

Abstract
Death-associated protein (DAP)–kinase is a calcium/calmodulin regulated serine/threonine kinase that carries ankyrin repeats, a death domain, and is localized to the cytoskeleton. Here, we report that this kinase is involved in tumor necrosis factor (TNF)-α and Fas-induced apoptosis. Expression of DAP-kinase antisense RNA protected cells from killing by anti–Fas/APO-1 agonistic antibodies. Deletion of the death domain abrogated the apoptotic functions of the kinase, thus, documenting for the first time the importance of this protein domain. Overexpression of a fragment encompassing the death domain of DAP-kinase acted as a specific dominant negative mutant that protected cells from TNF-α, Fas, and FADD/MORT1–induced cell death. DAP-kinase apoptotic function was blocked by bcl-2 as well as by crmA and p35 inhibitors of caspases, but not by the dominant negative mutants of FADD/MORT1 or of caspase 8. Thus, it functions downstream to the receptor complex and upstream to other caspases. The multidomain structure of this serine/threonine kinase, combined with its involvement in cell death induced by several different triggers, place DAP-kinase at one of the central molecular pathways leading to apoptosis.
Keywords: DAP-kinase, tumor necrosis factor-α, Fas, death domain, apoptosis
Apoptosis (programmed cell death) is an important regulatory mechanism that eliminates unwanted cells during development and maintenance of tissue homeostasis. Over the past few years several apoptotic genes have been identified. One approach consisted of the isolation of proteins that are recruited to the intracellular domains of cytokine receptors belonging to the tumor necrosis factor (TNF)1 family. The yeast two-hybrid system served as a major tool for the isolation of these cytoplasmic proteins that sequentially bind to the intracellular part of TNF-α receptor (p55 TNF-R), the Fas receptor, and to each other. This includes a number of adaptor molecules (TRADD, FADD/MORT-1, RAIDD, MADD, TRAF-1 or -2, and FLIP), proteins with enzymatic activity (RIP, caspase 8 and 10, NIK), and death inhibitory molecules (c-IAPs) (for review see Salvesen and Dixit 1997; Cryns and Yuan 1998; Wallach et al. 1998). The interaction between these proteins is mediated by several structural motifs, i.e., the death domain (DD), the death effector domain (DED), and the caspase-recruiting domain (CARD).
Genetic screens performed in lower invertebrates provided a second fruitful approach for rescuing central apoptotic genes. The rescue of ced-3 from Caenorhabditis elegans and the identification of its mammalian homologues as cysteine proteases (named caspases, more than 14 family members) established that a major arm of the death-promoting pathways involves protease activation. In mammals, the activation of the proteolytic activity can be initiated by different mechanisms and at different intracellular sites. One takes place at the receptor proximal level through adaptor-mediated recruitment of the pro-caspases to the death-inducing signaling complex named DISC (e.g., by binding of pro-caspase 8 or 10 to FADD/MORT-1 that in turn binds to Fas) (Kischkel et al. 1995; Medema et al. 1997). A mitochondrial-based mechanism involving ced-4 and ced-9 homologues (Apaf-1 and members of the bcl-2 family, respectively) forms together with cytochrome C another caspase-activating complex termed the apoptosome (Green and Kroemer 1998). These regulatory caspases function by cleaving the terminator caspases. The spectrum of proteins that are cleaved by terminator caspases is broad, including various structural proteins (e.g., nuclear lamins, Gas2, and gelsolin; Brancolini et al. 1995; Lazebnik et al. 1995; Kothakota et al. 1997), as well as enzymes whose activity is regulated directly or indirectly by the proteolytic cleavage (e.g., PAK2/PAK65 protein kinase, the PKC isoforms δ and φ, MEKK-1, PITSLRE protein kinase, and the endonuclease CAD) (Cryns and Yuan 1998; and references within). As the multiplicity of mechanisms of caspase activation becomes apparent, it is becoming clear that membrane signals formed by ligand–receptor interactions must diverge into distinct biochemical pathways. These branches may each contribute a distinct effect to the apoptotic phenotype or alternatively may be redundant in their final cellular effects.
