeled with NH2-terminal Cx43 antibody (1:8,000) detected with goat anti–rabbit peroxidase antibody (1:10,000). To detect mutant and WTCx43 expression (second panel), membranes were labeled with Cx43 CT antibody (1:8,000; Sigma-Aldrich) detected with goat anti–rabbit peroxidase antibody (1:10,000). (C) Compilation of trials in which WT fibroblasts were transfected with either vector alone, mutant, or WT Cx43 and GJ assembly monitored without (A) or with (B) 8Br-cAMP.
As enhancement of GJ assembly by 8Br-cAMP did not occur in null cells expressing M257Cx43, we also questioned whether coexpression of truncated Cx43 with native WTCx43 would allow for enhanced assembly. Because M257Cx43 formed minimal GJs in stably transfected KO fibroblasts of low passage number, it was expected that transient expression of M257Cx43 in WT fibroblasts might also allow for the formation of homomeric and/or heteromeric GJs. However, although high levels of M257Cx43 expression were achieved within 48 h of transfection (Fig. 6 B, arrowhead), communication between M257Cx43/WT fibroblasts in assembly assays was either decreased below control levels (two out of four experiments) or unaffected (two out of four experiments). Because of the mixed results, and because a longer assembly time of 15 min was utilized, these data are not included in Fig 6 C. Assuming that both homomeric (M257Cx43/M257Cx43) and heteromeric (M257Cx43/WTCx43) connexons were formed and functional in the M257Cx43 transfectants, an increase in control levels of GJ communication should have occurred. This was not the case, and a decrease was observed in two instances, so it is likely that the truncation mutant does not traffic normally in the fibroblast background and may even inhibit trafficking of endogenous Cx43. GJ assembly was still enhanced by 8Br-cAMP in the presence of M257Cx43 expression, and to the same extent as cells transfected with vector alone (approximately twofold), suggesting that primarily homomeric connexons of native WTCx43 were responding to cAMP.
Discussion
The COOH-terminal domain of Cx43 is required for efficient GJ assembly
A need for Cx43 to regulate function in the diverse environments of the different tissues in which it is expressed may explain the multiple regulatory sites and domains on its relatively long CT. Thus, it is not surprising that the regulation of GJs formed by truncated M257Cx43 is defective in certain cell types. GJs formed by the M257Cx43 in the Xenopus oocyte system are not sensitive to pH, whereas those formed by WTCx43 are quite sensitive (Ek-Vitorin et al., 1996), and the expression of M257Cx43 in 3T3 A31 fibroblasts prevents PDGF-induced inhibition of intercellular communication and appears to alter growth (Moorby and Gherardi, 1999). Our findings depicting a lack of enhanced assembly between cells expressing M257Cx43 further demonstrate that the CT is critical for Cx43 GJ regulation by cAMP.
Most of the regulatory domains within the CT must not be required for trafficking to the cell interface or for successful docking of channels in adjacent cells, as M257Cx43 can assemble into functional GJs in at least some cell types. One would expect this to be the case, considering that Cx26, the smallest Cx identified to date, has a cytoplasmic tail of only 15–18 amino acids in length. Further truncation of the Cx43 tail from amino acids 257 to 245 also does not appear to prevent the protein from reaching cell membranes and forming channels with single channel conductances not unlike Cx43 (Dunham et al., 1992; Fishman et al., 1991). Truncation of Cx43 has been utilized in vitro to obtain a more ordered array of assembled hexamers for electron cryocrystallography studies (Unger et al., 1999), showing that the tail is not needed for the aggregation of Cx43 hemichannels.
Using freeze fracture microscopy, we have found that M257Cx43 GJ assembly proceeds more slowly than WTCx43 GJ assembly, with fewer unaggregated particles detected in GJ formation plaques in spite of similar levels of protein expression (unpublished data). Although trafficking and assembly of M257Cx43 occurs, these processes appear to be less effective upon loss of the CT. There are reports that COOH-terminal domains of certain other proteins are also required for regulated intracellular trafficking. For example, the endosomal recycling of endothelial-derived G protein–coupled receptor or the subendosomal trafficking of Glut4 are inhibited by truncation of the CT or removal of certain CT residues (Liu et al., 1999; Cope et al., 2000). Indeed, a recent report shows that the CT of Cx43 interacts with microtubules in Rat-1 cells (Giepmans et al., 2001). As nocodazole treatment inhibits de novo GJ assembly in the WTCx43/KO fibroblast by 60% (unpublished data), it is likely that microtubules are also involved with assembly in the fibroblast system. The CT may be required for interaction with the microtubule and efficient targeting of Cx43 to the GJ.
