

Abstract
Mice heterozygously deficient in the p0 gene (P0+/−) are animal models for some forms of inherited neuropathies. They display a progressive demyelinating phenotype in motor nerves, accompanied by mild infiltration of lymphocytes and increase in macrophages. We have shown previously that the T lymphocytes are instrumental in the demyelination process. This study addresses the functional role of the macrophage in this monogenic myelin disorder.
In motor nerves of P0+/− mice, the number of macrophages in demyelinated peripheral nerves was increased by a factor of five when compared with motor nerves of wild-type mice. Immunoelectron microscopy, using a specific marker for mouse macrophages, displayed macrophages not only in the endoneurium of the myelin mutants, but also within endoneurial tubes, suggesting an active role in demyelination. To elucidate the roles of the macrophages, we crossbred the myelin mutants with a spontaneous mouse mutant deficient in macrophage colony-stimulating factor (M-CSF), hence displaying impaired macrophage activation. In the P0-deficient double mutants also deficient in M-CSF, the numbers of macrophages were not elevated in the demyelinating motor nerves and demyelination was less severe. These findings demonstrate an active role of macrophages during pathogenesis of inherited demyelination with putative impact on future treatment strategies.
Keywords: macrophage, macrophage colony-stimulating factor, myelin degeneration, Schwann cell, inherited neuropathies
Introduction
Inherited neuropathies are chronic disorders of the peripheral nervous system with a prevalence of 1:2,500 (Skre 1974) including Charcot-Marie-Tooth disease (CMT), hereditary neuropathy with liability to pressure palsies, Dejerine-Sottas syndrome, and congenital hypomyelination. The majority of CMT patients suffer from a demyelinating neuropathy with juvenile or adult onset. There are now seven identified genes with various mutations associated with the family of CMT-like disorders. Historically, the peripheral myelin protein 22 gene was the first identified as the culprit gene for CMT1A (Suter and Snipes 1995); and myotubularin-related protein-2, N-myc downstream-regulated gene 1, and neurofilament-light are the most recently discovered genes related to CMT4C, hereditary motor and sensory neuropathy-Lom, and to a distinct variant of CMT2, respectively (Bolino et al. 2000; Kalaydjieva et al. 2000; Mersiyanova et al. 2000). The remaining genes related to CMT-like disorders encode the gap-junction protein connexin 32, the transcription factor designated early growth response 2 gene, and the cell adhesion molecule P0 (for reviews see Nelis et al. 1999; Martini 2000). A good model for some CMT-like disorders are mice heterozygously deficient in the p0 gene. We have shown recently that in this model immune cells are involved in demyelination that is primarily caused by an abnormal dosage of P0. The experimental proof that lymphocytes are not only innocent bystanders, but functionally involved in demyelination was provided by crossbreeding P0 mutants (P0+/−) with mice deficient in recombination activating gene-1 that lack mature T and B lymphocytes. These double mutants showed a clear amelioration of the neuropathological changes in peripheral nerves and of nerve conduction properties compared with P0 mutants having wild-type T lymphocytes (Schmid et al. 2000). The same effect was observed in mice deficient in the α-subunit of the T cell receptor, reflecting a selective involvement of T cells in the pathogenesis of this inherited demyelinating disease (Schmid et al. 2000). Based on these observations, we proposed that a secondary immunopathological mechanism may contribute to nerve pathology (Schmid et al. 2000).
In the present study, we focussed on the potential pathogenic impact of macrophages in inherited neuropathies. We demonstrate that in the animal models macrophages are present in a pattern reminiscent of chronic human and experimental inflammatory neuropathies (Schmidt et al. 1996; Ho et al. 1998; Gold et al. 1999). Moreover, myelin mutants with an additional defect in microglia/macrophage activation show less nerve pathology than P0+/− mice with an intact macrophage system. These findings are potentially important in the context of the understanding of pathomechanisms in hereditary neuropathies and designing disease modifying treatments.
Materials and Methods
Animals and Determination of Genotypes
P0+/− mice backcrossed to the C57/Bl6 background have been taken from our own breeding colony (Schmid et al. 2000). Osteopetrosis is a spontaneous autosomal recessive mutation with the homozygous mice (op/op) lacking biologically active M-CSF due to a single base insertion (T), creating a premature stop codon (Yoshida et al. 1990). Lack of incisor tooth eruption is the most definitive phenotypic marker of the op/op genotype. The heterozygous op/wt mice are phenotypically normal and these mice were maintained as a breeding colony on a mixed C57/Bl6/C3 background. Double mutants were obtained by crossing op/wt with P0+/− mice, leading to ∼25% of the F1 progeny with the P0+/− op/wt genotype. Phenotypically normal op/wt, wt/wt, and osteopetrotic (op/op) animals with P0+/− genotype (P0+/− op/op) and normal P0+/+ were the F2 product of the double heterozygous mice and were investigated at the age of 6 mo. Later stages have not been investigated, since op/op mice often die at a premature age. To avoid effects of the different genetic backgrounds, only littermates were analyzed.
Genotypes of P0 mutants were determined by conventional PCR as described (Schmid et al. 2000). Mice of the op/op genotype were determined by their typical absence of incisivi (Yoshida et al. 1990) and by a PCR protocol generously provided by Dr. S. Oehen (University of Zürich, Zürich, Switzerland). Since op/wt mice display a normal phenotype and behavior, their genotype had to be determined. The presence of wild-type and mutant allele was detected by using oligonucleotides 5′-TGTGTCCCTTCCTCAGATTACA-3′ and 5′-GGTCTCATCTATTATGTCTTGTACCAGCCAAAA-3′, which introduce an additional BglI restriction site into the mutant allele. The PCR was performed in a final volume of 20 μl; 300 ng genomic DNA was used as template. The reaction product was denaturated at 95°C for 3 min, followed by 5 cycles (94°C, 1 min; 56°C, 45 s; 72°C, 45 s), 35 cycles (94°C, 30 s; 52°C, 30 s; 72°C, 30 s), and a final extension at 72°C for 10 min with a PE Biosystems thermal cycler. The PCR product was digested with BglI (20 IU; Boehringer) for 1 h at 37°C. After digestion, the characteristic fragments indicating the genotypes could be detected on a polyacrylamide gel by autoradiography. The wild-type was represented by a 99- and 96-bp fragment, op/wt mice by a 99-, 96-, 70-, and 30-bp fragment, and op/op-mice by a 96-, 70-, and 30-bp fragment.
