

Abstract
We report a cell-free system that measures transport-coupled maturation of carboxypeptidase Y (CPY). Yeast spheroplasts are lysed by extrusion through polycarbonate filters. After differential centrifugation, a 125,000-g pellet is enriched for radiolabeled proCPY and is used as “donor” membranes. A 15,000-g pellet, harvested from nonradiolabeled cells and enriched for vacuoles, is used as “acceptor” membranes. When these membranes are incubated together with ATP and cytosolic extracts, ∼50% of the radiolabeled proCPY is processed to mature CPY. Maturation was inhibited by dilution of donor and acceptor membranes during incubation, showed a 15-min lag period, and was temperature sensitive. Efficient proCPY maturation was possible when donor membranes were from a yeast strain deleted for the PEP4 gene (which encodes the principal CPY processing enzyme, proteinase A) and acceptor membranes from a PEP4 yeast strain, indicating intercompartmental transfer. Cytosol made from a yeast strain deleted for the VPS33 gene was less efficient at driving transport. Moreover, antibodies against Vps33p (a Sec1 homologue) and Vam3p (a Q-SNARE) inhibited transport >90%. Cytosolic extracts from yeast cells overexpressing Vps33p restored transport to antibody-inhibited assays. This cell-free system has allowed the demonstration of reconstituted intercompartmental transport coupled to the function of a VPS gene product.
Keywords: carboxypeptidase Y, lysosome, membrane fusion, Saccharomyces cerevisiae, vacuole
The secretory and endocytic pathways in eukaryotic cells comprise a series of intercompartmental transport events. The directed movement of protein and lipid from the endoplasmic reticulum to the plasma membrane or from the plasma membrane to the lysosome necessarily engages transfer between multiple organelles. In most cases, carrier vesicles mediate the traffic of protein and lipid cargo from one subcellular compartment to another.
Two integral approaches, genetics and biochemistry, continue to contribute preeminently in elucidating the molecular details of vesicle-mediated transport in the secretory and endocytic pathways. Mutant isolation screens and selections in such diverse organisms as Drosophila melanogaster (Swanson et al. 1998), Caenorhabditis elegans (Brenner 1974), and Saccharomyces cerevisiae (Novick and Schekman 1979; Deshaies and Schekman 1987) have not only uncovered hundreds of genes, but also helped reveal the ubiquitous nature of secretion and endocytosis among eukaryotic organisms. These genetic efforts were pioneered in yeast and include >20 secretion (sec) mutants (Novick et al. 1980), >40 mutants defective for vacuolar protein sorting (vps) (Robinson et al. 1988; Rothman et al. 1989), and >10 mutants defective for endocytosis (end) (Raths et al. 1993; Munn and Riezman 1994; Munn et al. 1995). Biochemical efforts using reconstitution assays have also uncovered many proteins involved in vesicle-mediated transport. These assays are focused on anterograde and retrograde transfer between the ER and Golgi complex (Balch et al. 1988; Baker and Schekman 1989; Balch 1989; Spang and Schekman 1998), intra-Golgi transport (Balch et al. 1984), fusion of secretory vesicles with the plasma membrane (Martin and Kowalchyk 1997), and recycling from late endosomes to the trans-Golgi network (Goda and Pfeffer 1988).
Despite the progress made in other vesicle-mediated events, a poorly understood intercompartmental step in eukaryotic cells continues to be transfer of proteins from prelysosomal compartments (PLC)1 to the lysosome. The PLC, or late endosome, plays a pivotal role in protein traffic since it is the organelle where the secretory and endocytic pathways converge. Resident lysosomal proteins pass through the PLC after being sorted away from secretory proteins in the trans-Golgi network (Pfeffer 1991). Similarly, cell surface proteins destined for degradation in the lysosome pass through the late endosome after endocytosis. Thus, eukaryotic cells must blend a variety of events for proper sorting, targeting, and delivery of proteins from the PLC. Several factors have concealed the molecular details of protein transport from the PLC to the lysosome. Perhaps the greatest source of confusion about the PLC revolves around whether carrier vesicles transport material to lysosomes or if the PLC undergoes a maturation process, changing into a lysosome (Futter et al. 1996). Evidence has been presented and interpreted to support both models. Even a hybrid of the vesicle-shuttle vs maturation models is suggested for PLC to lysosome transport in macrophages (Racoosin and Swanson 1993).
Saccharomyces cerevisiae contains not only a lysosome-like vacuole, but also a PLC-like prevacuolar compartment (PVC) (Raymond et al. 1990; Davis et al. 1993; Vida et al. 1993). The PVC is central to the function of nearly all VPS genes. The vps mutants either cause defects in anterograde or retrograde sorting/transport between the late Golgi complex and the PVC or in transport between the PVC and the vacuole (Bryant and Stevens 1998). Subsequent studies have focused on uncovering the function of VPS genes and are beginning to reveal aspects on the biochemistry of transport to and from the PVC because several of the gene products have biochemical activity in vitro. For example, the VPS1 gene product is a dynamin-like protein that can bind and hydrolyze (Vater et al. 1992). Likewise, the VPS21 gene encodes a protein that binds and hydrolyzes GTP but with similarity to the small G protein, Rab5 (Horazdovsky et al. 1994). The VPS4 gene product is an ATPase (Babst et al. 1997), VPS34 encodes a phosphatidylinositol-3 kinase (Stack and Emr 1994), and Vps15p is a serine/threonine protein kinase (Herman and Emr 1990; Stack et al. 1993). However, coupling the catalytic activity of a VPS gene product to intercompartmental protein transport in a reconstituted assay remains elusive. A permeabilized yeast spheroplast system (Vida et al. 1990) has not allowed analysis of VPS gene product function because the membranes are not depleted for any VPS protein that has been examined.
In this report, we describe an intercompartmental protein transport assay using partially purified organelles. This cell-free system measures proteolytic maturation of soluble vacuolar proenzymes such as carboxypeptidase Y and proteinase A after transfer from the PVC to the vacuole. The reaction is sensitive to membrane dilution, requires ATP, and cytosol. Importantly, cytosol made from a vps33Δ strain is deficient at stimulating transport in the new cell-free system. Furthermore, antibody raised against Vps33p can inhibit the assay >90% and cytosolic extracts made from strains overexpressing Vps33p can restore this inhibition. Thus, we have developed a transport-coupled assay for the function of a VPS gene product.
Materials and Methods
Media
Yeast strains were maintained on YPD media (1% yeast extract, 2% peptone, 2% dextrose, and 2.5% bacto-agar). Liquid media for radiolabeling and plasmid maintenance was Wickerham's minimal proline (WIMP) (Wickerham 1946) media supplemented with 0.5% yeast extract.
Strains and Plasmids
The yeast strains used in this study include BGY3300 (Gerhardt et al. 1998) MAT α ura3-52 leu2-3,112 his3-Δ200 trp-Δ901 lys2-801 suc2-Δ9 vps33Δ::HIS3. SEY6210 (Robinson et al. 1988) TVY614 MAT α ura3-52 leu2-3,112 his3-Δ200 trp 1-Δ901 lys2-801 suc2-Δ9 prc1Δ::HIS3 prb1Δ::hisG pep4Δ::LEU2; and TVY1 (Gerhardt et al. 1998) MAT α ura3-52 leu2-3,112 his3-Δ200 trp 1-Δ901 lys2-801 suc2-Δ9 pep4Δ::LEU2. Several constructs were made to put the VPS33 gene under control of the glyceraldehyde-3-phosphate dehydrogenase promoter (GPD1 pr). First, site-directed mutagenesis (Deng and Nickoloff 1992) was used to place a BamHI site at the second codon of the VPS33 gene and a SalI site ∼200 bp from the stop codon in pPRP33-100, which contains the complete VPS33 gene in pBluescript KS (Stratagene, Inc.). The resulting plasmid (pBG33BbSe) was digested with BamHI and SalI and subcloned into pGPD426 (Mumberg et al. 1995) to generate pGPD-BbSe-2. A six-histidine tag was placed at the NH2 terminus of VPS33 with the PCR using 5′-TACGGATCCATGAGAGGATCGCATCACCATCACCATCACGGTTCTAGATTTTGGAATACTAAG-3′ as the forward primer and 5′-CAAAAAAGCTTGCCTTTGTTGCAAAG-3′ as the reverse primer. The amplicon was digested with BamHI and ClaI and subcloned into pGPD-BbSe-2 to generate pGPDHIS633-2.
Antibody Production
Two previously described trpe-VPS33 fusion constructs (Banta et al. 1990) were expressed in E. coli and the insoluble fraction of cell lysates was prepared (Koerner et al. 1991). After SDS-PAGE, the trpe-Vps33p fusion proteins were cut out of the gel and the gel slice used as antigens in rabbits at Cocalico Biologicals, Inc. Antiserum against Vam3p was a generous gift of William Wickner (Dartmouth Medical School). A protein A–Sepharose column was used to purify total IgG from pre- and immune Vps33p rabbit sera.
Preparation of Cytosol
Yeast strain TVY614 was grown at 30°C in YPD (with 5% glucose) to an OD600 of 4–6 (usually 800–1,600 total OD600 units of cells were used). The cells were harvested with centrifugation at 5,000 rpm in a Beckman JA-14 rotor for 15 min. The cells were washed once with sterile distilled water (using 50% of the original volume of media) and harvested again as above. The washed cell pellet was resuspended in ∼35 ml of ice-cold 0.25 M sorbitol, 20 mM Hepes-KOH, 150 mM potassium acetate, and 5 mM magnesium acetate pH 7.0 (standard transport buffer, TB). The resuspended cells were transferred to a 50-ml conical tube and harvested via centrifugation at the highest setting (∼1,750 g) of a clinical centrifuge (International Equipment Co., Inc.) for 15 min at 4°C. The cells were resuspended in TB to 200 OD600/ml and transferred to 2-ml polypropylene tubes in 1-ml aliquots. Approximately 1 g of acid-washed glass beads (0.5 mm) was added and then agitated for three 30-s intervals in a Mini Bead-Beater (BioSpec Products, Inc.) at 4°C. All tubes were subjected to centrifugation at 1,500 g for 2 min. The supernatant was removed from each tube and the pellet was rinsed with 1 ml of TB, agitated briefly on a vortex mixer, and subjected to centrifugation at 1,500 g for 2 min. The second supernatant was pooled with the first supernatant and subjected to centrifugation in a Beckman TLA 100.3 rotor at 50,000 rpm (∼103,000 g average) for 10 min. The supernatant was removed, dispensed into small aliquots, and snap-frozen in liquid nitrogen. The protein concentration of all cytosolic extracts ranged from 25–50 mg/ml.
