

Abstract
The G protein–coupled thrombin receptor can induce cellular responses in some systems by transactivating the epidermal growth factor (EGF) receptor. This is in part due to the stimulation of ectoproteases that generate EGF receptor ligands. We show here that this cannot account for the stimulation of proliferation or migration by thrombin of Swiss 3T3 cells. Thrombin has no direct effect on the activation state of the EGF receptor or of its downstream effectors. However, thrombin induces the subcellular clustering of the EGF receptor at filamentous actin–containing structures at the leading edge and actin arcs of migrating cells in association with other signaling molecules, including Shc and phospholipase Cγ1. In these thrombin-primed cells, the subsequent migratory response to EGF is potentiated. Thrombin did not potentiate the EGF-stimulated EGF receptor phosphorylation. Thus, in Swiss 3T3 cells the G protein–coupled thrombin receptor can potentiate the EGF tyrosine kinase receptor response when activated by EGF, and this appears to be due to the subcellular concentration of the receptor with downstream effectors and not to the overall ability of EGF to induce receptor transphosphorylation. Thus, the EGF receptor subcellular localization which is altered by thrombin appears to be an important determinant of the efficacy of downstream EGF receptor signaling in cell migration.
Keywords: costimulation, rafts, G protein, actin arc, crosstalk
Introduction
Cell cycle progression and cell migration can be activated by hormone receptors through stimulation of signaling systems that are incompletely characterized. Such receptors include those that couple to heterotrimeric GTP-binding proteins and those with intrinsic tyrosine kinase activity. Among these, thrombin receptors are known to be capable of stimulating at least four G proteins, Gq, Gi, G12, and G13 (Crouch et al. 1990; LaMorte et al. 1993; Offermanns et al. 1994; Post et al. 1996; Verrall et al. 1997; Gohla et al. 1999), complicating our understanding of the mechanisms of stimulation of cell proliferation by this hormone. Our laboratory and others have shown that these G proteins convey receptor and downstream signals to distinct phases of the proliferative process. Gi has been shown to mediate at least part of the mitogenic response to thrombin (Verrall et al. 1997), and to also be transforming when constitutively activated (Pace et al. 1991; Gupta et al. 1992). We have shown that Gi signaling occurs in Swiss 3T3 cells late in the cell cycle where it regulates transition through M phase (Crouch and Simson 1997). In contrast, the G12/G13 pathway appears to regulate stimulation of actin polymerization and stress fiber formation (Buhl et al. 1995; Gohla et al. 1999). This pathway may not play a direct role in the cell cycle. Previous work has shown that activated mutants of G12 and G13 are transforming, and promote growth in soft agar (Voyno-Yasenetskaya et al. 1994), but such transfection alone did not confer stimulation of pathways such as mitogen-activated protein (MAP) kinase. Rather, constitutively activated G12 and G13 potentiated the stimulation of these pathways by growth factors (Voyno-Yasenetskaya et al. 1994). G12 and G13 may allow G protein–coupled receptor stimulation of tyrosine kinases and thus association of adapter proteins (Collins et al. 1997; Needham and Rozengurt 1998). Gq, in contrast, does appear to be necessary for activation of early aspects of the cell cycle, including DNA synthesis, probably through stimulation of PLC and generation of diacylglycerol, and activation of PKCs (De Vivo et al. 1992; Kalinec et al. 1992; Verrall et al. 1997). Therefore, the varied involvement of multiple G proteins in distinct aspects of receptor activation of the cell cycle may allow the full mitogenic stimulus to be presented to the cell in a coordinated fashion.
Recently, it has been found in some systems that at least part of the mitogenic stimulus of thrombin receptors and other G protein–coupled receptors can come from the transactivation of the EGF receptor (Daub et al. 1996, Daub et al. 1997; Vaingankar and Martins-Green 1998; Luttrell et al. 1999; O'Hackel et al. 1999). Thrombin, endothelin-1, muscarinic agonists, angiotensin II, and lysophosphatidic acid (LPA) each stimulated the tyrosine phosphorylation of the EGF receptor, and inhibition of EGF receptor function reduced the mitogenic stimulus of each G protein–coupled receptor. Other studies have shown varying degrees of such transactivation of receptor- and receptor-like tyrosine kinases by G protein–coupled receptors (Della Rocca et al. 1999). However, it has been proposed that transduction of a mitogenic response from a G protein–coupled receptor requires the EGF receptor and its intact tyrosine kinase domain (Carpenter 1999). Most recently, evidence has been presented to implicate G protein–coupled receptor–mediated activation of ectoproteases in release of EGF receptor ligands from the cell surface as a mechanism of EGF receptor transactivation (Prenzel et al. 1999).
In addition to the mitogenic actions of thrombin, this growth factor also strongly stimulates migration of Swiss 3T3 cells, a response that requires the activation by thrombin of p70 S6 kinase (p70S6k) (Crouch 1997; Berven et al. 1999) and phosphatidylinositol 3-kinase (Johanson et al. 1999). Cell migration is associated with dramatic changes in the structure of the cytoskeleton that accompany cellular polarization. These involve polymerization of actin and protrusion of microtubules at the leading edge of cells. The F-actin structures in migratory fibroblasts include stress fibers, the actin fringe at the anterior surface, and semicircular arcs (actin arcs), the latter moving posteriorly as the cell moves forward (Soranno and Bell 1982; Heath and Holifield 1993; Nabi 1999). The functions of these actin structures are thought to be largely structural, although those of the actin arc are unclear.
Here, we have further examined the signaling systems of the thrombin receptor in Swiss 3T3 cells, with particular relevance to the potential involvement of the EGF receptor in mitogenic and migratory stimulation. We have examined the interaction between signaling by endogenous thrombin and EGF receptors, so that promiscuous effects between pathways due to receptor overexpression are not a consideration. We show in our Swiss 3T3 cell clone that EGF receptor transactivation is not part of the initial cellular response activated by thrombin, which is independent of this receptor. However, our data show that thrombin can induce the subcellular redistribution and clustering of the EGF receptor which potentiates subsequent activation by EGF, and this is achieved in part by its receptor-mediated association with the actin arc.