A functional approach to gene cloning, based on transfections of HeLa cells with antisense cDNA libraries and subsequent isolation of the fragments that protected cells from interferon-γ (IFN-γ)–induced cell death, initiated an additional direction in the field (Deiss and Kimchi 1991; Deiss et al. 1995). This approach has recently led to the identification of several novel proteins that are part of the apoptotic pathways, called death-associated proteins (DAPs) (for review see Kimchi 1998). One of these isolated proteins, DAP-kinase, identified as a calcium/calmodulin–regulated serine/threonine protein kinase, associated with actin microfilaments (Deiss et al. 1995; Cohen et al. 1997). Its structure contains at least two additional domains that might mediate interactions with other proteins including ankyrin repeats and a typical death domain located at the COOH-terminal part of the protein (Feinstein et al. 1995). Overexpression of DAP-kinase in several cell lines resulted in cell death and this death-promoting property strictly depended on the intrinsic kinase activity. The latter emerged from the finding that a constitutively active kinase mutant, deleted of the calmodulin regulatory domain (ΔCaM), had stronger cell death effects than the wild-type kinase, whereas a catalytically inactive mutant (K42A) was not cytotoxic to cells (Cohen et al. 1997).
One of the surprising facets in the function of DAP-kinase relates to its antimetastatic activity, recently analyzed in animal model systems (Inbal et al. 1997). This feature was attributed to the finding that DAP-kinase conferred sensitivity to apoptotic stimuli encountered by the metastasizing cells, and opened a major question as to how broad is the spectrum of apoptotic signals that depend on DAP-kinase. In this work we focused specifically on apoptotic responses that are triggered by two cytokines belonging to the TNF family. We present several independent lines of evidence indicating that DAP-kinase is involved in cell death induced by TNF-α and Fas. The importance of the death domain in mediating the death-promoting function of DAP-kinase is documented here for the first time. Finally, by transfection-based functional analyses it is shown that DAP-kinase acts downstream to the DISC formation (i.e., FADD/MORT1 and caspase 8) and upstream of some other caspases, and that its death-promoting effects are counteracted by bcl-2.
Materials and Methods
Plasmids
All expression plasmids used in this work were constructed in pcDNA3 vector (Invitrogen Corp.). Construction of wild-type DAP-kinase and ΔCaM mutant was described before (Cohen et al. 1997). DAPk/ΔDD and ΔCaM/ΔDD were constructed by truncation of wild-type and ΔCaM DAP-kinase, respectively, at the HindIII site, thus, deleting the 152–COOH-terminal amino acids. DD-DAPk (amino acids 1,301–1,431), Flag-tagged at the NH2 terminus, was constructed by PCR. The DD/L1337N mutation was constructed by in vitro mutagenesis. Amino acid numbers in DAP-kinase are according to Swissprot accession number P53355. The luciferase gene was subcloned into pcDNA3 from pGL3-luciferase (Promega), and bcl-2 from pBluescript-bcl-2. The previously described MORT1 and its dominant negative mutant (DN-MORT1), DN-Caspase-8 (also named MACHα-C360S), p55-TNF-R, and p55/Fas chimera cloned into pcDNA3, were used (Boldin et al. 1996). CrmA and p35 cDNAs were previously described (Beidler et al. 1995; Tewari and Dixit 1995). pEGFP-NI was purchased from CLONTECH Laboratories (GFP, green fluorescent protein).
Cell Lines, Transfections, and Apoptotic Assays
The HeLa human epithelial carcinoma cells, 293 human embryonic kidney cells, and MCF7 human breast carcinoma cells were grown in DME (Biological Industries) with 10% FCS (Bio-Lab Scientific Ltd.). The HeLa-tTA clone (Gossen and Bujard 1992) was used for transient transfections because of its high transfectability and the fact that it undergoes apoptotic death in response to TNF-α and cycloheximide. All cells for transient transfection were seeded in a 6-well plate a day before transfection at density of 105 cells/well. Transfections were done by the calcium–phosphate method. For each well, we used a mixture containing 0.5 μg of cell death–inducing plasmid (either p55-TNF-R, p55/Fas chimera, MORT1, or ΔCaM DAPk mutant), 1.5 μg of a plasmid to be tested for cell death protection (DN-MORT, DN-Caspase-8, DD-DAPk, CrmA, p35, or luciferase as a control), and 0.5 μg of GFP plasmid. Cells were counted and photographed 24 h after transfection. In each transfection four fields, each consisting of at least 100 GFP-positive cells, were scored for apoptotic cells according to their morphology. All the experiments were repeated at least four times. When indicated, cell lysates were prepared from the transient transfection at 24 h. For the experiments in Fig. 3 e, cells were transfected solely with the DD-DAPk or DN-MORT and treated 24 h after transfection with human recombinant TNF-α (30 ng/ml; R&D Systems, Inc.) and cycloheximide (10 μg/ml) for 3 h. Stable transfections of HeLa cells and neutral red dye uptake assays were done as previously described (Deiss et al. 1995). Human recombinant IFN-γ (PeproTech) was added at 1,000 U/ml. For Fas killing of HeLa cells by agonistic antibodies, the cells were pretreated for 24 h with 25 U/ml of IFN-γ (to increase Fas expression) and exposed to 50 ng/ml of anti–Fas/APO-1 antibodies (IgG3; P.H. Krammer). The percentage of viability was calculated as a fraction of the values measured in the absence of treatment. For poly (ADP–ribose) polymerase (PARP) cleavage experiments, protein A (5 μg/ml; Sigma Chemical Co.) was added concomitantly with the anti-Fas agonistic antibodies and cell extracts were prepared after 4 h.