Cell Preparation for Donor and Acceptor Membranes
All steps are reported for the preparation of donor membranes from 25 OD600 units of cells. Whenever preparing more than this amount, volumes were scaled up proportionately. Yeast cells were grown in Wickerham's minimal proline (Wickerham 1946) media supplemented with 0.5% yeast extract at 30°C to an OD600 between 0.8–1.2. The cells were harvested with centrifugation at 1,500 g for 5 min and washed once with 25 ml of sterile distilled water. After harvesting the cells (1,500 g for 5 min), they were resuspended in 2.5 ml of 0.1 M Tris-HCl, pH 9.4, plus 10 mM DTT and incubated with shaking at 30°C for 15–30 min. After harvesting the cells (1,500 g for 5 min), they were resuspended in Wickerham's minimal proline media containing 0.2% glucose, 1.0 M sorbitol and 25 mM Tris-HCl, pH 7.5, to a total volume of 1 ml. The cells were converted to spheroplasts using 25 μg of Zymolyase 100T (Seikagaku America, Inc.) and 0.5% (vol/vol) glusulase (NEN Dupont) with gentle agitation for 20–30 min at 30°C. The cells were harvested (1,500 g for 5 min) and resuspended in 2.5 ml of Wickerham's minimal proline media containing 2% glucose and 1.0 M sorbitol. The cells were incubated with gentle shaking at 30°C for 15 min and then pulse-labeled with Tran35S-label (ICN, Inc.) at 200 μCi/ml for 5 min. After this pulse, methionine (5 mM final), cysteine (1 mM final), and yeast extract (0.5% final) were added and the cells were chased for 2 min. After the chase, the cells were transferred to 10 ml of ice-cold 1.0 M sorbitol, 20 mM Hepes-KOH, 150 mM potassium acetate, and 5 mM magnesium acetate (freezing buffer) and incubated on ice for 5 min. The cells were harvested (1,500 g for 5 min) and washed two times with 1 ml freezing buffer in a 1.7-ml polypropylene tube and a microcentrifuge (1 min at 16,000 g) at 4°C. Approximately 45 μl of freezing buffer was added to the washed cells. They were then resuspended, placed in a Nalgene™ 1°C cryofreezer jacketed with isopropanol (prechilled to 4°C), and the cryofreezer was incubated at −70°C for at least 45 min. All steps to prepare cells for acceptor membrane were identical to the above steps for donor membranes except for the following changes. Rich media, YPD, was used instead of WIMP and after cell wall removal, the spheroplasts were incubated at 30°C (at 10 OD600 units/ml) in YPD plus 1.0 M sorbitol for 60 min without shaking.
Preparation of Donor and Acceptor Membranes
If using frozen cells, they were thawed in a 25°C circulating water bath for 1 min and placed on ice. 600 μl of 0.6 M sorbitol with 5 mM Hepes-KOH, pH 7.0 (lysis buffer) was added and the cells were resuspended thoroughly. The cells were harvested by centrifugation for 1 min at 16,000 g and then resuspended to 8 OD600 units/ml in lysis buffer. The resuspended cells were pushed through a 13-mm polycarbonate filter (Nucleopore™; Corning) with 3-μm pores using a 3-ml syringe. The filter effluent was subjected to centrifugation at 440 g for 5 min to generate a P1 pellet and S1 supernatant fraction. The S1 supernatant was subjected to centrifugation at 15,000 g for 10 min to generate a P2 pellet (acceptor membranes) and S2 supernatant fraction. The S2 supernatant was subjected to centrifugation at 125,000 g for 10 min to generate a P3 pellet (donor membranes) and S3 supernatant fraction.
Cell-free Assays
Radiolabeled donor membranes and nonradiolabeled acceptor membranes were resuspended in TB. Standard conditions for assays were 50 μl total volume containing donor membranes (equivalent to 5 OD600 units of cells), 100–125 μg of acceptor membranes, 1 mM ATP, 40 mM creatine phosphate, 0.2 mg/ml creatine kinase, and 5 mg/ml cytosol. All reactions were assembled on ice and then incubated at 25°C in a circulating water bath for 60 min. To stop the reactions, 3 μl of 100 mM PMSF, 25 μl of 8.0 M urea, 5% SDS, and 5% NP-40 was added and they were boiled for 5 min. All samples were processed for immunoprecipitation, SDS-PAGE, and autoradiography as previously described (Vida et al. 1990). Autoradiograms were digitized with an Epson Expression 636 flatbed scanner and quantitation of the protein bands was done using NIH Image software (v 1.61).
Microscopy
All light microscopy images were obtained as previously described (Gerhardt et al. 1998). Yeast cells were stained with dichlorocarboxyfluorescein diacetate and FM4-64 (Molecular Probes, Inc., Eugene, OR) as previously described (Vida and Emr 1995). For electron microscopy, sample membrane pellets were fixed as previously described (Vida et al. 1993). After fixation, the samples were washed and treated with Millipore-filtered, cacodylate-buffered 0.1% tannic acid, postfixed with buffered 1% osmium tetroxide, and stained en bloc with Millipore-filtered aqueous 1% uranyl acetate. The samples were dehydrated in increasing concentrations of ethanol, infiltrated, and embedded in microcentrifuge tubes in Spurr's low viscosity medium. The samples were then polymerized in a 60°C oven for 2 d. Ultrathin sections were cut in an LKB Nova ultramicrotome (Leica), stained with uranyl acetate and lead citrate in an LKB Ultrostainer, and then examined in a JEOL 1200-EX transmission electron microscope at an acceleration voltage of 80 kV.
Results
A New Method to Lyse Yeast Spheroplasts
The usefulness of cell-free assays cannot be overstated in their contribution to our understanding of mechanisms in protein transport, secretion, and endocytosis. Since the development of a permeabilized cell assay for transport to the yeast vacuole (Vida et al. 1990), a longstanding goal has been to establish an intercompartmental transport assay using separate subcellular fractions in a cell-free system. The previous permeabilized cell assay is not cell-free per se because the organelles are housed in a broken plasma membrane, accessible only to exogenous cytosol and membrane impermeable compounds (Vida et al. 1990). This system poses severe limitations on assigning separate roles to individual membranes or for developing stage-specific subreactions. We began the effort for a truly cell-free assay by dissociating and dispersing intact organelles into separate membrane pellets that contained the donor organelle (PVC) and acceptor organelle (vacuole) from permeabilized cells using triethanolamine buffers. These conditions maintain organelle structural integrity while simultaneously dissociating membrane aggregates (Vida et al. 1993). However, these membranes were routinely devoid of transport activity (data not shown). We solved this problem and used another lysis method using polycarbonate filters with a defined pore size to gently shear away the plasma membrane.
The technique of passing cells through a small orifice to generate a crude lysate from shear forces was first used for mammalian cells. For example, cell homogenates have been prepared from PC12 cells by passing cell suspensions 15 times through a narrow clearance (10 μm) stainless steel ball homogenizer (Martin and Kowalchyk 1997). Stainless steel ball homogenizers have been instrumental at reconstituting several steps in the secretory pathway such as fusion of secretory vesicles with the plasma membrane and ER to Golgi transport (Balch and Rothman 1985). Rather than use a steel ball homogenizer, we used polycarbonate filters to shear yeast spheroplasts. Intact spheroplasts were suspended with an osmotic support of 0.6 M sorbitol giving them a diameter in the range of 5–8 μm. The cells were then forced through a 3-μm polycarbonate filter from a syringe (Fig. 1 A). Typically, in a single pass through the filter, >98% of the cells lysed to generate a crude homogenate.
Figure 1.
