

Abstract
The intermediate filament protein keratin 8 (K8) is critical for the development of most mouse embryos beyond midgestation. We find that 68% of K8−/− embryos, in a sensitive genetic background, are rescued from placental bleeding and subsequent death by cellular complementation with wild-type tetraploid extraembryonic cells. This indicates that the primary defect responsible for K8−/− lethality is trophoblast giant cell layer failure. Furthermore, the genetic absence of maternal but not paternal TNF doubles the number of viable K8−/− embryos. Finally, we show that K8−/− concepti are more sensitive to a TNF-dependent epithelial apoptosis induced by the administration of concanavalin A (ConA) to pregnant mothers. The ConA-induced failure of the trophoblast giant cell barrier results in hematoma formation between the trophoblast giant cell layer and the embryonic yolk sac in a phenocopy of dying K8-deficient concepti in a sensitive genetic background. We conclude the lethality of K8−/− embryos is due to a TNF-sensitive failure of trophoblast giant cell barrier function. The keratin-dependent protection of trophoblast giant cells from a maternal TNF-dependent apoptotic challenge may be a key function of simple epithelial keratins.
Keywords: keratin; placenta; TNF; concanavalin A; intermediate filament
Introduction
The first epithelial cells of the mammalian embryo, trophectoderm, and later trophoblast derivatives form the cellular interface between maternal and embryonic environments. Type II keratin 8 (K8)* and type I keratin 18 (K18) are the first intermediate filament proteins expressed during embryogenesis and are diagnostic of the first epithelial cells (Brulet et al., 1980; Jackson et al., 1980). Subsequently, K8 and K18 are expressed in trophoblast derivatives, embryonic and extraembryonic endoderm (Jackson et al., 1981; Oshima, 1981), and simple epithelia of adult organs, such as liver, lung, kidney, pancreas, gastrointestinal tract, and mammary gland (Oshima et al., 1996). At least 49 keratins constitute a family of obligate, heteropolymeric intermediate filament proteins expressed in pairs in various epithelia (Moll et al., 1982; Hesse et al., 2001).
Although the function of epidermal keratins in providing mechanical strength to the epidermis is well documented (for reviews see Fuchs and Weber, 1994; Ma et al., 2001), the functions of the keratins of simple epithelium, K7, K8, K18, K19, and K20, have been more elusive. Tissue culture experiments have suggested a role of simple epithelium keratins in invasion of extracellular matrix (Hendrix et al., 1996), drug resistance (Bauman et al., 1994; Parekh and Simpkins, 1995), and apoptosis (Caulin et al., 2000; Gilbert et al., 2001; Inada et al., 2001). K8, K18, and K19 knockout and dominant-negative K18 mutation mouse models have revealed functions during embryogenesis, in female reproduction, in colon homeostasis, and in providing protection against hepatotoxic drugs and other liver injuries (for review see Omary and Ku, 1997). The K8 knockout mutation results in embryonic lethality at midgestation in a C57BL/6;129X1/SvJ (B6;129) hybrid genetic background (Baribault et al., 1993), whereas in a FVB/N genetic background, half of K8 −/− mice survive into adulthood (Baribault et al., 1994). These (FVB/N) K8 −/− mice develop a severe colorectal inflammation and hyperplasia. (FVB/N) K8 −/− mice are also more susceptible to liver injuries induced by porphyrinogenic drugs (Zatloukal et al., 2000), microcystin-LR (Toivola et al., 1998), partial hepatectomy (Loranger et al., 1997), and hepatic apoptosis caused by concanavalin A (ConA) (Caulin et al., 2000). K8 −/− females are largely sterile. In contrast, K18 −/− and K19 −/− mice develop normally to adulthood and are fertile (Magin et al., 1998; Tamai et al., 2000). However, both K8−/− and K18−/− mice are more sensitive to ConA-induced apoptosis in the liver where K8 and K18 are the only expressed intermediate filament proteins (Caulin et al., 2000). Similarly, transgenic mice expressing a dominant-negative K18 gene have an increased susceptibility to liver injury (Ku et al., 1995, 1996). The phenotypes of compound, simple epithelial keratin knockouts reflect overlapping functions for K18 and K19 and the obligate heteropolymeric stabilization of keratin subunits. K18 −/−; K19 − / − compound homozygotes die at E9.5 associated with a disruption of the trophoblast giant cell layer (Hesse et al., 2000). This was interpreted as increased fragility of K8−/− trophoblast cells. Similarly, K8−/−; K19 −/− double mutants on an FVB/N background have trophoblast tissue abnormalities and develop hematoma beneath the yolk sac before death at about embryonic day (E)10.5 (Tamai et al., 2000).
We present three lines of evidence that support the view that K8 provides trophoblast giant cells resistance to a maternal challenge that results in failure of the trophoblast barrier function and eventually death of the embryo. The evidence includes the following: (1) the rescue of K8−/− lethality by tetraploid wild-type extraembryonic tissues; (2) the partial rescue of K8−/− embryos by the absence of maternal but not paternal TNF and TNF receptor (TNFR)2; and (3) the experimental induction of K8−/− trophoblast giant cell barrier function failure by treatment of mothers with ConA, a known nonspecific activator of the immune system. K8-deficient embryos die as a consequence of the failure of trophoblast giant cell barrier function.
Results
Nearly all (B6;129) K8 −/− embryos die in utero by E12.5 with internal hemorrhages, which seem to originate from embryonic liver tissues (Baribault et al., 1993). However, these changes may be caused by primary failure of extraembryonic tissues, which separate the embryo from the maternal environment and also ensure nutrient supply. For example, placental malfunction in retinoblastoma-deficient mice causes multiple indirect embryonic defects (Wu et al., 2003). We have tested whether the K8 −/− embryonic lethality originates from extraembryonic tissues by generating chimeric concepti in which the embryo proper was derived from k8-deficient embryos, and most extraembryonic tissues were derived from wild-type tetraploid cells.
Embryos resulting from heterozygous intercrosses were aggregated with tetraploid wild-type embryos and then reimplanted in the uteri of foster mothers. The concepti were allowed to develop to either E16.5 or to term. Tail samples were collected from E16.5 embryos and newborn pups for genotyping by PCR (Table I). 3 out of 19 E16.5 embryos (16%) were homozygous for the K8 knockout mutation, suggesting that the presence of K8 in extraembryonic tissues protected the embryos from dying at E12 (Table I). We confirmed the absence of K8 in all three E16.5 embryos proper, including in the liver and the intestine by immunofluorescence staining (unpublished data). Extraembryonic tissues, including the yolk sac and the placenta, stained positive for the presence of K8 (unpublished data). These three embryos appeared normal with no traces of internal bleeding. Three resorbing embryos were found in these experiments. These embryos were identified as wild-type and heterozygous by PCR. Thus, these resorptions appeared to be incidental to embryo manipulations rather than genotype related.
