

Abstract
The αvβ3 integrin has been shown to promote cell migration through activation of intracellular signaling pathways. We describe here a novel pathway that modulates cell migration and that is activated by αvβ3 and, as downstream effector, by cdc2 (cdk1). We report that αvβ3 expression in LNCaP (β3-LNCaP) prostate cancer cells causes increased cdc2 mRNA levels as evaluated by gene expression analysis, and increased cdc2 protein and kinase activity levels. We provide three lines of evidence that increased levels of cdc2 contribute to a motile phenotype on integrin ligands in different cell types. First, increased levels of cdc2 correlate with more motile phenotypes of cancer cells. Second, ectopic expression of cdc2 increases cell migration, whereas expression of dominant-negative cdc2 inhibits migration. Third, cdc2 inhibitors reduce cell migration without affecting cell adhesion. We also show that cdc2 increases cell migration via specific association with cyclin B2, and we unravel a novel pathway of cell motility that involves, downstream of cdc2, caldesmon. cdc2 and caldesmon are shown here to localize in membrane ruffles in motile cells. These results show that cdc2 is a downstream effector of the αvβ3 integrin, and that it promotes cell migration.
Keywords: cell adhesion; cyclin B2; caldesmon; prostate cancer; purvalanola
Introduction
Cell–ECM interactions are predominantly mediated by integrins (Hynes, 1999). The αvβ3 integrin is predominantly, although not exclusively, found in cancer cells and neovessels (for review see Byzova et al., 1998; Seftor et al., 1999). Expression of αvβ3 in tumor cells alters cell–ECM interactions and causes increased tumorigenicity (Felding-Habermann et al., 1992), as well as invasiveness of several cancer cells. αvβ3 has been shown to contribute to the establishment and growth of pulmonary metastatic melanoma lesions (Filardo et al., 1995), and to increased invasiveness of cutaneous melanomas from the epidermis to the dermis (Hsu et al., 1998) and of human breast cancer cells in nude mice (Felding-Habermann et al., 2001).
Cell migration mediated by integrins, a crucial step in in vivo metastasis establishment and growth, has been shown in vitro to be supported by multiple downstream signaling pathways (Schwartz et al., 1995). Although activation of these pathways is a prerequisite for cell migration, changes in gene expression are also likely to play a role in cell invasion.
As shown recently by several groups, alterations of gene expression occur in response to integrin binding to ECM proteins (Damsky and Werb, 1992; Ruoslahti and Reed, 1994; Juliano, 1996). Cell adhesion has been shown to increase the levels of cyclin A mRNA and protein (Guadagno et al., 1993), cyclin D1 mRNA and protein (Zhu et al., 1996), cyclin E-cdk2 kinase activity (Fang et al., 1996; Zhu et al., 1996), gelatinase in T cells becoming transmigratory (Romanic and Madri, 1994), metalloproteinases in fibroblasts (Werb et al., 1989; Huhtala et al., 1995), immediate–early response genes, as well as transcription factors in monocytes responding to injury or infection (for review see Juliano and Haskill, 1993), and more than 32 genes identified in salivary epithelial cells undergoing morphological differentiation (Lafrenie and Yamada, 1998; Lafrenie et al., 1998). In addition to a requirement for integrin engagement, it has been consistently highlighted that specific integrins uniquely affect gene expression. Among β1 integrins, it has been shown that α1 and α2, but not α6, integrins specifically regulate stromelysin-1 expression in mouse mammary carcinoma cells (Lochter et al., 1999). Similarly, αvβ3-induced gene expression in response to cell adhesion to αvβ3 ligands has been documented in several instances. Binding of denatured collagen to αvβ3 in smooth muscle cells induces tenascin-C expression during vascular remodeling (Jones et al., 1999). A different work showed that antibodies to αvβ3, but not to α5β1, increase the invasive ability of a melanoma cell line endogenously expressing αvβ3 concurrent with an induction of type IV collagenase mRNA and protein (Seftor et al., 1992). Furthermore, cell adhesion mediated by αvβ3 and α5β1, but not αvβ1, has been shown to regulate bcl-2 transcription (Matter and Ruoslahti, 2001).
The increased invasive behavior of neoplastic cells that occurs in response to αvβ3 integrin expression can be explained on the basis of a unique αvβ3-activated cellular response that may positively regulate cell migration. We searched for downstream effectors of αvβ3 in prostate cancer cells where αvβ3 expression correlates with a neoplastic and migratory phenotype (Zheng et al., 1999). Here, we show that αvβ3 integrin expression in LNCaP prostate cancer cells up-regulates cdc2 mRNA and protein levels, as well as cdc2 kinase activity. We demonstrate a new role for cdc2 in cell motility on integrin ligands and unravel a novel mechanism of cell motility mediated by cdc2, its cofactor cyclin B2 and, downstream of cdc2, caldesmon, a molecule known to be associated with the cytoskeleton. Together, these data show that cdc2 is a downstream effector of αvβ3 and that it promotes cell migration.
Results
α v β 3 integrin up-regulates cdc2 mRNA, protein, and kinase levels
In an attempt to determine genes regulated by the αvβ3 integrin, which contribute to this phenotype in cancer cells, a gene expression analysis was undertaken. As a model system, LNCaP prostate cancer cells were stably transfected with expression vector containing human β3 integrin cDNA, or empty expression vector (mock), or expression vector containing human ICAM-1 cDNA as a transfection control for the effects of ectopically expressing a cell surface protein. Expression of αvβ3 integrin in three different cell populations (β3-1, β3-2, and β3-3), as well as ICAM expression in two different populations, was confirmed by FACS® analysis (Fig. 1 A; unpublished data; Zheng et al., 1999).
Figure 1.



cdc2 mRNA, protein, and kinase levels are increased in β3-LNCaP cells. (A) Surface expression of ectopic αvβ3 integrin in LNCaP cell stable transfectants. Mock-, β3-1, β3-2, and β3-3 LNCaP cells were incubated with mAb to αvβ3 integrin (AP-3, thick line) or, as negative control, to the HA epitope (12CA5, thin line) followed by staining with FITC-conjugated goat anti–mouse Ig antibody and analysis using a FACScanTM flow cytometer. (B) cDNA expression array analysis. First strand cDNA probes prepared with either β3-1 or mock-LNCaP mRNA were hybridized to Atlas human cancer cDNA expression arrays. Shown are areas of section A of the cDNA array membrane. The arrow indicates the spots corresponding to cdc2 on the array. The brackets indicate other genes on the array that are not up-regulated in β3-1. (C–F) cdc2 protein and kinase levels are increased in β3-LNCaP cells grown in 2D (C and D) and 3D (E and F) cultures. (C) cdc2 protein levels in extracts from LNCaP cell transfectants grown in 2D culture were analyzed by IB with an antibody to cdc2. In C and E, antibody to ERK-1 was used for protein loading control. (D) Immunocomplexes precipitated from 75 μg of β3-1, β3-3, β3-2, ICAM-, or mock-LNCaP cell extracts using an mAb to cdc2 were used in a kinase reaction using histone H1 as a substrate. Consistent results were obtained in two additional experiments. (E) cdc2 protein levels in extracts from LNCaP cell transfectants grown in 3D Matrigel culture for 48 h were analyzed by IB with an antibody to cdc2. (F) Immunocomplexes precipitated from 75 μg of β3-1, β3-2, ICAM-, or mock-LNCaP cell extracts using an mAb to cdc2 were used in a kinase reaction using histone H1 as a substrate. Consistent results were obtained in two additional experiments.
First-strand cDNA of mRNA isolated from β3-LNCaP, ICAM-LNCaP, and mock-LNCaP cells were used as probes on cDNA array filters containing 588 human genes known to be disregulated in cancer. Only those genes that displayed at least a threefold difference in expression between β3- and mock-LNCaP cells (Fig. 1 B), or β3- and ICAM-LNCaP cells (unpublished data) were considered as legitimate targets of αvβ3 integrin–mediated expression. Among others, cdc2 was specifically up-regulated in β3-LNCaP cells (Fig. 1 B). Because cdc2 is a prognostic indicator of prostate tumor progression (Kallakury et al., 1997), further investigation of the expression of this gene was undertaken. Northern blot analysis was performed to verify the cDNA expression array results (unpublished data).
Using extracts prepared from β3-, ICAM-, and mock-LNCaP cells, we observed up-regulation of cdc2 protein in β3-LNCaP cells as compared with mock- and ICAM-LNCaP cells (Fig. 1 C). Also shown in Fig. 1 C are cdc2 levels in extracts from LNCaP cells stably transfected with an expression vector containing the human β6 integrin subunit cDNA (β6-LNCaP clones: β6-1 and β6-2). The β6 integrin subunit was chosen because, like β3, it is not expressed in LNCaP cells (unpublished data) and it also heterodimerizes with αv and shares several ligands with αvβ3 (Busk et al., 1992; Huang et al., 1998). The results show that cdc2 levels in two clones expressing αvβ6 are significantly lower than in β3-LNCaP cells. β3-LNCaP cells display increased cdc2 kinase activity compared with mock- and ICAM-LNCaP cells (Fig. 1 D), as well as β6-LNCaP cells (unpublished data). cdc2 protein and kinase levels were also increased in three-dimensional (3D)* Matrigel cultures in β3-LNCaP cells compared with mock- and ICAM-LNCaP cells (Fig. 1, E and F, respectively).
Overall, the results show that the αvβ3 integrin expression in LNCaP cells specifically up-regulates cdc2 mRNA, protein, and kinase levels.