Immunohistochemistry and Immunoelectron Microscopy
For histological analyses, we have chosen the two major branches of the femoral nerve, the quadriceps muscle nerve, and the cutaneous saphenous nerve of the mouse. In some experiments, sciatic nerves have been investigated at the level of the sciatic notch. In addition, we investigated the spinal roots of the third and fourth lumbar segment.
For detection and quantification of T lymphocytes, antibodies to CD8 (a gift from R. Zinkernagel, University of Zürich, Zürich, Switzerland) have been used on serial nerve cryosections as described previously (Schmid et al. 2000).
For detection of macrophages, antibodies to mouse F4/80 (1:300; Serotec) were applied overnight on 14-μm-thick serial sections from fresh frozen femoral nerves and spinal roots. To visualize primary antibodies, a biotinylated secondary antibody to rat Igs was applied for 1 h, followed by avidin/biotin reagent (Dako) and staining with diaminobenzidine-HCl and H2O2. For negative control, the primary antibody was omitted. In femoral nerves, immunoreactive profiles were counted on serial cryosections as described (Schmid et al. 2000). In addition, immunolabeling of single fiber preparations of spinal roots from transcardially perfused mice was performed on free-floating tissue as described (Guénard et al. 1996), with the only modification that primary antibodies were detected by biotinylated secondary antibodies, followed by avidin/biotin reagent as indicated above. In some experiments, immunolabeled fibers were embedded on slides and investigated under the light microscope. In other experiments, the immunolabeled fibers were processed for electron microscopy as described (Guénard et al. 1996).
For double immunofluorescence with the monoclonal rat antibodies to mouse F4/80 and mouse major histocompatibility complex (MHC) class II, fresh frozen sections of femoral nerves and spinal roots were first incubated with the MHC class II antibodies (rat anti–mouse I-Ab, 1:400, 1 h; BD PharMingen), followed by incubation with anti–rat Ig antibodies coupled to FITC (1:200, 30 min; Leinco Technologies). Then, incubation with a blocking reagent followed to avoid nonspecific binding of biotin/avidin system reagents (Vector-Sp-2001, 30 min; Vector Laboratories). As a next step, sections were incubated with a biotinylated F4/80 antibody (1:300, 1 h; Serotec), which was visualized with streptavidin coupled to the red fluorescent dye Cy3 (30 min; Dianova). Control experiments were performed omitting primary antibodies, resulting in complete absence of immunoreactivity.
Double immunofluorescence with polyclonal antibodies to neural cell adhesion molecule from rabbit (1:500; Carenini et al. 1999) and with monoclonal antibodies to mouse MHC class I (ER-HR 52, 1:100; Dianova) from rat was performed by simultaneous incubation of primary antibodies for 1.5 h, followed by simultaneous incubation of secondary antibodies (goat anti–rabbit Cy3, 1:500; Dianova; goat anti–rat FITC, 1:500; Leinco Technologies) for 1.5 h. Control experiments were performed omitting primary antibodies resulting in complete absence of immunoreactivity.
For localizing M-CSF receptor expression, double immunofluorescence using rabbit anti–mouse M-CSF receptor antibody (06-175, 1:5,000; UBI) and the macrophage- and microglia-specific rat antibody to mouse αMβ2 integrin (5C6, 1:5,000; Serotec) has been performed as described (Raivich et al. 1998), with the only exception that teased ventral root fibers instead of free-floating brain sections have been used. Digital micrographs of the FITC and Cy3 fluorescence were taken using a Leica TCS 4D confocal laser microscope with a 63× objective in an 8-bit greyscale (b/w), 1,024 × 1,024 pixel format as described in previous studies (Raivich et al. 1998; Kloss et al. 1999). 10 consecutive, equidistant levels spacing 12 μm were recorded and condensed to a single bitmap using the MaxIntens algorithm.
Tissue Preservation for Light and Conventional Electron Microscopy and Morphometry
Femoral nerves and spinal roots of mice were processed for light and electron microscopy as described previously (Schmid et al. 2000). Analysis of the g-ratio (diameter of axons/diameter of fiber) of randomly selected nerve fibers was performed on electron micrographs of the quadriceps nerve and of the ventral roots at a final magnification of 1,600× using Scion Image Software (Scion Corp.).
Statistics
Statistical analysis was performed by using Student's t test or Mann-Whitney U test, when appropriate. P < 0.05 was considered statistically significant.
Results
Immunohistochemistry and Quantification of F4/80- and αMβ2-positive Macrophages
In nerve cross sections of normal mice, the F4/80 immunoperoxidase-positive macrophages were usually ramified or showed a few slender processes (Fig. 1A and Fig. B). Most, if not all, F4/80-immunoreactive macrophages in the quadriceps nerve and ventral root were MHC class II–positive, as revealed by double immunofluorescence (not shown). By contrast, MHC class I immunoreactivity was confined to nonmyelinating Schwann cells, as revealed by double immunofluorescence with antibodies to neural cell adhesion molecule, recognizing nonmyelinating Schwann cells in peripheral nerves (not shown).
Figure 1.