S364 resides in a domain of the Cx43 CT that is critical to enhanced assembly
Although S364 is phosphorylated in “resting” cells, its phosphorylation is not necessary for GJ communication, as GJ assembly still occurs between both the S364ACx43 and S364PCx43/ KO fibroblasts, and with a time course like that seen between WTCx43/KO transfectants. However, phosphorylation of this site does appear to be necessary for cAMP stimulation of GJ assembly, as S364ACx43 and S364PCx43/KO fibroblasts do not display enhanced assembly in response to 8Br-cAMP. That the S364ECx43/KO fibroblasts do display enhanced assembly in response to cAMP is consistent with glutamic acid resembling a phosphate in charge.
Replacement of S364 with P not only blocks phosphorylation of the mutated site, but also likely alters the native conformation of the Cx43 by inducing an obligate bend at the proline ring (Eyles and Gierasch, 2000). This alteration in conformation could interfere with the function of other regions on the molecule, such as phosphorylation sites or protein-binding domains, that are critical for enhanced assembly. As it is likely that heteromeric channels are formed with nativeWTCx43 in the transiently transfected WT fibroblasts, the proline substitution of the S364PCx43 might also interfere with the function or phosphorylation of WTCx43 in heteromeric connexons through steric hindrance.
Importantly, GJ assembly between either the S364PCx43/KO or the S364PCx43/WT fibroblast transfectants is not enhanced following the elevation of cAMP, strongly suggesting that S364PCx43 acts as a dominant negative, with respect to enhancement of GJ assembly. It could be that S364ACx43 did not inhibit enhanced assembly when expressed in the WT fibroblast because its conformation is more similar to that of WTCx43. Phosphorylation of only some connexon subunits of WTCx43 may be sufficient for interactions that take place during enhanced assembly. We can conclude from this work that S364 is either a critical site or resides in an important domain of the Cx43 CT.
Stimulation of GJ assembly by cAMP requires activation of PKA but not increased Cx43 synthesis
Cx43 synthesis is stimulated by extended exposure to cAMP-elevating agents in certain cell systems, resulting in elevated levels of communication (Mehta et al., 1992; Darrow et al., 1995). Such increased synthesis in the WT fibroblast may occur prior to an increase in trafficking (Hendrix et al., 1995). Importantly, 8Br-cAMP treatment did not increase the synthesis of WTCx43 or of mutant Cx43 in transfected KO fibroblasts, showing that elevation of Cx43 synthesis is not required for enhanced assembly. Interestingly, treatment with cycloheximide for recovery and reaggregation blocked cAMP-enhanced assembly between WTCx43/KO fibroblasts and between WT fibroblasts (e.g., at 7-, 15-, and 60-min reaggregation times). This was in spite of significant levels of dye coupling and Cx43 detected by Western and immunoblotting after a 1-h assembly (Fig. 2). In sum, these results suggest that another, short-lived protein participates in the assembly process.
Because inhibition of PKA by H89 blocks 8Br-cAMP–stimulated GJ assembly when added immediately prior to cell reaggregation, the enhancement of GJ assembly is probably not mediated by an increase in Cx43 synthesis (8Br-cAMP is added at the beginning of recovery, so stimulation of Cx43 synthesis would have already occurred). Blockage of PKA with H89 inhibited both enhanced and control levels of GJ assembly in the WT fibroblast, so PKA may regulate even basal levels of GJ formation. However, the Cx43 CT was not a substrate for PKA, and the Cx43 phosphorylation molar ratio was not increased during enhanced assembly. Therefore, it is unlikely that phosphorylation of Cx43 by PKA mediates enhanced assembly. In spite of this, either a negative charge at 364 (e.g., glutamic acid) or a serine (which was found to be phosphorylated in untreated cells) allowed for elevation of GJ assembly by 8Br-cAMP. Mutation of S364 did not obviously affect control levels of GJ communication, whereas treatment with the PKA inhibitor did. Therefore, it appears that phosphorylation at S364 by an unknown kinase is necessary but not sufficient for enhanced assembly to occur, and that PKA activation is sufficient for enhanced assembly given phosphorylation at S364.
An increase in the size and number of GJ punctae at interfaces between WTCx43-expressing WT fibroblasts was seen with 8Br-cAMP treatment (Fig. 2 B), suggesting that connexon trafficking or GJ assembly is enhanced. However, we cannot exclude the possibility that 8Br-cAMP works in part by indirect effects on Cx43 channel gating (e.g., open probability) in this system. Further analysis will be required to determine whether PKA impacts enhanced assembly through phosphorylation of a separate protein(s) that interacts with Cx43. Such a phosphoprotein could be required for the trafficking of Cx43 or recruitment of hemichannels into GJs at the cell surface. Regulation of trafficking by PKA is observed with several other proteins (Brignoni et al., 1995; Howard et al., 2000; Zhou et al., 2000), indicating a general control mechanism. For example, the transport of vesicular stomatitis virus G glycoprotein to the cell surface is accelerated by the activation of PKA and inhibited by H89 (Muniz et al., 1996).