Materials and Methods
Materials
Except as otherwise stated, materials were derived as described elsewhere (Crouch 1997; Berven et al. 1999; Johanson et al. 1999). The p70S6k substrate peptide (AKRRRLSSLRA) was synthesized by Dr. Peter Milburn and coworkers (John Curtin School of Medical Research, Australian National University). Antibodies to Shc, Grb2, EGF receptor, and phosphotyrosine (4G-10) were purchased from Upstate Biotechnology, p42/p44 kD MAP kinase antibodies from Santa Cruz Biotechnology, Inc., phospho-specific p42/p44 kD MAP kinase antibodies from New England Biolabs, Inc., AG1478 from Calbiochem-Novabiochem, and Texas red-X phalloidin and carboxy fluorescein diacetate, succinimidyl ester AM (CFSE) from Molecular Probes.
Swiss 3T3 Cell Culture, DNA Synthesis, p70S6k Activity Measurements, Immunoprecipitation, Western Blotting, Immunohistochemistry, and Confocal Microscopy
The above measurements and experimental approaches were carried out as described previously (Crouch 1997; Berven et al. 1999; Johanson et al. 1999).
Cell Fractionation
Swiss 3T3 cells were fractionated into membrane/cytosol, cytoskeletal, and nuclear fractions using Triton X-100 lysis buffer and differential centrifugation, as described previously (Crouch 1997; Berven et al. 1999; Johanson et al. 1999).
Cell Migration Assay
Cell migration was assessed using CFSE-labeled cells in transwell chambers. Cells were grown to confluency in tissue culture flasks in DME with 10% FCS. Cells were then rinsed twice with Hank's balanced salt solution and incubated in this for 20 min with CFSE (10 μM). This medium was then discarded, and cells were washed once with DME with 10% FCS, once with DME alone, and finally incubated overnight in DME alone. The following day, cells were harvested with trypsin (Crouch 1997; Berven et al. 1999; Johanson et al. 1999) and resuspended in DME. Cells (350 μl) were aliquoted into transwell inserts (8 μm, Falcon HTS Fluoroblok; Becton Dickinson) and agonists or inhibitors were added as indicated in the figure legends. The insert was then loaded into the culture plate, which contained DME (800 μl) in each well with or without agonist, as indicated. Migration of cells through the optically opaque insert membrane to the lower culture plate well was assessed by measurement of cellular CFSE fluorescence using a Cytofluor II fluorescence plate reader (PerSeptive Biosystems) with excitation and emission wavelengths of 485 and 530 nm, respectively. Readings were taken immediately after addition of cells and at hourly or half-hourly intervals thereafter.
Results
Thrombin and EGF Activate DNA Synthesis: Differential Dependence on EGF Receptor Tyrosine Kinase Activity
Both thrombin and EGF stimulated the synthesis of DNA in Swiss 3T3 cells, but thrombin was a much more effective stimulus than EGF for this response (Fig. 1). To examine the requirement of the EGF receptor tyrosine kinase in each of these responses, cells were preincubated for 1 h with or without the EGF receptor kinase inhibitor AG1478 (0.5 μM). AG1478 completely inhibited DNA synthesis stimulated by EGF. The thrombin-induced response was resistant to inhibition by AG1478, being only partially inhibited.
Figure 1.

Thrombin-induced DNA synthesis is not dependent on EGF receptor tyrosine kinase activity in Swiss 3T3 cells. The abilities of thrombin and EGF to induce DNA synthesis of Swiss 3T3 cells in the presence or absence of the EGF receptor kinase inhibitor AG1478 were measured by the incorporation of [3H]thymidine. Cells were left inactive or stimulated with dilutions of thrombin (diluted as indicated from a maximum of 6 U/ml) or EGF (maximum of 60 nM) for 24 h in the presence of [3H]thymidine. Thrombin was a more effective agonist than EGF but EGF was much more sensitive to inhibition by AG1478 (0.5 μM) than thrombin. Results are the mean ± SEM of 8–12 observations from two experiments.
EGF but Not Thrombin Stimulates EGF Receptor Tyrosine Phosphorylation and Association of Shc with Phosphotyrosine-containing Proteins
To examine whether the partial inhibitory effect of AG1478 on thrombin-stimulated DNA synthesis was nonspecific or due to the inhibition of EGF receptor activation by thrombin, we have examined the phosphorylation status of the EGF receptor.
Cells stimulated for 5 min with EGF showed enhanced tyrosine phosphorylation of the EGF receptor, as seen by blotting EGF receptor immunoprecipitates with phosphotyrosine antibodies (Fig. 2 A). This was completely inhibited by preincubation of cells with AG1478 (0.5 μM). Thrombin, in contrast, at either 1 or 5 min of stimulation, had no influence on the tyrosine phosphorylation state of the EGF receptor (Fig. 2 A), or after longer time periods (not shown). Consistent with these observations, EGF and not thrombin induced the association of Shc with phosphotyrosine immunoprecipitates (Fig. 2 B). Both the 66- and 52-kD forms of Shc were found in these immunoprecipitates, and this EGF-induced association was blocked in cells preincubated with AG1478 (Fig. 2 B).
Figure 2.

Thrombin does not activate the EGF receptor in Swiss 3T3 cells. Swiss 3T3 cells were left inactive (0) or were stimulated for 5 min with EGF (E5, 10 nM) or thrombin (1 U/ml) for 1 min (T1) or 5 min (T5) in the presence or absence of AG1478 (0.5 μM). Cells were lysed and extracted proteins were immunoprecipitated with (A) EGF receptor antibodies or (B) phosphotyrosine antibodies. Proteins were separated by SDS-PAGE and Western blotted for phosphotyrosine (A) and Shc (B). It can be seen that the EGF receptor was tyrosine phosphorylated when cells were acutely activated with EGF (5 min). Thrombin had no effect on the phosphorylation state of the EGF receptor (A). In all lanes there were equal amounts of EGF receptor immunoprecipitated (not shown). Similarly, EGF brought about the association of 52- and 66-kD forms of Shc with phosphotyrosine immunoprecipitates, but thrombin had little such effect (B). The heavy chain (HC) of the immunoprecipitating antibodies is indicated, as are positions of molecular weight standards (shown in kD) in B. Qualitatively identical results were seen in four separate preparations.
Thrombin and EGF both Stimulate p70S6k: Differential Dependence on EGF Receptor Tyrosine Kinase Activity
Thrombin and EGF both stimulated p70S6k activity, although EGF was a much more potent agonist (Table ). Pretreatment of cells for 1 h with AG1478 (0.5 μM) totally inhibited the EGF response while only causing a partial inhibition of that to thrombin. This effect on thrombin may be nonspecific, as a similar percentage reduction in the basal activity of p70S6k was observed when unstimulated cells were treated with AG1478 (Table ). Thus, thrombin stimulates p70S6k independently of the EGF receptor.