Figure 3.

Expression of DD-DAPk protects from TNF-α– and Fas-induced cell death. (a) Expression of endogenous DAP-kinase in cell lines. Western blotting analysis of extracts of indicated cells, using anti–DAP-kinase antibodies. (b) Expression of recombinant DD-DAPk. Extracts from cells transfected with DD-DAPk construct were immunoprecipitated by anti-Flag antibodies and analyzed by Western blotting using anti-Flag. (c) Transient transfection of 293, HeLa, or MCF7 cells with vectors encoding p55-TNF-R, GFP, and either luciferase (Luc), death domain of DAP-kinase (DD-DAPk), or dominant negative mutant of FADD/MORT1 (DN-MORT). The percentage of apoptotic cells was calculated as described in Materials and Methods. (d) Same as c, except that p55/Fas chimera was used instead of p55-TNF-R. (e) Transient transfection of HeLa cells with vectors encoding GFP and either luciferase, DD-DAPk, or DN-MORT. Cells were treated 24 h after transfection with a combination of TNF-α and cycloheximide. The number of apoptotic cells was scored under fluorescent microscopy 3 h after treatment.
Immunoblot Analysis
Western analysis was done as described before (Cohen et al. 1997) using anti–DAP-kinase mAbs (1:2,000; Sigma Chemical Co.) or anti-PARP antibodies (1:5,000; BIOMOL). For detection of Flag-tagged DD-DAPk, 4 × 106 cells were lysed in protein sample buffer and boiled. The clear supernatant was diluted eightfold with protein lysis buffer (Cohen et al. 1997) and 20 μl of anti-Flag mAbs coupled to agarose beads (M2 affinity gel; IBI, Kodak) were added for 3 h of incubation at 4°C. After three washes with protein lysis buffer, proteins were eluted with sample buffer and boiled. Samples were analyzed by 15% SDS-PAGE, transferred to nitrocellulose, and subjected to Western blotting analysis with anti-Flag mAbs (1:200; IBI, Kodak).
Results
DAP-kinase Is Involved in FAS and TNF-α–induced Apoptosis
The involvement of DAP-kinase in Fas-induced cell death was first analyzed in HeLa cells, and compared in the same assays to the well established involvement of DAP-kinase in IFN-γ responses. For this purpose, a previously described polyclonal population of HeLa cells, which stably expresses high levels of DAP-kinase antisense RNA from an Epstein-Barr virus-based vector (Deiss et al. 1995), was used. As a control, we used another polyclonal population of HeLa cells that was transfected with a nonrelevant vector carrying the dihydrofolate reductase gene (Deiss et al. 1995). The viability of the cells was assessed using neutral red dye uptake assay. The DHFR-transfected HeLa cells were efficiently killed by IFN-γ as well as by the agonistic antibodies against Fas/APO-1, which trigger Fas signaling by inducing oligomerization of the receptors (Fig. 1 a). The antisense DAP-kinase transfectants, however, displayed reduced cell death sensitivity to both IFN-γ and Fas signaling. The extent of protection from IFN-γ and Fas-induced cell death was similar and in both cases cell viability in treated cultures remained ∼50–55% (Fig. 1 a). The difference in sensitivity between the two cell populations was also prominent when PRAP cleavage, indicative of caspase activation, was measured in response to increasing concentrations of the anti–Fas/APO-1 agonistic antibodies (Fig. 1 b). Thus, a reduction in the levels of endogenous DAP-kinase protein by antisense RNA (Deiss et al. 1995) relieves not only cell death responses to IFN-γ, the feature that served as the basis for the original selection, but also cell death responses to Fas. This suggests that DAP-kinase may be a common mediator in both cell death scenarios.