Correlation of cdc2 expression and cell migration
Because several molecules known to affect cell cycle progression or proliferation such as FAK and PI3K are positive modulators of cell migration (Cary et al., 1996; Keely et al., 1997; Shaw et al., 1997), it was hypothesized that the increased cdc2 protein and kinase levels might play a role in LNCaP cell migration. A correlation between cdc2 levels and more migratory phenotypes on integrin ligands was observed between a human fibrosarcoma cell line (HT1080) and a genetically modified variant cell line (HT2–19). In the latter, one cdc2 allele has been deleted, and the other placed under the control of an inducible promoter; removal of IPTG suppresses expression of the remaining allele (Itzhaki et al., 1997). Lower levels of cdc2 in HT2–19 cells correlated with a reduction in migration on fibronectin (FN; P = 2 × 10−8), under conditions in which they attached equally well to FN (Fig. 2). When HT2–19 cells were cultured in the absence of IPTG to suppress cdc2 expression, these cells' migration was further reduced (67% less than in the presence of IPTG, P = 2 × 10−5; Fig. 2). The results show that increased levels of cdc2 correlate with a more migratory phenotype on integrin ligands.
Figure 2.
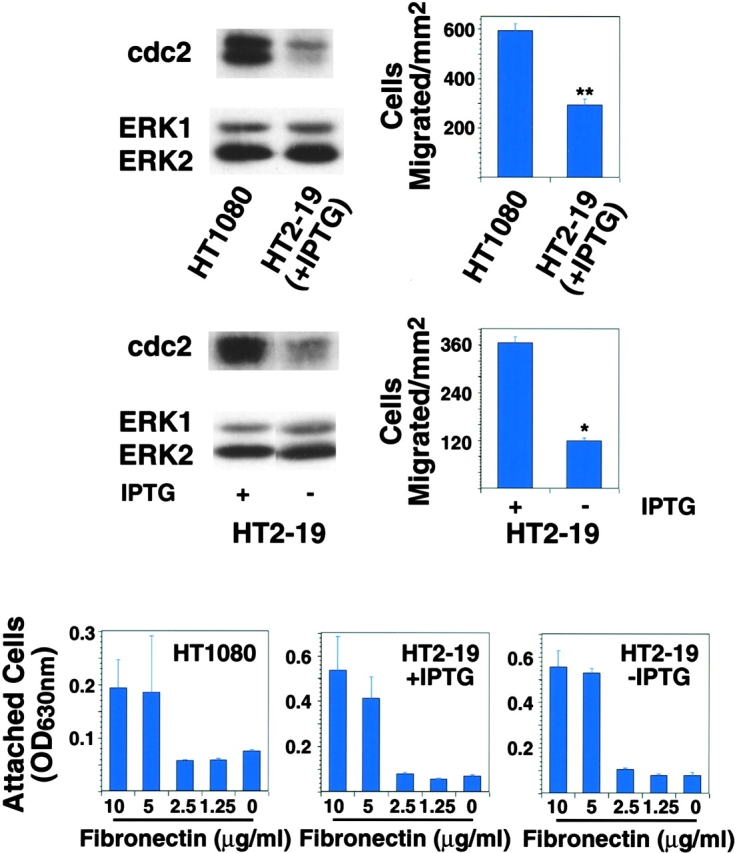
cdc2 levels correlate with cell migration. cdc2 protein levels in extracts from HT1080 and HT2–19 cells were analyzed by IB with an antibody to cdc2 (top left: HT1080 and HT2–19 +IPTG; middle left: HT2–19 +/−IPTG). For migration assays (top and middle right), 50,000 cells were seeded for 4.5 h, in serum-free medium on 5 μg/ml FN-coated transwell insert filters. After 3.5 h, cells were fixed and stained with crystal violet and the cells on the bottom of the filter were counted. The mean and SEM of 10 random fields is shown (** P = 2 × 10−8 for HT1080 compared with HT2–19 cells +IPTG; * P = 2 × 10−5 for HT2–19 +IPTG compared with HT2–19 cells −IPTG). For adhesion assays (bottom), 50,000 cells were seeded in serum-free medium in a 96-well plate coated with increasing concentrations of FN. After 2 h, plates were washed two times with PBS, fixed, and stained with crystal violet.
Inhibition of cdc2 kinase prevents LNCaP cell migration
Transient expression of a dominant-negative variant of cdc2 (cdc2dn) was used to determine whether cdc2 had an effect on migration of LNCaP cells (Fig. 3 A). The data show that cdc2dn significantly inhibits cell migration on FN. Comparable results were obtained using cells suspended in 0.5% (Fig. 3 A) or 10% FBS (Fig. 3 B); therefore, 0.5% or 10% FBS were interchangeably used in our paper. Ectopic expression of cdc2dn or wild-type cdc2 (cdc2wt) affected migration of β3-LNCaP cells on FN (Fig. 3 B) and vitronectin (VN; unpublished data), but had no effect on adhesion (unpublished data). To establish whether these results were due to cdc2 effects on cell proliferation, β3-LNCaP cells transfected with cdc2dn were tested in migration assays in the presence or absence of an inhibitor of cell proliferation, mitomycin C. As shown in Fig. 3 B, mitomycin C inhibited cell proliferation, but had no effect on migration in cells transfected with cdc2dn. Ectopic expression of cdc2dn in LNCaP cells was efficiently achieved by lipofection (unpublished data).
Figure 3.

cdc2 affects migration of β3-LNCaP cells. (A) β3-LNCaP (β3-2) cells cotransfected with pCMVβgal and pcDNA-3 or pCMVcdc2dn for 60 h were harvested and seeded in medium containing 0.5% FBS on 10 μg/ml FN-coated transwell insert filters. After 6 h, cells were fixed and stained for βgal activity and counted as in Materials and methods. (B) β3-LNCaP (β3-2) cells cotransfected with pCMVβgal and pcDNA-3 or pCMVcdc2dn for 60 h were incubated in the presence or absence of 1 μg/ml mitomycin C for 16 h, and then harvested and seeded in medium containing 10% FBS with or without 1 μg/ml mitomycin C on 10 μg/ml FN-coated transwell insert filters or on 10 μg/ml FN-coated 96-well plates with 1 μCi [3H]thymidine per well. After 6 h, cells were fixed and stained for βgal activity. The number of transfected cells that migrated and cells that proliferated was calculated as described in Materials and methods. (C; top) Purvalanol A prevents β3- LNCaP cell migration. β3-LNCaP cells were incubated for 2 h at 37°C in the presence or absence of purvalanol A and 200,000 cells were seeded in medium containing purvalanol A at the indicated concentrations on 10 μg/ml FN-coated transwell insert filters. After 6 h, cells were fixed and stained with crystal violet and the cells on the bottom of the filter were counted. The mean and SEM per square millimeter is shown. (Bottom) Purvalanol A does not affect β3-LNCaP cell adhesion to FN. β3-LNCaP cells incubated at 37°C in the presence or absence of purvalanol A for 2 h were trypsinized, washed, and 50,000 cells were seeded in medium containing purvalanol A at the indicated concentrations in a 96-well plate coated with FN. After 2 h, plates were washed two times with PBS, fixed, and stained with crystal violet. Purv A, purvalanol A.
Two potent and specific inhibitors of cdc2 kinase, purvalanol A (Gray et al., 1998) and alsterpaullone (Schultz et al., 1999), were tested to confirm a role for cdc2 in cell migration. After a 2-h exposure to 0.2 or 1 μM purvalanol A, migration was reduced by more than 30 and 70%, respectively (Fig. 3 C). Migration was reduced by ∼50% in cells cultured in the presence of 1.32 μM alsterpaullone for 2 h (unpublished data). Neither adhesion nor cell morphology was significantly or noticeably affected by these concentrations of inhibitors, as determined by adhesion assays and microscopical analysis, respectively (Fig. 3 C; unpublished data). In conclusion, inhibition of cdc2 kinase activity prevents cell migration without affecting cell adhesion.
cdc2 modulates migration of HeLa cells
To investigate whether cdc2 regulates migration in cells other than LNCaP, cdc2dn and cdc2wt were ectopically expressed in HeLa cells and their effects on migration and cell cycle were determined over time. At 24 h, there is a modest reduction of HeLa cell migration on FN by cdc2dn and a twofold increase in migration by cdc2wt (Fig. 4 A). Because cdc2 activity is regulated by cyclin levels, we tested whether increased levels of cdc2 would be sufficient to increase kinase levels. Immunoprecipitation of cdc2 from cells transfected with vector alone or cdc2wt determined that ectopic expression of cdc2wt increased cdc2 kinase activity (Fig. 4 B). At 48 h, cdc2dn reduces HeLa cell migration more than threefold, and cdc2wt increases migration modestly (Fig. 4 A); neither adhesion nor cell cycle profile was affected by either cdc2dn or by cdc2wt at these time points (unpublished data). HeLa cells cultured in the presence of micromolar concentrations of purvalanol A for 2 h show a dose-dependent reduction of migration on FN with a negligible effect on adhesion at 8 μM (Fig. 4 C). The results show that expression of cdc2wt and cdc2dn affects HeLa cell migration before a significant effect on cell cycle can be observed and that treatment with cdc2 inhibitors blocks HeLa cell migration.
Figure 4.
