

Abstract
We have analyzed the subnuclear position of early- and late-firing origins of DNA replication in intact yeast cells using fluorescence in situ hybridization and green fluorescent protein (GFP)–tagged chromosomal domains. In both cases, origin position was determined with respect to the nuclear envelope, as identified by nuclear pore staining or a NUP49-GFP fusion protein. We find that in G1 phase nontelomeric late-firing origins are enriched in a zone immediately adjacent to the nuclear envelope, although this localization does not necessarily persist in S phase. In contrast, early firing origins are randomly localized within the nucleus throughout the cell cycle. If a late-firing telomere-proximal origin is excised from its chromosomal context in G1 phase, it remains late-firing but moves rapidly away from the telomere with which it was associated, suggesting that the positioning of yeast chromosomal domains is highly dynamic. This is confirmed by time-lapse microscopy of GFP-tagged origins in vivo. We propose that sequences flanking late-firing origins help target them to the periphery of the G1-phase nucleus, where a modified chromatin structure can be established. The modified chromatin structure, which would in turn retard origin firing, is both autonomous and mobile within the nucleus.
Keywords: DNA replication, nuclear organization, in vivo green fluorescent protein imaging, origins of replication, nuclear envelope
Introduction
One characteristic feature of eukaryotic genomes, as opposed to bacterial genomes, is that each chromosome contains multiple origins of replication capable of initiating DNA replication at different times during S phase. In mammalian cells, the timing of replication of a given chromosomal domain often correlates with the transcriptional activity of nearby genes. More precisely, it was shown that the late-replicating Giemsa Dark bands contain tissue-specific genes that are repressed in many tissues, while Giemsa-light, or R-bands, replicate early and contain most ubiquitously expressed housekeeping genes (Holmquist 1987). Similarly, in yeast, origins of replication in the transcriptionally silent subtelomeric regions fire either late or not at all, although origin recognition complex and other components of the pre-Replicative Complex appear to assemble at these origins normally (Santocanale and Diffley 1996). Thus, despite correlations between a lack of transcriptional inactivity and a delay in origin firing, little is known about the molecular mechanisms that establish and regulate this latter event.
In addition to having a precise temporal pattern for origin activation, DNA replication has been shown to occur at a limited number of discrete foci within the nucleus in both yeast and higher eukaryotic cells (Nakamura et al. 1986; Pasero et al. 1997). Studies with synchronized mammalian cells suggest that the pattern of active replication foci changes in an ordered and reproducible fashion during S-phase progression (Nakayasu and Berezney 1989; van Dierendonck et al. 1989; Fox et al. 1991; Manders et al. 1992; O'Keefe et al. 1992). Consistently, using a proliferating cell nuclear antigen–green fluorescent protein (GFP) fusion protein to identify replication foci in situ, it was shown that the transition from one pattern to the next is not due to the movement of foci, but to their gradual assembly and disassembly at discrete sites (Leonhardt et al. 2000). The reproducible positioning of early- and late-replicating foci led to the proposal that origin location within the nucleus might not only correlate with, but also regulate, the time at which origins fire. Indeed, euchromatic regions are found throughout the nucleus and replicate early, while mid- to late-S phase foci tend to reflect heterochromatic regions that localize either at the nuclear periphery or near nucleoli (Ma et al. 1998).
The spatial positioning of late-replicating DNA within the mammalian nucleus seems to be determined in early G1 phase (Dimitrova and Gilbert 1999). Using a pulse-chase technique in which isolated mammalian nuclei are stimulated to synthesize DNA in a Xenopus egg extract, it was shown that a characteristic subnuclear pattern was established for late-replicated chromosomal regions roughly 2 h after the completion of anaphase (Dimitrova and Gilbert 1999). The colocalization of signals from a first and second round of replication suggests that both the timing and the characteristic subnuclear localization of a late-replicating region is retained through mitotic division. However, since very few mammalian origins of replication have been mapped, the subnuclear positioning of specific origins during cell-cycle progression could not be addressed in these studies of cultured cells.
Budding yeast has provided an excellent model system to identify the molecular mechanisms that control origin activation, thanks to a detailed understanding of the sequences and proteins responsible for origin function (reviewed in Campbell and Newlon 1991; Newlon 1997). In yeast, as in mammalian cells, each origin of replication has a characteristic time of activation in S phase (reviewed in Fangman and Brewer 1992). Two mechanisms for keeping late origins from firing early have been suggested: the first proposes that chromatin structure regulates origin activation by limiting accessibility to common factors, and the second proposes that timing reflects the action of a specific molecular regulator. The two mechanisms are not mutually exclusive. For instance, compact nucleosomal arrays may limit or modify access to the origin complex for components of the replication machinery or for specific initiation-promoting factors. One such limiting factor might be the cyclin B/Cdk1 kinase, for it has been shown that Cyclin B5 (Clb5p) is required for the firing and proper timing of both early and late origins, while Clb6p activates early origins only (Donaldson et al. 1998b). It is not known whether these complexes associate with late-firing origins early in S phase or only when initiation actually occurs. In addition to Clb5/Cdc28p, a second Ser/Thr kinase, encoded by CDC7, is necessary for both early and late origin firing (Bousset and Diffley 1998; Donaldson et al. 1998a). This complex appears to be bound to at least a subset of origins in late G1 (Pasero et al. 1999).
In addition to cell-cycle controlling kinases, the conserved checkpoint kinases encoded by MEC1 and RAD53 play a role in regulating the firing of late origins when replication fork progression is blocked by hydroxyurea (HU) or methylmethanesulfonate (Santocanale and Diffley 1998; Shirahige et al. 1998). Mutation of the Rad53 kinase or a component of the origin recognition complex results in the premature firing of late origins even in the absence of HU, suggesting that this kinase may also be constitutively involved in delaying the activation of certain origins until late in S phase (Shirahige et al. 1998). Rad53p interacts both genetically and physically with Dbf4p, the regulatory subunit of the Cdc7 kinase (Dohrmann et al. 1999), and, in HU-arrested cells, Dbf4p becomes hyperphosphorylated in a Rad53-dependent manner (Weinreich and Stillman 1999). The modification of Dbf4p results in its release from chromatin, suggesting that Rad53p helps regulate origin firing through this kinase complex (Pasero et al. 1999; Weinreich and Stillman 1999).
Evidence that chromosomal context plays an important role in replication timing in budding yeast comes from the analysis of timing of specific origin activation (reviewed in Fangman and Brewer 1992). These authors found that origins near yeast telomeres generally fire late in S phase, while those near centromeres fire early (McCarroll and Fangman 1988). The silent chromatin found at yeast telomeres also exerts a position effect, ensuring that origins in subtelomeric repeats are repressed in a manner dependent on structural proteins of silent chromatin [silent information regulators (SIR) 2, 3, and 4; Stevenson and Gottschling, 1999]. Although the timing of activation of a given origin in its genomic location is highly reproducible, this timing does not depend on the minimal core sequence of the origin, which contains the autonomously replicating sequence (ARS) consensus. More specifically, the transfer of a minimal origin domain (or ARS) to a plasmid, confers an early replication status on the episome, even though the origin may be late firing in its chromosomal context. Thus, it appears that the activation of origins in early S phase is the default state. Furthermore, DNA sequences that flank the late-firing origin on chromosome XIV (ARS1412) can render a plasmid-borne origin late firing (Friedman et al. 1996). It has been suggested that these “late elements” act by nucleating a specific chromatin structure that prevents initiation in early S phase, although the genes flanking ARS1412 are not transcriptionally silent. An alternative, but not mutually exclusive, mechanism for ensuring late initiation would be to sequester or target the origin to a specific subcompartment of the nucleus, which would delay initiation. Such a mechanism is consistent with the correlation found in many organisms between transcriptionally repressed chromatin, late-firing origins, and their localization near the nuclear periphery.
As in mammalian cells, the establishment of a late-firing origin in budding yeast requires events that occur between mitosis and the subsequent G1 phase (Raghuraman et al. 1997). This may correlate either with the formation of a repressed chromatin structure following DNA replication (Taddei et al. 1999), with the assembly of the pre–regulatory complex, or with establishment of a G1-specific nuclear organization, as proposed by Dimitrova and Gilbert 1999. To investigate the importance of the nuclear positioning on origin timing in yeast, we have localized early- and late-activated origins by a combined immunofluorescence/in situ hybridization (IF/FISH) protocol in fixed cells and by the lac operator/GFP-lac repressor recognition system in live cultures. In this paper, we demonstrate by time-lapse microscopy that in G1-phase nuclei there is a significant enrichment of late-activated origins in a zone immediately adjacent to the nuclear envelope, despite their oscillatory subnuclear movement. Using site-specific recombination to excise a chromosomal origin, we present data suggesting that local chromatin structure is dominant over nuclear positioning in the regulation of origin timing in S phase, although the establishment of a late replication state of chromatin in G1 phase correlates with its transient enrichment at the nuclear periphery.
Materials and Methods
Yeast Strains, Plasmids and Growth Conditions
Diploid strain GA-116 (MATa/MATα cdc4-3/cdc4-3 ade1/ADE1 ade2/ade2-R8 met/Met ura1/URA1 tyr1/TYR1 his7/HIS7 lys2/LYS2 can1-11/CAN1; formerly 212-10, gift from G. Simchen, The Hebrew University of Jerusalem, Jerusalem, Israel) was grown 14 h at 23°C to ∼5 × 106 cells/ml in YPD, synchronized in late G1 for approximately one generation (105 min) at 36°C, and subjected to IF/FISH. YPD medium supplemented with 40 mg/liter adenine, synthetic medium lacking uracil for selection of p12 and p12ARS plasmids (Friedman et al. 1996) were prepared and standard genetic techniques were used as described (Rose et al. 1990). p12 and p12ARS were introduced into GA-1190 (MATa/MATα cdc4-3/cdc4-3 ade2/ade2 leu2/leu2 ura3/ura3 trp1/trp1) by lithium acetate transformation, and cells were grown as described above. Haploid strain GA-1205 (MATa, cdc7-1, bar1 his6 leu2-3,112 trp1-289 ura3-52, formerly R3ZXVR2, described in Raghuraman et al. 1997) contains a 30-kb ARS501-origin excision cassette defined by two target sites for the “R” recombinase from Zygosaccharomyces rouxii, and three copies of the recombinase gene integrated in tandem under the control of the GAL1 UAS at the LEU2 locus. The conditions are modified from Raghuraman et al. 1997 as follows: cells were grown 14 h in 2% lactose, 3% glycerol containing YP-medium to 0.5 × 106 cells/ml and arrested in G1 by treatment with α-factor (40 nM) for approximately one generation (3.2 h). To excise the ARS501 cassette in G1, the culture was split in half and galactose was added to 2% to one portion, while the other half served as the uninduced control. After 4 h, 2% glucose was added to both, and, 30 min later, cultures were centrifuged, washed, and resuspended in YPD. Cultures were then shifted to 37°C and treated with pronase (50 μg/ml) to degrade α-factor. Cells accumulated at the CDC7 execution point (G1/S boundary) were subjected to IF/FISH. For excision at the nocodazole block, cells were grown and synchronized with α-factor as described above, and were released from α-factor and arrested as large-budded cells in the presence of 10 μg/ml nocodazole. The culture was split, 2% galactose was added to one half, and after 4 h, 2% glucose was added to both subcultures. Synchronization at the G1/S boundary was achieved by a shift to 37°C, as above, except pronase was omitted. Samples were collected throughout the procedure and analyzed by flow cytometry to confirm cell-cycle arrest (data not shown).