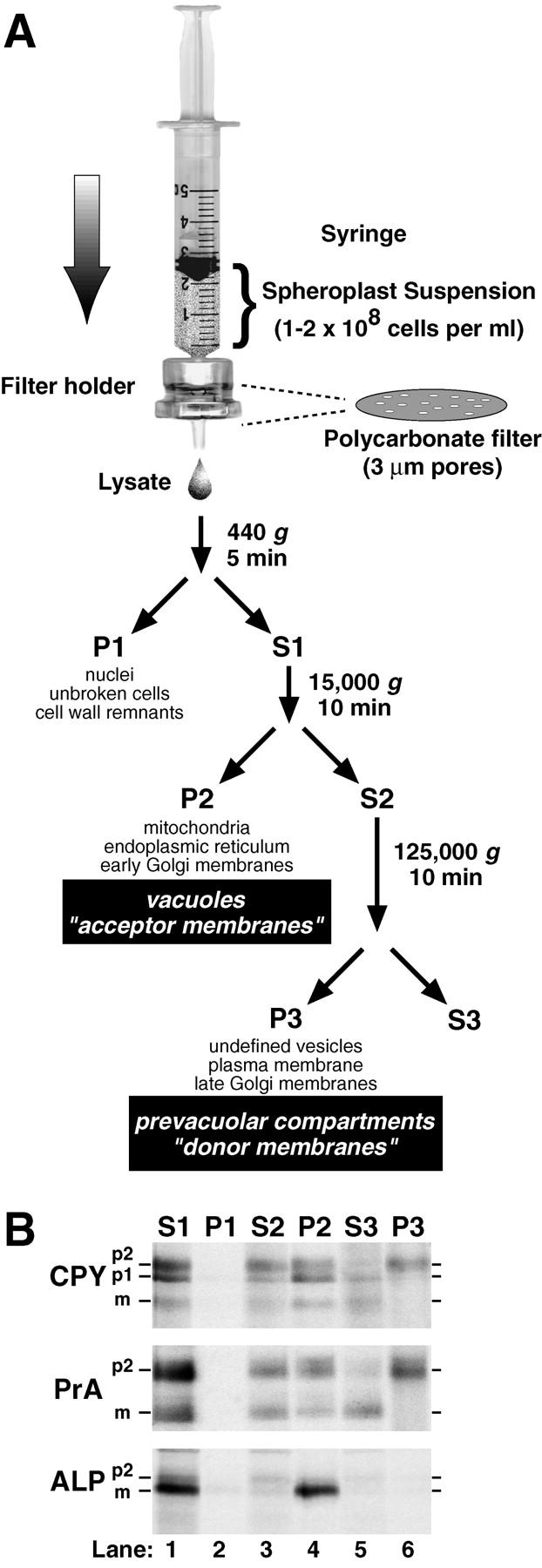
Lysis of yeast spheroplasts via extrusion through a polycarbonate filter. (A) Schematic diagram for polycarbonate filter lysis and subsequent differential centrifugation. After pushing a yeast spheroplast suspension in a syringe through a polycarbonate filter with 3-μm pores, the crude cell lysate was subjected to differential centrifugation using the indicated g forces and times. The P1, P2, and P3 pellets were enriched in the indicated organelles. (B) Fractionation of vacuolar marker proteins. Wild-type yeast spheroplasts (SEY6210) were radiolabeled with Tran35S-label for 5 min and chased with methionine and cysteine for 2 min at 30°C. The cells were subjected to lysis through a polycarbonate filter with subsequent differential centrifugation. Each supernatant and pellet from the 440 (S1 and P1), 15,000 (S2 and P2), and 125,000 g (S3 and P3) centrifugation steps was sequentially immunoprecipitated with antisera against carboxypeptidase Y (CPY), proteinase A (PrA), and alkaline phosphatase (ALP). Each immunoprecipitate was subjected to SDS-PAGE and autoradiography. The mid and late Golgi complex–modified precursor zymogen is designated p2, the endoplasmic reticulum and early Golgi complex precursor is designated p1 (CPY only), and the mature hydrolase is designated m for each protein.
We performed centrifugation techniques on crude lysates after extrusion through polycarbonate filters and examined each supernatant and pellet fraction for marker proteins. Simple differential centrifugation allows separation of a variety of yeast organelles and membranes (Bowser et al. 1992; Vida et al. 1993; Rieder and Emr 1997). Subjecting the lysate to 440 g produced a pellet (P1) containing insignificant amounts of CPY, PrA, and ALP (Fig. 1 B). Although not shown in this experiment, <2% of a cytosolic marker protein (glucose 6-phosphate dehydrogenase) fractionated with the P1 pellet, indicating that polycarbonate filter lysis was very efficient. Subjecting the postnuclear supernatant (S1) to 15,000 g produced a pellet (P2) containing ∼5% of the total p1CPY, ∼50% of p2CPY, and ∼95% of the sedimentable mCPY, suggesting the presence of ER, early Golgi membranes (p1CPY), and intact vacuoles (mCPY, Fig. 1 B). The majority (>90%) of the p2CPY in the postvacuolar supernatant (S2) was found in the pellet (P3) after subjecting the postvacuolar supernatant to centrifugation at 125,000 g. Although the late Golgi complex marker, Kex2p, also fractionated with the 125,000-g membrane pellet, <10% of p2CPY cofractionated with Kex2p activity on sucrose gradients (data not shown). This suggested that very little p2CPY was localized in the late Golgi complex and most likely resided in the PVC as previously shown using comparable pulse–chase radiolabeling conditions (Vida et al. 1993). Overall, these fractionation characteristics of membranes obtained from extrusion through polycarbonate filters were very similar to those observed after dissociating permeabilized cells (Vida et al. 1993).
Various steps from the filter lysis procedure were also examined with microscopy. To follow the vacuole, we prestained yeast cells with FM4-64 and CDCFDA. As expected, the P1 pellet was devoid of unbroken cells and was enriched in cell wall remnants (Fig. 2). As expected from the marker protein analysis, the P2 pellet was enriched in intact vacuoles, since many FM4-64–stained membranes containing CDCFDA were observed (Fig. 2). In contrast, the 125,000-g P3 pellet was devoid of vacuoles and instead was enriched for very small particulate structures. Importantly, if cells were stained with FM4-64 at 15°C, many of the small particulate structures in the 125,000-g pellet exhibited fluorescence (Fig. 2, inset). Additionally, membrane fluorescence in the 15,000-g P2 pellet was markedly reduced at 15°C (Fig. 2, inset). Since FM4-64 is kinetically trapped in prevacuolar compartments at 15°C (Vida et al. 1993), the membrane fluorescence in the 125,000-g pellet suggests that these differential centrifugation conditions separated vacuoles from prevacuolar compartments. Although not shown in this experiment, the P1 pellet also was enriched with intact nuclei after first staining yeast cells with DNA dyes (i.e., 4′,6-diamidino-2-phenylindole, dihydrochloride, DAPI). The P2 pellet was enriched for mitochondria after first staining cells with the mitochondrial vital dye 2,4-(4-(dimethylamino)styryl)-N-methylpyridinium iodide (DASPMI). We also examined the P2 and P3 pellets with electron microscopy. The P2 pellet comprised numerous electron-dense 1,000–1,500-nm membrane-delineated structures, which was consistent with the size expected for vacuoles (data not shown). In contrast, the P3 pellet was devoid of the relatively large, electron-dense membranes and instead was composed of 50–100 nm and 250–400 nm membrane-delineated structures (data not shown).
Figure 2.
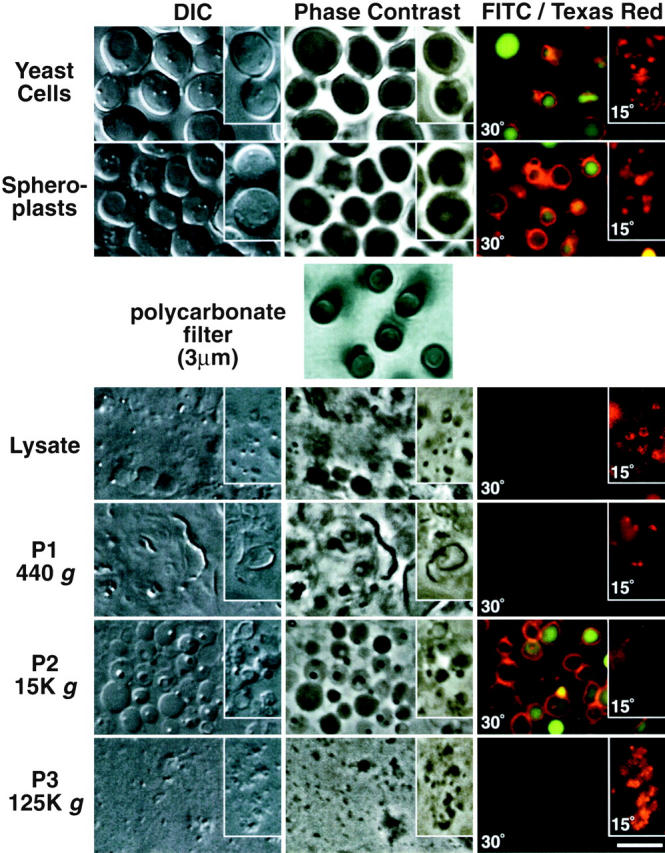
Light microscopy of cell-free membrane pellets after polycarbonate filter lysis. Wild-type yeast cells (as in Fig. 1) were first stained with FM4-64 (15-min pulse, 45-min chase) followed with dichlorocarboxyfluorescein diacetate (15 min at pH 4.0) to mark the vacuole membrane and lumen, respectively. The double-stained cells were then enzymatically converted to spheroplasts at 30 or 15°C (as indicated). The spheroplasts were extruded through a polycarbonate filter with 3-μm pores (as described in Fig. 1 A). The lysate, P1, P2, and P3 pellets (as indicated) were then examined under a light microscope with differential interference contrast (DIC), phase contrast, and epifluorescence optics using a FITC and Texas red filter set (as indicated). The fluorescence images were digitally overlaid for a composite. The cells at 15°C (inset) were stained with just FM4-64 for a 30-min pulse. Bar, 5 μm.
Reconstitution of p2CPY Maturation after Mixing the Donor and Acceptor Membrane Pellets
The polycarbonate filter lysis technique and simple differential centrifugation cleanly separated membranes containing vacuolar precursor proteins, the P3 pellet, from membranes containing vacuoles, the P2 pellet. These conditions set up the ability to use the P3 pellet as a donor membrane fraction and the P2 pellet as an acceptor membrane fraction. We prepared a P3 pellet after radiolabeling yeast spheroplasts with Tran35S-label (5-min pulse, 2-min chase) and incubated the membranes under various conditions of ATP, cytosol, and acceptor membranes. Each reaction was sequentially immunoprecipitated for both CPY and proteinase A (PrA), subjected to SDS-PAGE, and autoradiography. The level of both p2CPY and p2PrA remained unchanged after incubating the donor membranes with buffer, ATP alone, cytosol alone, or with ATP plus cytosol (Fig. 3, lanes 1–5). Even after adding back P2 acceptor membranes (made from unlabeled spheroplasts), alone and with cytosol, the amount of both p2CPY and p2PrA also remained constant (Fig. 3, lanes 6 and 8). Importantly, when ATP, cytosol, and unlabeled acceptor membranes were added back to the P3 radiolabeled donor membranes, ∼50% of the p2CPY and ∼60% of the proPrA (e.g., p2PrA) were converted to their mature, active forms (Fig. 3, lane 9). The cytosolic extract stimulated their maturation just over twofold compared with incubating the donor and acceptor membranes with ATP alone (Fig. 3, compare lane 7 with 9). However, this cytosol stimulation increased to at least 40-fold after incubating the donor and acceptor membranes for 15 min on ice and reharvesting them with centrifugation before setting up the reactions. (Fig. 3, lanes 10 and 11). This argued that some activity(ies) associated with either the donor membranes, acceptor membranes, or both was removed, rendering the p2CPY and p2PrA maturation completely dependent on exogenous cytosol.