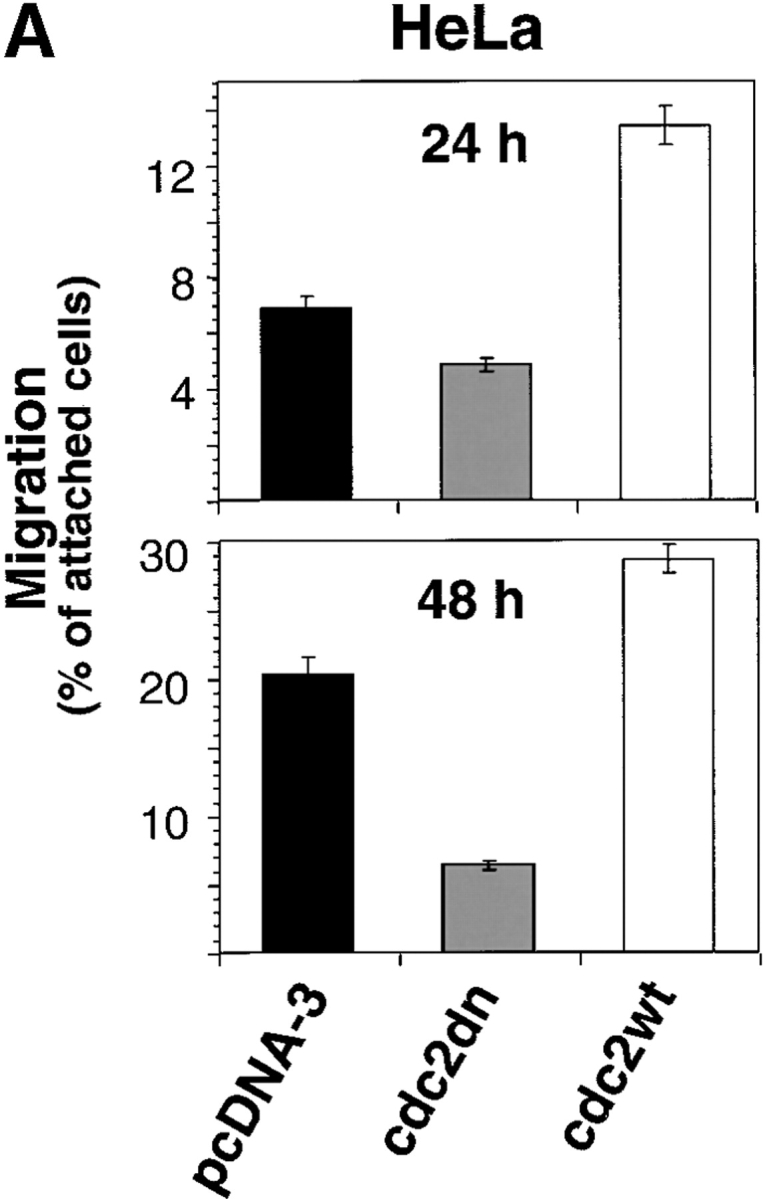
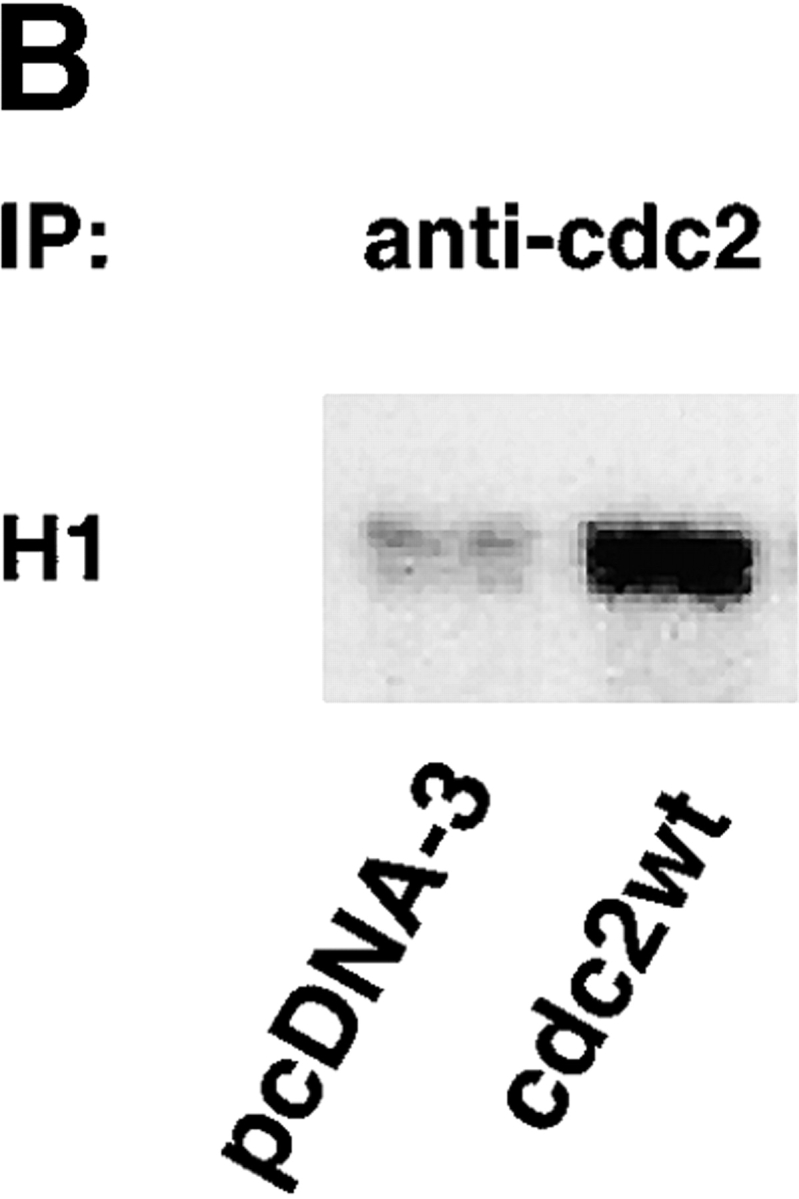
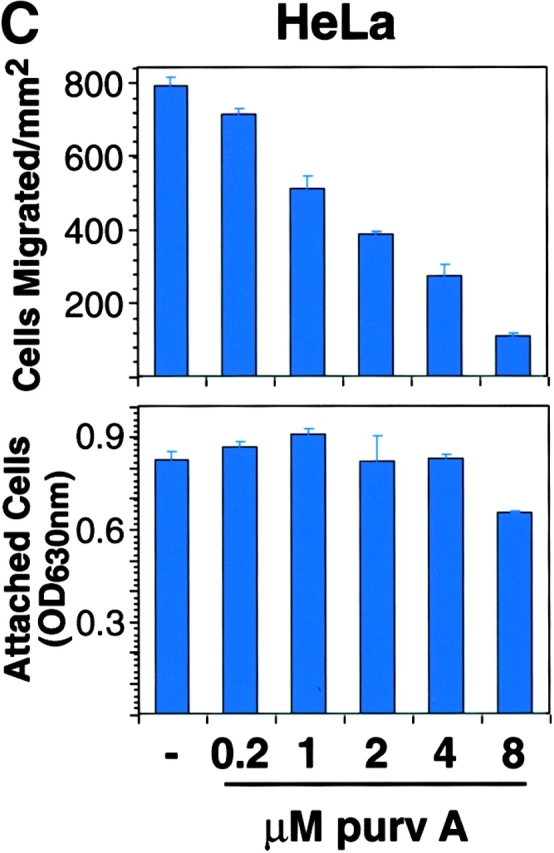
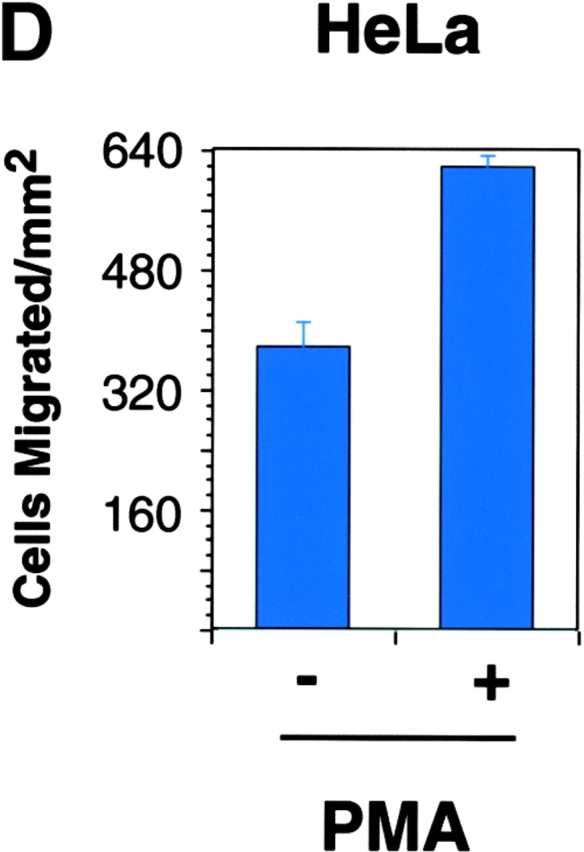
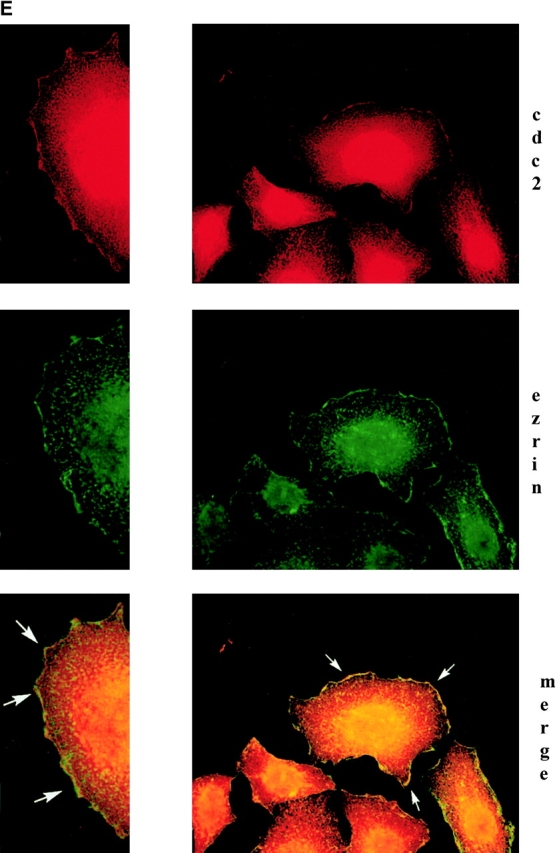
cdc2 regulates migration of HeLa cells. (A) Expression of cdc2dn or cdc2wt affects migration of HeLa cells. HeLa cells cotransfected with pCMVβgal and pcDNA-3, pCMVcdc2dn, or pCMVcdc2wt were trypsinized at two different time points (24 and 48 h) after transfection and cells were seeded in medium containing 10% FBS on 10 μg/ml FN-coated plates. After 16 h, cells were fixed and stained for βgal activity. The mean and SEM of 10 random fields is shown. (B) Expression of cdc2wt increases cdc2 kinase activity. Immunocomplexes precipitated from 60 μg cell extracts of HeLa cells transfected with pcDNA-3 or cdc2wt using an mAb to cdc2 were used in a kinase reaction using histone H1 as a substrate. (C) Purvalanol A inhibits HeLa cell migration without affecting cell adhesion. Migration assays (top): HeLa cells were incubated for 2 h at 37°C in the presence or absence of purvalanol A and 90,000 HeLa cells were seeded in medium containing purvalanol A at the indicated concentrations on FN-coated transwell insert filters. After 16 h, cells were fixed and stained with crystal violet and the cells on the bottom of the filter were counted. The mean and SEM of 10 random fields is shown. Adhesion assays (bottom): 100,000 HeLa cells grown in the presence or absence of purvalanol A for 2 h were seeded in medium containing purvalanol A at the indicated concentrations in a 96-well plate coated with FN. After 2 h, plates were washed two times with PBS, fixed, and stained with crystal violet. The results show the mean and standard deviation of duplicate observations. Purv A, purvalanol A. (D) PMA increases HeLa cell migration. HeLa cells were allowed to attach for 1 h to filters coated with 10 μg/ml FN and then were incubated in the presence or absence of 100 nM PMA for 3 h and processed as described above. The mean and SEM of 10 random fields is shown. (E) cdc2 is enriched in peripheral areas in motile cells. HeLa cells plated on 10 μg/ml FN-coated coverslips and incubated in the presence of 100 nM PMA were stained with cdc2 mAb (red) and ezrin mAb (green); image-merging analysis of ezrin and cdc2 is shown. (Arrows) Peripheral areas of the cell where ezrin and cdc2 colocalize.