A SacI/BamHI fragment containing the NUP49GFP fusion was inserted into pRS406 from pUN100-NUP49GFP (provided by V. Doye, Institut Curie, Paris, France; described in Belgareh and Doye 1997), to allow integration of the NUP49-GFP gene into GA-180 (W303-1a) by gene replacement (Rose et al. 1990). NheI-cut AFS135 (provided by A. Straight, Harvard University, Cambridge, MA) carrying the lac repressor-GFP fusion under the HIS3 promotor was integrated into the HIS3 locus in this same strain creating GA-1320. For lac repressor site integrations, PCR products of roughly 1 kb containing genomic DNA from regions several kilobases from a given origin (see below) were cloned into pAFS52 such that the region flanks the 256 multimerized lac operator repeats (see Straight et al. 1996). Unique sites were used to linearize the plasmids for integration, and insertion into the correct genomic loci was verified by PCR. GA-1323 contains the insert 10-kb upstream of ARS1413, 240 kb from the left telomere of ChrXIV. GA-1325 contains the insert near origin ChrIV-908, positioned ∼905 kb from the left telomere of ChrIV. Tagged strains were grown 14 h at 30°C, in SD-histidine medium to 5 × 106 cells/ml, and were spread on thin 1.4% agarose patches in SD-histidine (with 4% glucose) on slides with a circular depression (Merck Eurolab) for microscopy.
In Situ Hybridization Probes
Probes for FISH are labeled by nick-translation with Digitonin-dUTP (Roche Diagnostics), Alexa 488-5-dUTP, or Alexa 546-14-dUTP (Molecular Probes) as described in Gotta et al. 1999, with the following exceptions: the optimal amount of DNaseI (Amersham Pharmacia Biotech) was determined empirically for each batch (typically 1–10 ng/ml) to obtain DNA fragments in the range of 200–500 bp. Free label was removed directly after the nick-translation reaction by the use of spin columns (High Pure PCR Product Purification Kit; Roche Diagnostics).
The source of DNA templates used to make probes and the chromosomal position of the corresponding homologous sequences were as follows (chromosomal positions are expressed in kb downstream of the corresponding left telomere): XbaI fragments of plasmids p12 and p13 (Friedman et al. 1996) were ligated into XbaI cut pGEM3Zf(−), creating pARS1412-8844 and pARS1412-2400, and these were used for the ARS1412 probe (centered at position 204 kb on ChrXIV). pARS1413-4219, pARS1413-3929, and pARS1413-2684 served as template for the ARS1413 probe (centered at position 250 kb on ChrXIV). pEL42H10 contains the subtelomeric Y′ element and a short TG1-3 repeat (provided by E.J. Louis, Oxford University, Oxford, UK; described in Gotta et al. 1996) and plasmid J10A (provided by C.S. Newlon, University of Medicine and Dentistry of New Jersey, Nutley, NJ) for HML (centered at position 20 kb from the left telomere of ChrIII; Newlon et al. 1991). pBR322 was used as probe to hybridize to the plasmid backbone of p12 and p12ARS. Four cosmids were used for centromeric FISH, pUKG047 (CENXI, a gift from H. Scherthan, Kaiserslauten, Germany), and c8270, c9780, and 14-20 (70921 and 71205; American Type Culture Collection; CENXIII, CENVIII, and CENXIV, respectively, gifts from P. Philippsen, Basel, Switzerland).
For all other probes, appropriate primers were chosen to amplify genomic DNA regions of 6–8 kb using genomic DNA as a template. PCR-amplified probes extend to either side of the indicated origin, with the center of the probe at the chromosomal position relative to the left telomere indicated in parentheses. For the chromosomal origins: ARS1 (462 kb, or ∼17 kb downstream of CEN4); chromosome V, ARS501 (548 kb; Ferguson et al. 1991); chromosome VI, ARS603 (65 kb), ARS606 (168 kb), and ARS607 (200 kb; described in Friedman et al. 1997; Shirahige et al. 1993), the timing, position, and efficiency of firing were characterized by Meselson-Stahl CsCl-density gradient experiments on synchronized cells (McCarroll and Fangman 1988) and by two-dimensional gels (Brewer and Fangman 1987). Origins ChrIV-210, ChrIV-257, ChrIV-908, ChrIV-1153, ChrX-305, and ChrX-613 were identified as part of a genome-wide survey of replication timing in yeast. For these origins, the number after the dash indicates the position of the probe center from the left telomere of the given chromosome. The timing of these origins has been confirmed by CsCl-density gradient experiments (Table ). Probes to localize Tel V-R (562 kb) and the 2-μm circle were also obtained by PCR of genomic DNA.
Table 1.
The Timing of Firing of Selected Origins
| Early origins | R.I. | Late Origins | R.I. |
|---|---|---|---|
| ARS305 | 0 | ARS603 | 0.71 |
| ARS607 | 0.02 | ARS1413 | 0.76 |
| ChrIV-908 | 0.07 | ARS1412 | 0.77 |
| ChrIV-1153 | −0.05 | ChrX-305 | 0.88 |
| ChrX-613 | 0.21 | ChrIV-210 | 0.67 |
| ARS1 | 0.27 | ChrIV-257 | 0.87 |
| ARS606 | 0.28 | R11 | 1 |
The timing of activation of six early- and six late-activated origins is expressed as a replication index as determined by CsCl density transfer experiments (R.I.; Friedman et al. 1996). The R.I. ranges from the very early replicated ARS305 to the very late replicating DNA fragment R11 or RAP1 (=1, both shown in bold). The last number separated by a dash indicates the distance in kilobases from the left telomere.
Combined Immunofluorescence and In Situ Hybridization on Yeast Cells
Primary antibodies used include monoclonal antibodies against the yeast nuclear pore protein p62 (Mab414; Babco) and Spc98 (anti–p90, a gift of J. Kilmartin, Medical Research Council, Cambridge, UK) to stain the spindle pole body. Secondary antibodies were Cy5™ or Cy3™-conjugated Goat anti–mouse IgG (Jackson ImmunoResearch Laboratories) and fluorescein-derivatized sheep anti–digoxigenin F(ab) fragments (Roche Diagnostics). Secondary antibodies are preabsorbed against fixed yeast spheroplasts before use, and no cross reactivity among these reagents has been detected.
Cells were prepared for immunofluorescence and FISH following the standard fixation method described in Gotta et al. 1999, except that cells were fixed in growth medium for 10 min at 30°C in 4% paraformaldehyde before spheroplasting. Fixed cells were washed three times and the conversion to spheroplasts was done with 300 U/ml of lyticase and 0.6–1.2 mg/ml of Zymolyase (20T) for ∼15 min. The arrest of cdc4-3 and cdc7-1 strains at the restrictive temperature results in more fragile cell walls and requires use of less lyticase. For alternative preparations (see Fig. 1), cells were fixed in the presence of 0.1% Triton X-100 or fixed after exposure to detergent as described (Klein et al. 1992). To visualize DNA, we use POPO-3, YOYO-3, or TOTO-3 (Molecular Probes) as indicated.
Figure 1.

Three-dimensional reconstitution of confocal scans monitor nuclear integrity after combined IF/FISH labeling. (a–b) Double labeling of diploid budding yeast cells (GA-1190) by fluorescence in situ hybridization with a digoxigenin-derivatized subtelomeric repeat probe (Y′, green) and nuclear pore immunostaining (Mab414, red). (a) Cells synchronized at the G1/S border were fixed in the absence of detergents before processing for FISH. They remain structurally intact as judged by the presence of three to eight clusters of the subtelomeric Y′ element signals and their peripheral localization. Confocal sections were taken along the z axis of the nucleus and were reconstituted and deconvolved in 3-D (Imaris®). Three different views of the reconstituted nucleus are shown in the lower part of each panel, corresponding to the x–y, x–z, and y–z plane. (b) Cells were fixed in the presence of 0.1% Triton X-100 (see Materials and Methods). The nuclei are flattened (compare x–z with y–z planes) as nuclear organization is gradually destroyed. (c) Cells were fixed after exposure to 0.1% Triton X-100, a technique called nuclear spreading (Klein et al. 1992). In the samples shown in c, nuclear pore staining is routinely lost, probably due to solubilization of the pore protein. In this case, to visualize the nucleus, DNA was stained with POPO-3 (shown in red). (d and e) Double staining of cells that were fixed as in a. In this case, the DNA is visualized with YOYO-3 (green) and the nuclear pore is red (Mab414, see above). (f) A line profile histogram shows the DNA distribution along a line for the nucleus shown in e combined with the nuclear pore staining. a–c were collected on an LSM 410 confocal microscope and d–e on an LSM 510 confocal microscope (Carl Zeiss, Inc.). Scale bars: 2 μm.
Confocal images were obtained as described in Gotta et al. 1996, using an Axiovert 100 microscope (Carl Zeiss, Inc.) equipped with the Laser Scanning Microscope 410 or 510 (Carl Zeiss, Inc.), with a 63× or 100× Plan-Apochromat objective (1.4 oil). Nuclei were reconstituted in three dimensions by typically taking 30 stacks of images along the z axis (100-nm steps) and performing computational deconvolution using Imaris® and Huygens® Software (Bitplane).
Quantitation of Fluorescent Signals
Distance-to-edge Measurements.
FISH signals were localized relative to the nuclear periphery by the use of concentric circles that either define five regions within the nucleus of fixed area or two zones of equal area. For the latter, an internal circle of radius 0.71 divides the surface area into two equal parts. A second method for scoring perinuclear localization was performed by image processing as follows. The antinuclear pore signal was computationally expanded to occupy a zone that contains 50% of the surface area of the nucleus, while leaving the FISH signals unmodified. FISH signals overlapping >50% with the enlarged pore zone were scored as peripheral. Finally, to analyze the position of the ARS501-excision cassette or GFP-tagged origins relative to either the telomere proximal locus on chromosome V or to the nuclear periphery, the line profile tool of the 510 Confocal software version 2.5 was used. If the minimal distance between the FISH signal and the nuclear pore was <0.29×, the nuclear radius, it was scored as peripheral. In three-dimensional reconstituted nuclei, the presence of signals within two equal volumes defining a peripheral and an internal sphere was quantified. FISH signals were scored on orthogonal representations of the nucleus. Centromere FISH signals were scored as clustered when found within a circle containing 16% of the nuclear surface area. Standard χ2 tests were performed to determine the statistical significance of differences between the FISH signal distribution in the nucleus and a random distribution. Following the convention to avoid frequencies less than five in any given group, frequencies in zones 4 and 5 (see Fig. 3 a) have been combined for the χ2 test.
Figure 3.