Figure 1.
DAP-kinase antisense RNA expression protects HeLa cells from Fas-induced cell death. (a) HeLa cells were stably transfected with DAP kinase antisense cDNA fragment or with DHFR cDNA as a nonrelevant control. Viability was measured by neutral red dye uptake assay and was calculated as described in Materials and Methods either after 40 h of exposure to anti–Fas/APO-1 agonistic antibodies (50 ng/ml) or after 8 d of IFN-γ treatment (1,000 U/ml). The results represent an average of three independent experiments performed in quadruplet. (b) PARP cleavage analysis of the transfected HeLa cells treated with the indicated amount of anti–Fas/APO-1 antibodies. The upper band is the 116-kD noncleaved PARP and the lower band is the typical 85-kD cleaved product.


Another link between DAP-kinase and cytotoxic cytokines was found in our lab by a set of experiments that undertook an opposite approach. We reintroduced a DAP-kinase expression construct into DAP-kinase null cells and assayed whether it affected the cells' sensitivity to TNF-α. Expression of DAP-kinase enhanced the number of apoptotic nuclei as compared with cells transfected with an empty vector (Inbal et al. 1997). Thus, restoration of DAP-kinase into cells that are DAP-kinase negative accelerated TNF-α–induced cell death.
The Death Domain Is Essential for the Death-promoting Function of DAP-kinase and Displays Dominant Negative Features
To study the role of the death domain, it was first tested whether its deletion may reduce the death-inducing functions of DAP-kinase in transiently transfected 293 human embryonic kidney cells. A constitutively active mutant of DAP-kinase (ΔCaM) in which the catalytic activity is no longer dependent on calcium/calmodulin was employed. This gain-of-function mutant was previously shown to be an effective inducer of cell death when transfected on its own into cells (Cohen et al. 1997). To quantitate the number of apoptotic cells, we cotransfected the ΔCaM mutant with a vector expressing the GFP protein. The latter was used as a marker to visualize the transfected cells and to assess the apoptotic frequency among the transfectants according to morphological alterations. Apoptotic cells were scored after 24 h. Overexpression of the ΔCaM mutant of DAP-kinase resulted in massive apoptotic cell death (Fig. 2, a and d). Most of the GFP positive green cells rounded up and shrunk, some of them showed cytoplasmic blebs, and some were further fragmented into apoptotic bodies. In contrast, when the cells were transfected with the ΔCaM mutant deleted of its death domain (Fig. 2 c, ΔCaM/ΔDD), apoptotic cells were much less abundant (23% apoptotic cells compared with 68% in ΔCaM transfections; see Fig. 3 d). Similar results were obtained upon transfections of these constructs into MCF7 human breast carcinoma cells (data not shown). The two recombinant proteins were expressed to comparable levels in these transient transfection assays (Fig. 2 b). Deletion of the death domain from the wild-type DAP-kinase, which as expected is a less effective killer than the constitutively active kinase, also reduced its ability to induce cell death (14.5% apoptotic cells compared with 32% in DAP-kinase transfections; Fig. 2 d). Therefore, it is concluded that the death domain contributes to the death-inducing function of DAP-kinase.
Figure 2.