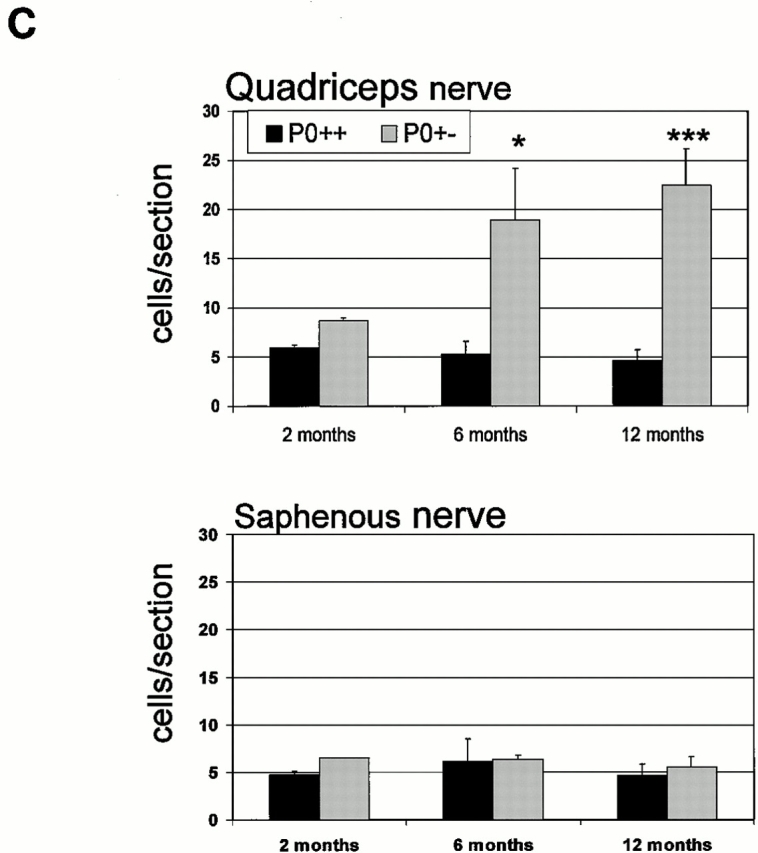
(A and B) Immunhistological localization of macrophages in quadriceps nerves of P0+/+ (A) and P0+/− mice (B) at the age of 6 mo using antibodies to F4/80. (A) In quadriceps nerves of P0+/+ mice some resident macrophages are detectable in the endoneurium. (B) In quadriceps nerves of P0+/− mice the number of macrophages is clearly elevated when compared with P0+/+ mice. Note the larger size of the cells and the close vicinity of two cells to an endoneurial blood vessel. (C) Quantification of F4/80-positive macrophages in quadriceps and saphenous nerves of P0+/+ (black bars) and P0+/− mice (gray bars) at the age of 2 (n = 2 for P0+/+ and P0+/−), 6 (n = 3 for P0+/+; n = 4 for P0+/−), and 12 mo (n = 4 for P0+/+ and P0+/−). Note elevated number of macrophages in quadriceps nerves of 6- and 12-mo-old P0+/− mice. A slight elevation of macrophage numbers is already seen in quadriceps nerves of 2-mo-old P0+/− mice. In nondemyelinating saphenous nerves, the number of macrophages is not increased at any age investigated. Bars represent mean values ± SD. *P < 0.05; ***P < 0.001. Bar, 50 μm.
Localization of the M-CSF receptor was performed on teased fibers from ventral roots of 6-mo-old P0+/+ and P0+/− mice by double immunofluorescence using an M-CSF receptor–specific antibody and the macrophage-specific marker αMβ2. Labeling for M-CSF receptor was exclusively associated with macrophages, whereas αMβ2-negative cells, such as Schwann cells and endoneurial fibroblasts, were never labeled with antibodies to M-CSF receptor (Fig. 2). Macrophages of the P0+/+ mice showed a weak labeling for M-CSF receptor (Fig. 2). Interestingly, P0+/− mice showed some very strongly labeled macrophages in addition to weakly labeled macrophages (Fig. 2). These data show that M-CSF receptor expression is confined to macrophages and that expression levels are increased in some macrophages of the P0+/− mice.
Figure 2.

Cellular localization of the M-CSF receptor (MCSFR, red) immunoreactivity in teased fiber preparations from ventral roots of P0+/+ and P0+/− mice using antibodies to αMβ2 integrin (green) as a marker for peripheral nerve macrophages. αMβ2-negative cells, such as the adjacent Schwann cells, were never labeled. Note the particularly strongly MCSFR-immunoreactive macrophage in the P0+/− mutant (arrow). Bar, 50 μm.
We quantified the macrophages in muscular and cutaneous branches of the femoral nerve and in spinal roots of P0+/+ and P0+/− mice by immunohistochemistry using the F4/80 antibody. In the quadriceps muscle nerves of the P0+/+ mice, approximately five F4/80-positive macrophages per nerve section were found at 2, 6, and 12 mo (Fig. 1 C). In contrast to the normal wild-type mice, the number of macrophages in the quadriceps nerve of heterozygous P0+/− mice was slightly elevated at 2 mo, increased ∼3.5-fold at 6 mo, and 5-fold at 12 mo of age (P < 0.001; Fig. 1 C).
Macrophage counts in the cutaneous saphenous nerve of P0+/+ mice revealed a similar number as that in the normal, wild-type quadriceps nerves at any age investigated (Fig. 1 C). The number of macrophages in the P0+/− saphenous nerves was normal and did not show an age-dependent increase, a finding in line with the still unexplained lack of demyelinating neuropathy in the sensory nerves of P0+/− mice (Martini et al. 1995; Shy et al. 1997) and of some other myelin mutants (Martini 1997). This divergence between muscular and cutaneous nerves in the P0+/− animals also extended to spinal roots, with the macrophages consistently more numerous in the ventral roots than in the dorsal, or in any nerves from the normal, P0+/+ mice. In addition to differences in number, the F4/80-immunoreactive cells in the quadriceps nerves and ventral roots of P0+/− mice also appeared larger compared with saphenous nerves and dorsal roots of the same genotype, or with spinal roots and femoral nerves of P0+/+ mice (Fig. 1 B). Some of these macrophages from the P0+/− nerves were also laden with lipid vacuoles or with material reminiscent of myelin debris, a finding that we did not observe in the P0+/+ mice or in the P0+/− sensory nerves.
Conventional Electron Microscopy and Immunoelectron Microscopic Localization of Macrophages in Peripheral Nerves of P0+/− Mice
To investigate the spatial relationship of macrophages to demyelinating fibers, we performed conventional electron microscopy and immunoelectron microscopy with F4/80 antibodies in quadriceps nerves and ventral roots of 6-mo-old P0+/− mice.
In ultrathin sections of conventionally prepared quadriceps nerves and ventral roots, endoneurial cells laden with lipid vacuoles or myelin debris were identified as macrophages. They were often found in the endoneurium and were characterized by polymorphic nuclei, abundant heterochromatin, and microvillus-like processes that extended from the surface. Occasionally, such cells were found in the endoneurial tubes, i.e., within the Schwann cell basal laminae (Fig. 3A and Fig. B). In very rare cases, macrophage-like cells were seen just penetrating the basal lamina with a cellular process, reflecting the invasion or leave of the endoneurial tube (Fig. 3 B). However, since Schwann cells can also phagocytose myelin and penetrate basal laminae under pathological conditions, morphological criteria alone appeared insufficient to characterize unequivocally the spatial relationship between macrophages and nerve fibers. Therefore, we performed immunoelectron microscopy using the macrophage-specific antibody F4/80.