Table I. Genotype analysis of the progeny resulting from tetraploid-diploid aggregation experiments.
| Type of transplanted embryos | Stage of analysis | K8+ /+ | K8− /+ | K8− /− |
|---|---|---|---|---|
| Tetraploid-diploid aggregates | ||||
| E16.5 | 4 | 12 | 3 | |
| Birth | 4 | 10 | 3 | |
| Diploid embryos | ||||
| E16.5 | 4 | 5 | 0 | |
| Birth | 9 | 12 | 0 |
Preimplantation diploid embryos were collected from (B6;129)K8+/− intercrosses and aggregated with wild-type tetraploid four-cell stage embryos. Chimeric embryos were allowed to develop to the blastocyst stage and were then transferred to the uteri of foster mothers. E16.5 embryos and newly born pups were genotyped. In a control experiment, morulae collected from (B6;129) heterozygous intercrosses were cultured overnight without aggregation with tetraploid embryos. These nonchimeric blastocysts were transferred to the uteri of a pseudopregnant female recipient. The resulting E16.5 embryos and newborn pups were genotyped. The numbers of K8+/+, K8+/−, and K8−/− embryos are presented.
When the chimeric concepti were allowed to develop to term, 3 out of 17 newly born pups (18%) were homozygous mutants (Table I). A total of 6 out of 36 mice (16.7%) of the mice resulting from tetraploid-diploid aggregation were K8−/−. This represents 68% of the expected frequency of K8−/− embryos. This reflects an excellent rate of rescue because of the random distribution of donor cells to the trophoblast layer. These results confirmed that K8 expression in extraembryonic tissues is necessary for the development of (B6;129) concepti, whereas K8 expression in the embryo proper is dispensable for its development to birth. The (B6;129)K8 −/− pups developed normally to adulthood without suffering anorectal prolapse as found for 88% of (FBV/N)K8 −/− mice up to 1 yr old (Baribault et al., 1994). This is consistent with the lack of anorectal prolapse previously found in six rare, spontaneous (B6)K8−/− escaper mice.
In control experiments, (B6;129)K8 −/− embryos resulting from heterozygous mating were cultured overnight and transferred to pseudopregnant female recipient without previous aggregation with tetraploid embryos (Table I). No homozygous mutant was identified among the resulting progeny either at E16.5 or at birth. These results confirmed that the rescue of the (B6;129)K8 −/− embryos was not due to the transfer to CD-1 recipients but to K8 expression in the extraembryonic tissues of chimeric concepti.
Hematoma formation in (B6;129)K8 −/− concepti
The extraembryonic tissues of additional mutant concepti (B6;129) from heterozygous intercrosses were analyzed histologically. Consistent with the functional demonstration that K8 −/− embryonic lethality originated from a defect in extraembryonic tissues, the dissection of the uterine wall of heterozygous females revealed partially coagulated blood between the decidua and the yolk sac attached to K8-deficient embryos starting at E10.5. The yolk sac, placenta, and embryo proper appeared normal at this stage (Fig. 1, A and B). Histological examination of five homozygous mutant concepti revealed the presence of massive hemorrhages of maternal blood confined between the decidua capsularis and the parietal yolk sac (Fig. 1, B and D). Resulting hematomas consisted of nonnucleated maternal erythrocytes and fibrin aggregates infiltrated with granulocytes. Hematomas in mutant concepti were apparently due to the disruption of trophoblast giant cell layer (Fig. 1 E) that normally forms a barrier between the maternal and embryonic compartments. At the site of disruption, trophoblast giant cells undergo degeneration (Fig. 1 F). The overall structure of placenta and the yolk sacs in K8 −/− concepti (Fig. 1, D and E) and the structure of embryo proper (unpublished data) at E10.5 was morphologically normal.
Figure 1.
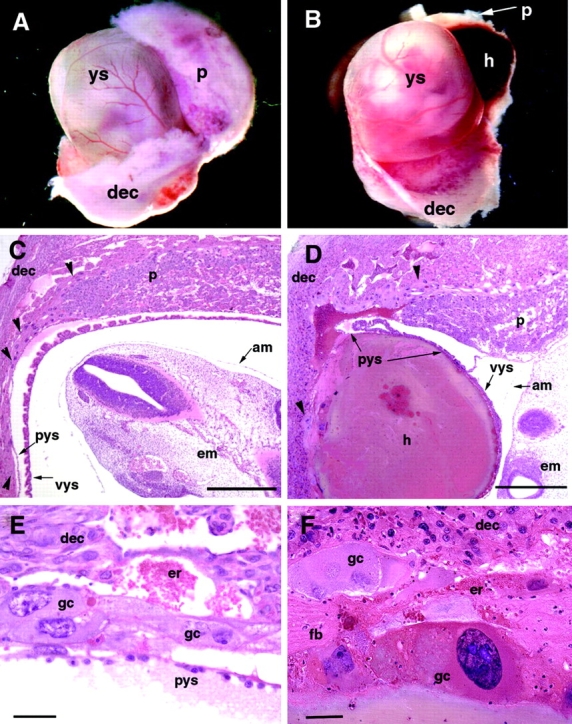
Morphological and histological analysis of (B6;129) wild-type and K8 − / − extraembryonic tissues. Wild-type (A) and K8 −/− (B) E10.5 concepti from which the uterine wall and part of decidua are removed. Note the hematoma (h) located between the decidual tissue (dec) and the yolk sac (ys) in the K8 −/− conceptus. In mutant conceptus, the placenta (p) is hidden behind the hematoma. (C and D) Sagittal histological sections of E10.5 wild-type (C) and K8 −/− (D) concepti. In the wild-type conceptus, the embryo proper (em) is surrounded by the amnion (am), visceral yolk sac (vys), parietal yolk sac (pys), the layer of trophoblast giant cells (arrowheads), and maternal decidual tissue (dec). In the K8 −/− conceptus, the hematoma (h) is located between the decidual tissue (dec) and parietal yolk sac (pys). p, placenta. Bar, 200 μm. (E and F) Trophoblast giant cells in E10.5 wild-type (E) and K8 − / − (F) concepti at higher magnification. In wild-type conceptus, the continuous layer of trophoblast giant cells (gc) divides maternal decidua (dec) from the parietal yolk sac (pys). Note the disrupted layer of trophoblast giant cells (gc), which undergo degeneration, maternal erythrocytes (er), and fibrin (fb), infiltrated with granulocytes in K8 − / − conceptus. Bar, 50 μm.