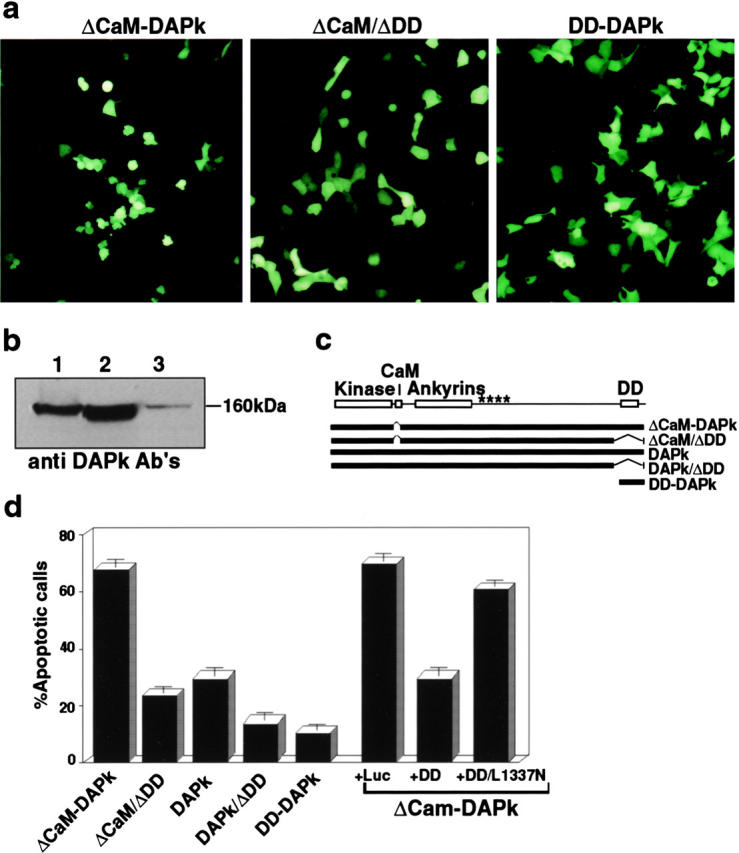
The death domain of DAP-kinase is important for its function in apoptosis. (a) Transient transfections of 293 cells with ΔCaM mutant, ΔCaM/ΔDD mutant, or with the DD-DAPk. Photographs were taken 24 h after transfection under fluorescent light microscope to visualize GFP positive cells. (b) Expression of ΔCaM and ΔCaM/ΔDD mutants in the transiently transfected 293 cells. Lane 1, expression of ΔCaM-DAPk; lane 2, expression of ΔCaM/ΔDD-DAPk; and lane 3, endogenous DAPk in cells transfected with a nonrelevant vector. Western blotting analysis was done with anti–DAP-kinase antibodies. (c) Schematic presentation of DAP-kinase mutant proteins used in these experiments. Kinase, kinase domain; CaM, calmodulin binding and regulatory domain; ankyrin, ankyrin repeats; and DD, death domain. Asterisks delineate the region that by deletion mapping was shown to be responsible for cytoskeletal binding. (d) Transfections as shown in a, with the indicated constructs or double transfections of ΔCaM mutant with the indicated constructs. The percentage of apoptotic cells was calculated as described in Materials and Methods.
Since death domains of other known proteins were shown to mediate protein–protein interactions, we postulated that the death domain of DAP-kinase (DD-DAPk), which contains all the functionally conserved regions (Feinstein et al. 1995), may also be involved in interactions with its specific partners, and, thus, may act in a dominant negative manner. For that purpose, the death domain fragment (Fig. 2 c, DD-DAPk) was subcloned into pcDNA3 expression vector. The DD-DAPk did not display any apoptotic activity when transfected into 293 cells (Fig. 2, a and d), although expression was detectable in the transiently transfected populations (Fig. 3 b). When the DD-DAPk was cotransfected with the ΔCaM mutant of DAP-kinase it reduced significantly cell death induced by DAP-kinase overexpression (Fig. 2 d). In contrast, a mutant death domain (DD/L1337N), which carries a mutation equivalent to the known inactivating lpr mutation in the Fas receptor death domain, failed to inhibit cell death induced by DAP-kinase (Fig. 2 d). Thus, the DD-DAPk can be used as a dominant negative fragment that blocks the action of the full-length protein, and, therefore, might be suitable for checking the involvement of DAP-kinase in cell death induced by TNF-α or Fas.
The Death Domain of DAP-kinase Can Protect from TNF-α– and Fas-induced Cell Death
TNF-α– and Fas-induced cell death was triggered by overexpressing the corresponding receptors in 293 and HeLa cervical carcinoma cells in transient transfection assays. Both cell lines express the endogenous DAP-kinase protein (Fig. 3 a). The receptor cDNAs were cotransfected with the vector expressing the GFP protein. Transfection of p55 TNF-R into 293 or HeLa cells resulted in massive cell death by 24 h (Fig. 3 c). We also confirmed that the observed cell death in these transient assays was caused by p55 TNF-R activation, by using the previously described dominant negative mutant of FADD/MORT-1 (called DN-MORT or DN-FADD) that is deleted of its death effector domain (Boldin et al. 1995; Chinnaiyan et al. 1995). DN-MORT abrogates the cytotoxic effects of the Fas ligand or TNF-α by preventing the endogenous adaptor protein from forming the signaling complexes at the receptor level (Chinnaiyan et al. 1996; Boldin et al. 1996). Cotransfection of DN-MORT with p55 TNF-R inhibited almost completely the induced cell death in 293 and HeLa cells: ∼90% of the transfected cells remained viable with normal flat morphology, thus, confirming the specificity as well as providing a positive death protective control in these transient assays (Fig. 3 c).