In summary, upregulation of GJ assembly by cAMP is mediated by a PKA-dependent stimulation of connexon trafficking that also requires microtubules (Paulson et al., 2000). Importantly, the CT of Cx43 is instrumental for the effects of cAMP, possibly by mediating an interaction with intracellular trafficking proteins. The site of such an interaction may lie in the region of the CT surrounding S364, the phosphorylation of which appears to be necessary for cAMP-enhanced GJ assembly.
Materials and methods
Cx43 antibodies
Cx43 polyclonal antibodies directed against residues 368–382 of human and rat Cx43 were provided by Dr. Alan Lau (University of Hawaii, Honolulu, Hawaii) and Sigma-Aldrich (#C6219). Antibody to the cytoplasmic loop of Cx43 was provided by Dr. Nalin Kumar (Scripps Institute, La Jolla, CA), and antibody specific for the NH2 terminus of Cx43 was provided by Dr. Dale Laird (University of Western Ontario, London, Ontario, Canada). Cx43IF1 and Cx43CT2 mouse monoclonal antibodies were generated against the last 23 amino acids of rat Cx43.
Cells
Several immortalized fibroblast cell lines were generously provided by Dr. Lau. Cells were cloned from the cardiac tissue of Cx43 KO fetal mice (KO fibroblasts) and normal fetal mice (WT fibroblasts) (Martyn et al., 1997). The KO fibroblasts and transfectants were propagated in low-glucose DME with Hepes (LG DMEM; Life Technologies) containing 10% fetal calf serum (Hyclone), 100 U/ml penicillin/streptomycin (Life Technologies), and selective antibiotics where indicated. WT fibroblasts were propagated in LG DMEM containing 10% fetal calf serum and penicillin/streptomycin. WTCx43- and M257Cx43-transfected N2A cells, provided by Dr. Steve Taffet (State University of NY, Syracuse, NY), were maintained in high-glucose DMEM containing 10% fetal calf serum and penicillin/streptomycin with 800 μg/ml geneticin (G418; Life Technologies). T51B rat epithelial cells were donated by Dr. A. Boynton (Northwest Hospital, Seattle, WA) and were propagated in LG DMEM with 5% FBS and penicillin/streptomycin. All cells were kept in a humidified 5% CO2 incubator at 37°C.
Transfectants
Stably transfected WTCx43/KO fibroblasts were generated using rat WTCx43 cDNA (Beyer et al., 1987) in pZeoSV (Invitrogen) and lipofectamine (Life Technologies), or Geneporter I (GTS) transfection protocols. WTCx43 fibroblast clones 22C-3 and 25C-2 were isolated by repeated dilution subcloning in the selective antibiotic zeocin (350 μg/ml). KO fibroblasts were similarly transfected with rat WTCx43 cDNA truncated at amino acid 257 (M257Cx43) subcloned into pZeoSV (M257Cx43/KO fibroblasts), or serine to glutamic acid, serine to alanine, and serine to proline 364 mutant rat Cx43 cDNAs (S364ECx43, S364ACx43 and S364PCx43, respectively) subcloned into pIRES1hyg (CLONTECH laboratories, Inc.). All mutant Cx43 cDNAs were provided by Dr. Taffet and were sequenced to confirm the introduced mutations and lack of extraneous mutations prior to transfection. Stably transfected clones were isolated by repeated dilution subcloning using progressively higher concentrations of the selective antibiotic hygromycin (150–500 μg/ml). Clones were established and maintained in 100 μg/ml endothelial cell growth supplement (ECGS; Becton Dickinson). ECGS was not included in the medium during experiments. At least three, usually more, different clones of WT and mutant transfectants were tested in assembly assays.
For transient transfections, rat M257Cx43, S364ECx43, S364ACx43, and S364PCx43 cDNAs in pIRES1hyg or, for confirmatory experiments, in pCDNA3.1, were transfected into 40–60% confluent WT fibroblasts using Fugene 6 reagent (Roche). Cells were utilized 48 h after transfection. All transfection efficiencies (ranging from 70–90%) were monitored using the Green Lantern plasmid (Life Technologies).