K8−/− death depends on maternal TNF
Some K8−/− epithelial cell lines are 100-fold more sensitive to TNF-induced apoptosis (Caulin et al., 2000). To test genetically whether TNF may contribute to the death of K8−/− embryos, we combined the K8 deficiency (FVB/N) with TNF deficiency (C57Bl/6;129). The FVB/N genetic background was chosen for the K8−/− mice to efficiently obtain adequate numbers of progeny for statistical analysis. Interbreeding of the two targeted alleles resulted in mixed background (FVB/N;B6;129) K8+/− parents with either TNF+/− or TNF−/− (Fig. 2, G3). The recovery of K8−/− progeny from three different crosses was measured. If TNF and the maternal immune system participated in the death of K8−/− embryos, a maternal dependence on TNF was expected. Approximately twice as many K8−/− progeny were recovered when the mother was TNF−/− (C1 and C2) than when the mother was TNF+/− (Fig. 3, A and B). The difference in K8−/− recovery from the C1 cross was significantly different from the reciprocal C3 cross in which the father, rather than the mother, was TNF+/− (Fig. 3 B). TNF deficiency of both the mother and father did not yield greater recovery of K8−/− progeny than when only the mother was deficient. Combining the results of both crosses in which the mothers were TNF−/− (C1 and C2) further reinforced the conclusion. However, a full recovery of K8−/− mice was not obtained. The recovery of K8−/− mice increased from ∼25% to >50% of the number expected for Mendelian inheritance (Fig. 2 C). The recovery of K8+/+ and K8+/− mice did not differ significantly from the expected number. Survival of TNF+/− and TNF−/− progeny in the C2 and C3 crosses were not statistically different (46 TNF+/− versus 51 TNF−/−). Thus, TNF deficiency of the embryo does not influence survival. These data indicate that maternally expressed TNF is deleterious to the survival of K8−/− embryos.
Figure 2.

Breeding strategy to test the influence of TNF on K8 −/− survival. Two rounds of matings (G1 and G2) were performed to obtain the mouse genotypes needed for the experimental matings (G3) that produced offspring of experimental matings C1, C2, and C3. All parents were K8+/−.
Figure 3.
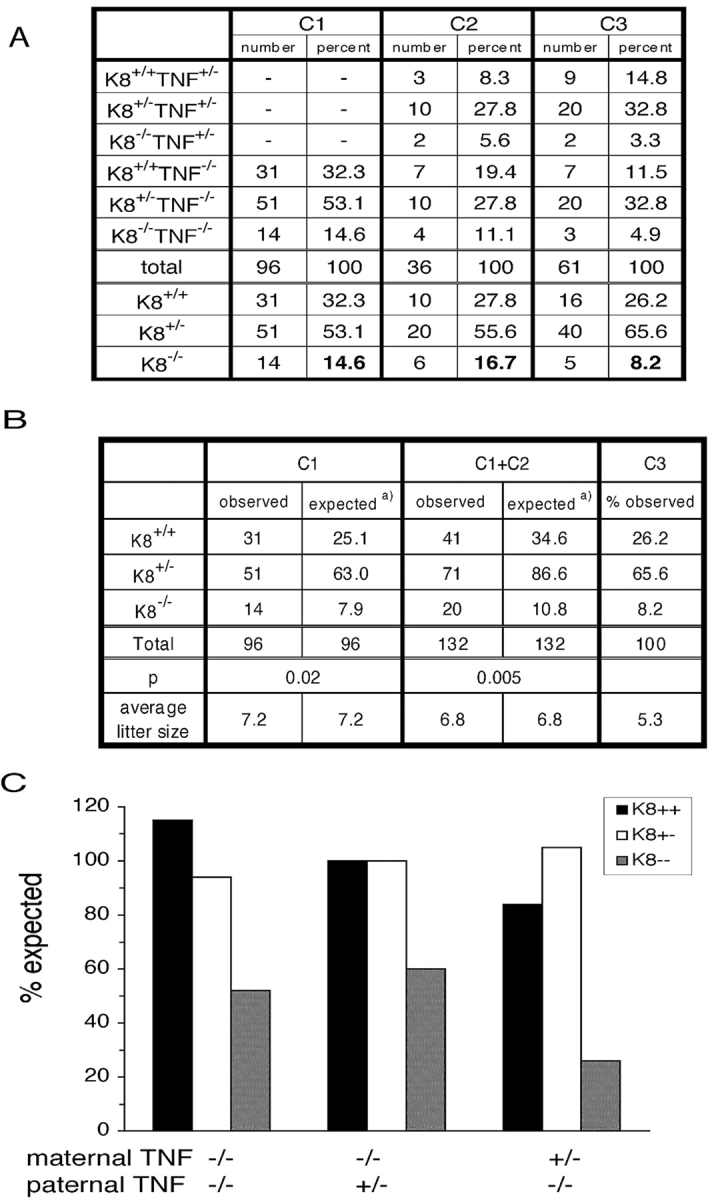
The absence of maternal TNF increases the recovery of K8 − / − mice. (A) The number of mice of each genotype from the crosses C1, C2, and C3 (Fig. 2) are shown. (B) Statistical analysis using the Chi squared test is presented. The observed number of K8+/+, K8+/−, and K8−/− mice from crosses C1 and C1 + C2 are compared with the numbers expected using the percentages from cross C3 as standard. (C) Graphic representation of the recovery of K8+/+, K8+/−, and K8−/− mice compared with expected Mendelian inheritance.
(FVB/N) K8−/− mice develop inflammatory bowel disease. Thus, it was of interest to determine if the deficiency in TNF, a common mediator of inflammation, altered this phenotype. 60% (12 out of 20) of informative K8−/−;TNF−/− mice survived for >250 d. Five animals that died prematurely were confirmed to have anorectal prolapse and colonic hyperplasia with inflammation (unpublished data). Similarly, 50% of the K8−/−;TNF+/− mice (two out of four) survived for >250 d, and the inflammatory bowel disease was confirmed in the two that died prematurely. Inflammatory bowel disease can develop in the absence of TNF in K8−/− mice.