The death assays showed that in 293 and HeLa cells, the DD-DAPk inhibited TNF-induced cell death by ∼50% (Fig. 3 c). To further assess the specificity of DD-DAPk inhibitory effect toward DAP-kinase, another cell line, MCF7 breast carcinoma, was used in these transfections. MCF7 cells were chosen since they do not express the endogenous DAP-kinase gene (Fig. 3 a), consistent with previous data (Kissil et al. 1997). DD-DAPk had no protective effect on p55 TNF-R–induced cell death in MCF-7 cells (Fig. 3 c), in spite of the fact that it was efficiently expressed in these assays (Fig. 3 b) and in contrast to the strong protection conveyed in these assays by DN-MORT. DD-DAPk by itself had no cytotoxic effects in MCF7 cells (data not shown), which is consistent with the results obtained in 293 cells (Fig. 2). The lack of any death protective effect in DAP-kinase negative cells provides strong evidence that DD-DAPk inhibits exclusively the function of the endogenous DAP-kinase, indicating, again, that this fragment can be used as a specific dominant negative mutant for DAP-kinase.
We used the same method of cotransfections to investigate the involvement of DAP-kinase in Fas-induced cell death. For this purpose, we used a chimeric receptor composed of the extracellular portion of TNF-R and the intracellular portion of Fas, which was known to be more effective in inducing cell death by self oligomerization than wild-type Fas receptors (Boldin et al. 1996). Its cotransfection with GFP into 293 or HeLa cells resulted in ∼80% apoptotic cells among the GFP positive cells. Cotransfection with DN-MORT reduced cell death to 10% and cotransfection with DD-DAP-kinase reduced cell death to 40% (Fig. 3 d). Again, DD-DAPk failed to protect the MCF7 cells that do not express endogenous DAP-kinase from Fas-induced cell death. Interestingly, the wild-type Fas receptor, which by itself is a weak inducer of cell death, when cotransfected with wild-type DAP-kinase yielded strong death responses (data not shown).
Finally, in another type of assay, apoptosis was induced by adding an external ligand (instead of receptor overexpression). To this aim, HeLa cells were treated with a combination of TNF-α and cycloheximide, which induced apoptosis in these cells, at 24 h after transfection with DD-DAPk, DN-MORT, or the luciferase control. DD-DAPk reduced apoptotic cell death by 60% (Fig. 3 e) and DN-MORT was more potent in reducing cell death. This assay demonstrated again that DAP-kinase participates in TNF-induced cell death. It also indicated that its function is not dependent on de novo protein synthesis. Altogether, these transient transfection assays provide additional support for DAP-kinase being a positive mediator in both TNF-α– and Fas-induced cell death.
Functional Position of DAP-kinase with Respect to the DISC formation, bcl-2, and Terminator Caspases
To place DAP-kinase along the apoptotic pathways of TNF-α and Fas, several known components of the system were assayed in cotransfection assays. First the 293 cells were transfected with a vector encoding the adaptor protein–FADD/MORT1 that recruits proteins such as caspase-8 to the vicinity of TNF-R and Fas. In agreement with results from other laboratories (Boldin et al. 1995; Chinnaiyan et al. 1995), overexpression of FADD/MORT1 efficiently induced cell death. Cotransfection of FADD/MORT1 with its previously mentioned dominant negative mutant DN-MORT1, significantly reduced cell death (Fig. 4 a). As expected, a dominant negative mutant of caspase-8 (DN-Caspase-8), in which the cysteine in the active site was substituted for serine (MACHα-C360S in Boldin et al. 1996), reduced cell death induced by MORT1. Interestingly, cotransfection of DD-DAPk together with FADD/MORT1 reduced very similarly the number of apoptotic cells, thus, placing DAP-kinase downstream to MORT1.
Figure 4.