Figure 3.
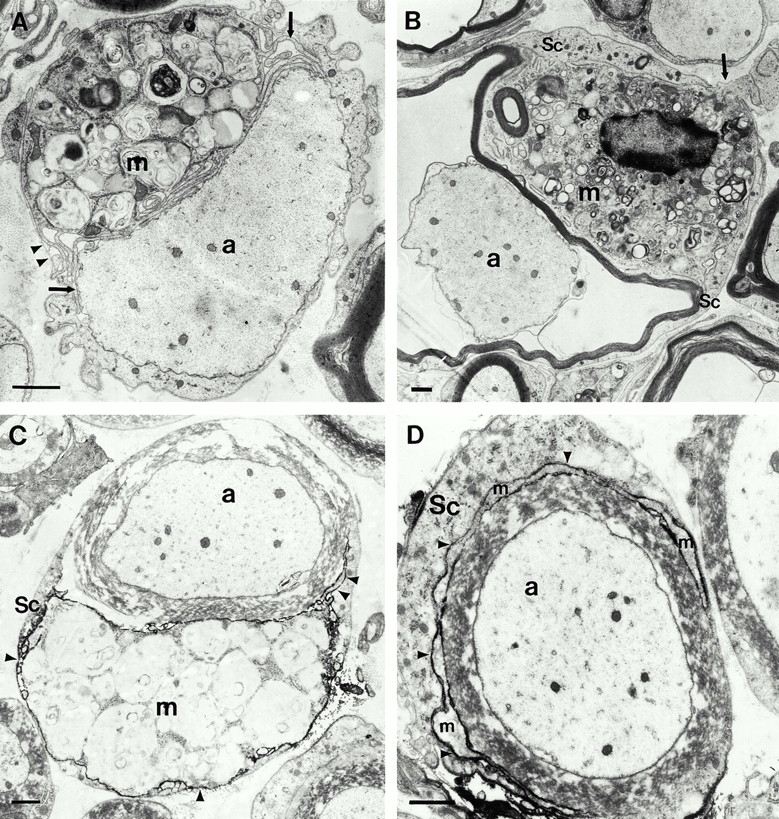
Conventional electron microscopy (A and B) and immunoelectron microscopy using the macrophage-specific antibody F4/80 (C and D) in quadriceps nerves of 6-mo-old P0+/− mice. (A) A putative macrophage (m) that is laden with myelin debris is closely apposed to a demyelinated axon (a). Note the slender processes of the macrophage (arrows) and the position of the cell within the endoneurial tube. The arrowheads demarcate the Schwann cell basal lamina. (B) A putative macrophage (m) is in close contact to a thin myelin sheath that is partially detached from the corresponding axon (a). The arrow demarcates a process of the putative macrophage that penetrates the Schwann cell basal lamina. Sc, Schwann cell. (C) An F4/80-positive macrophage (m) containing myelin debris is in close apposition to a myelin sheath. Arrowheads indicate electron-dense immunoreaction product. a, Axon; Sc, Schwann cell. (D) A slender process of a F4/80-positive macrophage (m) has penetrated in between the pericaryon of the Schwann cell (Sc) and its normal appearing myelin sheath. Arrowheads indicate electron-dense immunoreaction product. a, axon. Bars, 1.5 μm.
The majority of the F4/80-positive macrophages were found in the endoneurial space. Approximately 10–30% of the F4/80-positive cells were found within the endoneurial tubes, i.e., within the inner aspects of the basal laminae (Fig. 3C and Fig. D). Occasionally, the macrophages were in intimate association with myelin sheaths that appeared morphologically normal (Fig. 3C and Fig. D), suggesting an active role of macrophages during demyelination. In other cases, myelin appeared disorganized or was even absent and the demyelinated axons were in direct contact with the macrophages (not shown). No immunoreactivity was observed on axons or on morphologically unequivocally identified Schwann cells.
P0/op Double Mutants: Fewer Macrophages in Femoral Nerves and Less Severe Demyelination
In the current study, we used mice deficient for M-CSF (op/op) and heterozygous for P0 (P0+/−) to investigate the role of macrophages in genetically caused demyelination. Myelin mutant littermates with the normal M-CSF phenotype (P0+/− op/wt or P0+/− wt/wt) served as controls.
In the quadriceps nerve of M-CSF–deficient, P0 heterozygous mice (P0+/− op/op), the number of macrophages was not increased compared with mice with normal P0 dosage (P0+/+ op/wt and P0+/+ op/op; Fig. 4). In contrast, and similar to the findings obtained from P0+/− mice (Fig. 1 C), the number of macrophages in the quadriceps nerve was strongly elevated in P0+/− op/wt mice compared with op mutants with normal P0 gene dosage (P0+/+ op/wt and P0+/+ op/op mice; P = 0.01; Fig. 4). Thus, homozygous deficiency in M-CSF leads to a lack of an age-dependent macrophage increase in the motor nerves of P0+/− mutants.
Figure 4.

Quantification of F4/80-positive macrophages in quadriceps and saphenous nerves of P0+/+ op/wt (black bars, n = 5), P0+/+ op/op (white bars, n = 3), P0+/− op/wt (dark gray bars, n = 9), and P0+/− op/op mice (bright gray bars, n = 3) at the age of 6 mo. The op genotype has no influence on the number of resident macrophages in quadriceps nerves of P0+/+ mice. In quadriceps nerves of P0+/− op/wt mice, the number of macrophages is similarly increased, as shown in Fig. 1 C, whereas homozygosity for op prevents the increase of macrophage numbers in quadriceps nerves of the myelin mutants (P0+/− op/op). In the nondemyelinating saphenous nerves, the number of macrophages is never increased. Bars indicate mean values ± SD. * P < 0.05.