Maternal TNFR2 deficiency increases viability of K8−/− embryos
We also tested the influence of TNFR2 deficiency on K8−/− viability. K8+/−(FVB/N) mice were bred with TNFR2−/− (B6;129), and progeny were backcrossed with TNFR2−/−(B6;129) to generate K8+/−;TNFR2−/− males and either K8+/−;TNFR2−/− or K8+/−;TNFR2+/− mothers (analogous to C1 and C3 crosses of Fig. 2 A). The absence of TNFR2 in mothers resulted in a more than fourfold increase in the frequency of K8−/− progeny (Fig. 4 A). The difference between the recoveries of K8−/− progeny was statistically significant (Fig. 4 B). However, TNFR2 deficiency also failed to completely rescue K8−/− lethality (Fig. 4 C). TNFR2-deficient embryos were found as frequently as TNFR2 heterozygotes. Thus, both embryonic TNFR2 and TNF are dispensable for embryonic development. In the presence of maternal TNFR2, K8−/− embryos with an absence of TNFR2 were not recovered more frequently than K8−/− embryos with TNFR2 (Fig. 3 A, C3). Thus, the possible modulation of TNFR2 signaling by K8/K18 is not sufficient to ensure embryo survival. The recovery of K8+/+ and K8+/− animals was statistically not distinguishable from the expected number based on Mendelian inheritance (Fig. 3 C). The lower recovery of K8−/− embryos from the C3 cross with TNFR2− compared with the analogous C3 cross with TNF− may be due to differences in the contributions of the B6 and 129 genetic backgrounds in the TNF−/− and TNFR2−/− mouse strains. These results indicate that maternal TNFR2 contributes to the death of K8−/− embryos but provides no support for a role for embryonic TNFR2 in the death of K8−/− embryos.
Figure 4.
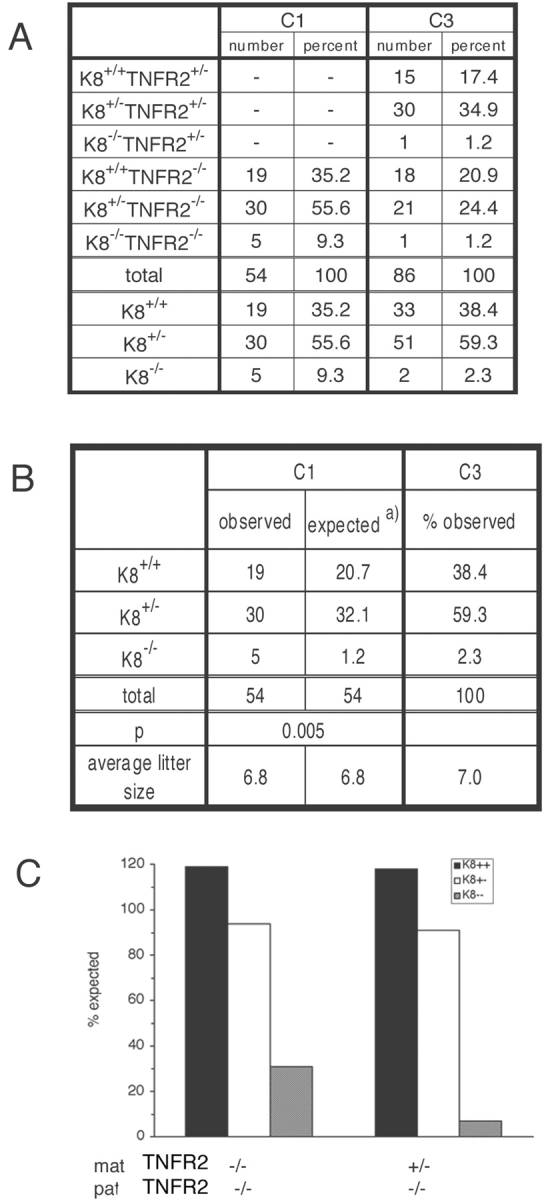
More K8 − / − mice are recovered in the absence of maternal TNFR2. The same strategy shown in Fig. 2 was used for the K8 × TNFR2 crosses except that cross C2 was not performed. (A) The number of mice obtained from the K8 × TNFR2 cross as determined by tail DNA PCR. (B) The observed number of K8 wild-type, K8 heterozygote, and K8 knockout mice from crosses C1 are compared with the percentage of recovery from cross C3 as standard using the Chi squared analysis. (C) Graphic representation of the recovery of K8 genotypes to that expected for Mendelian inheritance.
Injection of ConA activates lymphoid cells resulting in lymphoid cell–dependent liver apoptosis (Tiegs et al., 1992). TNF, TNFR1, TNFR2, fas, and other cytokines are involved in the hepatic damage (Gantner et al., 1995; Kusters et al., 1997). To challenge the trophoblast-derived cells of K8−/− conceptus, we treated E9.5 pregnant mothers with ConA and examined both the livers and concepti for pathological changes. ConA treatment of pregnant K8+/−; TNF+/− mothers resulted in the expected liver apoptosis. However, both the liver and concepti of a single available K8+/− TNF−/− mother were resistant to the effects of ConA (unpublished data) consistent with previous investigations of male mice.
ConA causes larger hematoma formation in K8−/− concepti
(FVB/N; B6;129) K8+/−; TNF+/− females were mated with (FVB/N; B6;129) K8−/−; TNF−/− males (both from the experiment shown in Fig. 2) to generate K8+/− and K8−/− embryos within a TNF+/− maternal environment. At E9.5, mothers were treated with ConA, and a portion of each embryo was dissected for PCR analysis, and the remaining concepti were fixed, bisected, and processed for paraffin sections. Embryos removed from the yolk sacs were all similar size and without obvious defects. Stained sections revealed hematoma formation in K8−/− extraembryonic tissues very similar to the phenotype of the spontaneous K8−/− concepti in the B6;129 genetic background at E10.5 (Fig. 5, A–F, and Fig. 1). Hematomas were commonly found between the trophoblast giant cell layer and the parietal yolk sac. Quantitation of bleeding in extraembryonic tissues revealed significantly increased bleeding in K8−/− extraembryonic tissues (Fig. 5, G–I). The average hematoma area for 16 concepti from three litters was 4,578 arbitrary units compared with 1,607 for 14 K8+/− concepti. (P = 0.007, two tailed t test). No significant correlation was found between the degree of bleeding and the TNF genotype of the embryos. Thus, K8−/−concepti are more sensitive to a ConA-induced, maternal TNF-dependent loss of trophoblast giant cells barrier function. This phenotype is similar to that observed for K8−/− concepti in a B6;129 genetic background without ConA treatment. ConA treatment of a single pregnant TNF−/−; K8+/− mother of the same mixed genetic background did not result in visible hematoma formation. This suggests that both liver apoptosis and concepti hematoma formation caused by ConA treatment of pregnant mothers is dependent on maternal TNF.
Figure 5.