Figure 3.

Vacuolar precursor proteins undergo maturation after incubating donor and acceptor membranes in a cell-free system. Wild-type yeast spheroplasts were radiolabeled (as in Fig. 1 B). The cells were subjected to lysis through a polycarbonate filter with subsequent differential centrifugation to generate a 125,000 g, P3 donor membrane pellet. The same yeast strain was used to make a 15,000 g, P2 acceptor membrane pellet from nonradiolabeled spheroplasts. The radiolabeled donor membranes (from ∼5 × 107 spheroplasts per reaction) were incubated at 25°C for 60 min with various combinations of buffer, ATP (plus a regeneration system), cytosol (5 mg/ml), and acceptor membranes (∼100 μg) in a total volume of 50 μl, as indicated. All reactions were sequentially immunoprecipitated for CPY and PrA, subjected to SDS-PAGE, and autoradiography. For the reactions in lanes 10 and 11, both donor and acceptor membranes were washed once with lysis buffer and reharvested before incubation with ATP and cytosol.
Characteristics of the Cell-free Assay
The characteristics of cell-free assays with P3 donor membranes and P2 acceptor membranes were examined to determine if they suggested that the reaction was intercompartmental. The first characteristic that we examined was dilution sensitivity. Normally, reactions were carried out in a 50-μl volume with the efficiency of p2CPY maturation ranging from 35 to 55%. To test the effect of dilution, the reaction volume was increased to dilute the concentration of donor/acceptor membranes while the concentration of ATP and cytosol was maintained at a constant level. An exponential decrease in p2CPY maturation efficiency was observed concomitant with an incremental increase in the reaction volume (Fig. 4 A). For example, a sixfold decrease in efficiency (38% vs 6%) took place with a 10-fold increase in reaction volume (from 50 to 500 μl). This suggested that the concentration of donor and acceptor membranes had a critical threshold for optimal reconstitution of p2CPY maturation. The second characteristic that we examined of the cell-free assay was the reaction kinetics. A prominent lag period was observed in the first 15–20 min (Fig. 4 B). A linear phase followed for the next 20 min and reached a plateau between 40 and 60 min (Fig. 4 B). Although not shown in this experiment, an increase in p2CPY maturation did not occur after a further 60 min incubation. This kinetic analysis suggested that a rate-limiting event(s) occurred early in the incubation, which might be the formation of a transport intermediate. The third characteristic that we examined of the cell-free assay was its temperature dependence. The maturation of p2CPY was undetectable when the incubation was carried out at 0 or 5°C (Fig. 4 C). The optimal efficiency occurred between 20 and 30°C and sharply tapered off at temperatures above 30°C (Fig. 4 C). Overall, the dilution sensitivity, kinetics, and temperature dependence of this new cell-free assay for p2CPY maturation indicated a complex event(s) was reconstituted after incubating P3 donor membranes and P2 acceptor membranes in the presence of ATP and cytosol.
Figure 4.
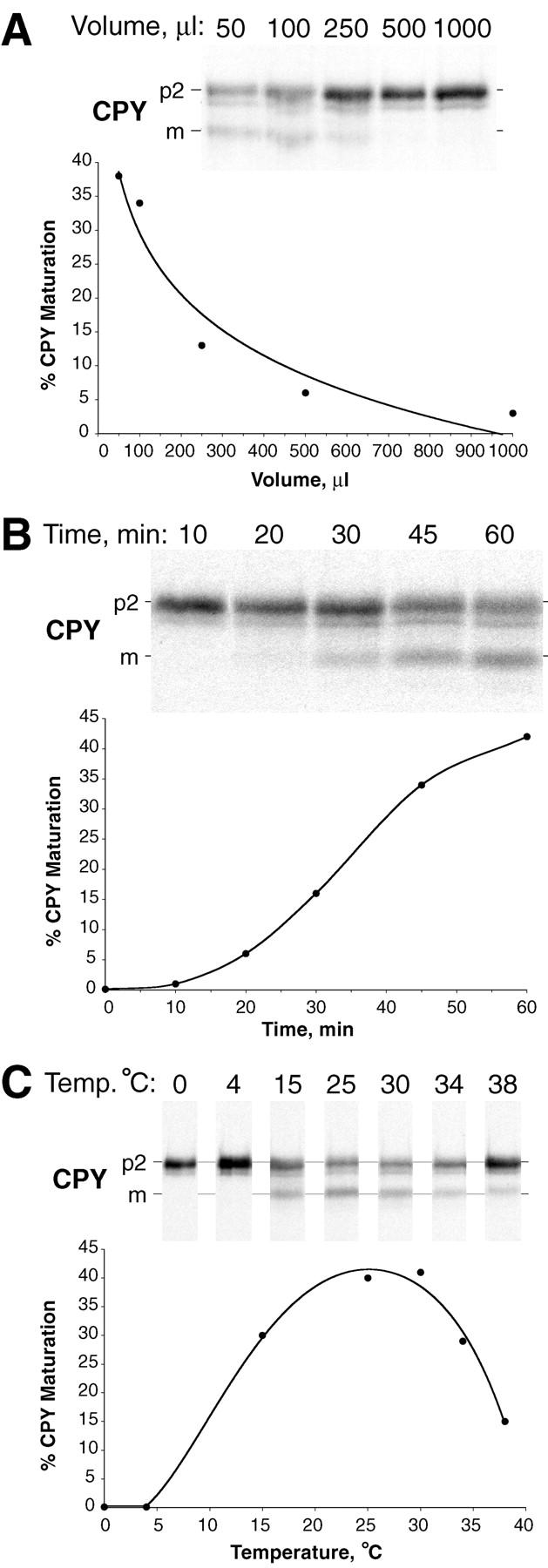
Characteristics of the cell-free system. Radiolabeled donor membranes and nonradiolabeled acceptor membranes were prepared and incubated (as described in Fig. 3) with modifications in reaction volume, time, or temperature. (A) Dilution sensitivity. All reactions were performed with a constant amount of donor and acceptor membranes while the reaction volume was increased (50–1,000 μl), as indicated. The concentration of ATP (plus regeneration components) and cytosol was equivalent in all reactions. (B) Maturation kinetics. A standard 50-μl reaction (see Fig. 3) was scaled up fivefold, incubated at 25°C, and aliquots were removed at the indicated times. (C) Temperature dependence. Standard 50-μl reactions were incubated at the indicated temperatures. All reactions were immunoprecipitated for CPY, subjected to SDS-PAGE, and autoradiography.
The Cell-free Assay Reconstitutes Intercompartmental Protein Transport
To truly determine if this new cell-free assay reconstituted intercompartmental protein transport, we performed reactions where the donor and acceptor fractions were prepared from yeast strains defective in vacuolar processing enzymes. A hallmark of most cell-free intercompartmental protein transport assays is using donor membranes deficient in the activity that marks the transport event. Two proteases are responsible for cleaving the propeptide from p2CPY, proteinase A (PEP4 gene) and proteinase B (PRB1 gene). In yeast strains mutant for the PEP4 gene (pep4-1, or pep4Δ), p2CPY travels to the vacuole but is not processed to the mature form of the protein (Stevens et al. 1982). In prb1 mutant strains, p2CPY also travels to the vacuole but instead of remaining unprocessed, active proteinase A (which can autoactivate) cleaves away a portion of the propeptide (Knop et al. 1993).
We took advantage of CPY processing characteristics in vivo (Fig. 5 A) using PEP4 and pep4Δ yeast strains as a source of both donor and acceptor membranes in vitro. To confirm the genotype of the strains, we performed pulse–chase analysis and compared CPY processing. Both PEP4 and pep4Δ strains showed no significant differences for p1 and p2CPY after a 5-min pulse (Fig. 5 B, lanes 1 and 3). However, after a 60-min chase the fate of the p1 and p2CPY precursors was different. The PEP4 strain produced mCPY and the pep4Δ strain did not produce any mCPY but the p2CPY precursor accumulated (Fig. 5 B, lanes 2 and 4). With these phenotypes established, we prepared radiolabeled P3 donor membranes and unlabeled P2 acceptor membranes from the wild-type PEP4 and the pep4Δ mutant strains. These membranes were then mixed and incubated for cell-free assays in all combinations. Importantly, the radiolabeled reaction product took on the processing phenotype of the unlabeled acceptor membranes, not the radiolabeled donor membranes. For instance, PEP4 acceptor membranes gave rise to mCPY even from pep4Δ donor membranes (Fig. 5 C; lanes 5 and 6). Acceptor membranes from the pep4Δ strain did not produce detectable p2CPY maturation (Fig. 5 C, lanes 7 and 8). The small amount of mCPY (∼9%) that occurred from mixing PEP4 donor membranes with pep4Δ acceptor membranes (Fig. 5 C, lane 7) was present in the reaction where no acceptor membranes were added back (Fig. 5 C, lane 3). This indicated that a trace amount of vacuoles contaminated the PEP4 donor membranes in this experiment. These reactions with donor and acceptor membranes from a strain deleted for the principal processing protease gene provided the strongest evidence that our new cell-free assay was indeed intercompartmental. This reconstitution was likely an intercompartmental transport process between the PVC and the vacuole.
Figure 5.

The cell-free assay reconstitutes intercompartmental transport between donor and acceptor membranes. (A) Strategy for using yeast strains deleted for processing protease genes to test for intercompartmental transport in vitro (see Results for details). (B) Processing of CPY in intact spheroplasts from wild-type strains and strains deleted for the proteinase A gene (pep4Δ). Yeast spheroplasts with the indicated genotypes were radiolabeled for 5 min with Tran35S-label (0 min) and chased with unlabeled methionine and cysteine for 60 min. After each time point, the cells were lysed, immunoprecipitated for CPY, subjected to SDS-PAGE, and autoradiography. (C) Cell-free assays with donor and acceptor membranes from wild-type strains and strains deleted for the proteinase A gene (pep4Δ). Donor membranes from radiolabeled, and acceptor membranes from nonradiolabeled yeast spheroplasts were prepared as previously described (Fig. 3) from strains with the indicated genotypes. Standard reaction conditions (Fig. 3) were used to incubate all combinations of donor and acceptor membranes (with and without ATP and cytosol), as indicated. All reactions were immunoprecipitated for CPY, subjected to SDS-PAGE, and autoradiography.