Table 1.
EGF Stimulation, but Not Thrombin Stimulation, of p70S6k Activity Is Abolished by AG1478
| Stimulus | p70S6k activity (percentage of unstimulated control) |
|---|---|
| Unstimulated control | 100 ± 2.7 |
| Unstimulated + AG1478 | 68 ± 4.2 |
| EGF | 678 ± 87 |
| EGF + AG1478 | 81 ± 3.7 |
| Thrombin | 231 ± 21 |
| Thrombin + AG1478 | 171 ± 15 |
Swiss 3T3 cells were left unstimulated or were activated with EGF (10 nM) or thrombin (1 U/ml) in the presence or absence of the EGF receptor kinase inhibitor AG1478 (0.5 μM) for 1 h. Cells were then lysed with Triton X-100–containing buffer and p70S6k was immunoprecipitated from the membrane plus cytosol fraction. The activity of p70S6k precipitated was quantitated by a kinase assay involving phosphorylation of an S6 kinase peptide substrate in the presence of [γ-32P]ATP. There was no difference in amount of p70S6k protein precipitated in the different samples (not shown). It can be seen that AG1478 totally inhibited EGF stimulation of p70S6k while only partially inhibiting the unstimulated value and that stimulated by thrombin. Results are the mean ± SE of six samples from two experiments.
Differential Sensitivity to AG1478 of EGF and Thrombin Stimulation of MAP Kinase
We have examined the effect of thrombin and EGF on the MAP kinase pathway by the use of phospho-MAP kinase (42- and 44-kD MAP kinase) antibodies. Stimulation of cells for 5 min with EGF induced the phosphorylation of both the MAP kinases extracellular signal–regulated kinase (ERK)1 and ERK2 (Fig. 3 A). This was inhibited by AG1478 (0.5 μM) pretreatment of cells. Thrombin was a weaker stimulus of activation of phosphorylation of either kinase and this stimulation was only partially inhibited by AG1478 (Fig. 3 A). The time courses of activation of MAP kinase were also different for thrombin and EGF. While thrombin stimulation of MAP kinase was fully desensitized after 15 min of activation, the EGF MAP kinase response was sustained at this time point (Fig. 3 B).
Figure 3.

Thrombin-induced MAP kinase activation is resistant to inhibition by AG1478. Swiss 3T3 cells were activated with (A) thrombin (T, 1 U/ml) or (B) EGF (E, 10 nM) for 1, 2, 5 or 15 min, as indicated, in the presence or absence of AG1478 (0.5 μM). Cells were lysed with SDS-PAGE sample buffer, and proteins were separated by SDS-PAGE, Western blotted, and probed with phospho-specific MAP kinase (MAPK) antibodies. It was found that EGF was a much more potent agonist for MAP kinase activation than thrombin, and that the EGF response was abolished by AG1478 treatment. While a weaker agonist, the thrombin stimulation of MAP kinase activation was only partially inhibited by AG1478. The time courses of activation of MAPK were different for thrombin and EGF. Whereas thrombin desensitized after 15 min (A), the response to EGF was maintained at this time point (B). The positions of 42- and 44-kD forms of MAP kinases are shown. The same effects were observed in two separate cell preparations.
Thrombin Induces the Clustering of EGF Receptors at the Actin Arc and Leading Edge
The data presented thus far show that the mitogenic signaling of the endogenous thrombin receptor of Swiss 3T3 cells is independent of the EGF receptor. We have also examined the potential crossover of these pathways in stimulation of cell migration. This included an examination of the effects of thrombin on the EGF receptor distribution in migrating cells.
Confocal microscopy of cells labeled with antibodies to the EGF receptor showed a differential distribution of the receptor depending on activation status of subconfluent migrating cells. Migrating cells were identified by their characteristic fan shape and polarized distribution of F-actin. In unstimulated cells there was little enrichment of EGF receptor staining within nonnuclear regions of the cell (Fig. 4 C). In contrast, cells activated with thrombin for 5 h and induced to migrate showed EGF receptor immunoreactivity toward the leading edge and on structures similar in appearance to the actin arc (Fig. 4 B). Double labeling of these cells with phosphotyrosine antibodies showed there to be only weak tyrosine phosphorylation coincident with the EGF receptor (Fig. 4 A), with the predominant phosphotyrosine staining on punctate structures (Fig. 4 A) that were coincident with the localization of focal adhesion kinase (FAK, not shown; see Fig. 6). This thrombin-stimulated clustering of EGF receptors was not inhibited by AG1478 (Fig. 4 D), showing a lack of requirement for EGF receptor kinase activity. This distribution of the EGF receptor was only seen in nonconfluent cells, and not in nonmigrating, contact-inhibited cells.
Figure 4.
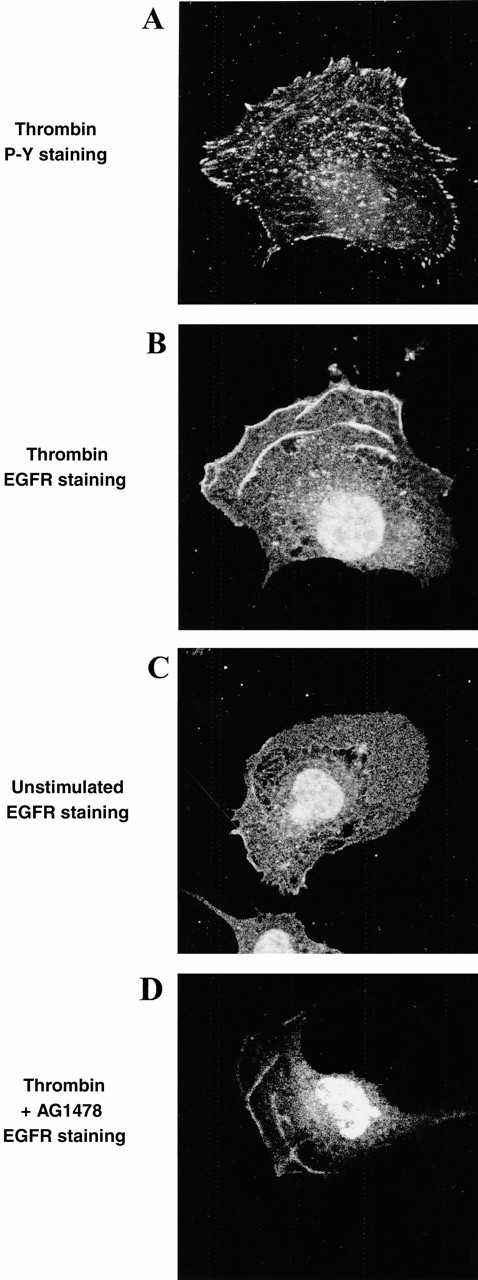
Thrombin induces the clustering of EGF receptors but not associated tyrosine phosphorylation. Swiss 3T3 cells were activated with thrombin for 5 h and then fixed and incubated with antibodies to the EGF receptor (EGFR) and/or phosphotyrosine (P-Y). Cells were then incubated with secondary fluorescent antibodies and visualized by confocal microscopy. A and B show the same cell which was double-labeled with P-Y and EGF receptor antibodies. It can be seen that the EGF receptor localized to ribbed structures at the anterior of the migrating cell, but that there was little tyrosine phosphorylation associated with the EGF receptor. C shows that in unstimulated cells there is little polarized accumulation of EGF receptor. Cells treated with AG1478 (D) still showed EGF receptor accumulation on actin arcs in thrombin-stimulated cells.