cdc2 is present in peripheral areas of the cell
We investigated whether endogenous cdc2 is associated with the cytoskeleton of adhering cells and in cells with a motile phenotype. HeLa cells, plated on FN for 3 h, were incubated in the presence or absence of 100 nM PMA for 30 min to induce a motile phenotype (Fig. 4 D; Besson et al., 2001). In PMA-treated cells, staining of HeLa cells with cdc2 and ezrin mAbs shows that cdc2 is concentrated in peripheral areas of the cell, specifically in lamellipodia, and that cdc2 and ezrin are colocalized (Fig. 4 E). Thus, cdc2 in motile cells is concentrated in peripheral areas where rapid actin reorganization occurs; in this cellular location, cdc2 may act on specific cytoskeleton proteins to modulate cell migration.
Cyclin B2 is the cyclin partner of cdc2 that modulates cell migration
Mammalian cdc2 is known to associate with cyclins A, B1, and B2 (for review see Kohn, 1999). Ectopic expression of cyclin B2, but not cyclin A or cyclin B1, increased β3-LNCaP and HeLa cell migration on FN (Fig. 5 A), without affecting cell adhesion to this substrate (unpublished data). The ectopically transfected cyclin B2 was able to form active kinase complex, as shown by immunoprecipitation kinase assays of nontransfected versus cyclin B2-transfected HeLa cells using a cyclin B2 antibody, as well as by immunoprecipitation kinase assay of cyclin B2 transfected HeLa cells using a c-myc antibody (Fig. 5 B).
Figure 5.
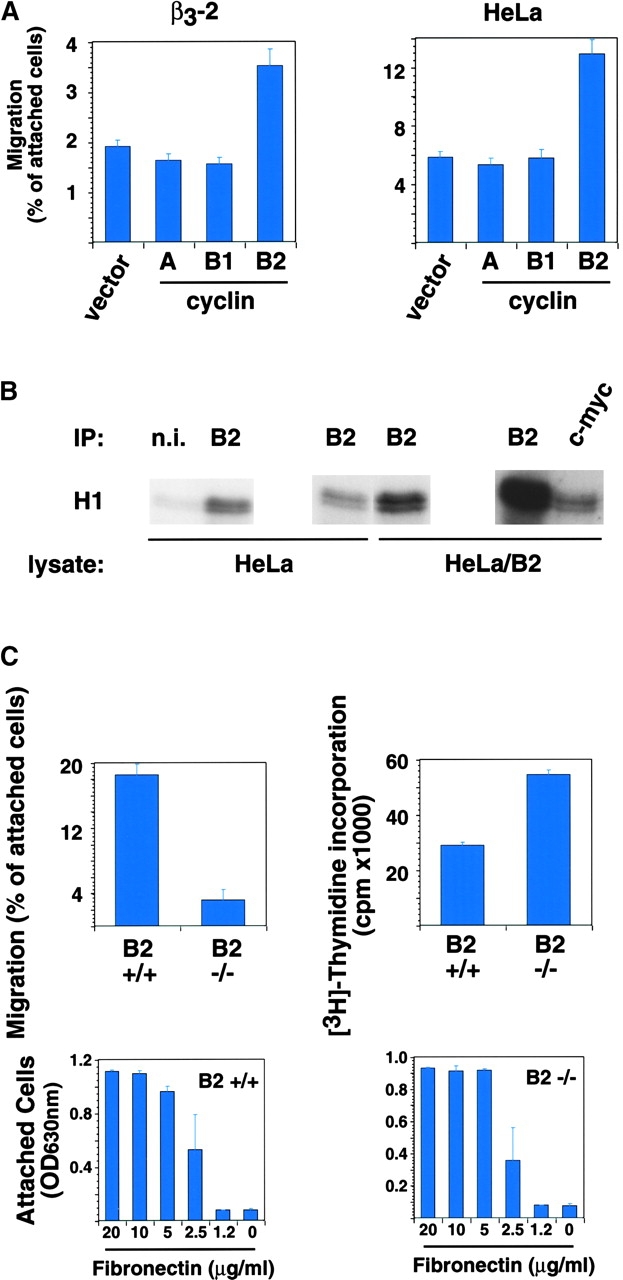
Expression of cyclin B2 increases cell migration. (A) β3-LNCaP (β3-2) and HeLa cells cotransfected with pCMVβgal and pcDNA-3 (vector), pCMXcyclin A (A), pCMVcyclin B1 (B1), or pCMVcyclin B2 (B2) were processed 24 h after transfection, as described in Figs. 3 and 4. The mean and SEM of 10 random fields is shown. (B) Ectopically expressed cyclin B2 forms active kinase complex. Immunocomplexes precipitated from HeLa and cyclin B2–transfected HeLa RIPA extracts using nonimmune rabbit serum (n.i.), or rabbit polyclonal antibody to cyclin B2 (B2) or a c-myc agarose-conjugated rabbit polyclonal antibody (c-myc) were used in kinase assays using histone H1 as a substrate. (C) Cyclin B2–null cells migrate poorly on FN. For migration assays (top left panel), 15,000, 30,000, or 60,000 cyclin B2–null (B2 −/−) and wt (B2 +/+) MEFs were seeded in serum-free medium on 5 μg/ml FN-coated transwell insert filters. After 4 h, cells were fixed and stained with crystal violet and the cells on the top and bottom of the filter were counted. The mean and SEM of 10 random fields is shown. For proliferation assays (top right panel), 10,000 cells were seeded in serum-free medium on 5 μg/ml FN-coated 96-well plates in the presence of 1 μCi [3H]thymidine per well. After 4 h, cells were processed to determine [3H]thymidine incorporation, as described in Materials and methods. For adhesion assays (bottom panels), 50,000 cells were seeded for 2 h in serum-free medium in a 96-well plate coated with increasing concentrations of FN as described in Materials and methods.
To confirm the role of cyclin B2 in cell migration, mouse embryonic fibroblasts (MEFs) derived from cyclin B2–null mice (Brandeis et al., 1998) were compared with their wild-type (wt) counterparts. Cyclin B2–null MEFs did not significantly migrate on FN (83% less than wt MEFs); adhesion to FN was similar for both cell types (Fig. 5 C). These differences in migration were not due to an increased proliferation capacity of wt MEFs as compared with cyclin B2–null MEFs because cyclin B2–null MEFs proliferate more than wt MEFs (Fig. 5 C). Together, these data implicate cyclin B2 as the specific cyclin partner of cdc2 that modulates cell migration.
Caldesmon is a downstream effector of cdc2 in the cell motility pathway
Caldesmon, a previously identified mitotic substrate of cdc2 (Mak et al., 1991; Yamashiro et al., 1991), appeared to be a reasonable candidate downstream of cdc2 in the cell motility pathway because it is found in membrane ruffles and its ability to bind actin is reduced upon phosphorylation by cdc2 (Huber, 1997). We tested the migration of HeLa cells ectopically expressing cdc2wt in conjunction with either caldesmon wt or a variant form of caldesmon containing all seven of its cdc2 phosphorylation sites mutated (“7th mutant;” Yamashiro et al., 2001). Although caldesmon wt and cdc2 increased cell migration, caldesmon 7th mutant and cdc2 did not have any effect (Fig. 6 A, top); indeed, caldesmon 7th mutant suppressed cdc2-mediated cell migration (Fig. 6 A, bottom) without affecting cell adhesion (unpublished data), indicating that the mutant caldesmon inhibits cdc2 activity by competing with the endogenous caldesmon. It should be noted that expression of exogenous caldesmon wt and cdc2 had a synergistic effect on cell migration: specifically, motility was increased from 19.4 ± 3% (cdc2 alone) to 33 ± 7% (cdc2 plus caldesmon). Expression of caldesmon wt and of the 7th mutant was confirmed by immunoblotting (IB) analysis (unpublished data).
Figure 6.