A Role for Vps33p in the Cell-free Reconstitution Assay
One difficulty in reconstituting an intercompartmental transport event in our previous permeabilized cell assay was incomplete removal of many cytoplasmic VPS gene products such as Vps33p (Vida et al. 1990). For example, no transport defect has been observed when a cytosolic extract devoid of Vps33p from a vps33 null strain (vps33Δ) was added back to wild-type permeabilized cells (data not shown). However, a significant defect was observed in vps33Δ cytosol when it was added back to the cell-free transport assay. The transport efficiency was decreased ∼2.5-fold compared with cytosol made from a wild-type VPS33 strain (Fig. 6, lanes 2 and 3, 5 and 6). Although the standard concentration of cytosol in our cell-free reactions was 5 mg/ml, these experiments also demonstrated that overall transport efficiency was remarkably consistent with the concentration of protein in crude, undiluted wild-type cytosol. For example, using extracts with a protein concentration of 50 mg/ml produced an average transport efficiency of 47.0% ± 1.3%  . We observed an average transport efficiency of 32.6% ± 2.5%
. We observed an average transport efficiency of 32.6% ± 2.5%  with an undiluted cytosolic protein concentration of 35 mg/ml. The 30% decrease in transport efficiency correlated well with the 30% decrease in protein concentration, which suggested that the level of a soluble protein factor(s) was critical for driving intercompartmental transport.
with an undiluted cytosolic protein concentration of 35 mg/ml. The 30% decrease in transport efficiency correlated well with the 30% decrease in protein concentration, which suggested that the level of a soluble protein factor(s) was critical for driving intercompartmental transport.
Figure 6.

Cytosolic extracts from a vps33 null strain are deficient in stimulating intercompartmental transport in the cell-free system. Radiolabeled donor membranes and nonradiolabeled acceptor membranes were prepared from wild-type yeast spheroplasts (SEY6210). Standard reaction conditions (Fig. 3) were used to incubate the donor and acceptor membranes with ATP (plus regeneration components) and cytosol (5 mg/ml) from the VPS33 or vps33Δ strains, as indicated. The reactions in lanes 4–6 contained donor and acceptor membranes that had been washed once with lysis buffer (similar to Fig. 3, lanes 10 and 11) while the reactions in lanes 1–3 contained unwashed membranes. The bar graph depicts the average transport efficiency from three independent determinations and is normalized to the percent of maximal transport.
Polyclonal antiserum (raised against Vps33p-trpe fusion proteins) also directly implicated Vps33p in playing a specific role during the cell-free assay. We prepared a new antiserum against Vps33p and it proved to be monospecific, recognizing a single polypeptide of ∼72 kD after immunoprecipitation of a total yeast cell lysate (Fig. 7 A, lane 2). The preimmune serum did not immunoprecipitate any proteins in this cell lysate (Fig. 7 A, lane 1). In pilot experiments, the Vps33p immune serum inhibited the cell-free assay while the preimmune had no effect. To avoid potential inhibitory problems from whole serum, we purified total IgG from both the preimmune and immune sera and measured the inhibition in titration experiments with the cell-free assay. As more of the immune IgG against Vps33p was added to the cell-free assay, we observed a proportional decrease in p2CPY transport (Fig. 7 B). At 128 μg and above, the immune IgG was able to block >90% of intercompartmental transport in the assay (Fig. 7 B, lane 8). Importantly, preimmune IgG was without any measurable inhibitory effect (Fig. 7 B, lanes 3–8).
Figure 7.

Polyclonal IgG against Vps33p inhibits cell-free reconstitution of intercompartmental transport between donor and acceptor membranes. (A) Specificity of the anti-Vps33p serum. Whole yeast cells (SEY6210) containing a plasmid with the VPS33 gene under control of the GPD1 promoter (pGPD426-33) were radiolabeled for 10 min (Tran35S-label) and chased (methionine and cysteine) for 30 min. After cell lysis, the extracts were immunoprecipitated with preimmune (P, lane 1) or immune (I, lane 2) serum against Vps33p, subjected to SDS-PAGE, and autoradiography. The positions of molecular weight standards are given in kD. (B) Purified immune IgG inhibits cell-free transport of CPY. Protein A–Sepharose was used to purify IgG from both preimmune and immune rabbit serum. Different amounts (as indicated) of the purified IgG  were used in standard cell-free reactions with donor and acceptor membranes. All components of the reactions were added and incubated on ice with the indicated amounts of preimmune and immune IgG for 15 min before the standard 60-min incubation at 25°C. All reactions were immunoprecipitated for CPY, subjected to SDS-PAGE, and autoradiography.
were used in standard cell-free reactions with donor and acceptor membranes. All components of the reactions were added and incubated on ice with the indicated amounts of preimmune and immune IgG for 15 min before the standard 60-min incubation at 25°C. All reactions were immunoprecipitated for CPY, subjected to SDS-PAGE, and autoradiography.
Using immune IgG against Vps33p as a specific inhibitor of its function, we determined where Vps33p was most active during the cell-free assay. To test this, we added back IgG against Vps33p at different points during a time course (similar to the kinetics in Fig. 4 B). After allowing antibody/antigen binding for 15 min on ice at each time point, we then continued the incubation for 60 min at 25°C (see Fig. 8 A). The results from this analysis suggested that the role of Vps33p in intercompartmental transport to the vacuole was executed at an early stage in the cell-free assay. For example, when the antibody was added back before incubation (at 0 min), >90% inhibition was observed (Fig. 8 C). Moreover, this inhibition was most effective during the first 10–15 min of the time course (Fig. 8 C). This interval of time in the cell-free assay was the latent period showing very little maturation of p2CPY (Fig. 8 B and 4 B). The inhibition from adding immune IgG against Vps33p during the cell-free assay time course was significantly less at the 15 min time point and beyond (Fig. 8 C). For example, at 15 min only 10% of intercompartmental transport took place (Fig. 8 B) and the inhibition was only 40% (Fig. 8 C). This effect was more notable at the 30 min time point where ∼60% (Fig. 8 B) of intercompartmental transport occurred but the inhibition was only 10% (Fig. 8 C).
Figure 8.
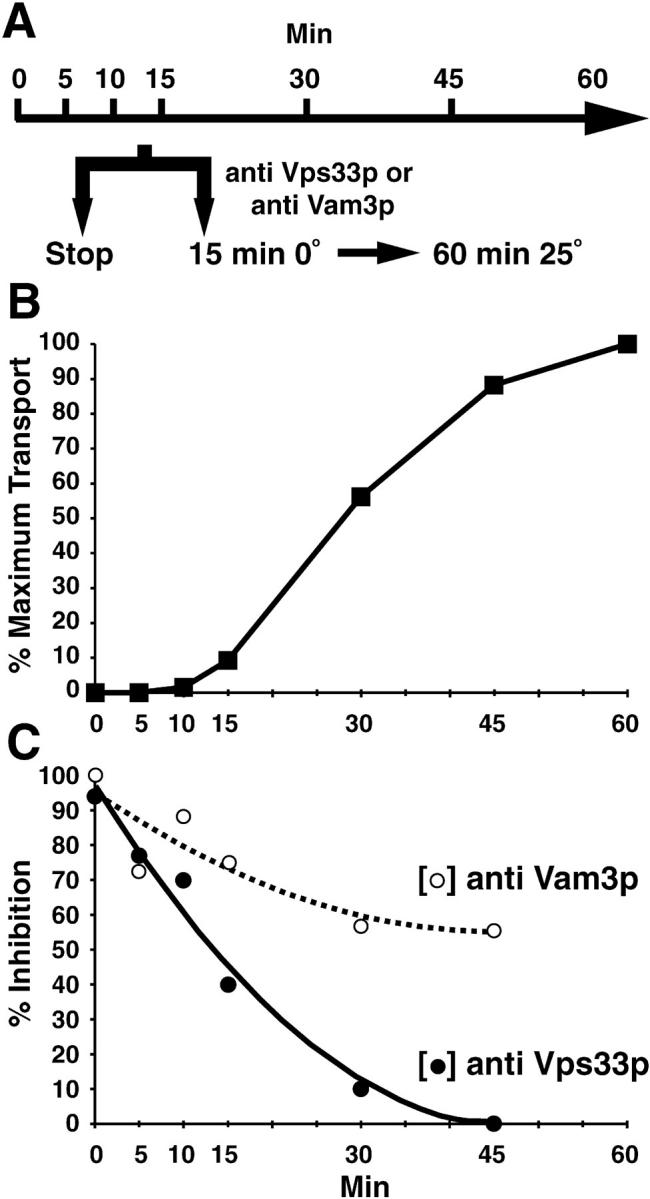
The function of Vps33p is required early during the cell-free reconstitution system and precedes the function of Vam3p. (A) Experimental strategy. (B) Transport kinetics. A standard 50-μl reaction (see Fig. 3) was scaled up, incubated at 25°C, and then aliquots were removed and stopped at the indicated times. (C) Antibody inhibition. Aliquots from the same reactions as in A were also removed and received either 128 μg of immune IgG against Vps33p or 5 μl of antiserum against Vam3p (as indicated), incubated 15 min on ice, and then shifted to 25°C for 60 min. All reactions were immunoprecipitated for CPY, subjected to SDS-PAGE, and autoradiography. Nonimmune serum was added to control for the effect of anti-Vam3p serum at each time point and did not inhibit the assay. A 2nd degree polynomial was used to fit the curve for the inhibition data points (C).