Figure 6.
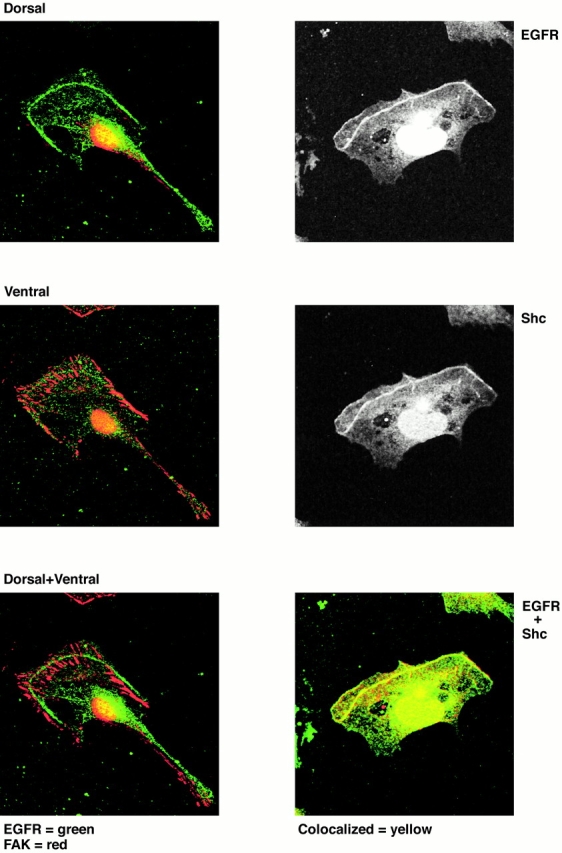
Shc, but not FAK, colocalizes with the EGF receptor on the actin arc in thrombin-stimulated cells. Subconfluent Swiss 3T3 cells were serum starved overnight and then reactivated with thrombin for 5 h and fixed for immunohistochemistry. Cells were double-labeled with EGF receptor antibodies (EGFR) and FAK or Shc antibodies. Antibody localization was examined by confocal microscopy using appropriate fluorescently tagged secondary antibodies. The left three panels show in each case double staining for both EGF receptor and FAK. The top left panel shows that at the dorsal aspect of migrating cells there is EGF receptor presence on the actin arc (green) with very little FAK staining (red). The middle left panel shows that at the ventral cell surface FAK is found abundantly, but there is an absence of EGF receptor. In neither of these panels is there evidence of coincident staining, which would be yellow. The bottom left panel shows the dorsal EGF receptor staining overlaid on the ventral FAK staining to emphasize that the EGF receptor is physically displaced from the ventral surface and focal adhesions. The right panels show the staining of Shc and EGF receptor in the same cell. It can be seen that both proteins localize to the actin arc, and overlay of these images in the bottom right panel shows the staining to be coincident. EGF receptor is stained green, Shc is red, and coincident staining is yellow. Such coincident staining was seen in all cells examined.
To confirm the site of accumulation of the EGF receptor in thrombin-stimulated cells as the actin arc, cells were double-labeled with EGF receptor antibodies and with fluorescent phalloidin to label F-actin (Fig. 5). It can be clearly seen that staining with these two agents was coincident on the actin arc. Both dorsal and ventral views of a representative cell are shown. There is enrichment of EGF receptor dorsally on the actin arc, less staining ventrally, and only weak staining on stress fibers. Thus, there is a degree of selectivity with regard to the actin-based structures with which the EGF receptor associates.
Figure 5.
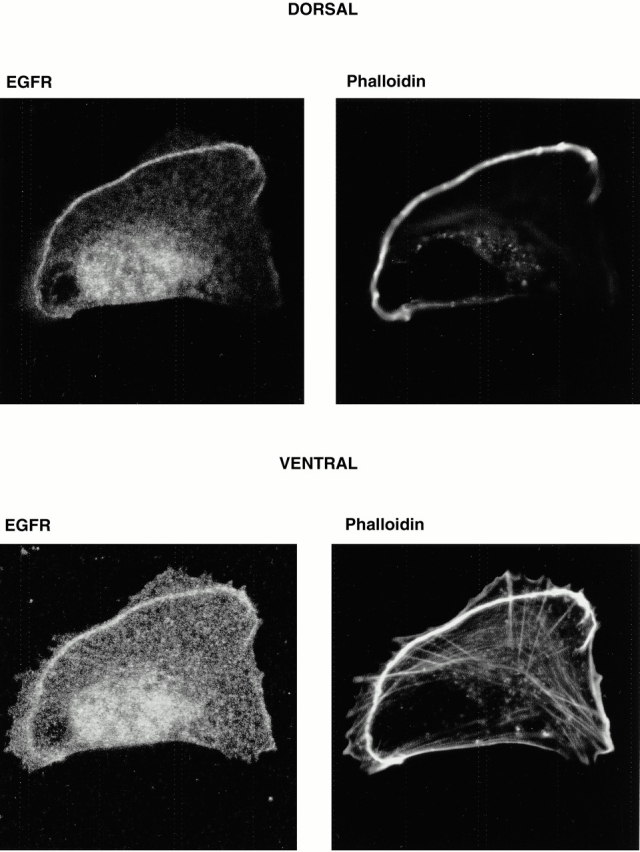
Thrombin stimulates the localization of EGF receptors to the actin arc in migrating Swiss 3T3 cells. Subconfluent Swiss 3T3 cells were serum starved overnight and then reactivated for 5 h with thrombin (1 U/ml). Cells were fixed and incubated with antibodies to the EGF receptor (EGFR) followed by secondary FITC-labeled antibodies and Texas red-X–labeled phalloidin to show F-actin structures. Cells were then visualized by confocal microscopy. The top panels show that the EGF receptor colocalizes with the phalloidin-stained actin arc on the dorsal aspect of the cell. The bottom panels show that this EGF receptor staining is maintained ventrally on the actin arc but that there is little staining of actin stress fibers. Such association of EGF receptors with the actin arc was seen in all thrombin-activated migrating cells examined.