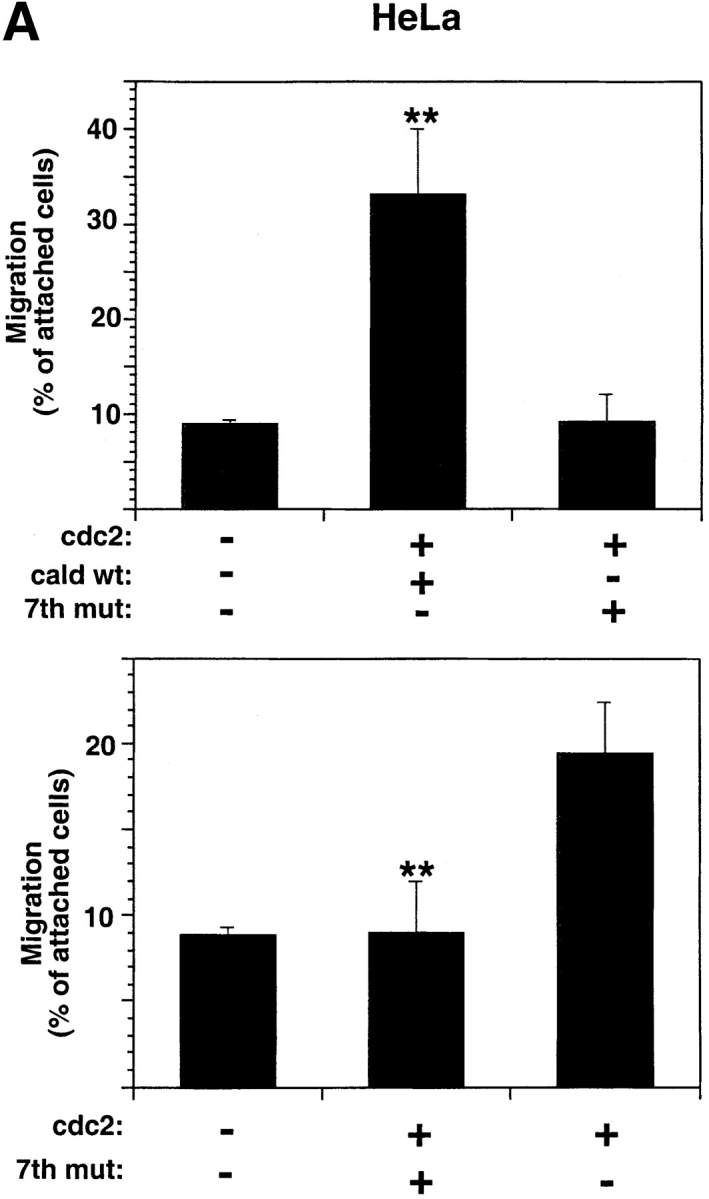


Caldesmon is a downstream effector of cdc2 in the cell motility pathway. (A) Coexpression of cdc2 and caldesmon wt, but not caldesmon 7th mutant, increases HeLa cell migration (top). Caldesmon 7th mutant inhibits cdc2-mediated HeLa cell migration (bottom). HeLa cells cotransfected with pCMVβgal and empty vector, or pCMVcdc2wt and pCMV myc-tagged caldesmon wt (cald wt) or pCMVcdc2wt and pCMV myc-tagged caldesmon with seven mutated cdc2 phosphorylation sites (7th mut; top) or pCMVcdc2wt alone or pCMVcdc2wt and pCMV myc-tagged caldesmon 7th mutant (7th mut; bottom) were processed as above, 24 h after transfection (** P = 2 × 10−5, top; ** P = 10−4 bottom). The mean and SEM of 10 random fields is shown. (B) Caldesmon is a substrate of cdc2/cyclin B2. (Left) Caldesmon immunoprecipitated from HeLa cells or H1 was used as substrate of either recombinant cdc2/cyclin B1 or cyclin B2 immunocomplexes in kinase assays. (Middle) An equivalent amount of caldesmon immunoprecipitate used in the kinase assay, and 3 μg histone H1, were separated using SDS-PAGE and stained with Coomassie blue. (Right) Immunoblot of caldesmon immunoprecipitate using SM12 mAb to caldesmon. Cald IP, caldesmon immunoprecipitate. (C) HeLa cells plated on 10 μg/ml FN-coated coverslips and incubated in the presence or absence of 100 nM PMA were stained with caldesmon mAb and cdc2 mAb. Image-merging analysis of caldesmon (green) and cdc2 (red) indicates yellow patches where superposition of the two molecules is particularly noted (see also arrows). (Arrows) Peripheral areas of the cell where caldesmon and cdc2 colocalize.
Next, we confirmed that caldesmon is a substrate of cdc2/cyclin B2 by immunoprecipitation kinase assay using caldesmon immunoprecipitate (Fig. 6 B, cald IP) as a substrate for cyclin B2 immunocomplexes (Fig. 6 B, B2 IP). These results implicate cdc2 phosphorylation as an important mechanism by which caldesmon functions to modulate migration. This conclusion is also supported by the finding that cdc2 and caldesmon colocalize in all cells where membrane ruffles were observed; neither cdc2 nor caldesmon was found by themselves in membrane ruffles (Fig. 6 C). In cells not treated with PMA, the two molecules did not colocalize (Fig. 6 C); caldesmon was found in stress fibers, in agreement with previous studies (Huber, 1997; Yamashiro et al., 2001; Fig. 6 C). Together, these results show that caldesmon is a downstream effector of cdc2 in the cell–motility pathway.
Discussion
In this paper, we show that cdc2 is a downstream effector of the αvβ3 integrin, and that it controls cell migration. Evidence is provided that exogenous expression of αvβ3 integrin up-regulates cdc2 mRNA and protein levels as well as cdc2 kinase activity and that cdc2 regulates cell migration on integrin ligands without affecting cell adhesion. It is also shown that cyclin B2 is the cdc2 cofactor that controls cell migration. Finally, it is shown that caldesmon, a cytoskeleton-associated molecule known to be phosphorylated by cdc2, is a substrate for cdc2–cyclin B2 in the migratory pathway, and colocalizes with cdc2 at the cell periphery and in membrane ruffles in motile cells.
This is the first paper that describes changes in cdc2 levels in response to integrin expression. Although regulation of cell cycle–related molecules in response to integrin engagement has been widely documented (Schwartz and Assoian, 2001), regulation of the levels of a cdk and of its kinase activity upon integrin expression has never been reported. Specifically, alterations of cdc2 protein levels in response to either integrin expression or integrin engagement have never been studied. In one work, cdc2 mRNA levels were shown to remain unchanged in response to cell adhesion (Guadagno et al., 1993). However, this analysis was performed in synchronized cells collected at times corresponding to transit through G1, whereas our work was performed in asynchronous cell populations. It should be noted that cdc2 protein levels, although increased in neoplastic cells (Kallakury et al., 1997), do not vary significantly throughout the cell cycle (Draetta and Beach, 1988; McGowan et al., 1990; Dalton, 1992; Welch and Wang, 1992), indicating that the rise of cdc2 seen in asynchronous populations of β3-LNCaP cells reflects a fundamental shift in cdc2 regulation by αvβ3.
Although another member of the cdk family, cdk5, has been shown to play a role in neuronal migration (Homayouni and Curran, 2000), this is the first paper identifying cdc2 as a modulator of cell motility. cdc2 is best characterized for its role in promoting cell cycle progression through the G2/M phase (Pines and Rieder, 2001). In our paper, experimental conditions where either cdc2dn or cdc2 inhibitors affected cell migration, but did not affect cell proliferation, were established in all cell types; the long doubling time of LNCaP cells facilitated the initial discrimination between cell migration and cell proliferation.
We demonstrate that the mechanism by which cdc2 regulates cell migration is via its specific association with cyclin B2. This claim is based on several experimental findings: first, cdc2 is localized in lamellipodia of motile cells; this is a novel finding because previous reports had analyzed cdc2 distribution in nonmotile cells and shown at interphase to be distributed both in the nucleus and the cytoplasm (Bailly et al., 1989; Pockwinse et al., 1997). Second, ectopic expression of cyclins B1, B2, and A, all known to associate with cdc2 (for review see Pines, 1999) shows that only cyclin B2 has the ability to increase cell migration. Finally, cyclin B2–null cells display significantly reduced cell migration, although their proliferation rates are not reduced. Indeed, subcellular localization of mitotic cdc2 by cyclins B1 and B2 has been shown to confer substrate specificity (Draviam et al., 2001). Thus, it is conceivable that the cdc2–cyclin B2 complex will also provide specificity for the substrate(s) that modulates a nonmitotic event like cell migration. We reasoned that the cdc2 substrate that is likely to mediate cdc2's effect on cell motility would be colocalized with cdc2. Among the mitotic substrates for cdc2 that are known to be associated with the cytoskeleton: dynein, caldesmon, plectin, and zyxin (Mak et al., 1991; Yamashiro et al., 1991; Malecz et al., 1996; Dell et al., 2000; Hirota et al., 2000), caldesmon was a strong candidate to mediate cdc2's effect on cell migration for two main reasons. First, caldesmon is associated with cytoskeletal structures such as stress fibers and membrane ruffles, and has been shown to interfere with focal contact formation; second, its actin binding ability is reduced upon phosphorylation by cdc2 (Yamakita et al., 1992; Yamashiro et al., 1995, 2001; Huber, 1997; Helfman et al., 1999). Our paper shows that caldesmon, indeed, modulates cell motility downstream of cdc2 and that phosphorylation by cdc2 is a crucial step in this motility pathway. It also shows that caldesmon and cdc2 colocalize in membrane ruffles, sites of rapid reorganization of actin. In these sites, cdc2 phosphorylation of caldesmon may affect actin reorganization during cell migration by modulating caldesmon's actin-binding ability (Yamashiro et al., 1990, 1991, 1995; Yamakita et al., 1992) and, potentially, focal contact formation (Helfman et al., 1999).
Previous observations showed that αvβ3 expression is detected only in prostate cancer, but not in normal prostate epithelial cells (Zheng et al., 1999). Our data suggest that αvβ3 integrin and its downstream effector, cdc2, may be important mediators of prostate cancer progression toward an aggressive metastatic phenotype. This conclusion is supported also by data reported by Kallakury et al., indicating that cdc2 is expressed in a majority of prostatic adenocarcinomas and correlates with high Gleason's grade, advanced pathologic stage, and metastatic adenocarcinomas (Kallakury et al., 1997). Changes in gene expression during prostatic metastatic spread in vivo are likely to occur as recently shown for RhoC in melanoma cells (Clark et al., 2000). In conclusion, the functional role of cdc2 in prostate cancer in vivo may be different than once thought; it may reflect the migratory, rather than the proliferative, ability of these cells.