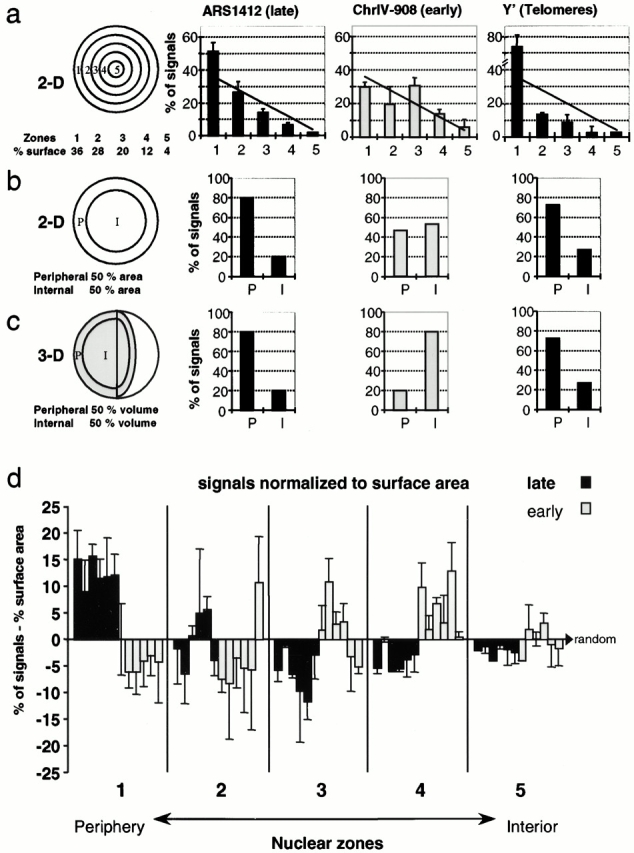
Late replication origins are enriched near the nuclear periphery. FISH signals were quantified on equatorial confocal sections of diploid strain GA-116, for ARS1412, ChrIV-908, and Y′-containing telomeres (Fig. 2b, Fig. c, and Fig. e) relative to the zones indicated in a–c. ARS1412 and the subtelomeric Y′ elements are late replicating, while ChrIV-908 is early. (a) Distance to edge is measured by the use of five zones, and data are represented in a bar graph as a percentage of signals per zone, each of which has a different area. The line corresponds to theoretical values for randomly distributed signals and is proportional to the surface area of each zone. From left to right (in brackets: n = signals, nuclei): ARS1412 (106, 76), ChrIV-908 (83, 64), and Y′ (264, 66). Each experiment has been performed at least twice and error bars represent standard deviations. Analysis using a standard χ2 test reveals a statistically significant nonrandom localization for ARS1412 (P < 0.05) and Y′ FISH probes (P < 0.001), but not for ChrIV-908 (P = 0.1). (b) The nuclei are divided into two zones of equal surface area: peripheral and internal. FISH signal localization was quantified on a computer graphic representation of 2-D confocal images in which the nuclear pore staining was enlarged to create a peripheral zone that comprises 50% of the original nuclear surface. FISH signals found within this zone were scored as peripheral. ARS1412 (43, 42), ChrIV-908 (59, 44), and Y′ (149, 44). ARS1412 and Y′ signals were scored using nuclei from multiple experiments. (c) 3-D reconstituted nuclei (as in Fig. 1 a) are divided in two spaces of similar volume and the frequency of FISH signals is scored. ARS1412 (5, 5), ChrIV-908 (10, 5), and Y′ (22, 7). (d) The localization of six late-activated (dark columns) and six early-activated (light columns) origins has been quantified by distance-to-edge measurements as described in a. For each zone, the theoretical random signal distribution, based on surface area, has been subtracted from the corresponding signal frequency. A value of 0 corresponds to random distribution of signals; numbers > and < 0 indicate enrichment and depletion, respectively. Columns from left to right (brackets: n = signals, nuclei): ARS1412 (106, 76), ARS1413 (143, 94), ChrIV-210 (112, 78), ChrIV-257 (111, 77), ARS603 (64, 54), ChrX-305 (189, 123) ARS1 (43, 32), ChrIV-908 (83, 64), ChrIV-1153 (123, 86), ARS606 (83, 66), ARS607 (47, 37), and ChrX-613 (81, 63). A χ2 test demonstrates that four out of six late origins and none of the early origin FISH signals differ significantly from a random distribution to P < 0.05. When taken together, the localization of the pooled late origins in zone 1 is significantly nonrandom and is significantly different from the localization of the pooled early origins (P < 0.001).
Quantitation of colocalization between different FISH signals was carried out on computer graphic representations of the confocal images of diploid cells in which at least two signals of one probe are detected. The threshold for contour tracing using Adobe Photoshop 4.0 software was set at 80 (from a gray scale maximum of 255) after normalization of each filter channel independently to give the same maximum signal. Overlap of >25% of the less abundant signal in a nucleus with an equal or more abundant signal was considered as colocalization. Chromatic aberration is corrected before image capture in all analyses by aligning the red and green channel signals from 0.1 and 0.2 μm Tetraspeck Microspheres (Molecular Probes).
In Vivo Imaging of Replication Origins
Cells with GFP-tagged origins (ChrIV-908 or ARS1413) were grown and mounted in media-containing agar as described above, and images were captured as cells progressed through the cell cycle at room temperature (25°C). Image capture was performed on an Olympus IX70 microscope with a Plan-Apochromat 60×/1.4 oil objective, a GFP filter (B51/DCLP500/LP515; Chroma), and 1.5× magnification using the Till Vision® imaging software (Till Photonics). Using monochromatic light at 475 nm to visualize GFP fluorescence, stacks of 14 images with a step size of 300 nm were taken along the z axis of the nuclei. To maximize resolution, only nuclei in which the GFP-tagged origin was detected in one of the three central focal planes were scored. Cell shape and bud size was determined using transmission light images. Categorization used the following criteria: G1, unbudded cell with undistorted round nuclear pore staining; early S, cells containing very small buds; mid S, cells with small to medium sized buds; late S/G2, cells with large buds and an undistorted round nucleus at the bud neck; M, cells with large buds and an elongated nuclear envelope staining entering the daughter cell. Images were pseudocolored and merged using Adobe Photoshop 4.0.
Online Supplementary Material
For time-lapse analysis, cells with GFP-tagged origins were grown and mounted in media-containing agar as described above. Focal stacks of three images (500-nm step size) were captured every minute during 5 h on cells carrying the tagged origin ChrIV-908 (50-ms exposure time) using an Olympus IX70 microscope with a Plan-Apochromat 60×/1.4 oil objective, a GFP filter (B51/DCLP500/LP515; Chroma), and 1.5× magnification using the Till Vision® imaging software. For each time point, a single picture was taken using infrared light to monitor the cellular shape. For short time-lapse photography, the LSM 510 (Carl Zeiss, Inc.) was used (one image every 612 ms for ChrIV-908, or every 336 ms for ARS1413) over a period of 100 s. The focal plane of the image was adjusted manually to maintain the tagged origin in focus. Available at http://www.jcb.org/cgi/content/full/152/2/385/DC1.
Results
To test the hypothesis that late replicating regions are targeted to specific subcompartments of the nucleus, we have localized early- and late-activated origins by combined IF/FISH in fixed diploid budding yeast cells. We have chosen to examine the position of origins before initiation, to avoid potential movements that may occur during replication. We therefore arrest cells late in G1 using the temperature-sensitive cdc4-3 mutation, which stabilizes the Clb/Cdk1 inhibitor p40SIC1 and arrests cells at the G1/S transition. The culture was then immediately fixed with formaldehyde and processed for in situ hybridization. To complement this approach, our analysis was extended to living cells and all phases of the cell cycle by the use of direct GFP tagging of chromosomal domains (see below).
Yeast Nuclear Structure Is Preserved after Combined IF/FISH Analysis
This study required that we optimize protocols for efficient in situ hybridization of DNA in yeast cells such that the three-dimensional (3-D) architecture of the nucleus was fully preserved. We have found that nuclear integrity is best monitored by confocal microscopy sectioning and 3-D reconstitution of the immunofluorescence signal from nuclear pore staining (Mab414, see Materials and Methods). To compare the degree to which different fixation techniques preserve nuclear organization, we combined antinuclear pore immunofluorescence with FISH using a subtelomeric Y′ sequence, which is a conserved element found on ∼75% of the telomeres of strain GA-1190 (derived from A364a background; Louis et al. 1994, Fig. 1, a–c, green). We have previously shown by FISH, IF, and live GFP-fluorescence (Palladino et al. 1993; Gotta et al. 1996; Martin et al. 1999; Laroche et al. 2000) that telomeres are localized in <10 clusters near the nuclear envelope in interphase yeast cells, although when cells are fixed in the presence of detergents a preparation that is commonly used for yeast FISH or IF analysis (Klein et al. 1992; Weiner and Kleckner 1994; Guacci et al. 1994; Loidl et al. 1998), subnuclear organization is lost and many dispersed telomeric signals can be detected. In situ hybridization is highly efficient on these spread nuclei, yet reliable data on the 3-D organization of chromosomal domains cannot be obtained. In contrast, efficient fixation before detergent treatment maintains the integrity of the nuclear pore staining, the clustering of telomeres, and allows us to monitor the diameter of the spherical nucleus in x–y, x–z and y–z planes after serial confocal image capture and reconstitution in 3-D (Fig. 1 a). Throughout this study, we use the combined nuclear pore IF/FISH method shown here, monitoring nuclear diameter and height for every cell (Ø < 2.5 μm for a diploid nucleus) as an internal control for structural preservation.
Because our aim is to probe for the position of specific DNA regions, it was important to demonstrate that the genome itself is evenly distributed throughout the nuclear volume, when optimal nuclear shape is preserved. To do this, pore staining was combined with the DNA stain YOYO-3. Intensity measurements reveal a fairly uniform distribution of DNA throughout the nuclear volume (Fig. 1, d–e, green is DNA; red is antinuclear pore), except for a region of weaker green staining that we could identify as the nucleolus by anti–Nop1 staining (data not shown). Note that in some nuclei a DNA loop projects into the nucleolus, apparently reflecting the tandem rDNA repeats on chromosome XII (Guacci et al. 1994). A scan profile shows that the periphery of the genomic DNA fluorescence typically extends to the midpoint of the nuclear envelope staining (Fig. 1e and Fig. f), allowing us to use the middle of the antipore ring to define the outer limit of genomic DNA distribution.
Late Activated Origins Are Enriched at the Nuclear Periphery
Short DNA probes were designed to recognize sequences flanking six late-firing origins, six early-firing origins, and the subtelomeric Y′ element that contains a silent origin (Fig. 2 a, and Table ). The regions chosen were based on published information (Shirahige et al. 1993; Friedman et al. 1996, Friedman et al. 1997) and on DNA microarray data that characterized the timing of replication and approximate origin positions on all 16 yeast chromosomes (Raghuraman, M.K., B. Brewer, and W. Fangman, personal communication). FISH probes are obtained by nick-translation of plasmid- or PCR-based DNA templates in the presence of derivatized nucleotides, designed to recognize from 6 to 8 kb of genomic DNA (see Materials and Methods). Diploid cdc4-3 yeast cells (GA-116) were synchronized in late G1 by shifting a culture in an exponential growth phase to restrictive temperature for one generation time (∼105 min). Conditions optimized for the maintenance of a spherical nuclear structure were used to label both nuclear pores and the indicated origins (Fig. 1 a and 2, b–f). Serial confocal microscopy images (z-scans) were performed in each experiment, followed by 3-D reconstruction to evaluate the quality of the nuclear preservation. For each probe, we performed distance-to-edge measurements of the FISH signals on 50–100 nuclei selected on the basis of having a bright undistorted nuclear pore staining that was either round or oval shaped. Typically, the efficiency of in situ hybridization is such that 40% of the cells with intact nuclear pore staining display at least one FISH signal on an equatorial focal section.
Figure 2.
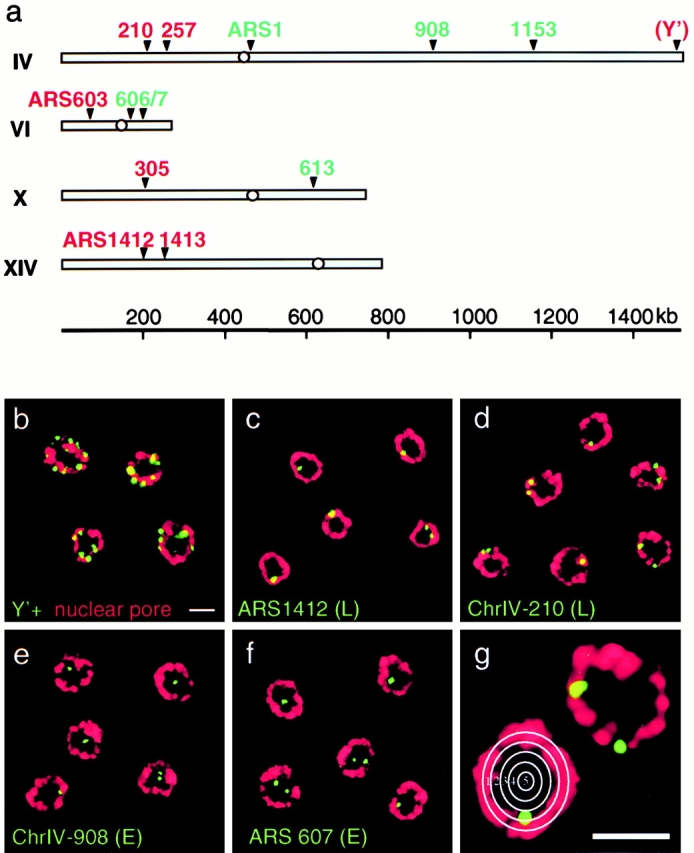
In situ hybridization of early- (E) and late- (L) firing origins of replication. (a) Origins for which the timing of replication has been determined by density gradient fractionation (Shirahige et al. 1993; Friedman et al. 1996) were chosen and appropriate FISH probes were prepared as described in Materials and Methods. Early and late origins are indicated in green and red, respectively (see Table ). The Y′ subtelomeric repeat element is not only found at the telomere of chromosome IV, but is at 70–80% of yeast telomeres (Louis et al. 1994). (b–g) IF/FISH has been performed on diploid GA-116 (cdc4-3/cdc4-3) cells that were synchronized in late G1 by a shift to restrictive temperature. Shown are representative examples of confocal images of the mid-cell plane showing nuclei stained for antipore (red) and hybridized with fluorescent probes (green) for the subtelomeric Y′ element, late-activated origins (ARS1412 and ChrIV-210), and early-activated origins (ChrIV-908 and ARS607). (g) Distance-to-edge measurements were performed by categorizing each FISH signal into one of five zones delineated by concentric circles. Images were collected on an LSM 410 confocal microscope. Scale bars: 2 μm.