Assembly assays
Cells were passaged two days before and fed 24 h before an experiment. For GJ assembly assays, 70–80% confluent cells were dislodged from 100-mm tissue culture dishes with 0.05% trypsin and 0.53 mM EDTA, and the neutralized cell suspension centrifuged at 800 g for 5 min. Cells were resuspended to 4–5 × 105 cells/ml with or without test reagents and filtered through a 35-μM pore filter to remove cell clumps. Where indicated, cycloheximide was added at a concentration in excess of that necessary to inhibit protein synthesis by at least 96% after 10 min (Musil et al., 2000). When possible, samples were coded for blind analysis. The single cell suspension was allowed to recover for at least 1.5 h in 60-mm dishes coated with nonadhesive poly(2-hydroxyethyl methacrylate) (1.5 mg/ml, POLYHEMA; Sigma-Aldrich) on a platform shaker at 37°C, 5% CO2 to insure that GJ remnants were internalized (Preus et al., 1981). Cells were then reaggregated to confluency by centrifugation at 500 g for 5 min. Samples were staggered to ensure that treatment times were as reported, and GJ permeability was evaluated by iontophoretic microinjection of 4.0% Lucifer yellow CH (Mr = 457) (Sigma-Aldrich) at the end of the assembly period. A 5-s pulse of 1.1 nA was used to inject dye, and the number of cells receiving dye was assessed 3 min later using a Zeiss IM35 fluorescence microscope with a mercury lamp. Images were captured using a DVC-1300 digital camera (DVC Company) and Image Pro 4.1 capture software (Media Cybernetics).
SDS-PAGE and Western immunoblotting
Cell homogenates were analyzed by SDS-PAGE and Western immunoblotting (TenBroek et al., 1994). Immunolabel was detected using enhanced chemiluminescence. Results were recorded on Kodak X-OMAT AR film during the first min of the reaction. Images for densitometry were collected and analyzed using a Quant Mac densitometer and Image Quant software. Variations in sample loading were corrected by calculating the ratio of the band intensities of the autoradiographs over intensities of the corresponding stained gel or by using a SDS and thiol reagent-compatible protein assay (Bio-Rad Laboratories).
In vitro phosphorylation
GST fusion proteins GST–Cx43CT (residues 236–382) and GST–Cx56CT (residues 255–510), a gift from Dr. Viviana Berthoud (University of Chicago, Chicago, IL) were prepared as previously described (Berthoud et al., 1997; Lampe et al., 2000). GST–Cx43CT was phosphorylated by PKC, provided by Dr. Mohammad Bazzi (University of Minnesota, St. Paul, MN). For phosphorylation of GST–Cx43CT and GST–Cx56CT with PKA, final reaction concentrations were 0.25 μg/ml fusion protein, 5 μg/ml PKA (Sigma P5511; Sigma-Aldrich), 5 μM cAMP, 10 mM MgCl2, 100 μM ATP ([32P]-ATP - Amersham PB170, diluted with cold ATP to 0.3 Ci/mmol; Amersham Pharmacia Biotech), and 40 mM Tris-HCl (pH 8.0). The phosphorylation reactions were initiated by the simultaneous addition of the ATP and MgCl2. After 30–60 min the reactions were terminated by the addition of Laemmli sample buffer, and fusion protein was analyzed using SDS-PAGE (10.0%).
Determination of the phosphorylation status of S364
For nonradioactive determination of phosphopeptides in the P2 isoform, Cx43 was immunoprecipitated from eight 150-mm dishes of T51B cells (Lampe et al., 2000). Briefly, the cells were rinsed in PBS, lysed in RIPA buffer (100 mM NaCl, 50 mM NaF, 500 μM Na3VO4, 2 mM PMSF, 0.5% Triton X-100, 0.4% SDS, 25 mM Tris-HCl, pH 7.2), clarified, and immunoprecipitated with a mixture of polyclonal and monoclonal antibody to Cx43 (Sigma #C6219 and Cx43CT2, respectively; Sigma-Aldrich) that had been crosslinked to protein A Sepharose. After SDS-PAGE and Coomassie blue staining of the gel, the P2 mobility form of Cx43 was excised, diced, washed in 25 mM ammonium bicarbonate, and digested overnight with sequencing grade trypsin at 37°C (Promega) as previously described (Shevchenko et al., 1996). After elution, the phosphorylated Cx43 peptides were separated from nonphosphorylated by immobilized metal affinity chromatography (Neville et al., 1997). The mixture of phosphopeptides was examined by MALDI-TOF MS at the Fred Hutchinson Cancer Research Center's MS facility (Seattle, WA). To confirm the identity of the mass species, the tryptic fragments (both phosphorylated and nonphosphorylated) were also analyzed via liquid chromatography/electrospray–tandem mass spectrometry (Cooper et al., 2000).