Materials and methods
Cells
LNCaP cell populations stably transfected with the pRc/CMV expression vector alone (mock) or containing human β3 integrin cDNA (β3-1) or ICAM-1 cDNA (ICAM) have been described (Zheng et al., 1999). In this paper, two additional β3 populations (β3-2 and β3-3) and two β6 LNCaP clones transfected with human β6 integrin cDNA (Sheppard et al., 1990) were generated. Expression of αvβ6 integrin was confirmed by FACS® analysis using the 10D5 mAb; mouse IgG was used as a negative control. HeLa and HT1080 cells were obtained from American Type Culture Collection. HT2–19 cells (Itzhaki et al., 1997) were a gift of Dr. A.C.G. Porter (Medical Research Council, London, UK). Cyclin B2–null and wt MEFs (Brandeis et al., 1998) were provided by Dr. T. Hunt (Imperial Cancer Research Fund, South Mimms, Herts, UK). For 3D cultures, cells were resuspended in Matrigel at a cell density of 106 cells/ml. For biochemical analysis, 3D cultures were detached from Matrigel by incubating with Matrisperse. For biochemical analysis or gene expression profiles, two-dimensional (2D) cultures were detached by trypsinization or, in parallel with the 3D cultures, by using Matrisperse (1 h on ice). Alternatively, 2D cultures were lysed directly on the plate.
Cell adhesion assays
Adhesion assays were performed as described previously (Languino et al., 1989).
RNA isolation and analysis
Gene expression profiles of β3-1, ICAM, and mock LNCaP cells were generated using Atlas Human Cancer cDNA Expression arrays (CLONTECH Laboratories, Inc.) according to the manufacturer's instructions. Northern blot analysis was performed using total RNA from 2D and 3D cell cultures isolated using TRIzol Reagent (GIBCO BRL). The 231-bp cdc2 cDNA fragment corresponding to the fragment on the cDNA array was generated by PCR. The cDNA fragment was amplified using the primers 5′-GGGTCAGCTCGTTACTCAACTCCAG-3′ and 5′-GACATGGGATGCTAGGCTTCCTGGT-3′ and human cdc2 cDNA as template. A 780-bp human GAPDH cDNA was excised from pGEM-3zf (+) with BamHI and PstI.
Immunoblotting and in vitro kinase assays
Cells were lysed using RIPA buffer (with 50 mM sodium fluoride for lysates to be used in in vitro kinase assays), either directly on the plate after washing with PBS, or after detaching with Matrisperse, as described for the 3D cultures above. For HT1080 and HT2–19 cells, cells were lysed as described (Itzhaki et al., 1997).
Primary antibodies to cdc2 (mAb, sc-54), to c-myc (mAb 9E10 and rabbit polyclonal agarose conjugate) and to ERK-1 (rabbit polyclonal that cross reacts with ERK-2, sc-94) were from Santa Cruz Biotechnology, Inc. Rabbit antiserum to cyclin B2 was a gift from J. Pines (Wellcome/Cancer Research Campaign Institute and Department of Zoology, Cambridge, UK). Caldesmon mAb SM12 was a gift from F. Matsumura (Rutgers University, Piscataway, NJ). Immunoprecipitation of cdc2 and in vitro kinase assays were performed essentially as described (Draetta and Beach, 1988; Morla et al., 1989; Pines and Hunter, 1989; Yu et al., 1998). 60 or 75 μg precleared cell lysate was used for immunoprecipitation using 2 μg mAb sc-54 or mouse IgG for 1 h, followed by protein A–Sepharose (Sigma Fast Flow) for 1 h. After two washes with lysis buffer and one wash with kinase buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 10 mM MgCl2, 0.5 mM DTT, 0.2 mM sodium orthovanadate, 0.1 mM sodium fluoride), immunoprecipitates were incubated in 20 μl kinase buffer with 250 μg/ml histone H1, 25 μM ATP, 62.5 μCi/ml [γ-32P]ATP for 30 min at 30°C, stopped by the addition of loading buffer, heated at 98°C for 5 min, separated on 12% SDS-PAGE, and visualized by autoradiography.
For cyclin B2 immunoprecipitation-kinase assays, cyclin B2 was immunoprecipitated from HeLa RIPA extracts with a cyclin B2 rabbit polyclonal antibody as described (Jackman et al., 1995) and incubated with histone H1 as described in the preceding paragraph, or with caldesmon immunoprecipitated from HeLa cells. Caldesmon was immunoprecipitated as described (Wang et al., 1999) except that, because caldesmon is heat stable, the lysate was boiled and clarified by centrifugation before immunoprecipitation by mAb SM12. Specifically, 5 × 106 HeLa cells were lysed with 50 mM Hepes, pH 7.5, 1% Triton X-100, 1% NP-40, 0.5% deoxycholate, 50 mM NaCl, 5 mM EDTA, 0.1 mM sodium vanadate, 1 mM PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, centrifuged 14,000 g for 10 min. The supernatant was then boiled for 5 min, cooled on ice for 30 min, and centrifuged 14,000 g for 10 min. An equal volume of immunoprecipitation buffer A (2.5% Triton X-100, 50 mM Tris-HCl, pH 7.4, 6 mM EDTA, 190 mM NaCl) was added to the supernatant, which was then precleared with protein A–Sepharose. Caldesmon mAb SM12 was added to the precleared lysate. After 1 h on ice, protein A–Sepharose was added and samples were rocked at 4°C for 1 h. Immunoprecipitates were washed three times with buffer B (150 mM NaCl, 10 mM Tris-HCl, pH 9, 5 mM EDTA, 0.1% Triton X-100) and once with kinase buffer (described in the preceding paragraph). Caldesmon immunoprecipitates were then used as substrate for either cyclin B2 immunocomplexes or recombinant cdc2/cyclin B1 (New England Biolabs, Inc.).
Migration assays
β3-LNCaP, β6-LNCaP, and HeLa cells were transiently cotransfected with a 1:7 ratio of pCMV-βgal and pcDNA-3 (empty vector), pCMVcdc2dn-HA, or pCMVcdc2wt-HA (van den Heuvel and Harlow, 1993). β3-LNCaP and HeLa cells were also transfected with pCMV-βgal and pCMX cyclin A, pCMV cyclin B1, or pCMV cyclin B2. HeLa cells were also transfected with pCMV-βgal and pCMVcdc2wt-HA and either 3 μg pCMV rat nonmuscle caldesmon wt or 3 μg pCMV rat nonmuscle caldesmon 7th mutant (Yamashiro et al., 2001). Lipofectamine 2000 (GIBCO BRL) was used as the transfection reagent. 1–3 d after transfection, the cells were seeded on 8-μm pore-sized transwell filter inserts (Costar) coated with 5 or 10 μg/ml FN or 3 μg/ml VN. In parallel, transiently transfected cells were also seeded on FN, VN, and poly-l-lysine–coated plates to measure their ability to adhere to these substrates. After 6 h, cells were fixed with 0.2% glutaraldehyde, washed with TTBS, and stained for βgal using x-gal as substrate (400 μg/ml x-gal, 0.5 mM K4Fe[CN]6, 0.5 mM K3Fe[CN]6, 1 mM MgCl2 in PBS), at 37°C for 2 h. The number of transfected cells in 10 random fields on the top and the bottom were counted for each filter. The percentage (average and SEM) of the attached transfected cells (βgal-positive cells on the top and bottom of the filter) that migrated (βgal-positive cells on the bottom of the filter) was calculated.
β3-LNCaP, β6-LNCaP, HT1080, HT2–19 cells, cyclin B2–null, and wt MEFs were seeded on 5-μm (HT1080, HT2–19), 8-μm (β3-LNCaP, β6-LNCaP), or 12-μm (cyclin B2–null MEFs, wt MEFs) pore-sized transwell filter inserts coated with 5 or 10 μg/ml FN or 3 μg/ml VN. After 4 h (HT1080, HT2–19, cyclin B2–null MEFs, wt MEFs) or 6 h (β3-LNCaP, β6-LNCaP), cells were fixed with 3% PFA/PBS, stained with crystal violet, and the number of cells per square millimeter on the bottom were counted (average and SEM of 10 random fields).
For cells cultured in the presence or absence of alsterpaullone and purvalanol A (Calbiochem) for 2 h, cells were seeded on filters as above in the absence or presence of alster or purvalanol A, for 6 h (β3-LNCaP) or 16 h (HeLa), and counted as described in the preceding paragraph. In parallel, cell adhesion assays in the presence of alster or purvalanol A were performed; cells were seeded in 96-well plates coated with 1–10 μg/ml FN or 3 μg/ml VN for 2 h, fixed with 3% PFA/PBS, stained with crystal violet, and the absorbance at 630 nm measured.
For cells cultured in the presence of mitomycin C (Sigma-Aldrich; 16-h incubation), cells were trypsinized and seeded on filters as above in the absence or presence of mitomycin C. After 6 h, cells were stained for βgal and the number of cells per square millimeter on the top and bottom were counted (average and SEM of 10 random fields).
Proliferation assays
For cells cultured in the presence of mitomycin C (Sigma-Aldrich; 16-h incubation), cells were trypsinized and seeded on FN-coated 96-well plates, in triplicate, in the presence of 1 μCi [3H]thymidine in 100 μl medium per well (in the presence or absence of mitomycin C), and incubated for 6 h. Medium was then removed, and cells were washed three times with PBS, solubilized with 10% SDS, and the amount of [3H]thymidine incorporated by the cells was quantitated by scintillation counting.
Immunofluorescence microscopy
HeLa cells were seeded on 10 μg/ml FN-coated coverslips, incubated in the presence or absence of 100 nM PMA and fixed with 3% PFA, 2% sucrose, pH 7.6 for 10 min at room temperature. Coverslips were stained with mAb to cdc2 (A17, IgG2kappa; Zymed), mAb to l-caldesmon (Clone 8, IgG1; BD Biosciences) and mAb to ezrin (Clone 18, IgG1; BD Biosciences). Secondary antibodies were class-specific goat anti–mouse IgGs coupled to either FITC or Texas red (Southern Biotechnology Associates, Inc.). In some single staining experiments, we also used a different mAb to caldesmon (SM12) and obtained essentially identical results. Nonbinding mouse IgGs were used as a control. Nuclear staining was performed with Hoechst 33342. 200 cells were systematically analyzed and ∼30% cells showed changes in morphology upon PMA treatment.