Fig. 2 shows confocal images of the midsection of yeast nuclei selected from five different reactions with digoxigenin-labeled DNA probes specific for telomeres (Y′; b) for two late activated origins (ARS1412 in c and ChrIV-210, an origin located 210 kb from the left telomere of chromosome IV, in l d) and for two early activated origins (ChrIV-908, which lies 908 kb from the left telomere of chromosome IV, and ARS607 in e and f, respectively). These are visualized as green foci, while the nuclear pore staining is red.
We quantify the localization of FISH signals in relation to the nuclear periphery with three different methods (see Materials and Methods). In the first, the surface of a 2-D equatorial nuclear section is divided into five different zones by concentric circles of ∼0.25 μm in width, a value that is significantly larger than the resolution limit in the x–y plane in high resolution confocal microscopy images (∼180 nm in x–y; Webb 1999). The frequency of a randomly localized signal in any given zone would be proportional to the surface area, which is indicated by a straight line on each graph (Fig. 3 a). In the second method, the nucleus is divided in two zones of equal surface area on 2-D sections and signals are scored as either internal (I) or peripheral (P). The third method scores for signals within inner and outer shells of equal volumes in 3-D space-filling models reconstituted from stacks of confocal images through individual nuclei (Fig. 3 c, see Materials and Methods).
Confirming previous results, all three methods demonstrate a pronounced bias of the telomere-associated Y′ sequences and their associated ARS elements for the nuclear periphery. Applying the five-zone method on midfocal sections, we obtain 74 ± 7.6% of the Y′ signals in the most peripheral zone, zone 1, which contains 36% of the nuclear surface area (Fig. 3 a; n = 264 signals; 66 nuclei). Similarly, the nontelomeric late-firing origin ARS1412 is preferentially detected in the most peripheral region (51 ± 5.4% in zone 1; Fig. 3 a; n = 106 signals; 76 nuclei), while the early origin ChrIV-908 is not (30 ± 3%, Fig. 3 a; n = 83 signals; 64 nuclei). A χ2 analysis performed on these results confirms that the distribution of ARS1412 is significantly nonrandom (P < 0.01), with an enrichment at the nuclear periphery. In contrast, the distribution of the ChrIV-908 early firing origin among the five zones is not significantly different from random (P = 0.1). The frequency with which telomeric signals are detected at the nuclear periphery is even higher than the ARS1412 with statistical significance to P < 0.001. All three modes of evaluation led to a similar conclusion with respect to late origins and telomeres.
Additional early- and late-firing origins were scored on 30–120 2-D images, and the distance-to-edge measurements were classified into the five concentric zones, allowing us to take advantage of the better optical resolution in the x–y plane (∼180 nm in x–y, compared with ∼500 nm along the vertical z axis in the confocal images; Webb 1999). The summary of the values for six late and six early origins are presented in Fig. 3 d. To compensate for the different surface areas of each zone, we have subtracted from each the value expected for a distribution proportional to the surface area. Each bar represents the results from two to three independent experiments for each DNA probe, and the standard deviation is indicated. A statistically significant nonrandom distribution was obtained for four of the six late-firing origins (P < 0.05), while all the early-firing origins were scored as random. When the values obtained for the six late-firing origins in zone 1 were pooled and their frequency compared with either that of early origins in zone 1 or with a random distribution profile, the enrichment of late origins in the outermost zone was highly significant (P = 0.001).
Flanking Sequences Determine both the Timing and the Peripheral Location of a Plasmid-borne Late Firing Yeast Origin
Studies to determine elements responsible for the late activation of origins suggest that the flanking DNA plays an important role in establishing and maintaining late initiation (Friedman et al. 1996). Notably, a plasmid bearing only the core of the late-firing origin ARS1412 (p12ARS) is early replicating, while the same plasmid carrying an additional 16 kb of flanking DNA (p12) replicates almost as late as the origin in its genomic location. Based on our previous results, we asked whether these “late DNA elements,” which appear to determine the timing of replication, also determine nuclear localization of the plasmids. Both plasmids are detected by FISH in diploid cdc4-3 cells blocked in late G1 phase. Although the early replicating p12ARS is randomly localized (37 ± 0.5% in zone 1, Fig. 4 a), we again observed an enrichment in peripheral regions of the nucleus for the late-replicating plasmid p12 (49 ± 4.8% in zone 1, Fig. 4 b). This was scored for a large number of signals (n = 300 total) and the χ2 analysis again confirms that the peripheral enrichment of the late-replicating plasmid is highly significant (P < 0.01). These results are consistent with a model in which flanking sequences, or proteins bound to them, are responsible for targeting the late-replicating plasmid to the nuclear periphery.
Figure 4.
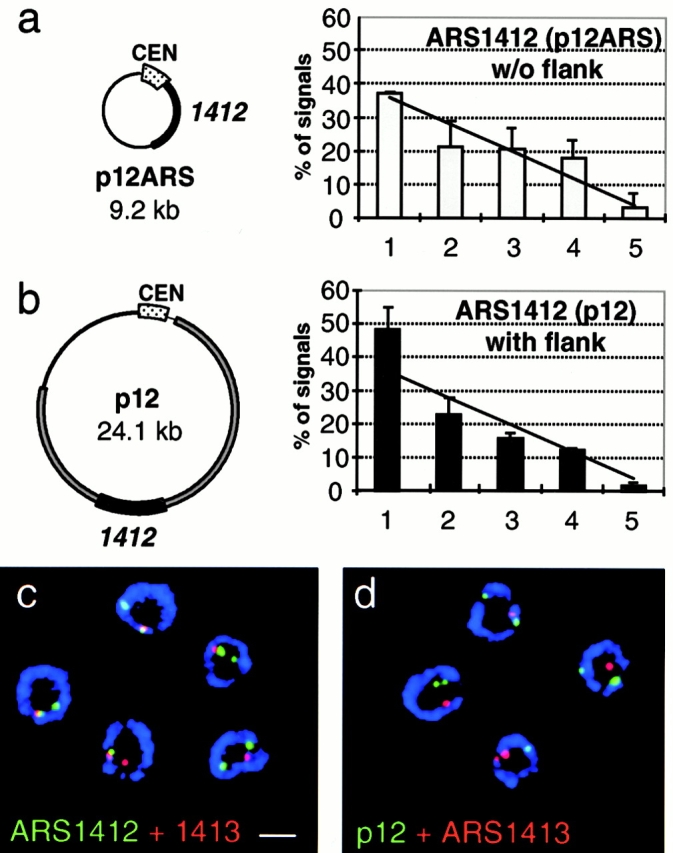
The late replicating plasmid p12 is enriched at the nuclear periphery, but does not colocalize with genomic ARS1412. (a and b) The plasmid p12ARS carries ∼1.5 kb of the core ARS1412 sequence, which fires late in its genomic location but becomes activated early on p12ARS (Friedman et al. 1996). The plasmid p12 bears an additional 17 kb of flanking sequences up- and downstream of the core ARS1412 and maintains a late activation in S phase. Subnuclear localization based on distance-to-edge measurements was quantified for p12ARS and p12, which were probed in diploid cdc4-3 cells that were carrying the appropriate plasmid and were blocked in G1 by a shift to restrictive temperature. Quantification of the FISH signal distribution was performed as described in Fig. 3 a. Numbers are based on two experiments for each plasmid and error bars represent standard deviations. p12ARS (n = 81 signals, 58 nuclei), p12 (n = 219 signals, 143 nuclei). A χ2 test shows that p12 is significantly enriched in the nuclear periphery to P < 0.01, while p12ARS is randomly distributed (P = 0.5). (c and d) Shown are representative confocal images of diploid cdc4-3 cells (GA-1190), blocked in late G1, and labeled with pairs of FISH probes and antinuclear pore (blue). In c, FISH probes recognize the genomic ARS1412 (green) and ARS1413 (red) loci, which colocalize. In d, cells carrying the late replicating plasmid p12 were probed for the plasmid (green) and the genomic ARS1413 (red). The signals were quantified for colocalization and the result is presented in Table . Images were collected on an LSM 410 confocal microscope. Scale bar: 2 μm.
Do the flanking sequences also position the plasmid adjacent to the genomic ARS1412 origin? To examine this question, we differentially labeled probes specific for the late-replicating plasmid p12, the genomic ARS1412 locus, and the adjacent late-replicating ARS1413 origin. Using different combinations of probes, we first determined the minimal distance we can optically resolve between two differentially labeled loci on the same chromosome. By measuring the resolution between the centers of FISH signals of different pairs of probes, we found that the minimal distance between two probes that allows reproducible signal separation is ∼50 kb (data not shown). Using the LSM410 confocal microscope (Zoom 3, 63×/1.4 oil objective; Carl Zeiss, Inc.), this represents an optical separation of three pixels or roughly 250 nm. Analysis of the resolution of signals from two adjacent chromosomal origins, ARS1412 and ARS1413 (separated by 50 kb), shows at least 25% signal overlap in 70% of the cells (Fig. 4 c and Table II; see Materials and Methods). In contrast, late-replicating plasmid p12 was in close proximity to the genomic ARS1413 in only 16% of the cells (Fig. 4 d). This result argues that late origin flanking sequences do not target a chromosome-specific subnuclear position, although the late-replicating plasmid is indeed enriched in this perinuclear zone.
Telomeres and Late Firing Origins Do Not Colocalize at the Nuclear Periphery
Since both internal late-activated origins and telomeres are enriched at the nuclear periphery in G1, we asked whether they localize to common sites by using codetection of differentially labeled FISH probes. As a control for multiple signals that we do not expect to colocalize, we first quantified the frequency with which two probes that recognize the centromere on chromosome VIII (CEN8) and the Y′ subtelomeric region overlap by ≥25% (Fig. 5 d, Table ). Using this criterion, 27% of the centromere signals were found to coincide with Y′ signals. In contrast, 88% colocalization was obtained using two differently labeled Y′ probes. This latter value indicates the experimental maximum for identical signals, while 27% most likely reflects the stochastic coincidence. When nontelomeric, late firing origins were tested in combination with the same Y′ probe, we found that only 33% of the signals coincide, a background equivalent to that obtained with CEN8. Similar frequencies of overlap were calculated for the late firing origins in combination with the Y′ probe (see probes ARS603, 1412, 1413, and ChrIV-257; Fig. 5, a and c, and Table ), indicating that in general, nontelomeric, late origins are not associated with telomeres, despite the fact that both are enriched in a perinuclear zone. Recent findings suggest that telomeres associate with the nuclear envelope through yKu (Laroche et al. 1998) and the large coiled-coil proteins Mlp1/2 (Galy et al. 2000). The lack of coincidence of Y′ elements and late origins, suggests that the latter associate with the nuclear periphery in a manner distinct from that which tethers telomeres.
Figure 5.