To investigate whether compromised macrophage increase in P0+/− op/op mice has an influence on the number of T lymphocytes, we also investigated the number of CD8-positive cells in 6-mo-old P0+/+ op/wt, P0+/− op/wt, and P0+/− op/op mice. Due to the low amount of tissue available from the limited number of mutants, only sciatic nerves could be investigated (n = 3 for each genotype). In the sciatic nerves of P0+/+ op/wt mice, 1.7 ±0.2 T lymphocytes/mm2 could be detected. There was a clear trend to higher values in P0+/− op/wt mice (2.6 ± 0.4 cells/mm2), but not in the P0+/− op/op mice (1.9 ± 0.3 cells/mm2). The differences between P0+/− op/wt and P0+/− op/op mice failed to reach statistical significance (P = 0.06), possibly due to the previously discussed heterogeneous distribution of the lymphocytes (Schmid et al. 2000).
Comparison of semithin sections of quadriceps nerves and ventral roots from 6-mo-old P0+/− op/wt mice with those from the P0+/− op/op littermates revealed that all mice with P0+/− genotype showed pathological alterations indicative of demyelination. However, the severity of the pathological alterations was clearly reduced in mice with the op/op genotype. Thus, an investigator who was not aware of the genotype (R. Martini) could unequivocally distinguish the nerves from P0+/− op/wt mice from those of P0+/− op/op mice.
The thickness of myelin was a particularly striking parameter in the presence or absence of M-CSF. Motor nerves from P0+/− op/op mice always had thicker myelin sheaths than those from the P0+/− op/wt littermates. This difference was strongest in the ventral roots (Fig. 5A and Fig. B), but substantial differences in myelin thickness between P0+/− op/wt and P0+/− op/op mice were also obtained for the quadriceps nerves (not shown). Semithin sections of quadriceps nerves and ventral roots of P0+/+ op/wt and P0+/+ op/op mice were also investigated at the age of 6 mo. These nerves were of normal phenotype with thick myelin and absence of features indicative of demyelination independent of the op genotype (not shown).
Figure 5.

(A and B) Electron microscopy of ventral roots of 6-mo-old P0+/− op/wt (A) and P0+/− op/op mice (B). Myelin thickness in P0+/− op/wt mice is profoundly reduced compared with P0+/− op/op littermates. Note that in P0+/− op/wt mice (A) both small (asterisk) and large caliber fibers (double asterisk) are associated with abnormally thin myelin. The presence of fibers with thicker and thinner myelin in one and the same section in A represents segmental demyelination as has been described previously in longitudinal views of teased fibers (Martini et al. 1995). (C) Quantitative analysis of g-ratios of myelinated fibers in ventral roots of P0+/− op/wt (dark gray bars, n = 3), P0+/− op/op (bright gray bars, n = 3), and P0+/+ mice (black bars, n = 3) at the age of six months. Note the reduction in g-ratios in roots from P0+/− op/op mice reflecting thicker myelin sheaths, when compared with values from P0+/− op/wt mice. *P < 0.05. P0+/+ mice display the thickest myelin sheaths. Bars, 5 μm.
To quantify the less reduced myelin thickness in quadriceps nerves and ventral roots of P0+/− op/op mice versus in P0+/− op/wt mice, we determined the g-ratio of the myelin sheaths at the electron microscopic level. In agreement with the light microscopic findings, the g-ratios were significantly lower in quadriceps nerves (0.78 in P0+/− op/op mice versus 0.82 in P0+/− op/wt mice, P < 0.05, n = 3). Similar differences in g-ratios were found in ventral roots of P0+/− op/op mice versus P0+/− op/wt mice (Fig. 5), thus reflecting thicker myelin in the myelin mutants with compromised macrophage function. Analysis of g-ratios of smaller (<6 μm) and larger (>6 μm) caliber fibers of ventral roots revealed that in P0+/− op/op mice, the g-ratios of both fiber groups were decreased, representing rescue of myelin thickness in both smaller and larger caliber fibers (P < 0.05 for both size groups; Fig. 5A and Fig. B). Although in P0+/− op/op mice myelin thickness was clearly increased compared with P0+/− op/wt mice, normal myelin thickness was not reached as reflected by a lower g-ratio in ventral roots of P0+/+ mice (Fig. 5 C).
Discussion
This study focused on the functional roles of macrophages in an established animal model for CMT1B, the P0 heterozygous mice. We found that the number of macrophages was strongly increased and that they showed a frequent association with demyelinating fibers. When the P0+/− mice were crossbred with spontaneous mouse mutants deficient in M-CSF, the resulting double mutants lacked the increase in number of macrophages and showed less severe pathological alterations. Our findings show that macrophages are actively involved in demyelination in a model for inherited neuropathies.
Functional Role of Macrophages in P0+/− Mice
Previous studies in M-CSF–deficient (op/op) mice revealed an impaired survival, development, and differentiation of the monocyte hematopoietic cell lineage in the absence of M-CSF (Felix et al. 1990; Metcalf 1991; Witmer-Pack et al. 1993; Stanley et al. 1997; for review see Teitelbaum 2000). In the central nervous system of the op/op mice, microglial activation is severely impaired under various pathological conditions (Raivich et al. 1994; Berezovskaya et al. 1995). In the peripheral nervous system of the osteopetrotic P0+/+ mice, the number of resident macrophages was normal. However, the number of macrophages did not increase in the myelin-deficient osteopetrotic double mutants, which is reminiscent of findings in the injured central nervous system of osteopetrotic single mutants (Raivich et al. 1994). This lack of macrophage reaction upon the diseased peripheral nervous tissue in op/op mice must be considered to be due to a direct and exclusive impairment of the macrophage response, since we show that in P0+/− mice M-CSF receptor was exclusively found on macrophages, a situation similar to the normal and injured central nervous system (Raivich et al. 1998). Thus, our combined findings not only demonstrate that macrophages are pivotal cellular mediators of myelin damage in this primarily genetically caused demyelinating neuropathy, but also that M-CSF is a critical molecule for the activation of these myelin-phagocytosing cells.
Macrophages play important roles under several pathological conditions, including inflammatory disorders of the nervous system (Hartung et al. 1998; Ho et al. 1998; Kiefer et al. 1998; Gold et al. 1999). In the peripheral nervous system, these functions are particularly well characterized in experimental autoimmune neuritis, the animal model for the Guillain-Barré syndrome. In this disease, macrophages present antigen to autoimmune lymphocytes in the context of MHC class II, which results in activation and proliferation of T lymphocytes (Hartung et al. 1998; Gold et al. 1999). Interestingly, in P0+/− mice the previously reported increase of the number of T lymphocytes (Schmid et al. 2000) appears to be lacking when macrophage activation is compromised due to the absence of M-CSF. Since T lymphocytes do not carry M-CSF receptors (Raivich et al. 1998), one has to postulate that the reduced number of antigen-presenting and/or cytokine-secreting macrophages might be the reason for the accompanying lack of increase of T lymphocytes.