We also find EGF receptor immunoreactivity in the nucleus (Fig. 4, Fig. 5, and Fig. 6). However, this appears to be due to reactivity with low molecular mass proteins of ∼20 kD and not the EGF receptor (not shown). There is no blocking antigen for the mouse EGF receptor antibody used in this study. However, to be confident that the EGF receptor immunoreactivity at the actin arc and leading edge is actually the EGF receptor protein, we have carried out long-term cell activation with EGF to downregulate receptor expression. After 4–6 h of stimulation with EGF, but not with thrombin or insulin, the EGF receptor immunoreactivity is selectively lost at the actin arc and leading edge (Soranno and Bell 1982; Crouch et al. 2000). Thus, we are confident that the EGF receptor immunoreactivity we see at these cellular sites in response to thrombin is due to EGF receptor accumulation. The fluorescence within the nucleus is unaffected by long-term EGF stimulation, supporting the identity of this reactivity being due to non-EGF receptor protein. Additionally, as shown above, the EGF receptor antibody selectively recognized the EGF receptor protein in the nonnuclear fraction on Western blots (Fig. 2).
EGF Receptors and Shc Colocalize on the Actin Arc and Not at Focal Adhesions
As EGF induced the enrichment of Shc in phosphotyrosine immunoprecipitates of the cytoskeletal fraction (not shown) as well as the membrane/cytosol fraction of Swiss 3T3 cells, we examined whether Shc was also present on the actin arc. We found that Shc and EGF receptors colocalized in all thrombin-stimulated cells where an actin arc was present (Fig. 6). As described above, the EGF receptor was found mainly near the dorsal surface of the cell on the actin arc. To further verify this, we double-labeled cells with EGF receptor antibodies and FAK antibodies. FAK is well recognized to localize to sites of focal adhesions on the ventral surface. As can be clearly seen in Fig. 6, the EGF receptor was found dorsal to the localization of FAK, and the two proteins were totally separable in their cellular distribution.
EGF Receptors Induced to Aggregate by Thrombin on the Actin Arc Are Activatable by EGF
To examine the functionality of aggregated EGF receptors, migration of Swiss 3T3 cells was determined in cells with or without prior stimulation with thrombin for 7 h, after which cells were left with no further addition or were activated secondarily for 5 min with EGF. Staining for the EGF receptor revealed the characteristic accumulation of EGF receptors at the leading edge of cells activated by thrombin (Fig. 7). In the absence of further additions, there was only a relatively small amount of tyrosine phosphorylation of proteins at the actin arc (Fig. 7). However, cells treated additionally for 5 min with EGF showed a marked tyrosine phosphorylation not only at the actin arc but also at focal adhesions (Fig. 7). As we could not observe EGF receptors at focal adhesions, this indicates that signaling has occurred from the actin arc and potentially other EGF receptors so as to induce tyrosine phosphorylation some distance from the site of receptor activation. The results strongly suggest that EGF receptors aggregated at the actin arc by thrombin are functional.
Figure 7.
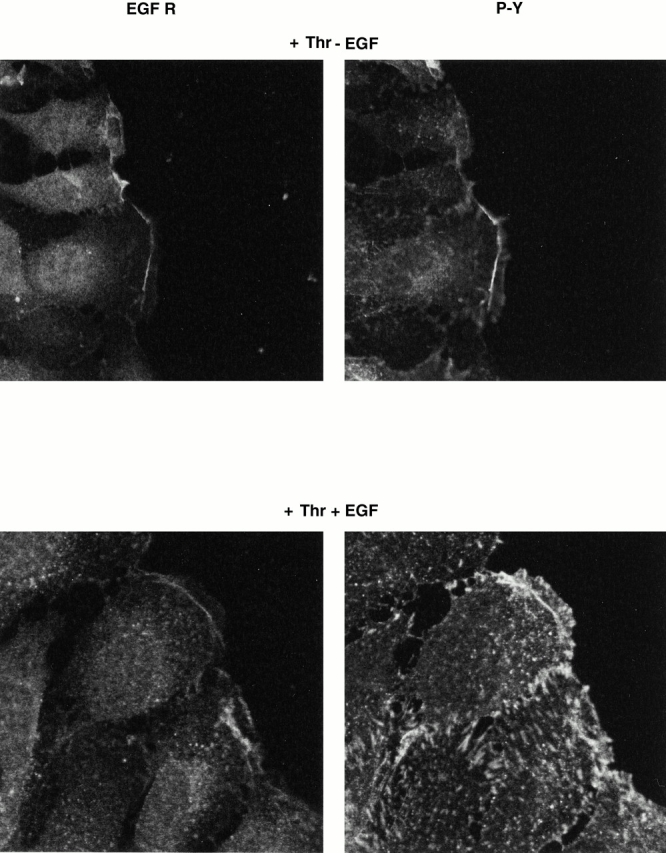
EGF receptors at the leading edge of thrombin-stimulated cells are responsive to EGF. Swiss 3T3 cells were grown to confluency, serum deprived overnight, and the monolayer was “wounded” with a 1-mm-wide blunt scraper. Cells were reactivated with thrombin (Thr, 1 U/ml) for 4 h and then fixed or were subsequently activated for 5 min with EGF (10 nM) and then fixed. Cells were processed for immunohistochemistry and then incubated with EGF receptor antibodies (EGFR) and phosphotyrosine antibodies (P-Y). As seen previously, thrombin induced the accumulation of EGF receptors at the leading edge of cells beginning migration and that there was only a small amount of tyrosine phosphorylation associated with the actin arc (top right). However, activation of these cells for 5 min with EGF induced a large increase in tyrosine phosphorylation both at the actin arc and at focal adhesions (bottom right).