Late replicating origins and telomeres do not colocalize at the periphery. Representative confocal images of the midsection of fixed yeast cells hybridized with FISH probes for the subtelomeric Y′ element and either (a) ARS1412, (b) the silent mating type locus HML, or (c) ChrIV-210. In d controls for maximal colocalization and stochastic coincidence are shown. For the latter, detection of a Y′ probe (red) was combined with the centromere of chromosome 8 (CEN8, green). For maximal colocalization values, cells were probed with identical Y′ DNAs labeled with dig-dUTP (green) or Alexa546-dUTP (red). The different combinations of probes were quantified for colocalization and results are shown in Table . Images were collected on an LSM 410 confocal microscope. Scale bar: 2 μm.
Table 2.
Nontelomeric Late Origins Do Not Colocalize with Telomeres
| Probe 1(Dig) | Probe 2(Alexa-546) | Colocalization | n (signal pairs, nuclei) |
|---|---|---|---|
| ARS1412 | ARS1413 | 70% | 33, 27 |
| p12 | ARS1413 | 16% | 49, 36 |
| ARS1412 | Y' | 32% | 41, 32 |
| Y′ | ARS1413 | 33% | 71, 56 |
| Y′ | ChrIV-257 | 30% | 27, 20 |
| Y′ | ARS603 | 27% | 30, 23 |
| HML | Y' | 68% | 69, 60 |
| CEN8 | Y' | 27% | 60, 42 |
| Y′-tel | Y'-tel | 88% | 34, 23 |
Colocalization of FISH signals representing subtelomeric regions (Y′), the indicated late-firing ARS elements, the centromeric region from chromosome VIII (CEN8), and a region flanking the HML locus on the left arm of chromosome III: data are taken from experiments shown in Fig. 4c and Fig. d, and Fig. 5, a–d, and from additional experiments in which the fluorescent markers were switched such that the same probe was not always labeled with Dig-dUTP (see column labeled Probe 1/Dig) or with Alexa-546-dUTP (Probe 2/Alexa-546). For quantifying signals, midcell focal planes were examined for the presence of at least one signal of each labeled probe. Cells with more than two of each were omitted from evaluation, unless the more abundant signal was the subtelomeric Y′ probe. Cells displaying two signals from each probe were scored as two signal pairs. Overlap of signal areas >25% above a given threshold was defined as colocalization (see Materials and Methods).
Interestingly, a probe specific for a 10-kb DNA sequence 3-kb upstream of the silent mating type locus HML on chromosome III colocalizes efficiently with the Y′ probe (68%; Fig. 5, Table ). As the left telomere of chromosome III does not contain a Y′ element in the strain we use (data not shown), the coincidence of signals is not due to a lack of resolution along one chromosome arm, but probably reflects the clustering of different regions that are repressed in a SIR-dependent manner. Colocalization may also result from interactions between the left telomere of chromosome III (12 kb from HML) with telomeres of other chromosomes.
A Nonperipheral Localization for Early Replicating Centromeres and 2-μm Circles
We next asked whether the characteristic pattern of subnuclear localization of early- and late-firing origins applies to other DNA elements. We have chosen centromeres and the extrachromosomal 2-μm circle, both of which replicate early in S phase and have unusually stable segregation properties (Zakian et al. 1979; McCarroll and Fangman 1988). Centromere organization is of particular interest because FISH studies performed on nuclear spreads have suggested that they cluster close to the spindle pole body (SPB), which itself is embedded in the nuclear membrane in yeast (Jin et al. 1998). Our experiments are performed on diploid cdc4-3 cells blocked in late G1 phase, using four different probes recognizing the centromeres of chromosomes VIII, XI, XIII, and XIV (see Materials and Methods). We clearly observe the clustering of centromeres in a subcompartment of the nucleus, scored here as 16% of the surface area (Fig. 6 a, circle). Moreover, codetection of the CEN-probe FISH with anti–SPB immunofluorescence (anti–Spc98, see Materials and Methods) shows that the clustering occurs around the spindle pole body, forming a ring-like structure (Fig. 6, a and b; see also Jin et al. 2000). However, centromere signals do not coincide with the SPB (Hayashi et al. 1998) and distance-to-edge measurements indicate that the centromeres are enriched in zones 2 and 3, but not zone 1, which is most peripheral (Fig. 6, a and c). This distribution of centromeres is significantly nonrandom (P < 0.05), unlike most genomic early origins.
Figure 6.

The localization of yeast centromeres and the 2-μm circle. Centromeres cluster close to the spindle pole body in late G1, but do not localize to the extreme nuclear periphery. Diploid GA-1190 cells with the cdc4-3 allele synchronized in late G1 were fixed and subjected to IF/FISH. (a) Four different centromeres, those of ChrVIII, XI, XIII, and XIV, have been detected by FISH and the signals were analyzed for cluster formation and nuclear localization. Centromere probes, green; nuclear pore, red. (Inset) Centromere probes, green; anti–p90 (Spc98, which localizes to the spindle pole body), red; DNA stain (TOTO-3; Molecular Probes), blue. (b) Quantification of the clustering of centromere signals. The criteria for scoring clustering was that the labeled centromeres fall within a circle containing 16% of the nuclear surface at the midsection focal plane (a). (c) Distance-to-edge measurements for the centers of the centromere cluster are displayed in five different nuclear zones (n = 546 signals, 208 nuclei). (d) The early replicating, endogenous 2-μm plasmid clusters in internal regions of the nucleus. Cells were probed for the 2-μm circle (red) and the subtelomeric Y′ element. Nuclear pore, blue. (e) Quantification of the number of 2-μm foci found in 2-D sections of the nuclei. (f) Distance-to-edge measurements (n = 541 signals, 109 nuclei). A χ2 test revealed a significant nonrandom distribution for centromeres (P < 0.05) and a highly significant enrichment of the 2-μm signals at the nuclear interior (P < 0.001). Images were collected on an LSM 410 confocal microscope. Scale bars: 2 μm.
The 2-μm circle is a 6-kb endogenous, early-replicating plasmid that distributes symmetrically between mother and daughter cells despite its lack of a centromere. It overcomes the maternal inheritance bias by the use of a partitioning system that involves a cis-acting stability locus STB, and the 2-μm circle-encoded proteins Rep1p and Rep2p (Hadfield et al. 1995). Immunolocalization studies of these two proteins that bind the STB locus show a staining of about four to eight foci in the nucleus, although the 2-μm circle is usually maintained at a high copy number of 30–100 per cell (Scott-Drew and Murray 1998). We performed FISH experiments with a probe for the 2-μm circle and found them to be clustered in 4–10 foci in late G1 phase (Fig. 6d and Fig. e). Strikingly, the 2-μm clusters are virtually excluded from the nuclear periphery with a very strong enrichment in internal nuclear zones 3 and 4 (Fig. 6 f). χ2 analysis indicates that this internal localization is highly significant (P < 0.001).
Late Firing ARS501 Distributes Randomly Away from Tel V-R When Excised after Establishment of its Late-firing State
It has been shown for a telomere-proximal late-firing origin that the establishment of the late replication program in yeast is cell-cycle dependent (Raghuraman et al. 1997). Using site-specific recombination to separate the late origin ARS501 from its associated telomere in vivo, excision of an origin at the G2/M transition was found to correlate with a switch from late to early firing, while excision in G1 allowed maintenance of the late-firing state. Thus, late initiation appeared to be determined by events that occur between mitosis and “Start” in mid-G1 phase. Using this system, we could ask whether or not the excised origin moves away from its adjacent telomere on chromosome V, once the late replication state is established in G1. With this analysis, we also determine whether a late-firing excised circle retains its perinuclear localization or is free to move throughout the nucleoplasm.
The yeast strain used for this study carries the temperature-sensitive cdc7-1 allele and direct repeats flanking ARS501 (GA-1205), enabling the excision of a 30-kb circle upon induction of a site-specific recombinase and subsequent synchronization immediately before initiation of DNA replication. Cells were first arrested either at G2/M with nocodazole or at Start in G1 with the pheromone α factor, and the recombinase was induced by the addition of galactose. After a 4-h incubation, release from the arrest points was achieved by transferring cells to inhibitor-free glucose-containing medium at 37°C. This results in two populations of cells synchronized just before initiation of DNA replication: one carries an ARS501 circle excised in midmitosis and the other excised in mid-G1 (see Materials and Methods). Cells were fixed and subjected to nuclear pore immunofluorescence and FISH with probes recognizing either ARS501 (red) or a nonexcised 5-kb sequence 14-kb closer to the right telomere of chromosome V (Fig. 7 a, green).
Figure 7.

Once the late timing is established, ARS501 requires neither telomere proximity nor a peripheral position to remain late firing. Haploid budding yeast cells with the temperature-sensitive cdc7-1 allele and the excisable 30-kb cassette bearing ARS501 (GA-1205; Raghuraman et al. 1997) were blocked with nocodazole at G2/M (c and e) or with α factor at Start in G1 (b and d). The site-specific recombinase was induced by addition of galactose. After a 4-h incubation, cells were released into glucose-containing medium at 37°C, which represses the recombinase and synchronizes cells at the G1/S boundary (see Materials and Methods). Cells were fixed and subjected to nuclear pore immunofluorescence (blue), FISH with ARS501 (red), and Tel V-R (green). In a, we show a scheme of the FISH probes recognizing 7 kb on the excisable ARS501 circle (red) and an adjacent 5-kb probe that is 14 kb closer to Tel V-R (green). The 30-kb ARS501 cassette is flanked by recombination sites specifically recognized by the R recombinase from Z. rouxii (Raghuraman et al. 1997). (b–e) Codetection of the two FISH probes has been performed as described above and representative confocal images of equatorial sections through hybridized nuclei are shown. In cells with preserved nuclear pore staining, subnuclear localization of the 30-kb ARS501 cassette was quantified for its position relative to either the telomere proximal locus on chromosome V or to the nuclear periphery using the line profile tool of LSM510 Confocal Software. Distance measurements between the maxima of two signals were categorized in groups from 0–99 nm = 100, 100–199 = 200 etc., and expressed in bar graphs in f (n nonexcised; excised = 42, 37) and g (n nonexcised; excised = 24, 22). The general position of the ARS501 cassette within the nucleus was analyzed by computing the ratio between the minimal distance from the pore of the FISH signal and the radius of the nucleus. Values <0.29 were scored as peripheral as in Fig. 3 b (see Materials and Methods). Percentages of peripheral (dark grey) or internal (light grey) localization are presented as bar graphs. Peripheral localization is expressed in percent in f (nnonexcised; excised = 48, 66) and g (nnon-excised; excised = 57, 48), insets. Analysis using a standard χ2 test revealed that the localization of ARS501 is significantly nonrandom before excision (P < 0.001), but not after (P = 0.35). Images were collected on an LSM 510 confocal microscope. Scale bar: 2 μm.
As expected, in control cells in which the recombinase is not induced, colocalization of the two probes is observed in >90% of the cells (Table , and Fig. 7, a–c), and these signals were found predominantly at the nuclear periphery even at the G1/S boundary (Fig. 7f and Fig. g, inset, −gal). Interestingly, in cells that had expressed recombinase, the two signals were separated regardless of whether excision took place in G1 phase or in mitosis (Fig. 7, d–g, +gal). We have scored the distance between the excised circle and Tel V-R and find that the distances between the signals range from 0.1 to 2.1 μm, spanning the nuclear diameter (Fig. 7, d–g). Thus, although association with the telomere in G1 is necessary to establish the late chromatin state, the 30-kb circle migrates rapidly from the telomere from which it was excised, regardless of whether it will fire early or late in S phase. By scoring the frequency with which the signals are found at the nuclear periphery, we detect no significant bias for a particular subnuclear position at the cdc7 arrest point, after excision (Fig. 7f and Fig. g, insets, and Table III; 56% of signals are peripheral). Statistical analyses confirm that the ARS501 signal is significantly peripheral at its chromosomal locus (P < 0.001), whereas its distribution is random when excised. In conclusion, excision either in mitosis or in G1 phase after establishment of the timing pattern releases the telomere-adjacent origin and its chromatin from the nuclear periphery. This also indicates that, once a late state has been established, association with the nuclear envelope is no longer necessary to maintain delayed initiation.