Comparison of the phosphorylation status of WTCx43- and S364PCx43-expressing cells
KO fibroblasts expressing WTCx43 or S364PCx43 were metabolically labeled with [32P]i (NEN, NEX-053) at 1.0 mCi/ml for 3 h in phosphate-deficient medium (GIBCO BRL). Cx43 was then immunoprecipitated and analyzed by SDS-PAGE as described above. After excision of only the phosphorylated species, the gel was diced, washed, and dehydrated (Shevchenko et al., 1996). The gel was then rehydrated in a solution containing 200 ng modified trypsin (Promega) and 2 μg of nonphosphorylated recombinant fusion protein, GST–Cx43CT (carrier peptide), in 50 mM ammonium bicarbonate, pH 8.0, and incubated overnight at 37°C. Tryptic fragments were then separated by reverse-phase HPLC on a Hewlett Packard 1050 LC using a Vydac C18 reverse phase column as previously described (Lampe et al., 2000). 1-min fractions were counted without fluor in a Wallac scintillation counter and MALDI-TOF MS was performed. Because equivalent tryptic fragments of Cx43 from the Cx43CT, WTCx43, or S364PCx43 were expected to elute in essentially the same fractions, nonphosphorylated carrier peptide GST–Cx43CT was included in the samples. The excess unlabeled GST–Cx43CT fragments facilitated the mass identification of the co-eluting WTCx43 or S364PCx43 phosphofragments. Although phosphorylation of a peptide can reduce the elution time of short peptides by affecting the absorption of the hydrophobic “foot” region of a peptide, V347-R366 and A371-I382 were of sufficient length that phosphorylation would not be expected to shift elution by more than a few seconds.
Statistics
Data are expressed as a sample mean ± SEM. Number of injections (n) are noted in the figures. Comparisons of sample means were made using the Student's t test and two-tailed probability (P). Each experiment contained control samples. Vector alone controls were included in transient transfections whenever possible. Although Figs. 5 C and 6 C represent a compilation of several experiments, individual experiments were analyzed separately.
Acknowledgments
We thank Judd Sheridan for helpful discussions on the manuscript and other members of our laboratories for their continued support.
This work was financed by National Institutes of Health grants GM46277 and GM55632, and Minnesota Affiliate American Heart Association Grant-in-Aid 96064340.
Footnotes
Abbreviations used in this paper: CT, COOH terminus; Cx, connexin; GJ, gap junction; GST, glutathione sulfotransferase; KO, knockout; MALDI-TOF, matrix-assisted laser desorption ionization–time of flight; MS, mass spectrometry; PKA, cAMP-dependent protein kinase; PKC, preotein kinase C; WT, wild-type.
References
- Atkinson, M.M., P.D. Lampe, H.H. Lin, R. Kollander, X.R. Li, and D.T. Kiang. 1995. Cyclic AMP modifies the cellular distribution of connexin43 and induces a persistent increase in the junctional permeability of mouse mammary tumor cells. J. Cell Sci. 108:3079–3090. [DOI] [PubMed] [Google Scholar]
- Beardslee, M.A., J.G. Laing, E.C. Beyer, and J.E. Saffitz. 1998. Rapid turnover of connexin43 in the adult rat heart. Circ. Res. 83:629–635. [DOI] [PubMed] [Google Scholar]
- Berthoud, V.M., M.B. Rook, O. Traub, E.L. Hertzberg, and J.C. Saez. 1993. On the mechanisms of cell uncoupling induced by a tumor promoter phorbol ester in clone 9 cells, a rat liver epithelial cell line. Eur. J. Cell Biol. 62:384–396. [PubMed] [Google Scholar]
- Berthoud, V.M., E.C. Beyer, W.E. Kurata, A.F. Lau, and P.D. Lampe. 1997. The gap-junction protein connexin 56 is phosphorylated in the intracellular loop and the carboxy-terminal region. Eur. J. Biochem. 244:89–97. [DOI] [PubMed] [Google Scholar]