EGF Receptors Aggregated at the Actin Arc Are Not Necessary for Thrombin-induced Cell Migration but Thrombin Potentiates EGF-induced Migration
To see if cell migration, as opposed to cell proliferation, stimulated by thrombin may require EGF receptor activity, we have carried out a cell migration assay of cells stimulated by thrombin or EGF in the presence and absence of AG1478 (Fig. 8 A). Both thrombin and EGF induced cell migration when added to the lower chamber of a transwell plate. Unstimulated cells showed little migration. Cells activated with EGF that were pretreated with AG1478 displayed a blockade of migration to levels of unstimulated cells. In contrast, there was no inhibition of migration of thrombin-stimulated cells (Fig. 8 A). Thus, the tyrosine kinase activity of EGF receptors is not required for thrombin-stimulated cell migration.
Figure 8.


Thrombin-induced migration of Swiss 3T3 cells does not require the EGF receptor but potentiates EGF receptor action. (A) The effect of the EGF receptor kinase inhibitor (AG1478) on thrombin- and EGF-induced cell migration was examined in a transwell migration assay. Cells were labeled with the fluorescent indicator CFSE and placed in an 8-μm transwell insert in the presence or absence of AG1478 (0.5 μM). The insert was then placed in a 24-well plate with DME alone or in wells to which was added thrombin (2.5 U/ml) or EGF (25 nM). Fluorescence measurements to assess migration of CFSE-labeled cells through the transwell membrane were then carried out at the intervals shown. The results are means of duplicate assays from a single experiment, and are representative of the same effect seen in three separate preparations. (B) Cell migration in response to EGF and thrombin was assessed as described above, except that these agonists were added alone or in combination either in the upper transwell insert chamber or in the lower chamber of the 24-well plate. It can be seen that the presence of thrombin in the upper chamber potentiated the migratory response to EGF in the lower chamber. This was highly selective, as other combinations of agonist addition did not have this effect. The results are the mean ± SEM of three samples from a single experiment and are representative of the same qualitative effect seen in two such experiments.
When added to the upper transwell chamber, neither EGF nor thrombin stimulated cell migration to the lower chamber (Fig. 8 B). EGF, in fact, was slightly inhibitory for migration under these conditions. However, stimulation of cells with thrombin in the upper chamber was able to enhance the subsequent migration of cells when presented with EGF in the lower chamber compared with stimulation with EGF in the lower chamber alone. This was not observed when both thrombin and EGF were placed in the upper chamber with the cells (Fig. 8 B), showing that there was a requirement for directionality in the EGF stimulation.
Discussion
The contribution of receptor tyrosine kinases to thrombin receptor–induced mitogenic stimulation has been examined in several cell lines, with the data suggesting a common requirement for such signaling by thrombin (Daub et al. 1996, Daub et al. 1997; Vaingankar and Martins-Green 1998; Carpenter 1999; Della Rocca et al. 1999; Luttrell et al. 1999; O'Hackel et al. 1999). However, we have found that this is not the case in Swiss 3T3 cells, as thrombin was found to signal independently of the EGF receptor in activating DNA synthesis, p70S6k, and cell migration. We find, though, that thrombin does potentiate migratory signaling by the EGF receptor and appears to do so by clustering the EGF receptor.
Thus, in our cell line, there was no evidence of an intracellular biochemical link from the thrombin receptor to phosphorylation and activation of the EGF receptor, or one involving the generation of extracellular EGF receptor ligands by proteolysis (Prenzel et al. 1999).
Further, we could detect no tyrosine phosphorylation of the EGF receptor when cells were stimulated with thrombin. Rather, we have shown a role for direct stimulation by thrombin of PLC, Ca2+ mobilization, and PKC activation in mitogenic signaling (Crouch et al. 2000). The small inhibition of thrombin responses by the EGF receptor kinase inhibitor AG1478 appears to be due to a partial nonspecific effect of this agent. Although not utilizing the EGF receptor pathway directly, thrombin-activated Swiss 3T3 cells are still highly responsive to EGF, as shown by tyrosine phosphorylation of both the receptor and focal adhesions upon addition of EGF. If an EGF receptor ligand was being produced by proteolysis, one would expect the activation of EGF receptors to have occurred. Therefore, it must be concluded that EGF receptor transactivation by the thrombin receptor is not a universal phenomenon, and that thrombin delivers a mitogenic stimulus in its own right in certain cellular contexts. In our cell line, this is also supported by the fact that thrombin is a stronger mitogen than EGF, indicating that it is unlikely that EGF is acting as the mediator of thrombin activation. The lack of effect of thrombin in inducing binding of Shc and Grb2 (not shown) to phosphotyrosine immunoprecipitates suggests that thrombin delivers little or no signal to any other receptor tyrosine kinase to induce its mitogenic response.
Despite our observation that the signaling pathways activated by thrombin do not converge to directly activate the EGF receptor, we found that thrombin does cause the subcellular clustering of EGF receptors. In the absence of added growth factors, there were no obvious accumulations of EGF receptors. However, after stimulation with thrombin for 4–7 h, EGF receptors could be seen to accumulate at the actin arc, a broad anterior structure important for cell migration (Soranno and Bell 1982; Heath and Holifield 1993; Nabi 1999). Such accumulations of the EGF receptor were not seen in confluent cells activated by thrombin, and were only seen at the leading edge of subconfluent cells in the direction of their migration. EGF itself was a potent downregulator of its receptor in cells exposed to EGF for such prolonged periods. However, accumulation of phosphotyrosine at the leading edge was observed in these cells, consistent with our concept that EGF may itself also be upregulating the accumulation of another tyrosine kinase receptor at the leading edge. We are further examining this question at present.
In cells induced to migrate by thrombin, the accumulated EGF receptors were clearly functional, as a subsequent 5-min activation with EGF of long-term thrombin-stimulated cells caused enhanced tyrosine phosphorylation at the actin arc as well as at sites of focal adhesions. However, the total EGF receptor tyrosine phosphorylation induced by EGF was not enhanced by thrombin prestimulation (not presented). Despite this, thrombin prestimulation potentiated the subsequent migration of cells induced by EGF. Thus, this enhancement of the EGF response did not appear to be due to an enhanced signaling at the level of receptor transphosphorylation, but we feel that the EGF receptor clustering induced by thrombin is an important mediator of this synergistic effect, particularly as thrombin also stimulates the coclustering of the downstream effectors Shc and PLCγ1 (Crouch et al. 2000). In vivo, cells are exposed to a large number of mitogens and growth factors capable of stimulating migration. Such heterologous receptor clustering may underlie the multiple synergistic interactions of such ligands.