We also determined the possible involvement of another protein in the cell-free assay, Vam3p. Vam3p is a Q-SNARE protein (Fasshauer et al. 1998) of the vacuole that is directly implicated in homotypic vacuole fusion (Nichols et al. 1997) and delivery of vacuolar proenzymes through the biosynthetic pathway (Darsow et al. 1997). In contrast to antibodies against Vps33p, antiserum against Vam3p inhibited the assay when added at any time during the entire 45-min incubation (Fig. 8 C). The inhibition by anti-Vam3p serum decreased only ∼25% in the first 15 min and remained ≥60% throughout the time course (Fig. 8 C). These results suggested that the function of both Vps33p and Vam3p was required for efficient transport in the cell-free assay. However, an early event(s) was more dependent on the function of Vps33p, particularly during the first 15 min, than later events and Vam3p appeared to be required both early and late in the assay. The ability to inhibit the assay with Vps33p-specific antibodies decreased nearly threefold faster than with antibodies against Vam3p during the first 15 min of the cell-free assay.
Biochemical Complementation of VPS33 Function
The cell-free assay has allowed us to directly implicate the function of a VPS gene product in a reconstituted intercompartmental transport event. The inefficient transport from vps33Δ cytosolic extracts and the inhibition by IgG against Vps33p demonstrate that the function of this protein is required during incubation of donor and acceptor membranes. We wanted to positively implicate the role of Vps33p in the cell-free assay, which would establish a transport-coupled biochemical assay for a VPS gene product.
To this end, we expressed Vps33p in bacteria and it was produced at high levels (data not shown). However, over a variety of induction conditions with changes in temperature, time, or inducer concentration, Vps33p repeatedly was insoluble in bacterial lysates (data not shown). To avoid the insolubility problems from overexpression in bacteria, we overexpressed the VPS33 gene in yeast. We placed Vps33p under control of the promoter for glyceraldehyde 3-phosophate dehydrogenase (GPD1 pr) because it is one of the strongest promoters in S. cerevisiae (Mumberg et al. 1995). Indeed, a 100–200-fold increase in the amount of GPD1 pr-Vps33p was observed compared with endogenous levels of the protein and the overexpressed Vps33p behaved like a soluble protein (data not shown). The high level overproduction of soluble Vps33p in yeast allowed us to determine if we could reverse the antibody inhibition of the cell-free reconstitution assay. After incubating the donor/acceptor assay with IgG against Vps33p, a transport efficiency of ∼50% maximum was observed with a cytosolic extract from a strain expressing GPD1 pr-VPS33 (Fig. 9, lane 4). A significant level of transport was not observed when a cytosolic extract from the vps33Δ strain was added back to the IgG-inhibited reaction (Fig. 9, lane 2). This suggested that restoration of p2CPY transport to the vacuole may be specific to Vps33p. A wild-type cytosol (i.e., VPS33 pr-VPS33) leads to a transport efficiency just under 20% maximum, further suggesting that reversal of inhibition reflected the level of Vps33p added back to the assay. The results of this experiment provide evidence for biochemical complementation of a Vps protein-dependent defect to the yeast vacuole.
Figure 9.
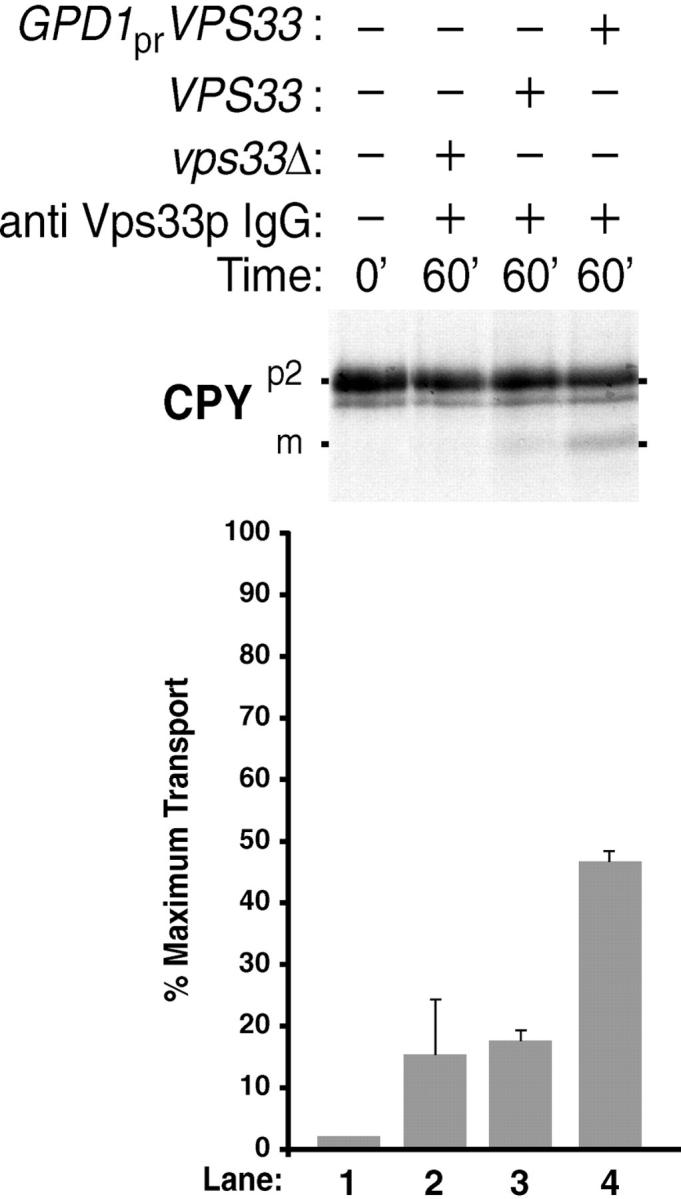
Biochemical reconstitution of Vps33p-dependent transport between donor and acceptor membranes. Radiolabeled donor membranes and nonradiolabeled acceptor membranes were prepared from wild-type yeast spheroplasts (Fig. 1 A). The donor and acceptor membranes were incubated for 15 min at 0°C with 128 μg of anti Vps33p IgG. Cytosol (5 mg/ml) was then added from a vps33Δ (lane 2), a VPS33 (lane 3), or a VPS33 strain containing pGPDHIS633-2 (lane 4), and incubated at 25°C for 60 min (as indicated). All reactions were immunoprecipitated for CPY, subjected to SDS-PAGE, and autoradiography. The bar graph depicts average transport efficiency from three independent determinations and normalized to the percent of maximal transport.
Discussion
Gaining access to the cell cytoplasm is essential for detailed understanding of intracellular transport between organelles. Genetics and molecular biological approaches are able to obtain entrance into cells with the manipulation of genes and gene products. Although this control can often be very thorough, it is also often limited without augmentation using biochemical approaches in parallel. The biochemistry of transport between organelles requires working in a cell-free system. Frequently, severe limitations to cell-free analyses are not only maintaining organelle structure, but also (and more importantly) organelle function. These are the two most important criteria in successful cell-free reconstitution of intercompartmental transport.
Lysing yeast spheroplasts by extrusion through polycarbonate filters maintains function of organelles in the yeast vacuolar system. Polycarbonate filters are hydrophilic and contain uniform cylindrical pores. The diameter of these pores can be carefully controlled via ion etching, which allows for an even distribution across one plane over the entire exposed membrane surface. The ability to change the overall diameter of a yeast spheroplast with osmotic forces permits swelling of the cells to just greater than the diameter of the polycarbonate filter pores. Thus, in one simple step, the plasma membrane can be gently sheared away from cells and most organelles are free to pass through with little damage. In fact, we have used this method of lysis on mammalian cells (Chinese hamster ovary), which required a simple increase in pore size (from 3 to 8 μm). The yeast vacuole does undergo some loss of luminal content during extrusion through polycarbonate filters as expected from its labile structure. This loss is most likely from leakage rather than lysis and is inconsequential because the amount of soluble proteases is sufficient for processing of propeptides from vacuolar zymogens.
Proteolytic maturation within the donor compartment, presumably the PVC, does not appear to be an efficient process in vitro. Another explanation for our cell-free assay that measures maturation of p2CPY could be intracompartmental activation of processing proteases. To a first approximation, proPrA is contained in the same compartments as p2CPY. Unlike proCPY, the proPrA precursor has the ability to autoactivate (Mechler et al. 1988, Mechler et al. 1987). Therefore, incubation of donor membranes with ATP and cytosol might lead to changes in luminal pH that would enhance autoactivation of proteinase A. Once proPrA becomes an active hydrolase it would begin a cascade of proteolytic events leading to the activation of Prb1p and ultimately maturation of p2CPY. However, under our cell-free assay conditions, maturation of p2CPY was undetectable after incubating donor membranes alone with ATP and cytosol. The inhibition of the cell-free assay after diluting the membranes further supports the conclusion that proteolytic processing of p2CPY does not occur within the donor membrane compartment. These points argue that the organelle containing p2CPY does not acquire the capacity to cleave propeptides, which indicates that organelle maturation may not be a prevalent mechanism to produce vacuoles/lysosomes.
This cell-free system is easily manipulated to show a near absolute requirement for exogenous cytosol. With this cytosol requirement, we can tentatively assign the location of Vps33p function to cycling from the cytosol to either the donor or acceptor membrane fractions. Although Vps33p is predominately localized to the cytosol, a fraction of the protein sediments with membranes (Gerhardt et al. 1998). Since Vps33p binds ATP and readily interchanges between soluble and insoluble forms in an energy-dependent fashion (Gerhardt et al. 1998), the cell-free system is the best way to understand how ATP influences its function in transport to the vacuole.
One prospect for functional interaction of Vps33p with an insoluble component(s) is a SNARE complex (Bennett 1995). The VPS33 gene product is a member of the Sec1p family of proteins (Pevsner 1996) and thus is expected to bind to a target SNARE protein on the vacuole such as Vam3p (Darsow et al. 1997; Wada et al. 1997). Our results suggest that Vps33p acts earlier than Vam3p in the cell free assay. This is distinct to the inhibition via antibodies against Vam3p, which still show a significant block late into the reaction. One possibility to explain these results is that the epitopes on Vps33p are exposed for antibody binding early during the assay but become inaccessible due to conformational changes later in the time course. On the other hand, Vps33p may have a catalytic or binding activity that is required early during intercompartmental transport but not at late stages. Although the precise cause for losing Vps33p function during intercompartmental transport to the vacuole is unknown, the results suggest that it may be independent of Vam3p function. This has implications on how we view the roles Sec1- and syntaxin-like proteins play in vesicle-mediated transport.