- Beyer, E.C., D.L. Paul, and D.A. Goodenough. 1987. Connexin43: a protein from rat heart homologous to a gap junction protein from liver. J. Cell Biol. 105:2621–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brignoni, M., O.P. Pignataro, M.L. Rodriguez, A. Alvarez, D.E. Vega-Salas, E. Rodriguez-Boulan, and P.J. Salas. 1995. Cyclic AMP modulates the rate of “constitutive” exocytosis of apical membrane proteins in Madin-Darby canine kidney cells. J. Cell Sci. 108:1931–1943. [DOI] [PubMed] [Google Scholar]
- Britz-Cunningham, S.H., M.M. Shah, C.W. Zuppan, and W.H. Fletcher. 1995. Mutations of the Connexin43 gap-junction gene in patients with heart malformations and defects of laterality. N. Engl. J. Med. 332:1323–1329. [DOI] [PubMed] [Google Scholar]
- Burghardt, R.C., R. Barhoumi, T.C. Sewall, and J.A. Bowen. 1995. Cyclic AMP induces rapid increases in gap junction permeability and changes in the cellular distribution of connexin43. J. Membr. Biol. 148:243–253. [DOI] [PubMed] [Google Scholar]
- Charollais, A., A. Gjinovci, J. Huarte, J. Bauquis, A. Nadal, F. Martin, E. Andreu, J.V. Sanchez-Andres, A. Calabrese, D. Bosco, et al. 2000. Junctional communication of pancreatic beta cells contributes to the control of insulin secretion and glucose tolerance. J. Clin. Invest. 106:235–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, C.D., J.L. Solan, M.K. Dolejsi, and P.D. Lampe. 2000. Analysis of connexin phosphorylation sites. Methods. 20:196–204. [DOI] [PubMed] [Google Scholar]
- Cope, D.L., S. Lee, D.R. Melvin, and G.W. Gould. 2000. Identification of further important residues within the glut4 carboxy-terminal tail which regulate subcellular trafficking. FEBS Lett. 481:261–265. [DOI] [PubMed] [Google Scholar]
- Darrow, B.J., J.G. Laing, P.D. Lampe, J.E. Saffitz, and E.C. Beyer. 1995. Expression of multiple connexins in cultured neonatal rat ventricular myocytes. Circ. Res. 76:381–387. [DOI] [PubMed] [Google Scholar]
- Debrus, S., S. Tuffery, R. Matsuoka, O. Galal, P. Sarda, U. Sauer, A. Bozio, B. Tanman, A. Toutain, M. Claustres, et al. 1997. Lack of evidence for connexin 43 gene mutations in human autosomal recessive lateralization defects. J. Mol. Cell. Cardiol. 29:1423–1431. [DOI] [PubMed] [Google Scholar]
- De Sousa, P.A., G. Valdimarsson, B.J. Nicholson, and G.M. Kidder. 1993. Connexin trafficking and the control of gap junction assembly in mouse preimplantation embryos. Development. 117:1355–1367. [DOI] [PubMed] [Google Scholar]
- Dunham, B., S. Liu, S. Taffet, E. Trabka-Janik, M. Delmar, R. Petryshyn, S. Zheng, R. Perzova, and M.L. Vallano. 1992. Immunolocalization and expression of functional and nonfunctional cell-to-cell channels from wild-type and mutant rat heart connexin43 cDNA. Circ. Res. 70:1233–1243. [DOI] [PubMed] [Google Scholar]
- Ek-Vitorin, J.F., G. Calero, G.E. Morley, W. Coombs, S.M. Taffet, and M. Delmar. 1996. PH regulation of connexin43: molecular analysis of the gating particle. Biophys. J. 71:1273–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyles, S.J., and L.M. Gierasch. 2000. Multiple roles of prolyl residues in structure and folding. J. Mol. Biol. 301:737–747. [DOI] [PubMed] [Google Scholar]
- Fishman, G.I., A.P. Moreno, D.C. Spray, and L.A. Leinwand. 1991. Functional analysis of human cardiac gap junction channel mutants. Proc. Natl. Acad. Sci. USA. 88:3525–3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giepmans, B.N., T. Hengeveld, F.R. Postma, and W.H. Moolenaar. 2000. Interaction of c-Src with gap junction protein connexin-43: role in the regulation of cell-cell communication. J. Biol. Chem. 276:8544–8549. [DOI] [PubMed] [Google Scholar]
- Giepmans, B.N., I. Verlaan, T. Hengeveld, H. Janssen, J. Calafat, M.M. Falk, and W.H. Moolenaar. 2001. Gap junction protein connexin-43 interacts directly with microtubules. Curr. Biol. 11:1364–1368. [DOI] [PubMed] [Google Scholar]
- Godwin, A.J., L.M. Green, M.P. Walsh, J.R. McDonald, D.A. Walsh, and W.H. Fletcher. 1993. In situ regulation of cell-cell communication by the cAMP-dependent protein kinase and protein kinase C. Mol. Cell. Biochem. 127–128:293–307. [DOI] [PubMed] [Google Scholar]