Acknowledgments
We are indebted to Drs. Hui Zhang (Yale University, New Haven, CT) for providing the human cdc2 cDNA and pCMX cyclin A construct; Dean Sheppard (University of California, San Francisco, San Francisco, CA) for the human β6 integrin cDNA; Jonathon Pines for the pCMV cyclin B1, pCMV cyclin B2 constructs, and cyclin B2 antibody; Sander van den Heuvel (Massachusetts General Hospital Cancer Center, Charlestown, MA) for the pCMV cdc2dn-HA and pCMV cdc2wt-HA constructs; Shigeko Yamashiro (Rutgers University) and Fumio Matsumura (Rutgers University) for the pCMV caldesmon wt and pCMV caldesmon 7th mutant constructs, as well as the SM12 mAb; Andrew Porter for the HT2-19 cells; and Tim Hunt for the cyclin B2–null and wt MEFs. A special acknowledgment to Dr. Cole Manes for critical comments on the manuscript; Dr. Mara Fornaro for insightful advice; Brian Dowd and Michael King for excellent and enthusiastic assistance; and Nancy Bennett for consistently excellent administrative assistance. Much gratitude to Michael King and Brian Paquin for assistance with graphics and to Zachary Pitluck for technical guidance.
This work was supported by grants from National Institutes of Health, R29 CA-71870 and RO1 CA-89720; from Army, PCRP DAMD17-98-1-8506 (to L.R. Languino); and DK-07556T32 training grant and Army DAMD-PC010384 fellowship (to T. Manes). P.C. Marchisio's work was supported by a grant from Associazione Italiana per la Ricerca sul Cancro and was performed in the frame of the Italian Ministry of Research Center of Excellence in Physiopathology of Cell Differentiation.
Amy S. Woodard's present name and address is Amy S. Woodard Freyman, Genetics Institute/Wyeth-Ayerst Research, 87 Cambridge Park Dr., Cambridge, MA 02140.
Footnotes
Abbreviations used in this paper: 2D, two dimensional; 3D, three dimensional; FN, fibronectin; IB, immunoblotting; MEF, mouse embryonic fibroblast; VN, vitronectin; wt, wild type.
References
- Bailly, E., M. Doree, P. Nurse, and M. Bornens. 1989. p34cdc2 is located in both nucleus and cytoplasm; part is centrosomally associated at G2/M and enters vesicles at anaphase. EMBO J. 8:3985–3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson, A., A. Davy, S.M. Robbins, and V.W. Yong. 2001. Differential activation of ERKs to focal adhesions by PKC epsilon is required for PMA-induced adhesion and migration of human glioma cells. Oncogene. 20:7398–7407. [DOI] [PubMed] [Google Scholar]
- Brandeis, M., I. Rosewell, M. Carrington, T. Crompton, M.A. Jacobs, J. Kirk, J. Gannon, and T. Hunt. 1998. Cyclin B2-null mice develop normally and are fertile whereas cyclin B1-null mice die in utero. Proc. Natl. Acad. Sci. USA. 95:4344–4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busk, M., R. Pytela, and D. Sheppard. 1992. Characterization of the integrin αvβ6 as a fibronectin-binding protein. J. Biol. Chem. 267:5790–5796. [PubMed] [Google Scholar]
- Byzova, T.V., R. Rabbani, S.E. D'Souza, and E.F. Plow. 1998. Role of integrin αvβ3 in vascular biology. Thromb. Haemost. 80:726–734. [PubMed] [Google Scholar]
- Cary, L.A., J.F. Chang, and J.-L. Guan. 1996. Stimulation of cell migration by overexpression of focal adhesion kinase and its association with Src and Fyn. J. Cell Sci. 109:1787–1794. [DOI] [PubMed] [Google Scholar]
- Clark, E.A., T.R. Golub, E.S. Lander, and R.O. Hynes. 2000. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 406:532–535. [DOI] [PubMed] [Google Scholar]
- Dalton, S. 1992. Cell cycle regulation of the human cdc2 gene. EMBO J. 11:1797–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damsky, C.H., and Z. Werb. 1992. Signal transduction by integrin receptors for extracellular matrix: cooperative processing of extracellular information. Curr. Opin. Cell Biol. 4:772–781. [DOI] [PubMed] [Google Scholar]
- Dell, K.R., C.W. Turck, and R.D. Vale. 2000. Mitotic phosphorylation of the dynein light intermediate chain is mediated by cdc2 kinase. Traffic. 1:38–44. [DOI] [PubMed] [Google Scholar]
- Draetta, G., and D. Beach. 1988. Activation of cdc2 protein kinase during mitosis in human cells: cell cycle-dependent phosphorylation and subunit rearrangement. Cell. 54:17–26. [DOI] [PubMed] [Google Scholar]
- Draviam, V.M., S. Orrechia, M. Lowe, R. Pardi, and J. Pines. 2001. The localization of human cyclins B1 and B2 determines cdk1 substrate specificity and neither enzyme requires MEK to disassemble the Golgi apparatus. J. Cell Biol. 152:945–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, F., G. Orend, N. Watanabe, T. Hunter, and E. Ruoslahti. 1996. Dependence of cyclin E-CDK2 kinase activity on cell anchorage. Science. 271:499–502. [DOI] [PubMed] [Google Scholar]
- Felding-Habermann, B., B.M. Mueller, C.A. Romerdahl, and D.A. Cheresh. 1992. Involvement of integrin α V gene expression in human melanoma tumorigenicity. J. Clin. Invest. 89:2018–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felding-Habermann, B., T.E. O'Toole, J.W. Smith, E. Fransvea, Z.M. Ruggeri, M.H. Ginsberg, P.E. Hughes, N. Pampori, S.J. Shattil, A. Saven, and B.M. Mueller. 2001. Integrin activation controls metastasis in human breast cancer. Proc. Natl. Acad. Sci. USA. 98:1853–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filardo, E.J., P.C. Brooks, S.L. Deming, C. Damsky, and D.A. Cheresh. 1995. Requirement of the NPXY motif in the integrin β3 subunit cytoplasmic tail for melanoma cell migration in vitro and in vivo. J. Cell Biol. 130:441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray, N.S., L. Wodicka, A.M. Thunnissen, T.C. Norman, S. Kwon, F.H. Espinoza, D.O. Morgan, G. Barnes, S. LeClerc, L. Meijer, et al. 1998. Exploiting chemical libraries, structure, and genomics in the search for kinase inhibitors. Science. 281:533–538. [DOI] [PubMed] [Google Scholar]
- Guadagno, T.M., M. Ohtsubo, J.M. Roberts, and R.K. Assoian. 1993. A link between cyclin A expression and adhesion-dependent cell cycle progression. Science. 262:1572–1575. [DOI] [PubMed] [Google Scholar]
- Helfman, D.M., E.T. Levy, C. Berthier, M. Shtutman, D. Riveline, I. Grosheva, A. Lachish-Zalait, M. Elbaum, and A.D. Bershadsky. 1999. Caldesmon inhibits nonmuscle cell contractility and interferes with the formation of focal adhesions. Mol. Biol. Cell. 10:3097–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota, T., T. Morisaki, Y. Nishiyama, T. Marumoto, K. Tada, T. Hara, N. Masuko, M. Inagaki, K. Hatakeyama, and H. Saya. 2000. Zyxin, a regulator of actin filament assembly, targets the mitotic apparatus by interacting with h-warts/LATS1 tumor suppressor. J. Cell Biol. 149:1073–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homayouni, R., and T. Curran. 2000. Cortical development: Cdk5 gets into sticky situations. Curr. Biol. 10:R331–R334. [DOI] [PubMed] [Google Scholar]
- Hsu, M.Y., D.T. Shih, F.E. Meier, P. Van Belle, J.Y. Hsu, D.E. Elder, C.A. Buck, and M. Herlyn. 1998. Adenoviral gene transfer of β3 integrin subunit induces conversion from radial to vertical growth phase in primary human melanoma. Am. J. Pathol. 153:1435–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X., J. Wu, S. Spong, and D. Sheppard. 1998. The integrin αvβ6 is critical for keratinocyte migration on both its known ligand, fibronectin, and on vitronectin. J. Cell Sci. 111:2189–2195. [DOI] [PubMed] [Google Scholar]
- Huber, P.A. 1997. Caldesmon. Int. J. Biochem. Cell Biol. 29:1047–1051. [DOI] [PubMed] [Google Scholar]
- Huhtala, P., M.J. Humphries, J.B. McCarthy, P.M. Tremble, Z. Werb, and C.H. Damsky. 1995. Cooperative signaling by α5β1 and α4β1 integrins regulates metalloproteinase gene expression in fibroblasts adhering to fibronectin. J. Cell Biol. 129:867–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes, R.O. 1999. Cell adhesion: old and new questions. Trends Cell Biol. 9:M33–M37. [PubMed] [Google Scholar]
- Itzhaki, J.E., C.S. Gilbert, and A.C. Porter. 1997. Construction by gene targeting in human cells of a “conditional” CDC2 mutant that rereplicates its DNA. Nat. Genet. 15:258–265. [DOI] [PubMed] [Google Scholar]
- Jackman, M., M. Firth, and J. Pines. 1995. Human cyclins B1 and B2 are localized to strikingly different structures: B1 to microtubules, B2 primarily to the Golgi apparatus. EMBO J. 14:1646–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, P.L., F.S. Jones, B. Zhou, and M. Rabinovitch. 1999. Induction of vascular smooth muscle cell tenascin-C gene expression by denatured type I collagen is dependent upon a β3 integrin-mediated mitogen-activated protein kinase pathway and a 122-base pair promoter element. J. Cell Sci. 112:435–445. [DOI] [PubMed] [Google Scholar]