Table 3.
Localization of an Excised Subtelomeric Origin
| Colocalization of ARS501 signals with Tel V-R | Localization of ARS501 | n(nuclei scored) | |||
|---|---|---|---|---|---|
| % peripheral | % internal | ||||
| α-factor | −gal | 90% | 73 | 27 | 42; 48 |
| α-factor | +gal | 11% | 56 | 44 | 37; 66 |
| nocodazole | −gal | 92% | 81 | 19 | 24; 57 |
| nocodazole | +gal | 18% | 56 | 44 | 22; 48 |
Subnuclear localization of the 30-kb ARS501 cassette was quantified for its position relative to either the telomere proximal locus on Chromosome V or to the nuclear periphery, as identified by antinuclear pore. Data were obtained from experiments like those described in Fig. 7b–e, in which the ARS501 cassette was excised. Distance measurements between the maxima of two signals were performed using the line profile tool of LSM510 Confocal Software. Signals ≤300 nm apart were scored as colocalizing. For subnuclear localization of the ARS501 signal, the nucleus was divided in two zones of similar area: peripheral and internal (Fig. 3 b, and see Materials and Methods). Right column: n colocalization, n peripheral internal. Analysis using a standard χ2 test revealed that the localization of ARS501 was significantly different from random localization before (P < 0.001) but not after excision.
Origin Localization Is Dynamic and Changes during the Cell Cycle
The FISH analysis of the recombinase-sensitive telomere-proximal origin suggests that it can diffuse rapidly from Tel V-R upon excision. To confirm this in living cells, we have tagged origins fluorescently to follow their localization in vivo, making use of a lac operator/GFP-lac repressor system (Robinett et al. 1996). A repeat of 256 lac operator sites has been integrated in haploid wild-type cells (GA-1320) at the late origin cluster on chromosome XIV adjacent to ARS1413, or next to the early-activated origin on chromosome IV, 908 kb away from the left telomere (Fig. 8, a and b). These cells carry both a GFP-tagged copy of the nuclear pore protein Nup49p and the GFP-lac repressor fusion protein. The binding of the lac repressor to the multimerized lac sites results in a strong signal that can easily be distinguished from the weaker GFP-nuclear envelope fluorescence, allowing us to monitor the relative position of the tagged origins with respect to the nuclear periphery. Control experiments confirm that the GFP tagging does not significantly alter the timing of replication of the modified region (F. Neumann, personal communication).
Figure 8.

Dynamics of origins in vivo: a nontelomeric late origin becomes increasingly nonperipheral in S and G2 phases. Origin localization has been followed in living cells by the use of the lac operator/lac repressor system. A repeat of 256 lac operator sites has been integrated in wild-type budding yeast cells (GA-1320), either at the late origin cluster on chromosome XIV, ∼10 kb upstream of ARS1413 (a), or ∼3 kb upstream of the early activated origin ChrIV-908 (d, see Materials and Methods). These cells also carry a GFP-Nup49p fusion protein to monitor the nuclear periphery. Log phase cells (<106 cells/ml) were concentrated by a short centrifugation and transferred to agar lacking histidine on a microscope slide. Transmission and fluorescence microscopy pictures were taken on an Olympus microscope coupled to the TillVision® imaging system. In nuclei where the GFP-tagged origin and the nuclear equator coincide in the same focal plane, origin position from the midpore signal was determined using the line profile tool of LSM510 Confocal Software. Distances were classified peripheral or internal using the 50% methods described in Fig. 3 b and 7. Cell-cycle stages were based on both cellular and nuclear shape and categorized as G1, early S, mid S, lateS/G2, and M phase (see Materials and Methods). Shown are results for the late-origin cluster on chromosome XIV (c; n = 161 nuclei) and the early activated origin ChrIV-908 (f; n = 165 nuclei). (b and e) One cell for each stage is shown as merged transmission/fluorescence images (top) and the fluorescence alone (bottom). A χ2 test demonstrates no significant difference from random localization for any origin at any cell-cycle phase, except for the late origin cluster on chromosome XIV in G1 (P < 0.001). Bar = 2 μm.
Cells growing exponentially were analyzed by fluorescence microscopy for origin positioning, and the distance between the origins and the nuclear pore fluorescence were measured on 2-D midcell confocal sections. Cells were categorized as G1, early S, mid-S, late S/G2, or M phase based on their cellular and nuclear morphology monitored by both transmission and fluorescence microscopy (see Materials and Methods). By classifying the distances into peripheral and internal zones, we find, in agreement with our FISH data, that the late replicating cluster on chromosome XIV is significantly enriched at the nuclear periphery in G1 phase (Fig. 8 c; P < 0.001 for ARS1413 in G1 phase cells), although this is lost as soon as cells progress into S phase. In contrast, the early replicating ChrIV-908 is randomly localized in both G1- and S-phase cells (Fig. 8 d). Intriguingly, the loss of the peripheral bias for the late-replicating ChrXIV cluster can be scored in cells with a very small bud, which is at least 20 min before the late ARS1413 fires (Fig. 8 c).
Time-lapse movies of these origin-tagged cells allow us to analyze origin movement as a single cell progresses through the cell cycle. These show that both early- and late-firing GFP-tagged origins are highly dynamic, particularly in G1 phase, revealing significant movements within the nucleoplasm (see online supplement). Two types of movements are observed, those that are small and oscillatory (≤0.3 μm), and others that reflect a shift of at least 0.5 μm, within a time scale of seconds (Laroche, T., P. Heun, and S.M. Gasser, unpublished results). The rapid oscillatory movement observed for both early- and late-firing origins suggests that the peripheral enrichment observed for the late-firing ARS1413 in G1 phase (Fig. 8 c) reflects a statistical probability of finding a given origin in this subnuclear region, and not the fact that a subpopulation of nuclei (75%) have a highly fixed peripheral position. Quantitative analysis will allow further characterization of the movement of these domains.
Discussion
In this study, we have examined the localization of early- and late-firing origins in budding yeast interphase nuclei, when they are present in their native chromosomal location, on autonomously replicating plasmids, or on an extrachromosomal element that results from a recombinase-mediated excision event. Using two independent, quantitative approaches (FISH signals on fixed cells, and the analysis of GFP-lac-repressor–tagged origins in living cells), we find that late-firing origins are detected preferentially at the nuclear periphery in G1 phase, while early-firing origins are randomly distributed. This is true both for subtelomeric late origins, which are peripheral due to their proximity to a telomere (e.g., ARS501), and for late origins that lie far from telomeres in linear distance along the chromosomal arm (e.g., ARS1413, ARS1412, ChrIV-210, etc.). The peripheral localization of late firing origins in G1 phase is not, however, as pronounced as that of telomeres, and time-lapse microscopy indicates that the perinuclear anchoring of late-firing origins is transient.
What could be the physiological significance of finding late-firing origins more frequently at the nuclear periphery in G1? First, it is important to note that this preferential positioning coincides with the window between mitosis and α-factor arrest (Start) when the timing of origin firing is established in yeast (Raghuraman et al. 1997). Moreover, it has been proposed that a local chromatin state, which relies on DNA sequences flanking an origin, is critical for the regulation of origin timing (Friedman et al. 1996). Indeed, we show here that a plasmid-borne origin (ARS1412) that contains the late-determining flanking sequences is found more frequently at the nuclear periphery in G1 phase, while early-replicating episomes containing the same origin without flanking DNA, as well as the 2-μm circle, are not. Since we find that a given nontelomeric late-firing origin is found near the nuclear periphery in 70–80% of the nuclei during this critical cell-cycle stage, we propose that enzymes or assembly factors important for establishing a particular chromatin structure are themselves perinuclear or at least function better in this subnuclear zone.
Three lines of evidence indicate that the positioning of late origins at the nuclear periphery is not stable, and that a peripheral attachment is not necessary to maintain the late-firing character in S phase. First, using time-lapse microscopy, we note a dramatic movement of GFP-tagged origins over distances as large as a third of the haploid nuclear diameter within seconds. This movement occurs independently of whether the origin fires early or late. The enrichment of late origins at the nuclear periphery therefore reflects an increased likelihood that a given origin will be present in this peripheral zone at a given moment in G1. Moreover, although ARS501 is clearly associated with the telomere of ChrV at the nuclear periphery in G1 phase, this same region diffuses rapidly away from the tethered telomere when it is excised from the chromosomal arm, whether it will fire early or late (Fig. 7). Finally, if we correlate the position of GFP-tagged origins with cell-cycle stages (G1, and early-, mid-, and late-S phase), we see that the preferential peripheral localization of late origins scored in G1 cells is largely lost in S phase, although the timing of firing is not compromised (Fig. 8).
Taken together, these results suggest a model in which the necessary prerequisites for late firing would be both contact with the nuclear periphery in G1 and the presence of “late determining DNA elements.” These would either help target the domain to the nuclear periphery in early G1, or allow assembly of a structure capable of retarding the initiation event, or both (Fig. 9). Once established, the modified late chromatin structure appears to be autonomous and mobile. Our data further suggest that a peripheral localization alone is not sufficient to confer delayed initiation, since early replicating origins are found with equal efficiency at internal sites as at the nuclear periphery using both FISH and GFP fluorescence.
Figure 9.

This model proposes that DNA elements flanking late firing origins promote association with the nuclear periphery in early G1 phase, an event necessary for the establishment of a late firing chromatin state. Our data suggest that it is not necessary to maintain perinuclear positioning to ensure that the origin fires late in S phase. Once formed, late chromatin may retard initiation by rendering the pre–regulatory complex less accessible to specific factors required for initiation, such as Cdc45p, Polα-primase, or either of the regulatory kinases Clb5/Cdc28 or Dbf4/Cdc7. The initiation of replication at early origins may also send signals through factors such as Rad53p to help repress initiation at late-activated origins.
The dynamic character of late origin localization contrasts with the more stable association of telomeres with components of the nuclear envelope (Gotta et al. 1996; Laroche et al. 1998), and suggests that there are at least two pathways for achieving perinuclear association. This is consistent with the stronger enrichment of subtelomeric Y′ elements at the extreme nuclear periphery as compared with other late origins (Fig. 3 a), and with the lack of colocalization between late-firing origins and telomeres (Fig. 5, and Table ).
What is the nature of this “late firing chromatin” state that appears to be assembled at the nuclear periphery? Here it helps to draw distinctions between three types of late origins. The first includes origins that are immediately adjacent to telomeres (Y′ or X elements). In these, the late firing character appears to result from the presence of SIR complexes that propagate along the nucleosomal fiber and restrict access of the DNA to large enzymes (e.g., RNA polymerases, DAM methyltransferase). Such origins show a SIR-dependent repression of firing (Stevenson and Gottschling 1999). The second class of late-firing origin is more internal, but still telomere-proximal, such as ARS501 (∼28 kb from the right telomere of ChrV). Although its late activation clearly depends on the presence of a nearby telomere (Ferguson and Fangman 1992), it is not affected by sir mutations (Stevenson and Gottschling 1999). ARS501 may nonetheless have an unusual nucleosomal ordering typical for chromosomal ends, since depletion of histone H4 affects gene expression of a large number of genes up to 20 kb from yeast telomeres, while sir mutations only affect genes immediately adjacent to the TG repeat (Wyrick et al. 1999). The third type includes internal late-firing origins such as ARS1412 and ARS1413, which are far from telomeres and fire late in a SIR-independent manner. Despite the fact that these origins are flanked by actively transcribed genes, they may still have characteristic patterns of histone modification that mark them for late initiation or for contact with the nuclear periphery in G1, without silencing transcription.