- Hendrix, E.M., L. Myatt, S. Sellers, P.T. Russell, and W.J. Larsen. 1995. Steroid hormone regulation of rat myometrial gap junction formation: effects on cx43 levels and trafficking. Biol. Reprod. 52:547–560. [DOI] [PubMed] [Google Scholar]
- Holm, I., A. Mikhailov, T. Jillson, and B. Rose. 1999. Dynamics of gap junctions observed in living cells with connexin43-GFP chimeric protein. Eur. J. Cell Biol. 78:856–866. [DOI] [PubMed] [Google Scholar]
- Howard, M., X. Jiang, D.B. Stolz, W.G. Hill, J.A. Johnson, S.C. Watkins, R.A. Frizzell, C.M. Bruton, P.D. Robbins, and O.A. Weisz. 2000. Forskolin-induced apical membrane insertion of virally expressed, epitope-tagged CFTR in polarized MDCK cells. Am. J. Physiol. Cell. Physiol. 279:C375–C382. [DOI] [PubMed] [Google Scholar]
- Krutovskikh, V., and H. Yamasaki. 2000. Connexin gene mutations in human genetic diseases. Mutat. Res. 462:197–207. [DOI] [PubMed] [Google Scholar]
- Kwak, B.R., M.M. Hermans, H.R. De Jonge, S.M. Lohmann, H.J. Jongsma, and M. Chanson. 1995. Differential regulation of distinct types of gap junction channels by similar phosphorylating conditions. Molec. Biol. Cell. 6:1707–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird, D.W., K.L. Puranam, and J.P. Revel. 1991. Turnover and phosphorylation dynamics of connexin43 gap junction protein in cultured cardiac myocytes. Biochem. J. 273:67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe, P.D. 1994. Analyzing phorbol ester effects on gap junction communication: a dramatic inhibition of assembly. J. Cell Biol. 127:1895–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe, P.D., and A.F. Lau. 2000. Regulation of gap junctions by phosphorylation of connexins. Arch. Biochem. Biophys. 384:205–215. [DOI] [PubMed] [Google Scholar]
- Lampe, P.D., W.E. Kurata, B.J. Warn-Cramer, and A.F. Lau. 1998. Formation of a distinct connexin43 phosphoisoform in mitotic cells is dependent upon p34cdc2 kinase. J. Cell Sci. 111:833–841. [DOI] [PubMed] [Google Scholar]
- Lampe, P.D., E.M. TenBroek, J.M. Burt, W.E. Kurata, R.G. Johnson, and A.F. Lau. 2000. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J. Cell Biol. 149:1503–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson, D.M., M.J. Wrobleski, G.D. Sagar, E.M. Westphale, and E.C. Beyer. 1997. Differential regulation of connexin43 and connexin37 in endothelial cells by cell density, growth, and TGF-beta1. Am. J. Physiol. 272:C405–C415. [DOI] [PubMed] [Google Scholar]
- Levin, M., and M. Mercola. 1998. Gap junctions are involved in the early generation of left-right asymmetry. Dev. Biol. 203:90–105. [DOI] [PubMed] [Google Scholar]
- Liu, C.H., S. Thangada, M.J. Lee, J.R. Van Brocklyn, S. Spiegel, and T. Hla. 1999. Ligand-induced trafficking of the sphingosine-1-phosphate receptor EDG-1. Mol. Biol. Cell. 10:1179–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo, C.W. 1999. Genes, gene knockouts, and mutations in the analysis of gap junctions. Dev. Genet. 24:1–4. [DOI] [PubMed] [Google Scholar]
- Martyn, K.D., W.E. Kurata, B.J. Warn-Cramer, J.M. Burt, E. TenBroek, and A.F. Lau. 1997. Immortalized connexin43 knockout cell lines display a subset of biological properties associated with the transformed phenotype. Cell Growth Differ. 8:1015–1027. [PubMed] [Google Scholar]
- Mehta, P.P., M. Yamamoto, and B. Rose. 1992. Transcription of the gene for the gap junctional protein connexin43 and expression of functional cell-to-cell channels are regulated by cAMP. Molec. Biol. Cell. 3:839–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorby, C.D., and E. Gherardi. 1999. Expression of a Cx43 deletion mutant in 3T3 A31 fibroblasts prevents PDGF-induced inhibition of cell communication and suppresses cell growth. Exp. Cell Res. 249:367–376. [DOI] [PubMed] [Google Scholar]
- Muniz, M., M. Alonso, J. Hidalgo, and A. Velasco. 1996. A regulatory role for cAMP-dependent protein kinase in protein traffic along the exocytic route. J. Biol. Chem. 271:30935–30941. [DOI] [PubMed] [Google Scholar]