K8-deficient concepti are more sensitive to ConA-induced hematoma formation. Three K8+/−TNF+/− females were bred to a K8−/−TNF−/− male. 10 d postcoitus, the mothers were subjected to ConA treatment. 5 h after injection, the mice were killed, and the concepti were removed for histological analysis. Hematoxylin and eosin–stained sections revealed different degrees of bleeding. (A–F) Red blood cells are rendered in black by digital color replacement. Note that most of the hematoma occurs opposite the placenta (E and F). The identity of each specimen is indicated by the litter number and embryo number. Bar, 1 mm. Quantitative measurements of the hemorrhage size for every embryo from the three litters are shown in G, H, and I. The numbers above the horizontal bar of each panel indicate the embryo identification number of that litter. A statistical analysis of these numbers confirms that there is a statistical significant difference in the amount of hemorrhage in K8+/− versus K8−/− embryos (P = 0.006), but there is no such difference between TNF+/− (black bars) and TNF−/− (gray bars) embryos (P = 0.08).
Apoptotic death of trophoblast giant cells
Immunohistochemical analysis of extraembryonic tissues of ConA-treated mice confirmed the expression of K8 in trophoblast derivatives, including spongiotrophoblast, giant cells, and epithelial cells, in the labyrinth region recognized by the presence of nucleated embryonic blood cells (Fig. 6 A). The placentas of E10 K8−/− embryos had a relatively normal structure but lacked K8 (Fig. 6 B). Staining nuclei for DNA nicks by the TUNEL method revealed apoptotic and degenerating giant cells near the large hematomas (Fig. 6, C–F). However, apoptotic giant cells were found in both K8+/− and K8−/− implantation sites, and it was not possible to distinguish the genotype based solely on apoptotic staining because of the relatively late stage at which hematoma formation best distinguished the K8-positive and -negative concepti. Nevertheless, these results confirmed apoptotic giant cells are common in ConA-treated mothers by the time large hematomas are detected.
Figure 6.
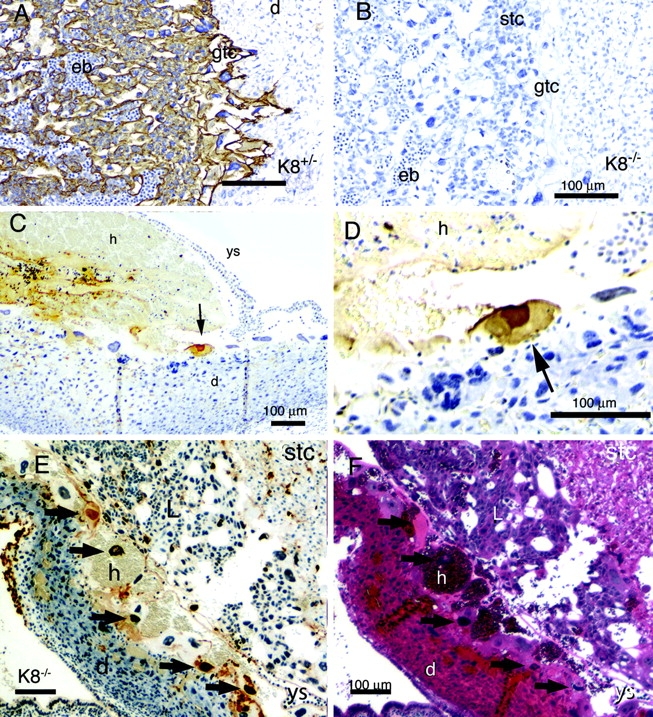
Apoptotic trophoblast giant cells in ConA-treated mice. Sections of the placenta (A and B) and uterine wall (C–F) were stained for K8 (A and B) and apoptotic nuclei by the TUNEL method (C–E). F shows a neighboring section of E stained with hematoxylin and eosin. Tissues with K8−/− concepti are shown in B–F. The tissues are labeled: d, decidua; eb, embryonic blood; gtc, giant trophoblast cell; h, hemorrhage; L, labyrinth region; stc, spongiotrophoblast cell; r, Reicharts membrane; ys, yolk sac. The black arrows in C–F indicate apoptotic giant cells. Bar, 100 μm. The tissue shown in E and F represent an adjacent uterine wall region which has folded up against the placental region as revealed by tracing the folded yolk sac basement membrane. This results in the atypical juxtaposition of the failed trophoblast giant cell layer against the labyrinth region of the placenta. Note the apoptotic giant cell nuclei associate with the accumulated hematomas.
Discussion
The tetraploid embryo rescue of K8-deficient embryos demonstrates that defective extraembryonic tissues cause the midgestational death of K8−/−. Nearly 70% of the expected K8−/− embryos were recovered by aggregation with tetraploid embryos. Since the number of tetraploid cells associated with the trophoblast layer will vary randomly, the remaining aggregated K8−/− embryos most likely did not receive a sufficient contribution of tetraploid cells to survive. The accumulation of blood between the decidua capsularis and the parietal yolk sac is consistent with a trophoblast giant cell barrier function failure. The integrity of the Reichert's membrane/parietal yolk sac to delimit the hematomas and previous observations that extraembryonic endodermal cell formation and function from K8−/− ES cells was not impaired (Baribault and Oshima, 1991) suggests that the trophoblast giant cell layer, not the extraembryonic endoderm, is defective in K8−/− concepti. Trophoblast giant cells are derivatives of mural extraembryonic ectoderm (primary giant cells) and the ectoplacental cone (secondary giant cells). They form an epithelial boundary between the maternal tissue and the embryonic environment (Cross et al., 1994; Cross, 2000; Ward and Devor-Henneman, 2000). Trophoblast giant cells also interact actively with maternal tissues, including the immune system, and produce several angiogenic factors and hormones (Cross, 2000).
The phenotype observed for dying (B6;129) K8−/− concepti is similar to that found when K8 and K19 or K18 and K19 deficiencies in FVB/N or mixed backgrounds are combined (Hesse et al., 2000; Tamai et al., 2000). These experiments indicate functional compensation between different simple epithelial keratin pairs (Oshima, 2002). Maternal TNF and TNFR2 deficiencies result in an increase in viable K8−/− progeny. This implicates TNF in the embryonic lethality of K8−/− embryos. Although maternal TNF deficiency results in a twofold increase in K8−/− progeny, it does not completely compensate. Other members of the TNF family or other cytokines may also be involved in the early death of K8−/− trophoblast cells. Alternatively, TNF and TNFR2 may be necessary for the activation of lymphoid cells or cytokine effectors. Circulating TNF and uterine natural killer cells are among possible direct effectors. Hepatic natural killer cells have been implicated in the ConA-induced liver pathology (Takeda et al., 2000). Natural killer cells also reside in the uterine environment of both rodents and humans (Moffett-King, 2002) where their cytolytic activity is inhibited. ConA stimulation may bypass the normal inhibition of uterine natural killer cells cytolytic activity (Muller et al., 1999). Our results are the first to associate the embryonic death of keratin-deficient concepti with TNF. The significant immunological differences between mice of different genetic backgrounds may contribute to the genetic background dependence of K8−/− embryonic lethality.