The presence of tightly positioned nucleosomes in the regions flanking late origins may favor the formation of a compact or folded chromatin structure that could also delay initiation. Such patterns have been observed at the recombination enhancer on ChrIII, which regulates recombination between the MAT locus with HM loci at a distance (Weiss and Simpson 1998). The resulting higher-order folding of the chromatin fiber in this region could interfere sterically with one or more components of the machinery required for initiation. Candidates for excluded factors include Cdc45p, Polα-primase, RFA, RFC, or regulatory kinases such as the Clb5/Cdc28 or Dbf4/Cdc7 complexes. Alternatively, the prereplication complex itself may have a different composition at the late firing origins, again possibly due to its assembly near the nuclear periphery. Finally, unidentified components that modulate initiation may be present at late-firing origins, or known components could be modified in a characteristic manner (e.g., ubiquitination, phosphorylation, methylation, or acetylation). Since it is unlikely that we will be able to biochemically purify early- and late-firing origins in significant amounts, distinguishing between these models will require the use of genetics and chromatin immunoprecipitation analyses in mutant cells.
How do our findings relate to the control of early- and late-DNA replication in mammalian cells? It is well-documented that heterochromatin replicates late and has a characteristic perinuclear and/or perinucleolar position in mammalian cells (O'Keefe et al. 1992; Li et al. 1998; Ma et al. 1998; Croft et al. 1999). The formation of this characteristic pattern of chromatin distribution has been shown to be established in early G1, roughly 2 h after mitosis (Dimitrova and Gilbert 1999), and correlates well with the observation described in this paper. Although several studies suggest that DNA regions move little during interphase in mammalian cells (confined to <0.3 μm; Shelby et al. 1996; Abney et al. 1997; Marshall et al. 1997; Zink et al. 1998; Manders et al. 1999), large-scale chromosomal movements have been observed during interphase in Drosophila (Csink and Henikoff 1998). The more highly compacted nature of mammalian heterochromatin may render it less dynamic. Moreover, mammalian nuclei have elements of nuclear substructure that are absent or modified in yeast, such as the nuclear lamina (Moir et al. 2000), NuMA (Harborth and Osborn 1999), and the tetratricopeptide repeat (TPR) filaments that appear to extend into the nuclear interior (Cordes et al. 1997; Bangs et al. 1998; Strambio-de-Castillia et al. 1999). In contrast to higher eukaryotic cells, the equivalent of the TPR fibers (Myosin-like proteins 1 and 2) are perinuclear in budding yeast (Galy et al. 2000).
Most studies on late mammalian origins have examined the late replication of satellite or repeated DNA elements, which are probably most comparable with subtelomeric regions in yeast. It remains to be seen whether there are late-firing origins in transcriptionally active regions of the mammalian genome, and whether these have a unique position in the interphase nucleus. An intriguing study by Bridger et al. 2000 has shown that the late-replicating chromosome 18 is largely peripheral in the nuclei of cultured human cells, although it loses this position when the cells entered quiescence. Upon restimulation of growth, the chromosome remains late-replicating, despite loss of its peripheral location. This is highly reminiscent of our observations in yeast, which suggest that the peripheral association is not necessary in S phase, but may be needed to establish the late-firing state in G1. Parallels such as these suggest that studies in yeast on the character of late-firing origins will provide useful paradigms for identifying the cause of late replication in more complex nuclei.
Supplemental Material
Acknowledgments
The data on origin timing were generously provided from a manuscript in preperation by M.K. Raghuraman, D. Collingwood, W. Winzeler, S. Hunt, L. Wodicka, A. Conway, D. Lockhart, R.W. Davis, B. Braurer, and W. Fangman. We thank Marc Gasser and Anya Wilson for their technical assistance, Frank Neumann for determining the timing of replication on tagged ARS1413 and ARS1, and Dr. J.-P. Antonietti (University of Lausanne, Lausanne, Switzerland) for help with statistical analyses. We thank Drs. C. Newlon, E. Louis, V. Doye, G. Simchen, and M. Peter (ISREC) for strains and plasmids. We thank Dr. Philippe Pasero (Centre National de la Recherche Scientifique, Montpellier, France) for suggesting this project and for helpful discussions, and Drs. Bernard Duncker, Moira Cockell, and Joachim Lingner (ISREC) for critically reading the manuscript.
This study was supported in part by a predoctoral fellowship from Boehringer Ingelheim Foundation (P. Heun), and the Roche Foundation. The Gasser laboratory is funded by grants from the Swiss National Science Foundation and the Swiss Cancer League.
Footnotes
The online version of this article contains supplemental material.
Abbreviations used in this paper: 3-D, three-dimensional; ARS, autonomously replicating sequence; GFP, green fluorescent protein; IF/FISH, immunofluorescence/in situ hybridization.
References
- Abney J.R., Cutler B., Fillbach M.L., Axelrod D., Scalettar B.A. Chromatin dynamics in interphase nuclei and its implications for nuclear structure. J. Cell Biol. 1997;137:1459–1468. doi: 10.1083/jcb.137.7.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangs P., Burke B., Powers C., Craig R., Purohit A., Doxsey S. Functional analysis of Tpridentification of nuclear pore complex association and nuclear localization domains and a role in mRNA export. J. Cell Biol. 1998;143:1801–1812. doi: 10.1083/jcb.143.7.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgareh N., Doye V. Dynamics of nuclear pore distribution in nucleoporin mutant yeast cells. J. Cell Biol. 1997;136:747–759. doi: 10.1083/jcb.136.4.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousset K., Diffley J.F. The Cdc7 protein kinase is required for origin firing during S phase. Genes Dev. 1998;12:480–490. doi: 10.1101/gad.12.4.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer B.J., Fangman W.L. The localization of replication origins on ARS plasmids in S. cerevisiae. Cell. 1987;51:463–471. doi: 10.1016/0092-8674(87)90642-8. [DOI] [PubMed] [Google Scholar]
- Bridger J.M., Boyle S., Kill I.R., Bickmore W.A. Re-modelling of nuclear architecture in quiescent and senescent human fibroblasts. Curr. Biol. 2000;10:149–152. doi: 10.1016/s0960-9822(00)00312-2. [DOI] [PubMed] [Google Scholar]
- Campbell J.L., Newlon C.S. Chromosomal DNA replication. In: Broach J.R., Pringle J., Jones E., editors. The Molecular Biology of the Yeast Saccharomyces cerevisiaeGenome Dynamics, Protein Synthesis and Energetics.Vol. 1. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1991. pp. 41–146. [Google Scholar]
- Cordes V.C., Reidenbach S., Rackwitz H.R., Franke W.W. Identification of protein p270/Tpr as a constitutive component of the nuclear pore complex-attached intranuclear filaments. J. Cell Biol. 1997;136:515–529. doi: 10.1083/jcb.136.3.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft J.A., Bridger J.M., Boyle S., Perry P., Teague P., Bickmore W.A. Differences in the localization and morphology of chromosomes in the human nucleus. J. Cell Biol. 1999;145:1119–1131. doi: 10.1083/jcb.145.6.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csink A.K., Henikoff S. Large-scale chromosomal movements during interphase progression in Drosophila . J. Cell Biol. 1998;143:13–22. doi: 10.1083/jcb.143.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova D.S., Gilbert D.M. The spatial position and replication timing of chromosomal domains are both established in early G1 phase. Mol. Cell. 1999;4:983–993. doi: 10.1016/s1097-2765(00)80227-0. [DOI] [PubMed] [Google Scholar]
- Dohrmann P.R., Oshiro G., Tecklenburg M., Sclafani R.A. RAD53 regulates DBF4 independently of checkpoint function in Saccharomyces cerevisiae . Genetics. 1999;151:965–977. doi: 10.1093/genetics/151.3.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson A.D., Fangman W.L., Brewer B.J. Cdc7 is required throughout the yeast S phase to activate replication origins Genes Dev 12 1998. 491 501a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson A.D., Raghuraman M.K., Friedman K.L., Cross F.R., Brewer B.J., Fangman W.L. CLB5-dependent activation of late replication origins in S. cerevisiae Mol. Cell. 2 1998. 173 182b [DOI] [PubMed] [Google Scholar]
- Fangman W.L., Brewer B.J. A question of timereplication origins of eukaryotic chromosomes. Cell. 1992;71:363–366. doi: 10.1016/0092-8674(92)90505-7. [DOI] [PubMed] [Google Scholar]
- Ferguson B.M., Brewer B.J., Reynolds A.E., Fangman W.L. A yeast origin of replication is activated late in S phase. Cell. 1991;65:507–515. doi: 10.1016/0092-8674(91)90468-e. [DOI] [PubMed] [Google Scholar]
- Ferguson B.M., Fangman W.L. A position effect on the time of replication origin activation in yeast. Cell. 1992;68:333–339. doi: 10.1016/0092-8674(92)90474-q. [DOI] [PubMed] [Google Scholar]
- Fox M.H., Arndt-Jovin D.J., Jovin T.M., Baumann P.H., Robert-Nicoud M. Spatial and temporal distribution of DNA replication sites localized by immunofluorescence and confocal microscopy in mouse fibroblasts. J. Cell Sci. 1991;99:247–253. doi: 10.1242/jcs.99.2.247. [DOI] [PubMed] [Google Scholar]
- Friedman K.L., Brewer B.J., Fangman W.L. Replication profile of Saccharomyces cerevisiae chromosome VI. Genes Cells. 1997;2:667–678. doi: 10.1046/j.1365-2443.1997.1520350.x. [DOI] [PubMed] [Google Scholar]
- Friedman K.L., Diller J.D., Ferguson B.M., Nyland S.V., Brewer B.J., Fangman W.L. Multiple determinants controlling activation of yeast replication origins late in S phase. Genes Dev. 1996;10:1595–1607. doi: 10.1101/gad.10.13.1595. [DOI] [PubMed] [Google Scholar]
- Galy V., Olivo-Marin J.C., Scherthan H., Doye V., Rascalou N., Nehrbass U. Nuclear pore complexes in the organization of silent telomeric chromatin. Nature. 2000;403:108–112. doi: 10.1038/47528. [DOI] [PubMed] [Google Scholar]
- Gotta M., Laroche T., Formenton A., Maillet L., Scherthan H., Gasser S.M. The clustering of telomeres and co-localization with Rap1, Sir3, and Sir4 proteins in wild-type Saccharomyces cerevisiae . J. Cell Biol. 1996;134:1349–1363. doi: 10.1083/jcb.134.6.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotta M., Laroche T., Gasser S.M. Analysis of nuclear organization in Saccharomyces cerevisiae . Methods Enzymol. 1999;304:663–672. doi: 10.1016/s0076-6879(99)04040-9. [DOI] [PubMed] [Google Scholar]
- Guacci V., Hogan E., Koshland D. Chromosome condensation and sister chromatid pairing in budding yeast. J. Cell Biol. 1994;125:517–530. doi: 10.1083/jcb.125.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadfield C., Mount R.C., Cashmore A.M. Protein binding interactions at the STB locus of the yeast 2 microns plasmid. Nucleic Acids Res. 1995;23:995–1002. doi: 10.1093/nar/23.6.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harborth J., Osborn M. Does NuMA have a scaffold function in the interphase nucleus? Crit. Rev. Eukaryot. Gene Expr. 1999;9:319–328. doi: 10.1615/critreveukargeneexpr.v9.i3-4.160. [DOI] [PubMed] [Google Scholar]