- Musil, L.S., and D.A. Goodenough. 1991. Biochemical analysis of connexin43 intracellular transport, phosphorylation and assembly into gap junctional plaques. J. Cell Biol. 115:1357–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musil, L.S., and D.A. Goodenough. 1993. Multisubunit assembly of an integral plasma membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell. 74:1065–1077. [DOI] [PubMed] [Google Scholar]
- Musil, L.S., A.C. Le, J.K. VanSlyke, and L.M. Roberts. 2000. Regulation of connexin degradation as a mechanism to increase gap junction assembly and function. J. Biol. Chem. 275:25207–25215. [DOI] [PubMed] [Google Scholar]
- Neville, D.C., C.R. Rozanas, E.M. Price, D.B. Gruis, A.S. Verkman, and R.R. Townsend. 1997. Evidence for phosphorylation of serine 753 in CFTR using a novel metal-ion affinity resin and matrix-assisted laser desorption mass spectrometry. Protein Sci. 6:2436–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson, A., P. Lampe, R.A. Meyer, E. TenBroek, M.M. Atkinson, T.F. Walseth, and R.G. Johnson. 2000. Cyclic AMP and LDL trigger a rapid enhancement in gap junction assembly through a stimulation of connexin trafficking. J. Cell Sci. 113:3037–3049. [DOI] [PubMed] [Google Scholar]
- Plum, A., G. Hallas, T. Magin, F. Dombrowski, A. Hagendorff, B. Schumacher, C. Wolpert, J. Kim, W.H. Lamers, M. Evert, et al. 2000. Unique and shared functions of different connexins in mice. Curr. Biol. 10:1083–1091. [DOI] [PubMed] [Google Scholar]
- Preus, D., R. Johnson, and J. Sheridan. 1981. Gap junctions between Novikoff hepatoma cells following dissociation and recovery in the absence of cell contact. J. Ultrastruct. Res. 77:248–262. [DOI] [PubMed] [Google Scholar]
- Saez, J.C., A.C. Nairn, A.J. Czernik, G.I. Fishman, D.C. Spray, and E.L. Hertzberg. 1997. Phosphorylation of connexin43 and the regulation of neonatal rat cardiac myocyte gap junctions. J. Mol. Cell. Cardiol. 29:2131–2145. [DOI] [PubMed] [Google Scholar]
- Saez, J.C., M.C. Branes, L.A. Corvalan, E.A. Eugenin, H. Gonzalez, A.D. Martinez, and F. Palisson. 2000. Gap junctions in cells of the immune system: structure, regulation and possible functional roles. Braz. J. Med. Biol. Res. 33:447–455. [DOI] [PubMed] [Google Scholar]
- Shevchenko, A., M. Wilm, O. Vorm, and M. Mann. 1996. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68:850–858. [DOI] [PubMed] [Google Scholar]
- Simon, A.M., and D.A. Goodenough. 1998. Diverse functions of vertebrate gap junctions. Trends Cell Biol. 8:477–483. [DOI] [PubMed] [Google Scholar]
- TenBroek, E.M., R. Johnson, and C.F. Louis. 1994. Cell-to-cell communication in a differentiating ovine lens culture system. Invest. Ophthalmol. Vis. Sci. 35:215–228. [PubMed] [Google Scholar]
- Toyofuku, T., H. Zhang, Y. Akamatsu, T. Kuzuya, M. Tada, and M. Hori. 2000. c-Src regulates the interaction between connexin-43 and ZO-1 in cardiac myocytes. J. Biol. Chem. 276:1780–1788. [DOI] [PubMed] [Google Scholar]
- Unger, V.M., N.M. Kumar, N.B. Gilula, and M. Yeager. 1999. Expression, two-dimensional crystallization, and electron cryo-crystallography of recombinant gap junction membrane channels. J. Struct. Biol. 128:98–105. [DOI] [PubMed] [Google Scholar]
- Veenstra, R.D., H.Z. Wang, E.M. Westphale, and E.C. Beyer. 1992. Multiple connexins confer distinct regulatory and conductance properties of gap junctions in developing heart. Circ. Res. 71:1277–1283. [DOI] [PubMed] [Google Scholar]
- Zhang, M., and S.S. Thorgeirsson. 1994. Modulation of connexins during differentiation of oval cells into hepatocytes. Exp. Cell Res. 213:37–42. [DOI] [PubMed] [Google Scholar]
- Zhou, J., J. Yi, N. Hu, A.L. George, Jr., and K.T. Murray. 2000. Activation of protein kinase A modulates trafficking of the human cardiac sodium channel in Xenopus oocytes. Circ. Res. 87:33–38. [DOI] [PubMed] [Google Scholar]