The Sec1p family has many members, suggesting that these proteins function at every vesicle-mediated step in eukaryotic cells (Pevsner 1996; Pevsner et al. 1994). The Drosophila Sec1 homologue, ROP, and yeast Sly1p can negatively regulate neurotransmitter release (Schulze et al. 1994) and prevent v-SNARE/t-SNARE interactions in ER to Golgi transport, respectively (Lupashin and Waters 1997). Mammalian Sec1-like proteins bind to syntaxin with high affinity (Hodel et al. 1994; Pevsner et al. 1994). Although this protein–protein interaction is most likely to be physiologically relevant, its biological meaning is far from clear. For example, the direct physical interaction between Sec1p-like proteins and t-SNARE proteins has only been demonstrated in vitro (Hodel et al. 1994; Pevsner et al. 1994). Furthermore, attempts to coimmunoprecipitate a Sec1p homologue-syntaxin homologue complex from cell extracts have never been successful (Garcia et al. 1995; Wu et al. 1998), suggesting their interaction in vivo is transient, weak, or both. Nonetheless, simultaneous overexpression of syntaxin and ROP in Drosophila suppresses the defects in neurotransmission that are observed when either is overexpressed individually (Wu et al. 1998). Moreover, recent genetic evidence in yeast suggests that Vps33p interacts with a Q-SNARE protein of the vacuole membrane, Vam3p (Darsow et al. 1997). A haploid double mutant strain with temperature-sensitive alleles in both vps33 and vam3 shows a ∼50% defect in CPY maturation under conditions where the single haploid mutations are wild-type (Darsow et al. 1997). These are compelling examples that implicate a ROP/syntaxin and Vps33p/Vam3p functional interaction in vivo. However, studies involving overexpression or synthetic defects of two different genes are not of sufficient resolution to distinguish whether their products physically bind to one another or if they are part of a linear pathway. A direct physical interaction between ROP and syntaxin or Vps33p and Vam3p has not been demonstrated and instead has been inferred from analogy to studies with n-Sec1 and syntaxin (Pevsner et al. 1994). This inference may be appropriate for ROP because it is significantly more similar to n-Sec1 than is Vps33p. Indeed, Vps33p shows characteristics not shared among the other Sec1 members such as ATP binding and energy insolubility (Gerhardt et al. 1998). These differences suggest that Vps33p may play a distinct role in vesicle-mediate transport.
The advent of our cell-free assay will help uncover biochemical activities of VPS gene products. The majority of these proteins do not show sequence similarity to proteins of known biochemical properties, although several VPS gene products have activities in vitro. Without exception, the ability to design assays for detection of these catalytic activities arose from sequence similarities to proteins that had been subjected to previous biochemical characterization. This underscores the importance that biochemistry plays in elucidating gene function and discovering new activities should progress rapidly with our intercompartmental assay. This assay will allow us to define biochemical functions of cytosolic and membrane-associated factors necessary to execute transport between the PVC and vacuole. Our results with the cell-free assay imply that a vesicle intermediate may truly shuttle cargo between the PVC and the vacuole in yeast. Preincubation of donor membranes (in the absence of acceptor membranes) with ATP and cytosol gives rise to a fraction that contains p2CPY and separates from donor membranes. This membrane-enclosed compartment can be used as a functional intermediate in a second incubation with acceptor membranes (Gerhardt and Vida, manuscript in preparation). Many of the factors involved in this process are likely VPS gene products and may play a role in vesicle formation, transport, targeting, and fusion with the yeast vacuole/lysosome.
Acknowledgments
We thank Etienne-Pascal Journet (Laboratoire de Biologie Moleculaire des Relations Plantes-Microorganismes Castanet-Tolosan Cedex France) for showing one of us (T. Vida) the polycarbonate filter lysis technique before publication. We acknowledge the excellent expertise of Kenneth Dunner, Jr. for electron microscopy. We thank William Wickner (Dartmouth Medical School) for his generous gift of antiserum against Vam3p. We thank Jean Whitesell and Cocalico Biologicals, Inc. for expert polyclonal antiserum production against Vps33p. We are grateful to Anita Seibold for telling us about propanol-jacketed cryo freezers. We also thank Victoria Knutson, Andrew Bean, and Neal Waxham for valuable discussions, and Andrew Bean for critically reading the manuscript. B. Gerhardt thanks Dave Matthews for inspirational music.
These studies were supported by a grant to T. Vida from the National Institutes of Health-National Institute of General Medical Sciences (GM52092). We also acknowledge an Institutional Core Grant (no. CA16672) that maintains the High Resolution Transmission Electron Microscopy Facility (University of Texas M.D. Anderson Cancer Center).
Footnotes
1.used in this paper: CPY, carboxypeptidase Y; CDCFDA, dichlorocarboxyfluorescein diacetate; PLC, prelysosomal compartment; PrA, proteinase A; PVC, prevacuolar compartment
References
- Babst M., Sato T.K., Banta L.M., Emr S.D. Endosomal transport function in yeast requires a novel AAA-type ATPase, Vps4p. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:1820–1831. doi: 10.1093/emboj/16.8.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D., Schekman R. Reconstitution of protein transport using broken yeast spheroplasts. Methods Cell Biol. 1989;31:127–141. doi: 10.1016/s0091-679x(08)61605-2. [DOI] [PubMed] [Google Scholar]
- Balch W.E. Biochemistry of interorganelle transport. A new frontier in enzymology emerges from versatile in vitro model systems. J. Biol. Chem. 1989;264:16965–16968. [PubMed] [Google Scholar]
- Balch W.E., Beckers C.J., Keller D.S. Reconstitution of protein transport from the endoplasmic reticulum to the Golgi using a cell free system. Prog. Clin. Biol. Res. 1988;270:333–342. [PubMed] [Google Scholar]
- Balch W.E., Dunphy W.G., Braell W.A., Rothman J.E. Reconstitution of the transport of protein between successive compartments of the Golgi measured by the coupled incorporation of N-acetylglucosamine. Cell. 1984;39:405–416. doi: 10.1016/0092-8674(84)90019-9. [DOI] [PubMed] [Google Scholar]
- Balch W.E., Rothman J.E. Characterization of protein transport between successive compartments of the Golgi apparatusasymmetric properties of donor and acceptor activities in a cell-free system. Arch. Biochem. Biophys. 1985;240:413–425. doi: 10.1016/0003-9861(85)90046-3. [DOI] [PubMed] [Google Scholar]
- Banta L.M., Vida T.A., Herman P.K., Emr S.D. Characterization of yeast Vps33p, a protein required for vacuolar protein sorting and vacuole biogenesis. Mol. Cell. Biol. 1990;10:4638–4649. doi: 10.1128/mcb.10.9.4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett M.K. SNAREs and the specificity of transport vesicle targeting. Curr. Opin. Cell Biol. 1995;7:581–586. doi: 10.1016/0955-0674(95)80016-6. [DOI] [PubMed] [Google Scholar]
- Bowser R., Muller H., Govindan B., Novick P. Sec8p and Sec15p are components of a plasma membrane-associated 19.5S particle that may function downstream of Sec4p to control exocytosis. J. Cell Biol. 1992;118:1041–1056. doi: 10.1083/jcb.118.5.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans . Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant N.J., Stevens T.H. Vacuole biogenesis in Saccharomyces cerevisiaeprotein transport pathways to the yeast vacuole. Microbiol. Mol. Biol. Rev. 1998;62:230–247. doi: 10.1128/mmbr.62.1.230-247.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darsow T., Rieder S.E., Emr S.D. A multispecificity syntaxin homologue, Vam3p, essential for autophagic and biosynthetic protein transport to the vacuole. J. Cell Biol. 1997;138:517–529. doi: 10.1083/jcb.138.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis N.G., Horecka J.L., Sprague G.F., Jr. Cis- and trans-acting functions required for endocytosis of the yeast pheromone receptors. J. Cell Biol. 1993;122:53–65. doi: 10.1083/jcb.122.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W.P., Nickoloff J.A. Site-directed mutagenesis of virtually any plasmid by eliminating a unique site. Anal. Biochem. 1992;200:81–88. doi: 10.1016/0003-2697(92)90280-k. [DOI] [PubMed] [Google Scholar]
- Deshaies R.J., Schekman R. A yeast mutant defective at an early stage in import of secretory protein precursors into the endoplasmic reticulum. J. Cell Biol. 1987;105:633–645. doi: 10.1083/jcb.105.2.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasshauer D., Sutton R.B., Brunger A.T., Jahn R. Conserved structural features of the synaptic fusion complexSNARE proteins reclassified as Q- and R-SNAREs. Proc. Natl. Acad. Sci. USA. 1998;95:15781–15786. doi: 10.1073/pnas.95.26.15781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futter C.E., Pearse A., Hewlett L.J., Hopkins C.R. Multivesicular endosomes containing internalized EGF-EGF receptor complexes mature and then fuse directly with lysosomes. J. Cell Biol. 1996;132:1011–1023. doi: 10.1083/jcb.132.6.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia E.P., McPherson P.S., Chilcote T.J., Takei K., De Camilli P. rbSec1A and B colocalize with syntaxin 1 and SNAP-25 throughout the axon, but are not in a stable complex with syntaxin. J. Cell Biol. 1995;129:105–120. doi: 10.1083/jcb.129.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardt B., Kordas T.J., Thompson C.M., Patel P., Vida T. The vesicle transport protein Vps33p is an ATP-binding protein that localizes to the cytosol in an energy-dependent manner. J. Biol. Chem. 1998;273:15818–15829. doi: 10.1074/jbc.273.25.15818. [DOI] [PubMed] [Google Scholar]