- Juliano, R. 1996. Cooperation between soluble factors and integrin-mediated cell anchorage in the control of cell growth and differentiation. Bioessays. 18:911–917. [DOI] [PubMed] [Google Scholar]
- Juliano, R.L., and S. Haskill. 1993. Signal transduction from the extracellular matrix. J. Cell Biol. 120:577–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallakury, B.V., C.E. Sheehan, R.A. Ambros, H.A. Fisher, R.P. Kaufman, Jr., and J.S. Ross. 1997. The prognostic significance of p34cdc2 and cyclin D1 protein expression in prostate adenocarcinoma. Cancer. 80:753–763. [DOI] [PubMed] [Google Scholar]
- Keely, P.J., J.K. Westwick, I.P. Whitehead, C.J. Der, and L.V. Parise. 1997. Cdc42 and Rac1 induce integrin-mediated cell motility and invasiveness through PI(3)K. Nature. 390:632–636. [DOI] [PubMed] [Google Scholar]
- Kohn, K.W. 1999. Molecular interaction map of the mammalian cell cycle control and DNA repair systems. Mol. Biol. Cell. 10:2703–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafrenie, R.M., and K.M. Yamada. 1998. Integrins and matrix molecules in salivary gland cell adhesion, signaling, and gene expression. Ann. NY Acad. Sci. 842:42–48. [DOI] [PubMed] [Google Scholar]
- Lafrenie, R.M., S.M. Bernier, and K.M. Yamada. 1998. Adhesion to fibronectin or collagen I gel induces rapid, extensive, biosynthetic alterations in epithelial cells. J. Cell. Physiol. 175:163–173. [DOI] [PubMed] [Google Scholar]
- Languino, L.R., K.R. Gehlsen, E. Wayner, W.G. Carter, E. Engvall, and E. Ruoslahti. 1989. Endothelial cells use α2β1 integrin as a laminin receptor. J. Cell Biol. 109:2455–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochter, A., M. Navre, Z. Werb, and M.J. Bissell. 1999. α1 and α2 integrins mediate invasive activity of mouse mammary carcinoma cells through regulation of stromelysin-1 expression. Mol. Biol. Cell. 10:271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak, A.S., M. Carpenter, L.B. Smillie, and J.H. Wang. 1991. Phosphorylation of caldesmon by p34cdc2 kinase. Identification of phosphorylation sites. J. Biol. Chem. 266:19971–19975. [PubMed] [Google Scholar]
- Malecz, N., R. Foisner, C. Stadler, and G. Wiche. 1996. Identification of plectin as a substrate of p34cdc2 kinase and mapping of a single phosphorylation site. J. Biol. Chem. 271:8203–8208. [DOI] [PubMed] [Google Scholar]
- Matter, M.L., and E. Ruoslahti. 2001. A signaling pathway from the α5β1 and αVβ3 integrins that elevates bcl-2 transcription. J. Biol. Chem. 276:27757–27763. [DOI] [PubMed] [Google Scholar]
- McGowan, C.H., P. Russell, and S.I. Reed. 1990. Periodic biosynthesis of the human M-phase promoting factor catalytic subunit p34 during the cell cycle. Mol. Cell. Biol. 10:3847–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morla, A.O., G. Draetta, D. Beach, and J.Y. Wang. 1989. Reversible tyrosine phosphorylation of cdc2: dephosphorylation accompanies activation during entry into mitosis. Cell. 58:193–203. [DOI] [PubMed] [Google Scholar]
- Pines, J. 1999. Four-dimensional control of the cell cycle. Nat. Cell Biol. 1:E73–E79. [DOI] [PubMed] [Google Scholar]
- Pines, J., and T. Hunter. 1989. Isolation of a human cyclin cDNA: evidence for cyclin mRNA and protein regulation in the cell cycle and for interaction with p34cdc2. Cell. 58:833–846. [DOI] [PubMed] [Google Scholar]
- Pines, J., and C.L. Rieder. 2001. Re-staging mitosis: a contemporary view of mitotic progression. Nat. Cell Biol. 3:E3–E6. [DOI] [PubMed] [Google Scholar]
- Pockwinse, S.M., G. Krockmalnic, S.J. Doxsey, J. Nickerson, J.B. Lian, A.J. van Wijnen, J.L. Stein, G.S. Stein, and S. Penman. 1997. Cell cycle independent interaction of CDC2 with the centrosome, which is associated with the nuclear matrix-intermediate filament scaffold. Proc. Natl. Acad. Sci. USA. 94:3022–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanic, A.M., and J.A. Madri. 1994. The induction of 72-kD gelatinase in T cells upon adhesion to endothelial cells is VCAM-1 dependent. J. Cell Biol. 125:1165–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruoslahti, E., and J.C. Reed. 1994. Anchorage dependence, integrins and apoptosis. Cell. 77:477–478. [DOI] [PubMed] [Google Scholar]
- Schultz, C., A. Link, M. Leost, D.W. Zaharevitz, R. Gussio, E.A. Sausville, L. Meijer, and C. Kunick. 1999. Paullones, a series of cyclin-dependent kinase inhibitors: synthesis, evaluation of CDK1/cyclin B inhibition, and in vitro antitumor activity. J. Med. Chem. 42:2909–2919. [DOI] [PubMed] [Google Scholar]
- Schwartz, M.A., and R.K. Assoian. 2001. Integrins and cell proliferation: regulation of cyclin-dependent kinases via cytoplasmic signaling pathways. J. Cell Sci. 114:2553–2560. [DOI] [PubMed] [Google Scholar]
- Schwartz, M.A., M.D. Schaller, and M.H. Ginsberg. 1995. Integrins: emerging paradigms of signal transduction. Annu. Rev. Cell Dev. Biol. 11:549–599. [DOI] [PubMed] [Google Scholar]
- Seftor, R.E., E.A. Seftor, and M.J. Hendrix. 1999. Molecular role(s) for integrins in human melanoma invasion. Cancer Metastasis Rev. 18:359–375. [DOI] [PubMed] [Google Scholar]
- Seftor, R.E.B., E.A. Seftor, K.R. Gehlsen, W.G. Stetler-Stevenson, P.D. Brown, E. Ruoslahti, and M.J.C. Hendrix. 1992. Role of αvβ3 integrin in human melanoma cell invasion. Proc. Natl. Acad. Sci. USA. 89:1557–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw, L.M., I. Rabinovitz, H.H. Wang, A. Toker, and A.M. Mercurio. 1997. Activation of phosphoinositide 3-OH kinase by the α6β4 integrin promotes carcinoma invasion. Cell. 91:949–960. [DOI] [PubMed] [Google Scholar]
- Sheppard, D., C. Rozzo, L. Starr, V. Quaranta, D.J. Erle, and R. Pytela. 1990. Complete amino acid sequence of a novel integrin β subunit (β6) identified in epithelial cells using the polymerase chain reaction. J. Biol. Chem. 265:11502–11507. [PubMed] [Google Scholar]
- van den Heuvel, S., and E. Harlow. 1993. Distinct roles for cyclin-dependent kinases in cell cycle control. Science. 262:2050–2054. [DOI] [PubMed] [Google Scholar]
- Wang, Z., A.J. Danielsen, N.J. Maihle, and M.J. McManus. 1999. Tyrosine phosphorylation of caldesmon is required for binding to the Shc.Grb2 complex. J. Biol. Chem. 274:33807–33813. [DOI] [PubMed] [Google Scholar]
- Welch, P.J., and J.Y.J. Wang. 1992. Coordinated synthesis and degradation of cdc2 in the mammalian cell cycle. Proc. Natl. Acad. Sci. USA. 89:3093–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werb, Z., P.M. Tremble, O. Behrendtsen, E. Crowley, and C.H. Damsky. 1989. Signal transduction through the fibronectin receptor induces collagenase and stromelysin gene expression. J. Cell Biol. 109:877–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakita, Y., S. Yamashiro, and F. Matsumura. 1992. Characterization of mitotically phosphorylated caldesmon. J. Biol. Chem. 267:12022–12029. [PubMed] [Google Scholar]
- Yamashiro, S., Y. Yamakita, R. Ishikawa, and F. Matsumura. 1990. Mitosis-specific phosphorylation causes 83K non-muscle caldesmon to dissociate from microfilaments. Nature. 344:675–678. [DOI] [PubMed] [Google Scholar]
- Yamashiro, S., Y. Yamakita, H. Hosoya, and F. Matsumura. 1991. Phosphorylation of non-muscle caldesmon by p34cdc2 kinase during mitosis. Nature. 349:169–172. [DOI] [PubMed] [Google Scholar]
- Yamashiro, S., Y. Yamakita, K. Yoshida, K. Takiguchi, and F. Matsumura. 1995. Characterization of the COOH terminus of non-muscle caldesmon mutants lacking mitosis-specific phosphorylation sites. J. Biol. Chem. 270:4023–4030. [DOI] [PubMed] [Google Scholar]
- Yamashiro, S., H. Chern, Y. Yamakita, and F. Matsumura. 2001. Mutant caldesmon lacking cdc2 phosphorylation sites delays M-phase entry and inhibits cytokinesis. Mol. Biol. Cell. 12:239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, D., T. Jing, B. Liu, J. Yao, M. Tan, T.J. McDonnell, and M.C. Hung. 1998. Overexpression of ErbB2 blocks Taxol-induced apoptosis by upregulation of p21Cip1, which inhibits p34Cdc2 kinase. Mol. Cell. 2:581–591. [DOI] [PubMed] [Google Scholar]
- Zheng, D.Q., A.S. Woodard, M. Fornaro, G. Tallini, and L.R. Languino. 1999. Prostatic carcinoma cell migration via αvβ3 integrin is modulated by a focal adhesion kinase pathway. Cancer Res. 59:1655–1664. [PubMed] [Google Scholar]
- Zhu, X., M. Ohtsubo, R.M. Bohmer, J.M. Roberts, and R.K. Assoian. 1996. Adhesion-dependent cell cycle progression linked to the expression of cyclin D1, activation of cyclin E-cdk2, and phosphorylation of the retinoblastoma protein. J. Cell Biol. 133:391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]