Macrophages are also involved in the effector phase of the disorders (Craggs et al. 1984; Jung et al. 1993; Hartung et al. 1998; Gold et al. 1999). In electron microscopy studies of Guillain-Barré syndrome and experimental autoimmune neuritis, activated macrophages are seen in direct contact with myelin (Ballin and Thomas 1969; Lampert 1969; Ho et al. 1998; Smith 1999) which is similar to the macrophage–myelin apposition in our animal model for inherited neuropathies. Apart from the direct physical attack, macrophages also synthesize numerous factors, such as arachidonic acid metabolites, oxygen radicals, nitrous oxide, matrix metalloproteinases, and proinflammatory cytokines that can all act to jeopardize myelin stability and the normal function of the myelin-forming Schwann cell (Banati et al. 1993; Ho et al. 1998; Gold et al. 1999).
The Role of Macrophages in Human Inherited Neuropathies
Similar to our animal model, an increased number of endoneurial macrophages has also been reported in nerve biopsies of patients with inherited peripheral neuropathies (Sommer and Schröder 1995; Schmidt et al. 1996; Stoll et al. 1998). In many forms of CMT the active phase of demyelination occurs during childhood (Gabreëls-Festen et al. 1995; Garcia et al. 1998; Thomas 1999), and nerve biopsies were usually not performed. It is conceivable that circulating monocytes enter the peripheral nerve to clear the myelin debris that results from genetically mediated disintegration of myelin. However, macrophages may also actively help to destroy the myelin produced by the mutant Schwann cells. This notion is clearly supported by the data of the current study in P0 mutant mice, as was suggested previously by noting the close association of macrophages with the myelin at the ultrastructural level of inherited human neuropathy (Vital et al. 1992).
What are the possible implications of the current findings for our understanding of human inherited neuropathies? Only few inherited neuropathies are caused by an abnormal gene dosage of P0 (Nelis et al. 1999), but this does not argue against the possibility that similar mechanisms can take place in demyelinating CMT forms that are unrelated to P0 mutations. Indeed, a similar elevation of macrophage numbers as seen in P0+/− mice has recently been found in Cx32-deficient mice (Kobsar, I., M. Mäurer, and R. Martini, unpublished observations), a model for the X-linked form of CMT (Anzini et al. 1997; Scherer et al. 1998). Most interestingly, in some patients with CMT a rather rapid worsening or even overt inflammation (Gregory et al. 1993; Malandrini et al. 1999) with clinical response to corticosteroid treatment (Bird and Slaky 1991; Crawford and Griffin 1991; Dyck et al. 1982) or plasma exchange (Malandrini et al. 1999) has been described. It is possible that these clinical cases are at the extreme end of an immunopathologic and chronic disease process that may be unnoticed in the majority of cases. Future studies in the mouse models as well as in human biopsies using appropriate macrophage markers (Kiefer et al. 1998) are needed to substantiate our conclusions with the aim to develop treatment strategies that culminate in the impairment of the detrimental immune cells in peripheral nerves of patients.
Acknowledgments
The authors are grateful to Dr. Stephan Oehen for providing the PCR-based protocol to genotype op mutants and for initial help; to Dr. Ralf Gold for stimulating discussions; and to Carolin Kiesel and Andrea Koppius for excellent technical assistance.
The study was supported by the Deutsche Forschungsgemeinschaft (SFB 581 to R. Martini and K.V. Toyka; Priority Program “Microglia” MA1053/3-1 and 3-2 to R. Martini; and Ra 486/3-1 to G. Raivich.); Gemmeinnützige Hertie-Stiftung (GHS2/476/98 to R. Martini); by the German Ministry of Research (grant 01KO9703/3 to G. Raivich and Interdisziplinäres Zentrum für klinische Forschung, project C-6, to R. Martini); and by local research funds of the University of Würzburg (to R. Martini).
Footnotes
S. Carenini's present address is Novartis-Pharmanalytica, CH-6601 Locarno, Switzerland.
C. Schmid's present address is The Scripps Research Institute, Department of Molecular Biology, La Jolla, CA 92037.
Abbreviations used in this paper: CMT, Charcot-Marie-Tooth disease; MHC, major histocompatibility complex; M-CSF, macrophage colony-stimulating factor; op/op, osteopetrotic; P0+/−, P0 mutant.