- Hayashi A., Ogawa H., Kohno K., Gasser S.M., Hiraoka Y. Meiotic behaviours of chromosomes and microtubules in budding yeastrelocalization of centromeres and telomeres during meiotic prophase. Genes Cells. 1998;3:587–601. doi: 10.1046/j.1365-2443.1998.00215.x. [DOI] [PubMed] [Google Scholar]
- Holmquist G.P. Role of replication time in the control of tissue-specific gene expression. Am. J. Hum. Genet. 1987;40:151–173. [PMC free article] [PubMed] [Google Scholar]
- Jin Q., Fuchs J., Loidl J. Centromere clustering is a major determinant of yeast interphase nuclear organization. J. Cell Sci. 2000;113:1903–1912. doi: 10.1242/jcs.113.11.1903. [DOI] [PubMed] [Google Scholar]
- Jin Q., Trelles-Sticken E., Scherthan H., Loidl J. Yeast nuclei display prominent centromere clustering that is reduced in nondividing cells and in meiotic prophase. J. Cell Biol. 1998;141:21–29. doi: 10.1083/jcb.141.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein F., Laroche T., Cardenas M.E., Hofmann J.F., Schweizer D., Gasser S.M. Localization of RAP1 and topoisomerase II in nuclei and meiotic chromosomes of yeast. J. Cell Biol. 1992;117:935–948. doi: 10.1083/jcb.117.5.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laroche T., Martin S.G., Gotta M., Gorham H.C., Pryde F.E., Louis E.J., Gasser S.M. Mutation of yeast Ku genes disrupts the subnuclear organization of telomeres. Curr. Biol. 1998;8:653–656. doi: 10.1016/s0960-9822(98)70252-0. [DOI] [PubMed] [Google Scholar]
- Laroche T., Martin S.G., Tsai-Pflugfelder M., Gasser S.M. The dynamics of yeast telomeres and silencing proteins through the cell cycle. J. Struct. Biol. 2000;129:159–174. doi: 10.1006/jsbi.2000.4240. [DOI] [PubMed] [Google Scholar]
- Leonhardt H., Rahn H.P., Weinzierl P., Sporbert A., Cremer T., Zink D., Cardoso M.C. Dynamics of DNA replication factories in living cells. J. Cell Biol. 2000;149:271–280. doi: 10.1083/jcb.149.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., Sudlow G., Belmont A.S. Interphase cell cycle dynamics of a late-replicating, heterochromatic homogeneously staining regionprecise choreography of condensation/decondensation and nuclear positioning. J. Cell Biol. 1998;140:975–989. doi: 10.1083/jcb.140.5.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loidl J., Klein F., Engebrecht J. Genetic and morphological approaches for the analysis of meiotic chromosomes in yeast. Methods Cell Biol. 1998;53:257–285. doi: 10.1016/s0091-679x(08)60882-1. [DOI] [PubMed] [Google Scholar]
- Louis E.J., Naumova E.S., Lee A., Naumov G., Haber J.E. The chromosome end in yeastits mosaic nature and influence on recombinational dynamics. Genetics. 1994;136:789–802. doi: 10.1093/genetics/136.3.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H., Samarabandu J., Devdhar R.S., Acharya R., Cheng P.C., Meng C., Berezney R. Spatial and temporal dynamics of DNA replication sites in mammalian cells. J. Cell Biol. 1998;143:1415–1425. doi: 10.1083/jcb.143.6.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manders E.M., Kimura H., Cook P.R. Direct imaging of DNA in living cells reveals the dynamics of chromosome formation. J. Cell Biol. 1999;144:813–821. doi: 10.1083/jcb.144.5.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manders E.M., Stap J., Brakenhoff G.J., van Driel R., Aten J.A. Dynamics of three-dimensional replication patterns during the S-phase, analyzed by double labelling of DNA and confocal microscopy. J. Cell Sci. 1992;103:857–862. doi: 10.1242/jcs.103.3.857. [DOI] [PubMed] [Google Scholar]
- Marshall W.F., Straight A., Marko J.F., Swedlow J., Dernburg A., Belmont A., Murray A.W., Agard D.A., Sedat J.W. Interphase chromosomes undergo constrained diffusional motion in living cells. Curr. Biol. 1997;7:930–939. doi: 10.1016/s0960-9822(06)00412-x. [DOI] [PubMed] [Google Scholar]
- Martin S.G., Laroche T., Suka N., Grunstein M., Gasser S.M. Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell. 1999;97:621–633. doi: 10.1016/s0092-8674(00)80773-4. [DOI] [PubMed] [Google Scholar]
- McCarroll R.M., Fangman W.L. Time of replication of yeast centromeres and telomeres. Cell. 1988;54:505–513. doi: 10.1016/0092-8674(88)90072-4. [DOI] [PubMed] [Google Scholar]
- Moir R.D., Spann T.P., Lopez-Soler R.I., Yoon M., Goldman A.E., Khuon S., Goldman R.D. Reviewthe dynamics of the nuclear lamins during the cell cycle—relationship between structure and function. J. Struct. Biol. 2000;129:324–334. doi: 10.1006/jsbi.2000.4251. [DOI] [PubMed] [Google Scholar]
- Nakamura H., Morita T., Sato C. Structural organizations of replicon domains during DNA synthetic phase in the mammalian nucleus. Exp. Cell Res. 1986;165:291–297. doi: 10.1016/0014-4827(86)90583-5. [DOI] [PubMed] [Google Scholar]
- Nakayasu H., Berezney R. Mapping replicational sites in the eucaryotic cell nucleus. J. Cell Biol. 1989;108:1–11. doi: 10.1083/jcb.108.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newlon C.S. Putting it all togetherbuilding a prereplicative complex. Cell. 1997;91:717–720. doi: 10.1016/s0092-8674(00)80459-6. [DOI] [PubMed] [Google Scholar]
- Newlon C.S., Lipchitz L.R., Collins I., Deshpande A., Devenish R.J., Green R.P., Klein H.L., Palzkill T.G., Ren R.B., Synn S. Analysis of a circular derivative of Saccharomyces cerevisiae chromosome IIIa physical map and identification and location of ARS elements. Genetics. 1991;129:343–357. doi: 10.1093/genetics/129.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Keefe R.T., Henderson S.C., Spector D.L. Dynamic organization of DNA replication in mammalian cell nucleispatially and temporally defined replication of chromosome-specific alpha-satellite DNA sequences. J. Cell Biol. 1992;116:1095–1110. doi: 10.1083/jcb.116.5.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palladino F., Laroche T., Gilson E., Axelrod A., Pillus L., Gasser S.M. SIR3 and SIR4 proteins are required for the positioning and integrity of yeast telomeres. Cell. 1993;75:543–555. doi: 10.1016/0092-8674(93)90388-7. [DOI] [PubMed] [Google Scholar]
- Pasero P., Braguglia D., Gasser S.M. ORC-dependent and origin-specific initiation of DNA replication at defined foci in isolated yeast nuclei. Genes Dev. 1997;11:1504–1518. doi: 10.1101/gad.11.12.1504. [DOI] [PubMed] [Google Scholar]
- Pasero P., Duncker B.P., Schwob E., Gasser S.M. A role for the Cdc7 kinase regulatory subunit Dbf4p in the formation of initiation-competent origins of replication. Genes Dev. 1999;13:2159–2176. doi: 10.1101/gad.13.16.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghuraman M.K., Brewer B.J., Fangman W.L. Cell cycle-dependent establishment of a late replication program. Science. 1997;276:806–809. doi: 10.1126/science.276.5313.806. [DOI] [PubMed] [Google Scholar]
- Robinett C.C., Straight A., Li G., Willhelm C., Sudlow G., Murray A., Belmont A.S. In vivo localization of DNA sequences and visualization of large-scale chromatin organization using lac operator/repressor recognition. J. Cell Biol. 1996;135:1685–1700. doi: 10.1083/jcb.135.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose M.D., Winston F., Hieter P. Methods in Yeast Genetics 1990. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: pp. 198 pp [Google Scholar]
- Santocanale C., Diffley J.F. ORC- and Cdc6-dependent complexes at active and inactive chromosomal replication origins in Saccharomyces cerevisiae . EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:6671–6679. [PMC free article] [PubMed] [Google Scholar]
- Santocanale C., Diffley J.F. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature. 1998;395:615–618. doi: 10.1038/27001. [DOI] [PubMed] [Google Scholar]
- Scott-Drew S., Murray J.A. Localisation and interaction of the protein components of the yeast 2 mu circle plasmid partitioning system suggest a mechanism for plasmid inheritance. J. Cell Sci. 1998;111:1779–1789. doi: 10.1242/jcs.111.13.1779. [DOI] [PubMed] [Google Scholar]
- Shelby R.D., Hahn K.M., Sullivan K.F. Dynamic elastic behavior of alpha-satellite DNA domains visualized in situ in living human cells. J. Cell Biol. 1996;135:545–557. doi: 10.1083/jcb.135.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirahige K., Hori Y., Shiraishi K., Yamashita M., Takahashi K., Obuse C., Tsurimoto T., Yoshikawa H. Regulation of DNA-replication origins during cell-cycle progression. Nature. 1998;395:618–621. doi: 10.1038/27007. [DOI] [PubMed] [Google Scholar]
- Shirahige K., Iwasaki T., Rashid M.B., Ogasawara N., Yoshikawa H. Location and characterization of autonomously replicating sequences from chromosome VI of Saccharomyces cerevisiae . Mol. Cell. Biol. 1993;13:5043–5056. doi: 10.1128/mcb.13.8.5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson J.B., Gottschling D.E. Telomeric chromatin modulates replication timing near chromosome ends. Genes Dev. 1999;13:146–151. doi: 10.1101/gad.13.2.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straight A.F., Belmont A.S., Robinett C.C., Murray A.W. GFP tagging of budding yeast chromosomes reveals that protein–protein interactions can mediate sister chromatid cohesion. Curr. Biol. 1996;6:1599–1608. doi: 10.1016/s0960-9822(02)70783-5. [DOI] [PubMed] [Google Scholar]
- Strambio-de-Castillia C., Blobel G., Rout M.P. Proteins connecting the nuclear pore complex with the nuclear interior. J. Cell Biol. 1999;144:839–855. doi: 10.1083/jcb.144.5.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei A., Roche D., Sibarita J.B., Turner B.M., Almouzni G. Duplication and maintenance of heterochromatin domains. J. Cell Biol. 1999;147:1153–1166. doi: 10.1083/jcb.147.6.1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dierendonck J.H., Keyzer R., van de Velde C.J., Cornelisse C.J. Subdivision of S-phase by analysis of nuclear 5-bromodeoxyuridine staining patterns. Cytometry. 1989;10:143–150. doi: 10.1002/cyto.990100205. [DOI] [PubMed] [Google Scholar]
- Webb R.H. Theoretical basis of confocal microscopy. Methods Enzymol. 1999;307:3–20. doi: 10.1016/s0076-6879(99)07003-2. [DOI] [PubMed] [Google Scholar]
- Weiner B.M., Kleckner N. Chromosome pairing via multiple interstitial interactions before and during meiosis in yeast. Cell. 1994;77:977–991. doi: 10.1016/0092-8674(94)90438-3. [DOI] [PubMed] [Google Scholar]
- Weinreich M., Stillman B. Cdc7p-Dbf4p kinase binds to chromatin during S phase and is regulated by both the APC and the RAD53 checkpoint pathway. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:5334–5346. doi: 10.1093/emboj/18.19.5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss K., Simpson R.T. High-resolution structural analysis of chromatin at specific lociSaccharomyces cerevisiae silent mating type locus HMLalpha. Mol. Cell. Biol. 1998;18:5392–5403. doi: 10.1128/mcb.18.9.5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyrick J.J., Holstege F.C., Jennings E.G., Causton H.C., Shore D., Grunstein M., Lander E.S., Young R.A. Chromosomal landscape of nucleosome-dependent gene expression and silencing in yeast. Nature. 1999;402:418–421. doi: 10.1038/46567. [DOI] [PubMed] [Google Scholar]
- Zakian V.A., Brewer B.J., Fangman W.L. Replication of each copy of the yeast 2 micron DNA plasmid occurs during the S phase. Cell. 1979;17:923–934. doi: 10.1016/0092-8674(79)90332-5. [DOI] [PubMed] [Google Scholar]
- Zink D., Cremer T., Saffrich R., Fischer R., Trendelenburg M.F., Ansorge W., Stelzer E.H. Structure and dynamics of human interphase chromosome territories in vivo. Hum. Genet. 1998;102:241–251. doi: 10.1007/s004390050686. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.