It is clear that EGF stimulates tyrosine phosphorylation at focal adhesion sites. However, we have shown that localization of the EGF receptor in migrating cells is separable from that of FAK. Thus, second messenger systems must be involved in EGF signaling to FAK or associated tyrosine kinases. It has been shown previously that actin will also bind downstream signaling molecules such as Shc (Thomas et al. 1995). Our results support this for Swiss 3T3 cells, with Shc accumulation being observed at the actin arc. Similar to that found by Thomas et al. (1995) for EGF and nerve growth factor stimulation of PC12 cells, the thrombin-induced association of Shc with actin in Swiss 3T3 cells was independent of its association with phosphotyrosine-containing proteins, as thrombin did not evoke such phosphorylations. Our work extends these observations to show that Shc binds preferentially to a subset of actin within the cell, the actin arc, relative to other actin structures such as stress fibers. We are now examining the complement of signaling molecules present at the actin arc, as it appears that this structure may include a repository of signaling proteins involved in the activation of both migration and proliferation of cells. Such accumulations of signaling molecules may allow for efficient downstream signaling as a result of the high concentration of these proteins in this localized region of the cell. Further, in cells where thrombin can stimulate the extracellular production of EGF receptor ligands, this may represent a mechanism of greatly enhancing the effect of such ligands, as the cognate receptor would be concentrated and primed for activation.
It is likely that the accumulations of Shc and EGF receptor at the actin arc are induced by similar or identical signaling mechanisms. However, these molecules do not appear to interact directly in thrombin-activated cells. It is known that both EGF receptors and Shc bind actin directly (den Hartigh et al. 1992; Thomas et al. 1995). As there was little or no tyrosine phosphorylation that could account for Shc association with the EGF receptor in thrombin-activated cells, it seems likely that the affinities of these molecules for actin allows a signaling complex to be set up without direct coupling, such that a readily activated signaling module is present at the required site of the cell. A signal transduction “particle” associated with a microfilament cell fraction has been recently described in mammary adenocarcinoma cells (Carraway et al. 1999), although the precise subcellular distribution of the complex was not examined. This complex contained the EGF receptor family member, p185neu, MAP kinase, and upstream regulators. Our evidence that a clustering of signaling molecules occurs at the actin arc suggests that this may represent a common mechanism of concentration of signaling entities in activated cells. The thrombin-stimulated signaling systems that induce actin polymerization, which are likely to involve G12/13 (Buhl et al. 1995; Gohla et al. 1999), are therefore likely to be critical components that allow the sensitization of heterologous receptors on F-actin structures. Further studies into the intracellular signaling systems necessary for induction of receptor clustering may provide therapeutic targets in the treatment of diseases of cell proliferation and cell migration.
In conclusion, we have shown that the subcellular site and concentration of EGF receptors can be regulated by the thrombin receptor and this appears to modulate the efficacy of receptor signaling induced by EGF receptor ligands.
Footnotes
Abbreviations used in this paper: CFSE, carboxy fluorescein diacetate succinimidyl ester AM; FAK, focal adhesion kinase; MAP, mitogen-activated protein; p70S6k, 70-kD S6 kinase.
References
- Berven L.A., Frew I.J., Crouch M.F. Nitric oxide donors selectively potentiate thrombin-stimulated p70S6k activity and morphological changes in Swiss 3T3 cells. Biochem. Biophys. Res. Commun. 1999;266:352–360. doi: 10.1006/bbrc.1999.1833. [DOI] [PubMed] [Google Scholar]
- Buhl A.M., Johnson N.L., Dhanasekaran N., Johnson G.L. Gα12 and Gα13 stimulate Rho-dependent stress fiber formation and focal adhesion assembly. J. Biol. Chem. 1995;270:24631–24634. doi: 10.1074/jbc.270.42.24631. [DOI] [PubMed] [Google Scholar]
- Carpenter G. Employment of the epidermal growth factor receptor in growth factor–independent signaling pathways. J. Cell Biol. 1999;146:697–702. doi: 10.1083/jcb.146.4.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carraway C.A., Carvajal M.E., Carraway K.L. Association of the Ras to mitogen-activated protein kinase signal transduction pathway with microfilaments. Evidence for a p185neu-containing cell surface signal transduction particle linking the mitogenic pathway to a membrane-microfilament association site. J. Biol. Chem. 1999;274:25659–25667. doi: 10.1074/jbc.274.36.25659. [DOI] [PubMed] [Google Scholar]
- Collins L.R., Ricketts W.A., Olefsky J.M., Brown J.H. The G12 coupled thrombin receptor stimulates mitogenesis through the Shc SH2 domain. Oncogene. 1997;15:595–600. doi: 10.1038/sj.onc.1201220. [DOI] [PubMed] [Google Scholar]
- Crouch M.F. Regulation of thrombin-induced stress fibre formation in Swiss 3T3 cells by the 70-kDa S6 kinase. Biochem. Biophys. Res. Commun. 1997;233:193–199. doi: 10.1006/bbrc.1997.6419. [DOI] [PubMed] [Google Scholar]
- Crouch M.F., Simson L. The G-protein Gi regulates mitosis but not DNA synthesis in growth factor-activated fibroblastsa role for the nuclear translocation of Gi. FASEB J. 1997;11:189–198. doi: 10.1096/fasebj.11.2.9039962. [DOI] [PubMed] [Google Scholar]
- Crouch M.F., Belford D.A., Milburn P.J., Hendry I.A. Pertussis toxin inhibits EGF-, phorbol ester- and insulin-stimulated DNA synthesis in BALB/c3T3 cellsevidence for post-receptor activation of Giα. Biochem. Biophys. Res. Commun. 1990;7:1369–1376. doi: 10.1016/0006-291x(90)90674-c. [DOI] [PubMed] [Google Scholar]
- Crouch M.F., Davy D.A., Willard F.S., Berven L.A. Insulin induces epidermal growth factor (EGF) receptor clustering and potentiates EGF-stimulated DNA synthesis in Swiss 3T3 cellsa mechanism for costimulation in mitogenic synergy. Immunol. Cell Biol. 2000;78:408–414. doi: 10.1046/j.1440-1711.2000.00929.x. [DOI] [PubMed] [Google Scholar]