The molecular mechanism by which K8 provides protection to trophoblast cells from TNF remains to be determined. The protection that K8 and K18 heteropolymers provide cultured epithelial cell lines and hepatocytes against TNF- (Caulin et al., 2000; Inada et al., 2001) and fas- (Gilbert et al., 2001) mediated apoptosis may extend in vivo to derivatives of the first epithelial cells to form during development. The sequestration of the TRADD signal transduction adaptor protein by K18 provides an attractive model of keratin-dependent protection from apoptosis (Inada et al., 2001). It also remains to be determined if the absence of K8 alters the strength of desmosomal, intracellular adhesion of trophoblast cells (Huen et al., 2002).
(FVB/N) K8−/− mice develop colon hyperplastic disease, and anorectal prolapse TNF has been implicated in inflammatory bowel disease. Anorectal prolapsed is not observed in K8−/− mice in the B6 background. However, since the compound TNF; K8 homozygous deficiency is on a mixed genetic background and the recovery of viable K8−/−;TNF+/− was low, we cannot conclude whether the incidence of the disease is influenced by TNF deficiency. However, clearly colonic hyperplasia can develop in the absence of TNF in K8−/− mice. Thus, TNF is not essential for prolapse formation in K8−/−mice.
The use of ConA stimulation of pregnant TNF-positive mothers provided the opportunity of challenging K8+/− and K8−/− embryos in the same uterine environment. The ConA-induced hematomas of K8−/− concepti are similar to those observed for K8−/− concepti in the C57BL/6; 129 genetic background and that reported for K18−/−; K19−/− and K8−/−; K19−/− compound homozygote concepti. ConA-induced liver apoptosis can be conferred by adoptive transfer of lymphoid cells and is dependent on TNF, TNFR1, TNFR2 (Gantner et al., 1995; Kusters et al., 1997), and fas (Takeda et al., 2000) in different strains of mice. We speculate that the ConA sensitivity of the trophoblast barrier function reflects the action of the host immune system, just as it does in ConA-mediated hepatitis. Both K8−/− and K18−/− animals have previously been found to be sensitive to liver apoptosis induced by ConA. The increased sensitivity of K18−/− mice to ConA-induced hepatitis is likely due to the exclusive expression of K8 and K18 keratins in the liver, whereas K7, K8, K18, and K19 are expressed in trophoblast derivatives. The sensitivity of K8−/− giant cells to disruption after ConA treatment may reflect the high levels of K8 and modest levels of K7 expression (Tamai et al., 2000).
The tolerance of genetically dissimilar embryos within the maternal environment remains a fascinating and important question. Simple epithelial keratins may contribute to the protection of the barrier that separates immunologically distinct environments.
Materials and methods
ConA was purchased from Sigma-Aldrich. TROMA1 rat mAb against K8 was a gift from Rolf Kemler (Max-Planck-Institute für Immunbiologie, Freiburg, Germany).
Animal husbandry
Mice were housed under specific pathogen-free conditions with a 14/10 h light/dark cycle and standard temperature and humidity. They were fed standard rodent chow and water ad libitum. The generation of the K8-deficient mouse line and the PCR genotyping procedure have been described previously (Baribault et al., 1993). The resulting hybrid progeny was backcrossed further to either C57BL/6 mice or to 129X1/SvJ mice for several generations and for over 10 generations to FVB/N. In both the C57BL/6 and 129X1/SvJ genetic backgrounds, the K8 mutation caused embryonic lethality at midgestation. For the tetraploid embryo rescue experiments presented here, we have used these C57BL/6;129X1/SvJ hybrid mice. For simplicity purposes, these mice are designated B6;129 mice. The B6;129S6-TNF tm1Gkl (TNF) and B6;129S2-TNFrsf1b tm1Mwm (TNFR2) mouse lines were obtained from Jackson Laboratory. Genomic DNA was isolated from mouse tails by standard methods (Miller et al., 1988). About 1/100 of the DNA isolated from a 5-mm piece of tail was used in a three primer PCR assay to identify the genotype of TNF mice: primers U4390 (GGACTAGCCAGGAGGGAGAACAG) and L6065 (GTGTGAGGGTCTGGGCCATAG) amplified an 887-bp wild-type fragment. Primers U5485 (CCTTAATATGCGAAGTGGACCTG) and L6065 amplified the 601-bp mutant allele. For genotyping the TNFR2 mice, the following three primers were used: U5579 (AGAGCGCAGCTGAGGCACTAGAG) and L6079 (CACCCCTTCCCAGCCTCTGAG) amplified the 521-bp mutant allele, and U5579 and L5861 (ATCCCAACGGCCAGACCTCG) yielded the 302-bp wild-type allele.
Collection of two-cell embryos and electro fusion
Super ovulated CD-1 females were mated with FVB/N males. E1.5 embryos at the two-cell stage were flushed from the oviducts and collected in FHM medium (Specialty Media Inc.). After the equilibration in fusion solution (0.3 M mannitol, 0.1 mM MgSO4, 50 μM CaCl2, 3% BSA), embryos were placed between the electrodes of a BTX 2001 electro cell manipulator (Genetronics, Inc.). Embryos were aligned in a 7.0 V AC field and treated with a single 100-μs DC pulse of 1.5–1.7 kV/cm. Then, the embryos were transferred to micro drops of KSOM medium (Specialty Media Inc.) under mineral oil and incubated at 37°C in an incubator with 5% CO2 for 30–60 min at which time embryos with successfully fused blastomeres were considered as tetraploid and selected for overnight culture under the same conditions. During the afternoon of the next day, four-cell stage tetraploid embryos were used for aggregation.
Generation of tetraploid-diploid aggregates
Super ovulated (B6;129)K8 +/− females were mated to (B6;129)K8 +/− males. Diploid E2.5 embryos at six- to eight-cell stage were isolated from the oviducts and uterine horns. The zona pellucida in both diploid and tetraploid embryos was removed in acidic Tyrode's solution (pH 2.1). Two tetraploid embryos were aggregated with one diploid embryo (Nagy and Rossant, 1993). After a 24–26-h incubation, successfully aggregated embryos formed single blastocysts, which were transferred to the uterine horns of day 2.5 pseudopregnant CD-1 female recipients. Chimeric concepti were collected at E16.5, or alternatively, newborn pups were recovered by caesarian section at E18.5 and fostered by females who had delivered litters on the same day. Genotypes were determined on tail DNA by PCR analysis as described previously (Baribault et al., 1994). In control experiments, diploid embryos resulting from heterozygous (B6;129)K8 +/− mating were collected but were not aggregated with tetraploid embryos. The zona pellucida was removed from the diploid embryos. These embryos were cultured overnight under the same conditions as the diploid-tetraploid aggregates described above and subsequently transplanted into the uterine horns of pseudopregnant females.