References
- Anzini P., Neuberg D.H.H., Schachner M., Nelles E., Willecke K., Zielasek J., Toyka K.V., Suter U., Martini R. Structural abnormalities and deficient maintenance of peripheral nerve myelin in mice lacking the gap junction protein connexin 32. J. Neurosci. 1997;17:4545–4551. doi: 10.1523/JNEUROSCI.17-12-04545.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballin R.H., Thomas P.K. Electron microscope observations on demyelination and remyelination in experimental allergic neuritis. I. Demyelination. J. Neurol. Sci. 1969;8:1–18. doi: 10.1016/0022-510x(69)90037-9. [DOI] [PubMed] [Google Scholar]
- Banati R.B., Gehrmann J., Schubert P., Kreutzberg G.W. Cytotoxicity of microglia. Glia. 1993;7:111–118. doi: 10.1002/glia.440070117. [DOI] [PubMed] [Google Scholar]
- Berezovskaya O., Maysinger D., Fedoroff S. The hematopoietic cytokine, colony-stimulating factor 1, is also a growth factor in the CNScongenital absence of CSF-1 in mice results in abnormal microglial response and increased neuron vulnerability to injury. Int. J. Dev. Neurosci. 1995;13:285–299. doi: 10.1016/0736-5748(95)00013-7. [DOI] [PubMed] [Google Scholar]
- Bird S.J., Slaky J.T. Corticosteroid-responsive dominantly inherited neuropathy in childhood. Neurology. 1991;41:437–439. doi: 10.1212/wnl.41.3.437. [DOI] [PubMed] [Google Scholar]
- Bolino A., Muglia M., Conforti F.L., LeGuern E., Salih M.A.M., Georgiou A.-M., Christodoulou K., Hausmanowa-Petrusewicz I., Mandich P., Schenone A. Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2. Nat. Genet. 2000;25:17–19. doi: 10.1038/75542. [DOI] [PubMed] [Google Scholar]
- Carenini S., Montag D., Schachner M., Martini R. Subtle roles of N-CAM and MAG during Schwann cell spiralling in P0-deficient mice. Glia. 1999;27:203–212. [PubMed] [Google Scholar]
- Craggs R.I., King R.H., Thomas P.K. The effect of suppression of macrophage activity on the development of experimental allergic neuritis. Acta Neuropathol. 1984;62:316–323. doi: 10.1007/BF00687614. [DOI] [PubMed] [Google Scholar]
- Crawford T.O., Griffin J.W. Morphometrical and ultrastructural evaluation of the sural nerve in children with Charcot-Marie-Toothimplications for pathogenesis and treatment. Ann. Neurol. 1991;30:500. [Google Scholar]
- Dyck P.J., Swanson C.J., Low P.A., Bartleson J.D., Lambert E.H. Prednisone-responsive hereditary motor and sensory neuropathy. Mayo Clin. Proc. 1982;57:239–246. [PubMed] [Google Scholar]
- Felix R., Cecchini M.G., Hofstetter W., Elford P.R., Stutzer A., Fleisch H. Impairment of macrophage colony-stimulating factor production and lack of resident bone marrow macrophages in the osteopetrotic op/op mouse. J. Bone Miner. Res. 1990;5:781–789. doi: 10.1002/jbmr.5650050716. [DOI] [PubMed] [Google Scholar]
- Gabreëls-Festen A.A., Bolhuis P.A., Hoogendijk J.E., Valentijn L.J., Eshuis E.J., Gabreels F.J.M. Charcot-Marie-Tooth disease type 1Amorphological phenotype of the 17p duplication versus PMP22 point mutations. Acta Neuropathol. 1995;90:645–649. doi: 10.1007/BF00318579. [DOI] [PubMed] [Google Scholar]
- Garcia A., Combarros O., Calleja J., Berciano J. Charcot-Marie-Tooth disease type 1A with 17p duplication in infancy and early childhooda longitudinal clinical and electrophysiologic study. Neurology. 1998;50:1061–1067. doi: 10.1212/wnl.50.4.1061. [DOI] [PubMed] [Google Scholar]
- Gold R., Archelos J.J., Hartung H.P. Mechanisms of immune regulation in the peripheral nervous system. Brain Pathol. 1999;9:343–360. doi: 10.1111/j.1750-3639.1999.tb00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory R., Thomas P.K., King R.H.M., Hallam P.L.J., Malcolm S., Hughes R.A.C., Harding A.E. Coexistence of hereditary motor and sensory neuropathy type Ia and IgM paraproteinemic neuropathy. Ann. Neurol. 1993;33:649–652. doi: 10.1002/ana.410330615. [DOI] [PubMed] [Google Scholar]
- Guénard V., Montag D., Schachner M., Martini R. Onion bulb cells in mice deficient for myelin genes share molecular properties with immature, differentiated non-myelinating and denervated Schwann cells. Glia. 1996;18:27–38. doi: 10.1002/(SICI)1098-1136(199609)18:1<27::AID-GLIA3>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Hartung H.P., van der Meche F.G.A., Pollard J.D. Guillain-Barre syndrome, CIDP and other chronic immune-mediated neuropathies. Curr. Opin. Neurol. 1998;11:497–513. doi: 10.1097/00019052-199810000-00013. [DOI] [PubMed] [Google Scholar]
- Ho T.W., McKhann G.M., Griffin J.W. Human autoimmune neuropathies. Annu. Rev. Neurosci. 1998;21:187–226. doi: 10.1146/annurev.neuro.21.1.187. [DOI] [PubMed] [Google Scholar]
- Jung S., Huitinga I., Schmidt B., Zielasek J., Dijkstra C.D., Toyka K.V., Hartung H.P. Selective elimination of macrophages by dichlormethylene diphosphonate-containing liposomes suppresses experimental autoimmune neuritis. J. Neurol. Sci. 1993;119:195–202. doi: 10.1016/0022-510x(93)90134-k. [DOI] [PubMed] [Google Scholar]
- Kalaydjieva L., Gresham D., Gooding R., Heather L., Baas F., de Jonge R., Blechschmidt K., Angelicheva D., Chandler D., Worsley P. N-myc downstream-regulated gene 1 is mutated in hereditary motor and sensory neuropathy-Lom. Am. J. Hum. Genet. 2000;67:47–58. doi: 10.1086/302978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefer R., Kieseier B.C., Bruck W., Hartung H.P., Toyka K.V. Macrophage differentiation antigens in acute and chronic autoimmune polyneuropathies. Brain. 1998;121:469–479. doi: 10.1093/brain/121.3.469. [DOI] [PubMed] [Google Scholar]
- Kloss C.U., Werner A., Klein M.A., Shen J., Menuz K., Probst J.C., Kreutzberg G.W., Raivich G. Integrin family of cell adhesion molecules in the injured brainregulation and cellular localization in the normal and regenerating mouse facial motor nucleus. J. Comp. Neurol. 1999;411:162–178. doi: 10.1002/(sici)1096-9861(19990816)411:1<162::aid-cne12>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Lampert P.W. Mechanism of demyelination in experimental allergic neuritis. Electron microscopic studies. Lab. Invest. 1969;20:127–138. [PubMed] [Google Scholar]