- De Vivo M., Chen J., Codina J., Iyengar R. Activated Gqα potentiates platelet-derived growth factor-stimulated mitogenesis in confluent cell cultures. J. Biol. Chem. 1992;267:18263–18266. [PubMed] [Google Scholar]
- Daub H., Weiss F.U., Wallasch C., Ullrich A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature. 1996;379:557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- Daub H., Wallasch C., Lankenau A., Herrlich A., Ullrich A. Signal characteristics of G protein-transactivated EGF receptor. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:7032–7044. doi: 10.1093/emboj/16.23.7032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della Rocca G.J., Maudsley S., Daaka Y., Lefkowitz R.J., Luttrell L.M. Pleiotropic coupling of G protein-coupled receptors to the mitogen-activated protein kinase cascade. Role of focal adhesions and receptor tyrosine kinases. J. Biol. Chem. 1999;274:13978–13984. doi: 10.1074/jbc.274.20.13978. [DOI] [PubMed] [Google Scholar]
- den Hartigh J.C., van Bergen en Henegouwen P.M., Verkleij A.J., Boonstra J. The EGF receptor is an actin-binding protein. J. Cell Biol. 1992;119:349–355. doi: 10.1083/jcb.119.2.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohla A., Offermanns S.O., Wilkie T.M., Schultz G. Differential involvement of Gα12 and Gα13 in receptor-mediated stress fiber formation. J. Biol. Chem. 1999;274:17901–17907. doi: 10.1074/jbc.274.25.17901. [DOI] [PubMed] [Google Scholar]
- Gupta S.K., Gallego C., Lowndes J.M., Pleiman C.M., Sable C., Eisfelder B.J., Johnson G.L. Analysis of the fibroblast transformation potential of GTPase-deficient gip2 oncogenes. Mol. Cell. Biol. 1992;12:190–197. doi: 10.1128/mcb.12.1.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath J.P., Holifield B.F. On the mechanisms of cortical actin flow and its role in cytoskeletal organisation of fibroblasts. Symp. Soc. Exp. Biol. 1993;47:35–56. [PubMed] [Google Scholar]
- Johanson S.O., Naccache P.A., Crouch M.F. A p85 subunit-independent p110α PI 3-kinase colocalizes with p70S6k on actin stress fibers and regulates thrombin-stimulated stress fiber formation in swiss 3T3 cells. Exp. Cell Res. 1999;248:223–233. doi: 10.1006/excr.1999.4405. [DOI] [PubMed] [Google Scholar]
- Kalinec G., Nazarali A.J., Hermouet S., Xu N., Gutkind J.S. Mutated α subunit of the Gq protein induces malignant transformation in NIH 3T3 cells. Mol. Cell. Biol. 1992;12:4687–4693. doi: 10.1128/mcb.12.10.4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMorte V.J., Harootunian A.T., Spiegel A.M., Tsien R.Y., Feramisco J.R. Mediation of growth factor induced DNA synthesis and calcium mobilization by Gq and Gi2. J. Cell Biol. 1993;121:91–99. doi: 10.1083/jcb.121.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttrell L.M., Daaka Y., Lefkowitz R.J. Regulation of tyrosine kinase cascades by G-protein-coupled receptors. Curr. Opin. Cell Biol. 1999;11:177–183. doi: 10.1016/s0955-0674(99)80023-4. [DOI] [PubMed] [Google Scholar]
- Nabi I.R. The polarization of the motile cell. J. Cell Sci. 1999;112:1803–1811. doi: 10.1242/jcs.112.12.1803. [DOI] [PubMed] [Google Scholar]
- Needham L.K., Rozengurt E. Gα12 and Gα13 stimulate Rho-dependent tyrosine phosphorylation of focal adhesion kinase, paxillin, and p130 Crk-associated substrate. J. Biol. Chem. 1998;273:14626–14632. doi: 10.1074/jbc.273.23.14626. [DOI] [PubMed] [Google Scholar]
- Offermanns S., Laugwitz K.L., Spicher K., Schultz G. G proteins of the G12 family are activated via thromboxane A2 and thrombin receptors in human platelets. Proc. Natl. Acad. Sci. USA. 1994;91:504–508. doi: 10.1073/pnas.91.2.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hackel P., Zwick E., Prenzel N., Ullrich A. Epidermal growth factor receptorscritical mediators of multiple receptor pathways. Curr. Opin. Cell Biol. 1999;11:184–189. doi: 10.1016/s0955-0674(99)80024-6. [DOI] [PubMed] [Google Scholar]
- Pace A.M., Wong Y.H., Bourne H.R. A mutant alpha subunit of Gi2 induces neoplastic transformation of Rat-1 cells. Proc. Natl. Acad. Sci. USA. 1991;88:7031–7035. doi: 10.1073/pnas.88.16.7031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post G.R., Collins L.R., Kennedy E.D., Moskowitz S.A., Aragay A.M., Goldstein D., Brown J.H. Coupling of the thrombin receptor to G12 may account for selective effects of thrombin on gene expression and DNA synthesis in 1321N1 astrocytoma cells. Mol. Biol. Cell. 1996;7:1679–1690. doi: 10.1091/mbc.7.11.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prenzel N., Zwick E., Daub H., Leserer M., Abraham R., Wallasch C., Ullrich A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402:884–888. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- Soranno T., Bell E. Cytostructural dynamics of spreading and translocating cells. J. Cell Biol. 1982;95:127–136. doi: 10.1083/jcb.95.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D., Patterson S.D., Bradshaw R.A. Src homologous and collagen (Shc) protein binds to F-actin and translocates to the cytoskeleton upon nerve growth factor stimulation in PC12 cells. J. Biol. Chem. 1995;270:28924–28931. doi: 10.1074/jbc.270.48.28924. [DOI] [PubMed] [Google Scholar]
- Vaingankar S.M., Martins-Green M. Thrombin activation of the 9E3/CEF4 chemokine involves tyrosine kinases including c-src and the epidermal growth factor receptor. J. Biol. Chem. 1998;273:5226–5234. doi: 10.1074/jbc.273.9.5226. [DOI] [PubMed] [Google Scholar]
- Verrall S., Ishii M., Chen M., Wang L., Tram T., Coughlin S.R. The thrombin receptor second cytoplasmic loop confers coupling to Gq-like G proteins in chimeric receptors. Additional evidence for a common transmembrane signaling and G protein coupling mechanism in G protein-coupled receptors. J. Biol. Chem. 1997;272:6898–6902. doi: 10.1074/jbc.272.11.6898. [DOI] [PubMed] [Google Scholar]
- Voyno-Yasenetskaya T.A., Pace A.M., Bourne H.R. Mutant alpha subunits of G12 and G13 proteins induce neoplastic transformation of Rat-1 fibroblasts. Oncogene. 1994;9:2559–2565. [PubMed] [Google Scholar]