Histology
Extraembryonic and embryonic tissues were dissected from individual uterine implantation sites of pregnant (B6;129) K8+/− females mated to (B6;129) K8+/− males at various days postcoitus as described (Hogan et al., 1986) except that the embryos were not removed from their surrounding extraembryonic tissues. When genotyping was performed, a small portion of embryonic tail was collected for PCR analysis. In some cases, uterine horns were not dissected to preserve the natural position of decidual tissues and attached layer of trophoblast giant cells. The collected tissues were fixed in Bouin's fixative and processed according to standard histological procedures for paraffin sections stained with hematoxylin and eosin.
ConA treatment
Mice were injected with ConA (30 mg/kg) in 200 μl pyrogen-free saline 10 d postcoitus. 5 h after injection, the mice were killed, and liver, spleen, and individual implantation sites within the uterine horns were fixed in 4% PFA or 1% acetic acid in ethanol, bisected, and both halves processed for embedding in paraffin. The cut sides of both halves of each implantation site were oriented to the cutting face so that increasing depths of both halves of the same site were sampled. 5-μm-thick sections were stained with hematoxylin and eosin for detection of apoptosis with the Apop Tag kit (Intergen) according to the instructions of the manufacturer. Staining for K8 was performed using the Vectastain ABC Elite kit according to the instructions of the manufacturer. Additionally, the sections were counterstained with hematoxylin.
Measurement of hemorrhage size
Digital images of two hematoxylin and eosin–stained sections of individual uterine implantation sites were captured with a Spot digital camera and manipulated in Adobe Photoshop® using selective color replacement to enhance erythrocytes. After changing the picture to grayscale, a threshold was applied to produce a black and white picture. The pixel values were inverted, and the resulting black pixels, representing erythrocytes, were counted using NIH Image. Both halves of each implantation site were counted and averaged. All images of sections from the same litter were manipulated with the same threshold settings.
Acknowledgments
The authors are grateful to A. Gleiberman for scientific discussions and R. Newlin for technical help.
This work was performed at The Burnham Institute with the support of National Institutes of Health grants from National Institute of Arthritis and Musculoskeletal and Skin Diseases to H. Baribault, grant RO1 CA42302 from the National Cancer Institute to R.G. Oshima, and a Cancer Center grant P30 30199. D. Jaquemar was supported by a grant (823A-064686) from the Swiss National Science Foundation.
Footnotes
Abbreviations used in this paper: B6;129, C57bL/6;129X1/SvJ; ConA, concanavalin A; E, embryonic day; K8, mouse keratin 8 or endoA; K18, mouse keratin 18 or endoB; TNFR, TNF receptor.
References
- Baribault, H., and R.G. Oshima. 1991. Polarized and functional epithelia can form after the targeted inactivation of both mouse keratin 8 alleles. J. Cell Biol. 115:1675–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baribault, H., J. Price, K. Miyai, and R.G. Oshima. 1993. Mid-gestational lethality in mice lacking keratin 8. Genes Dev. 7:1191–1202. [DOI] [PubMed] [Google Scholar]
- Baribault, H., J. Penner, R.V. Iozzo, and M. Wilson-Heiner. 1994. Colorectal hyperplasia and inflammation in keratin 8-deficient FVB/N mice. Genes Dev. 8:2964–2973. [DOI] [PubMed] [Google Scholar]
- Bauman, P.A., W.S. Dalton, J.M. Anderson, and A.E. Cress. 1994. Expression of cytokeratin confers multiple drug resistance. Proc. Natl. Acad. Sci. USA. 91:5311–5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brulet, P., C. Babinet, R. Kemler, and F. Jacob. 1980. Monoclonal antibodies against trophectoderm-specific markers during mouse blastocyst formation. Proc. Natl. Acad. Sci. USA. 77:4113–4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulin, C., C.F. Ware, T.M. Magin, and R.G. Oshima. 2000. Keratin-dependent, epithelial resistance to tumor necrosis factor–induced apoptosis. J. Cell Biol. 149:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross, J.C. 2000. Genetic insights into trophoblast differentiation and placental morphogenesis. Semin. Cell Dev. Biol. 11:105–113. [DOI] [PubMed] [Google Scholar]
- Cross, J.C., Z. Werb, and S.J. Fisher. 1994. Implantation and the placenta: key pieces of the development puzzle. Science. 266:1508–1518. [DOI] [PubMed] [Google Scholar]
- Fuchs, E., and K. Weber. 1994. Intermediate filaments: structure, dynamics, function, and disease. Annu. Rev. Biochem. 63:345–382. [DOI] [PubMed] [Google Scholar]
- Gantner, F., M. Leist, A.W. Lohse, P.G. Germann, and G. Tiegs. 1995. Concanavalin A-induced T-cell-mediated hepatic injury in mice: the role of tumor necrosis factor. Hepatology. 21:190–198. [DOI] [PubMed] [Google Scholar]
- Gilbert, S., A. Loranger, N. Daigle, and N. Marceau. 2001. Simple epithelium keratins 8 and 18 provide resistance to fas-mediated apoptosis. The protection occurs through a receptor-targeting modulation. J. Cell Biol. 154:763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix, M.J., E.A. Seftor, Y.W. Chu, K.T. Trevor, and R.E. Seftor. 1996. Role of intermediate filaments in migration, invasion and metastasis. Cancer Metastasis Rev. 15:507–525. [DOI] [PubMed] [Google Scholar]
- Hesse, M., T. Franz, Y. Tamai, M.M. Taketo, and T.M. Magin. 2000. Targeted deletion of keratins 18 and 19 leads to trophoblast fragility and early embryonic lethality. EMBO J. 19:5060–5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesse, M., T.M. Magin, and K. Weber. 2001. Genes for intermediate filament proteins and the draft sequence of the human genome: novel keratin genes and a surprisingly high number of pseudogenes related to keratin genes 8 and 18. J. Cell Sci. 114:2569–2575. [DOI] [PubMed] [Google Scholar]