- Goda Y., Pfeffer S.R. Selective recycling of the mannose 6-phosphate/IGF-II receptor to the trans Golgi network in vitro. Cell. 1988;55:309–320. doi: 10.1016/0092-8674(88)90054-2. [DOI] [PubMed] [Google Scholar]
- Herman P.K., Emr S.D. Characterization of VPS34, a gene required for vacuolar protein sorting and vacuole segregation in Saccharomyces cerevisiae . Mol. Cell. Biol. 1990;10:6742–6754. doi: 10.1128/mcb.10.12.6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodel A., Schafer T., Gerosa D., Burger M.M. In chromaffin cells, the mammalian Sec1p homologue is a syntaxin 1A-binding protein associated with chromaffin granules. J. Biol. Chem. 1994;269:8623–8626. [PubMed] [Google Scholar]
- Horazdovsky B.F., Busch G.R., Emr S.D. VPS21 encodes a rab5-like GTP binding protein that is required for the sorting of yeast vacuolar proteins. EMBO (Eur. Mol. Biol. Organ.) J. 1994;13:1297–1309. doi: 10.1002/j.1460-2075.1994.tb06382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M., Schiffer H.H., Rupp S., Wolf D.H. Vacuolar/lysosomal proteolysisproteases, substrates, mechanisms. Curr. Opin. Cell Biol. 1993;5:990–996. doi: 10.1016/0955-0674(93)90082-2. [DOI] [PubMed] [Google Scholar]
- Koerner T.J., Hill J.E., Myers A.M., Tzagoloff A. High-expression vectors with multiple cloning sites for construction of trpE fusion genespATH vectors. Methods Enzymol. 1991;194:477–490. doi: 10.1016/0076-6879(91)94036-c. [DOI] [PubMed] [Google Scholar]
- Lupashin V.V., Waters M.G. t-SNARE activation through transient interaction with a Rab-like guanosine triphosphatase. Science. 1997;276:1255–1258. doi: 10.1126/science.276.5316.1255. [DOI] [PubMed] [Google Scholar]
- Martin T.F., Kowalchyk J.A. Docked secretory vesicles undergo Ca2+-activated exocytosis in a cell-free system. J. Biol. Chem. 1997;272:14447–14453. doi: 10.1074/jbc.272.22.14447. [DOI] [PubMed] [Google Scholar]
- Mechler B., Hirsch H.H., Muller H., Wolf D.H. Biogenesis of the yeast lysosome (vacuole)biosynthesis and maturation of proteinase yscB. EMBO (Eur. Mol. Biol. Organ.) J. 1988;7:1705–1710. doi: 10.1002/j.1460-2075.1988.tb02999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechler B., Muller H., Wolf D.H. Maturation of vacuolar (lysosomal) enzymes in yeastproteinase yscA and proteinase yscB are catalysts of the processing and activation event of carboxypeptidase yscY. EMBO (Eur. Mol. Biol. Organ.) J. 1987;6:2157–2163. doi: 10.1002/j.1460-2075.1987.tb02483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D., Muller R., Funk M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene. 1995;156:119–122. doi: 10.1016/0378-1119(95)00037-7. [DOI] [PubMed] [Google Scholar]
- Munn A.L., Riezman H. Endocytosis is required for the growth of vacuolar H(+)-ATPase-defective yeastidentification of six new END genes. J. Cell Biol. 1994;127:373–386. doi: 10.1083/jcb.127.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn A.L., Stevenson B.J., Geli M.I., Riezman H. end5, end6, and end7mutations that cause actin delocalization and block the internalization step of endocytosis in Saccharomyces cerevisiae . Mol. Biol. Cell. 1995;6:1721–1742. doi: 10.1091/mbc.6.12.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols B.J., Ungermann C., Pelham H.R., Wickner W.T., Haas A. Homotypic vacuolar fusion mediated by t- and v-SNAREs. Nature. 1997;387:199–202. doi: 10.1038/387199a0. [DOI] [PubMed] [Google Scholar]
- Novick P., Field C., Schekman R. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell. 1980;21:205–215. doi: 10.1016/0092-8674(80)90128-2. [DOI] [PubMed] [Google Scholar]
- Novick P., Schekman R. Secretion and cell surface growth are blocked in a temperature-sensitive mutant of Saccharomyces cerevisiae . Proc. Natl. Acad. Sci. USA. 1979;76:1858–1862. doi: 10.1073/pnas.76.4.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevsner J. The role of Sec1p-related proteins in vesicle trafficking in the nerve terminal. J. Neurosci. Res. 1996;45:89–95. doi: 10.1002/(SICI)1097-4547(19960715)45:2<89::AID-JNR1>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Pevsner J., Hsu S.C., Scheller R.H. n-Sec1a neural-specific syntaxin-binding protein. Proc. Natl. Acad. Sci. USA. 1994;91:1445–1449. doi: 10.1073/pnas.91.4.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer S.R. Targeting of proteins to the lysosome. Curr. Topics Microbiol. Immunol. 1991;170:43–65. doi: 10.1007/978-3-642-76389-2_2. [DOI] [PubMed] [Google Scholar]
- Racoosin E.L., Swanson J.A. Macropinosome maturation and fusion with tubular lysosomes in macrophages. J. Cell Biol. 1993;121:1011–1020. doi: 10.1083/jcb.121.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raths S., Rohrer J., Crausaz F., Reizman H. end3 and end4Two mutants defective in receptor-mediated and fluid-phase endocytosis in Saccharomyces cerevisiae . J. Cell Biol. 1993;120:55–65. doi: 10.1083/jcb.120.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond C.K., O'Hara P.J., Eichinger G., Rothman J.H., Stevens T.H. Molecular analysis of the yeast VPS3 gene and the role of its product in vacuolar protein sorting and vacuolar segregation during the cell cycle. J. Cell Biol. 1990;111:877–892. doi: 10.1083/jcb.111.3.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder S.E., Emr S.D. A novel RING finger protein complex essential for a late step in protein transport to the yeast vacuole. Mol. Biol. Cell. 1997;8:2307–2327. doi: 10.1091/mbc.8.11.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J.S., Klionsky D.J., Banta L.M., Emr S.D. Protein sorting in Saccharomyces cerevisiaeisolation of mutants defective in the delivery and processing of multiple vacuolar hydrolases. Mol. Cell. Biol. 1988;8:4936–4948. doi: 10.1128/mcb.8.11.4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman J.H., Howald I., Stevens T.H. Characterization of genes required for protein sorting and vacuolar function in the yeast Saccharomyces cerevisiae . EMBO (Eur. Mol. Biol. Organ.) J. 1989;8:2057–2065. doi: 10.1002/j.1460-2075.1989.tb03614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze K.L., Littleton J.T., Salzberg A., Halachmi N., Stern M., Lev Z., Bellen H.J. rop, a Drosophila homolog of yeast Sec1 and vertebrate n-Sec1/Munc-18 proteins, is a negative regulator of neurotransmitter release in vivo. Neuron. 1994;13:1099–1108. doi: 10.1016/0896-6273(94)90048-5. [DOI] [PubMed] [Google Scholar]
- Spang A., Schekman R. Reconstitution of retrograde transport from the Golgi to the ER in vitro. J. Cell Biol. 1998;143:589–599. doi: 10.1083/jcb.143.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack J.H., Emr S.D. Vps34p required for yeast vacuolar protein sorting is a multiple specificity kinase that exhibits both protein kinase and phosphatidylinositol-specific PI 3-kinase activities. J. Biol. Chem. 1994;269:31552–31562. [PubMed] [Google Scholar]
- Stack J.H., Herman P.K., Schu P.V., Emr S.D. A membrane-associated complex containing the Vps15 protein kinase and the Vps34 PI 3-kinase is essential for protein sorting to the yeast lysosome-like vacuole. EMBO (Eur. Mol. Biol. Organ.) J. 1993;12:2195–2204. doi: 10.1002/j.1460-2075.1993.tb05867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens T., Esmon B., Schekman R. Early stages in the yeast secretory pathway are required for transport of carboxypeptidase Y to the vacuole. Cell. 1982;30:439–448. doi: 10.1016/0092-8674(82)90241-0. [DOI] [PubMed] [Google Scholar]
- Swanson D.A., Steel J.M., Valle D. Identification and characterization of the human ortholog of rat STXBP1, a protein implicated in vesicle trafficking and neurotransmitter release. Genomics. 1998;48:373–376. doi: 10.1006/geno.1997.5202. [DOI] [PubMed] [Google Scholar]
- Vater C.A., Raymond C.K., Ekena K., Howald-Stevenson I., Stevens T.H. The VPS1 protein, a homolog of dynamin required for vacuolar protein sorting in Saccharomyces cerevisiae, is a GTPase with two functionally separable domains. J. Cell Biol. 1992;119:773–786. doi: 10.1083/jcb.119.4.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vida T.A., Emr S.D. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J. Cell Biol. 1995;128:779–792. doi: 10.1083/jcb.128.5.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vida T.A., Huyer G., Emr S.D. Yeast vacuolar proenzymes are sorted in the late Golgi complex and transported to the vacuole via a prevacuolar endosome-like compartment. J. Cell Biol. 1993;121:1245–1256. doi: 10.1083/jcb.121.6.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vida T.A., Graham T.R., Emr S.D. In vitro reconstitution of intercompartmental protein transport to the yeast vacuole. J. Cell Biol. 1990;111:2871–2884. doi: 10.1083/jcb.111.6.2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada Y., Nakamura N., Ohsumi Y., Hirata A. Vam3p, a new member of syntaxin related protein, is required for vacuolar assembly in the yeast Saccharomyces cerevisiae . J. Cell Sci. 1997;110:1299–1306. doi: 10.1242/jcs.110.11.1299. [DOI] [PubMed] [Google Scholar]
- Wickerham L.J. A critical evaluation of the nitrogen assimilation tests commonly used in the classification of yeasts. J. Bacteriol. 1946;52:293–301. doi: 10.1128/JB.52.3.293-301.1946. [DOI] [PubMed] [Google Scholar]
- Wu M.N., Littleton J.T., Bhat M.A., Prokop A., Bellen H.J. ROP, the Drosophila Sec1 homolog, interacts with syntaxin and regulates neurotransmitter release in a dosage-dependent manner. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:127–139. doi: 10.1093/emboj/17.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]