- Malandrini A., Villanova M., Dotti M.T., Frederico A. Acute inflammatory neuropathy in Charcot-Marie-Tooth disease. Neurology. 1999;52:859–861. doi: 10.1212/wnl.52.4.859. [DOI] [PubMed] [Google Scholar]
- Martini R. Animal models for inherited peripheral neuropathies. J. Anat. 1997;191:321–336. doi: 10.1046/j.1469-7580.1997.19130321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini R. Animal models for inherited peripheral neuropathieschances to find treatment strategies? J. Neurosci. Res. 2000;61:244–250. doi: 10.1002/1097-4547(20000801)61:3<244::AID-JNR2>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Martini R., Zielasek J., Toyka K.V., Giese K.P., Schachner M. P0-deficient mice show myelin degeneration in peripheral nerves characteristic of inherited human neuropathies. Nat. Genet. 1995;11:281–286. doi: 10.1038/ng1195-281. [DOI] [PubMed] [Google Scholar]
- Mersiyanova I.V., Perepelov A.V., Polyakov A.V., Sidnikov V.F., Dadli E.L., Oparin R.B., Petrin A.N., Evgrafov O.V. A new variant of Charcot-Marie-Tooth disease type 2 is probably the result of a mutation in the neurofilament-light gene. Am. J. Hum. Genet. 2000;67:37–46. doi: 10.1086/302962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf D. The Florey Lecture, 1991. The colony-stimulating factorsdiscovery to clinical use. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1991;333:147–173. doi: 10.1098/rstb.1991.0065. [DOI] [PubMed] [Google Scholar]
- Nelis E., Haites N., Van Broeckhoven C. Mutations in the peripheral myelin genes and associated genes in inherited peripheral neuropathies. Hum. Mutat. 1999;13:11–28. doi: 10.1002/(SICI)1098-1004(1999)13:1<11::AID-HUMU2>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Raivich G., Moreno-Flores M.T., Möller J.C., Kreutzberg G.W. Inhibition of posttraumatic microglial proliferation in a genetic model of macrophage colony-stimulating factor deficiency in the mouse. Eur. J. Neurosci. 1994;6:1615–1618. doi: 10.1111/j.1460-9568.1994.tb00552.x. [DOI] [PubMed] [Google Scholar]
- Raivich G., Haas S., Werner A., Klein M.A., Kloss C., Kreutzberg G.W. Regulation of MCSF receptors on microglia in the normal and injured mouse central nervous systema quantitative immunofluorescence study using confocal laser microscopy. J. Comp. Neurol. 1998;395:342–358. doi: 10.1002/(sici)1096-9861(19980808)395:3<342::aid-cne6>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Scherer S.S., Xu Y.T., Nelles E., Fischbeck K., Willecke K., Bone L.J. Connexin32-null mice develop demyelinating peripheral neuropathy. Glia. 1998;24:8–20. doi: 10.1002/(sici)1098-1136(199809)24:1<8::aid-glia2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Schmid C.D., Stienekemeier M., Oehen S., Bootz F., Zielasek J., Gold R., Toyka K.V., Schachner M., Martini R. Immune deficiency in mouse models for inherited peripheral neuropathies leads to improved myelin maintenance. J. Neurosci. 2000;20:729–735. doi: 10.1523/JNEUROSCI.20-02-00729.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt B., Toyka K.V., Kiefer R., Full J., Hartung H.P., Pollard J. Inflammatory infiltrates in sural nerve biopsies in Guillain-Barre syndrome and chronic inflammatory demyelinating neuropathy. Muscle Nerve. 1996;19:474–487. doi: 10.1002/(SICI)1097-4598(199604)19:4<474::AID-MUS8>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Shy M.E., Arroyo E., Sladky J., Menichella D., Jiang H., Xu W., Kamholz J., Scherer S.S. Heterozygous P0 knock-out mice develop a peripheral neuropathy that resembles chronic inflammatory demyelinating polyneuropathy (CIDP) J. Neuropathol. Exp. Neurol. 1997;56:811–821. [PubMed] [Google Scholar]
- Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth's disease. Clin. Genet. 1974;6:98–118. doi: 10.1111/j.1399-0004.1974.tb00638.x. [DOI] [PubMed] [Google Scholar]
- Smith M.E. Phagocytosis of myelin in demyelinating diseasea review. Neurochem. Res. 1999;24:261–268. doi: 10.1023/a:1022566121967. [DOI] [PubMed] [Google Scholar]
- Sommer C., Schröder J.M. HLA-DR expression in peripheral neuropathiesthe role of Schwann cells, resident and hematogenous macrophages, and endoneurial fibroblasts. Acta Neuropathol. 1995;89:63–71. doi: 10.1007/BF00294261. [DOI] [PubMed] [Google Scholar]
- Stanley E.R., Berg K.L., Einstein D.B., Lee P.S., Pixley F.J., Wang Y., Yeung Y.G. Biology and action of colony-stimulating factor-1. Mol. Reprod. Dev. 1997;46:4–10. doi: 10.1002/(SICI)1098-2795(199701)46:1<4::AID-MRD2>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Stoll G., Gabreëls-Festen A.A.W.M., Jander S., Müller H.W., Hanemann C.O. Major histocompatibility complex class II expression and macrophage responses in genetically proven Charcot-Marie-Tooth type 1 and hereditary neuropathy with liability to pressure palsies. Muscle Nerve. 1998;21:1419–1427. doi: 10.1002/(sici)1097-4598(199811)21:11<1419::aid-mus9>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Suter U., Snipes G.J. Biology and genetics of hereditary motor and sensory neuropathies. Annu. Rev. Neurosci. 1995;18:45–75. doi: 10.1146/annurev.ne.18.030195.000401. [DOI] [PubMed] [Google Scholar]
- Teitelbaum S.L. Bone resorption by osteoclasts. Science. 2000;289:1504–1508. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- Thomas P.K. Overview of Charcot-Marie-Tooth disease 1A. Ann. NY Acad. Sci. 1999;833:1–5. [PubMed] [Google Scholar]
- Vital A., Vital C., Julien J., Fontan D. Occurrence of active demyelinating lesions in children with hereditary motor and sensory neuropathy (HMSN) type I. Acta Neuropathol. (Berl. 1992;84:433–436. doi: 10.1007/BF00227671. [DOI] [PubMed] [Google Scholar]
- Witmer-Pack M.D., Hughes D.A., Schuler G., Lawson L., McWilliam A., Inaba K., Steinman R.M., Gordon S. Identification of macrophages and dendritic cells in the osteopetrotic (op/op) mouse. J. Cell Sci. 1993;104:1021–1029. doi: 10.1242/jcs.104.4.1021. [DOI] [PubMed] [Google Scholar]
- Yoshida H., Hayashi S.-I., Kunisasa Z., Ogawa M., Nishikawa S., Okamura H., Sudo T., Shultz L.D., Nishikawa S. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. 1990;345:442–444. doi: 10.1038/345442a0. [DOI] [PubMed] [Google Scholar]