- Hogan, B., F. Costantini, and E. Lacy. 1986. Manipulating the Mouse Embryo: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Huen, A.C., J.K. Park, L.M. Godsel, X. Chen, L.J. Bannon, E.V. Amargo, T.Y. Hudson, A.K. Mongiu, I.M. Leigh, D.P. Kelsell, et al. 2002. Intermediate filament-membrane attachments function synergistically with actin-dependent contacts to regulate intercellular adhesive strength. J. Cell Biol. 159:1005–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inada, H., I. Izawa, M. Nishizawa, E. Fujita, T. Kiyono, T. Takahashi, T. Momoi, and M. Inagaki. 2001. Keratin attenuates tumor necrosis factor-induced cytotoxicity through association with TRADD. J. Cell Biol. 155:415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, B.W., C. Grund, E. Schmid, K. Burke, W. Franke, and K. Illmensee. 1980. Formation of cytoskeletal elements during mouse embryogenesis intermediate filaments of the cytokeratin type and desmosomes in preimplantation embryos. Differentiation. 17:161–179. [DOI] [PubMed] [Google Scholar]
- Jackson, B.W., C. Grund, S. Winter, W.W. Franke, and K. Illmensee. 1981. Formation of cytoskeletal elements during mouse embryogenesis: epithelial differentiation and intermediate-sized filaments in early postimplantation embryos. Differentiation. 20:203–216. [DOI] [PubMed] [Google Scholar]
- Ku, N.-O., S. Michie, R.G. Oshima, and M.B. Omary. 1995. Chronic hepatitis, hepatocyte fragility, and increased soluble phosphoglycokeratins in transgenic mice expressing a keratin 18 conserved arginine mutant. J. Cell Biol. 131:1303–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku, N.-O., S. Michie, R. Soetikno, E. Resurreccion, R.G. Oshima, and M.B. Omary. 1996. Susceptibility to hepatotoxicity in transgenic mice that express a dominant-negative human keratin 18 mutant. J. Clin. Invest. 98:1034–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusters, S., G. Tiegs, L. Alexopoulou, M. Pasparakis, E. Douni, G. Kunstle, H. Bluethmann, A. Wendel, K. Pfizenmaier, G. Kollias, and M. Grell. 1997. In vivo evidence for a functional role of both tumor necrosis factor (TNF) receptors and transmembrane TNF in experimental hepatitis. Eur. J. Immunol. 27:2870–2875. [DOI] [PubMed] [Google Scholar]
- Loranger, A., S. Duclos, A. Grenier, J. Price, M. Wilson-Heiner, H. Baribault, and N. Marceau. 1997. Simple epithelium keratins are required for maintenance of hepatocyte integrity. Am. J. Pathol. 151:1673–1683. [PMC free article] [PubMed] [Google Scholar]
- Ma, L.L., S. Yamada, D. Wirtz, and P.A. Coulombe. 2001. A “hot-spot” mutation alters the mechanical properties of keratin filament networks. Nat. Cell Biol. 3:503–506. [DOI] [PubMed] [Google Scholar]
- Magin, T.M., R. Schroder, S. Leitgeb, F. Wanninger, K. Zatloukal, C. Grund, and D.W. Melton. 1998. Lessons from keratin 18 knockout mice: formation of novel keratin filaments, secondary loss of keratin 7 and accumulation of liver-specific keratin 8–positive aggregates. J. Cell Biol. 140:1441–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, S.A., D.D. Dykes, and H.F. Polesky. 1988. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 16:1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffett-King, A. 2002. Natural killer cells and pregnancy. Nat. Rev. Immunol. 2:656–663. [DOI] [PubMed] [Google Scholar]
- Moll, R., W.W. Franke, D.L. Schiller, B. Geiger, and R. Krepler. 1982. The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells. Cell. 31:11–24. [DOI] [PubMed] [Google Scholar]
- Muller, H., B. Liu, B.A. Croy, J.R. Head, J.S. Hunt, G. Dai, and M.J. Soares. 1999. Uterine natural killer cells are targets for a trophoblast cell-specific cytokine, prolactin-like protein A. Endocrinology. 140:2711–2720. [DOI] [PubMed] [Google Scholar]
- Nagy, A., and J. Rossant. 1993. Production of completely ES cell-derived fetuses. Oxford University Press, Oxford, UK. 147–179 pp.
- Omary, M.B., and N.O. Ku. 1997. Intermediate filament proteins of the liver: emerging disease association and functions. Hepatology. 25:1043–1048. [DOI] [PubMed] [Google Scholar]
- Oshima, R.G. 1981. Identification and immunoprecipitation of cytoskeletal proteins from murine extra-embryonic endodermal cells. J. Biol. Chem. 256:8124–8133. [PubMed] [Google Scholar]
- Oshima, R.G. 2002. Apoptosis and keratin intermediate filaments. Cell Death Differ. 9:486–492. [DOI] [PubMed] [Google Scholar]
- Oshima, R.G., H. Baribault, and C. Caulin. 1996. Oncogenic regulation and function of keratin 8 and 18. Cancer Metastasis Rev. 15:445–471. [DOI] [PubMed] [Google Scholar]
- Parekh, H.K., and H. Simpkins. 1995. The differential expression of cytokeratin 18 in cisplatin-sensitive and -resistant human ovarian adenocarcinoma cells and its association with drug sensitivity. Cancer Res. 55:5203–5206. [PubMed] [Google Scholar]
- Takeda, K., Y. Hayakawa, L. Van Kaer, H. Matsuda, H. Yagita, and K. Okumura. 2000. Critical contribution of liver natural killer T cells to a murine model of hepatitis. Proc. Natl. Acad. Sci. USA. 97:5498–5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamai, Y., T.-O. Ishikawa, M.R. Bosl, M. Mori, M. Nozaki, H. Baribault, R.G. Oshima, and M.M. Taketo. 2000. Cytokeratins 8 and 19 in the mouse placental development. J. Cell Biol. 151:563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiegs, G., J. Hentschel, and A. Wendel. 1992. A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J. Clin. Invest. 90:196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toivola, D.M., M.B. Omary, N.O. Ku, O. Peltola, H. Baribault, and J.E. Eriksson. 1998. Protein phosphatase inhibition in normal and keratin 8/18 assembly-incompetent mouse strains supports a functional role of keratin intermediate filaments in preserving hepatocyte integrity. Hepatology. 28:116–128. [DOI] [PubMed] [Google Scholar]
- Ward, J.M., and D.E. Devor-Henneman. 2000. Gestational Mortality in Genetically Engineered Mice: Evaluating the Extraembryonal Embryonic Placenta and Membranes. Iowa State University Press, Ames, IA. 103–122 pp.
- Wu, L., A. de Bruin, H.I. Saavedra, M. Starovic, A. Trimboli, Y. Yang, J. Opavska, P. Wilson, J.C. Thompson, M.C. Ostrowski, et al. 2003. Extra-embryonic function of Rb is essential for embryonic development and viability. Nature. 421:942–947. [DOI] [PubMed] [Google Scholar]
- Zatloukal, K., C. Stumptner, M. Lehner, H. Denk, H. Baribault, L.G. Eshkind, and W.W. Franke. 2000. Cytokeratin 8 protects from hepatotoxicity, and its ratio to cytokeratin 18 determines the ability of hepatocytes to form Mallory bodies. Am. J. Pathol. 156:1263–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]