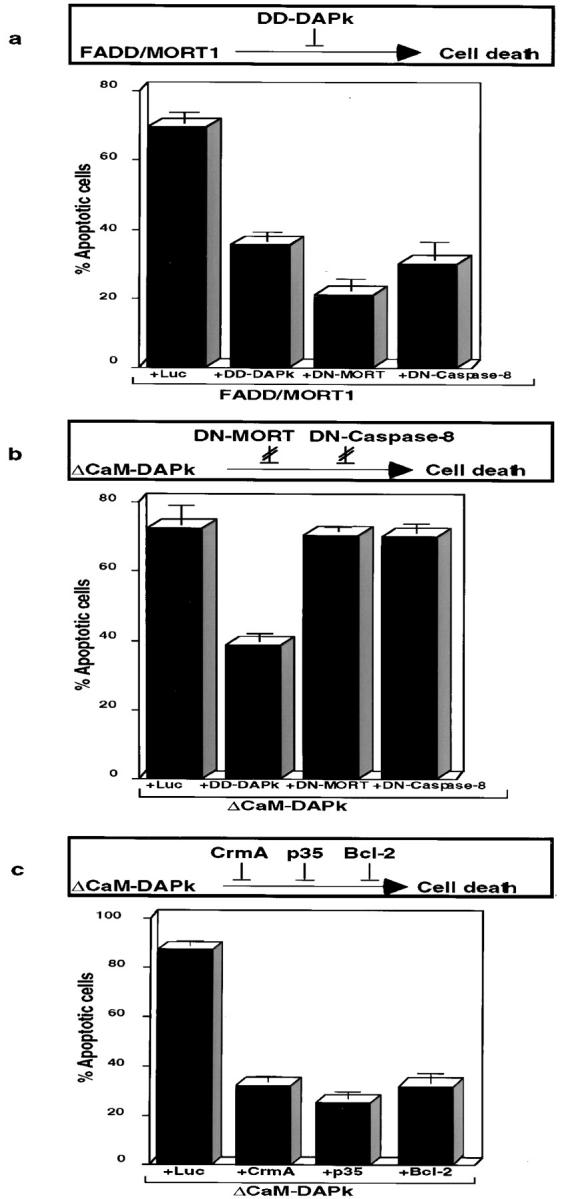
Functional positioning of DAP-kinase along TNF/Fas cell death pathways. Schematic representation of the results is depicted above each bar graph. (a) Transient transfections of 293 cells with vectors encoding FADD/MORT1 together with vectors encoding the indicated proteins. GFP plasmid is present in each of the transfections. (b and c) Transfections as in a, except that the ΔCaM mutant of DAP-kinase was used instead of FADD/MORT1 together with vectors encoding the indicated proteins.
The reciprocal approach was to test whether DN-MORT1, DN-Caspase-8, or DD-DAPk could rescue death imposed by ΔCaM-DAPk overexpression. Both DN-MORT1 and DN-Caspase-8 did not reduce cell death induced by activated DAP-kinase, whereas DD-DAPk served as a positive control to the experiment (Fig. 4 b). Thus, DAP-kinase functions downstream to FADD/MORT1 as well as to caspase-8, which are both recruited to the DISC formed at the cytoplasmic portions of the TNF-R or Fas. The inability of DN-MORT1, which is composed of the death domain of MORT1, to protect from DAP-kinase–induced death can serve as a control for specificity of overexpressed death domains. To address pathways involving mitochondria, the ability of bcl-2 to rescue death by activated DAP-kinase was tested. It was found that bcl-2 reduced death from 86 to 32% (Fig. 4 c), suggesting some functional interaction with mitochondrial-based events.
Finally, the possibility that other caspases may function as downstream mediators of DAP-kinase was tested by using two natural caspase inhibitors: CrmA, which is encoded by the cowpox virus genome; and p35, which is a baculovirus encoded protein (for review see Villa et al. 1997). These inhibitors were shown to inhibit the proteolytic activity of several caspases and, as a consequence, to block TNF-α– or Fas-induced cell death (Beidler et al. 1995; Enari et al. 1995; Tewari and Dixit 1995). Cotransfection of the ΔCaM mutant of DAP-kinase with either one of these inhibitors decreased significantly cell death (32% for CrmA, 26% for p35 compared with 86% without inhibitors) (Fig. 4 c). These results functionally place some members of the caspase family, probably other than caspase-8, downstream to DAP-kinase, along pathway(s) leading to cell death.
Discussion
The strategy of functional gene cloning, used for the rescue of DAP-kinase, was designed with the intention of isolating genes that lie downstream to the IFN-γ early JAK/STAT signaling and, therefore, probably common to various apoptotic systems. This was achieved by introducing an IFN-stimulated responsive element into the transcription cassette that drives the antisense RNA expression. The latter step in the construction of the antisense cDNA library guaranteed that the selection will depend on intact JAK/STAT signaling from IFN-γ receptors, thus, increasing the probability of hitting genes that lie further downstream (Deiss and Kimchi 1991; Deiss et al. 1995). Indeed, the present finding that DAP-kinase mediates TNF-α– and Fas-induced cell death, strongly supports the notion that a central death effector gene has been rescued, upon which various types of receptor signaling cascades eventually converge.
The involvement of DAP-kinase in TNF-α– and Fas-induced cell death is supported here by several independent lines of evidence. First, expression of the antisense RNA fragment of DAP-kinase protected HeLa cells from Fas-induced cell death (Fig. 1). Second, the death domain of DAP-kinase (DD-DAPk) protected 293 human embryonic kidney cells as well as HeLa cells from apoptosis triggered by overexpression of p55-TNF-R and Fas death receptors or by the TNF-α ligand (Fig. 3). Also, the previous data that restoration of DAP-kinase expression in D122 Lewis lung carcinoma cells, which do not express endogenous DAP-kinase, accelerated significantly the appearance of the apoptotic phenotype in response to TNF-α support this line (Inbal et al. 1997). Altogether, the data suggest that DAP-kinase functions as a positive mediator of these activated cytotoxic receptors belonging to the TNF receptor family.
The assays involving transfections with DD-DAPk support for the first time the notion that this region of the protein displays dominant negative features. Moreover, deletion of this region impaired the ability of DAP-kinase to induce cell death. In the yeast two-hybrid system, the death domain of DAP-kinase did not interact with itself (Feinstein, E., and A. Kimchi, unpublished data), suggesting that the death domain does not mediate homodimerization of DAP-kinase. Therefore, this domain could potentially mediate interactions with other proteins that are critical for the function of DAP-kinase in cell death, the nature of which is under current investigation.
The protection conveyed by the death domain of DAP-kinase was always partial (∼50%) and remained so even when the amount of DNA used for the transient transfections were significantly increased (data not shown). The effects of DD-DAPk were, therefore, clearly milder than the effects of the DN-MORT obtained in the same assays. This is not surprising considering the different functional position along the death pathways of the two proteins. FADD/MORT-1 acts in the proximity of Fas and TNF-α receptors and, therefore, DN-MORT mutant blocks early receptor-generated events, such as the recruitment of caspase-8 to the receptor complex. As a consequence, it efficiently prevents most intracellular responses. DAP-kinase, in contrast, is not part of the DISC, but rather functions further downstream. The downstream position with respect to the DISC was based on two lines of evidence. One showed that DD-DAPk protected from FADD/MORT1–induced cell death (Fig. 4 a). The other illustrated that the death-promoting effect of the ΔCaM gain-of-function mutant of DAP-kinase was clearly resistant to the dominant negative components of the DISC (e.g., DN-MORT and DN-Caspase 8) (Fig. 4 b). Also, when assayed by the yeast two-hybrid system, the death domain of DAP-kinase did not bind to the death domain of the Fas receptor (Feinstein, E., and A. Kimchi, unpublished data). Beyond the receptor complex, the death pathways may diverge to several branches, and the partial protections conveyed either by antisense DAP-kinase RNA (Fig. 1) or by DD-DAPk (Fig. 3) imply that DAP-kinase functions along some but not all these branches. Also, the finding that DAP-kinase negative cell lines, such as MCF7 or D122 (Inbal et al. 1997), can eventually be killed by TNF-α is consistent with the existence of DAP-kinase–dependent and –independent branches.
Virally produced inhibitors of caspases were used to show that members of the cysteine protease family are involved in DAP-kinase–induced cell death. Among the two inhibitors that were used, crmA is believed to be more specific to the subfamily of the interleukin 1β–converting enzyme (ICE)-like proteases, whereas p35 has a wider spectrum (for review see Villa et al. 1997) In our experiments, both inhibitors suppressed ΔCaM-DAPk–induced cell death to a similar extent. These results suggest that ICE-like proteases mediate the effect of DAP-kinase. The caspase family in general and the ICE-like subgroup in particular include several proteases acting at different positions along death pathways. Therefore, it is hard to speculate, at the present time, about the specific proteases that mediate the effect of DAP-kinase and their defined substrates.
It is well established that the fast track of apoptosis (comprising a direct cascade of caspase activation) is not an exclusive pathway in the Fas-induced signaling (Scaffidi et al. 1998). Mitochondrial-based events often provide a second route for caspase activation and cell death in these systems. In light of our findings that bcl-2 protected from cell death induced by the ΔCaM-DAPk mutant, one possibility is that DAP-kinase may be involved in one of these mitochondrial pathways. Alternatively, since DAP-kinase associates with the actin microfilament system (Cohen et al. 1997), it might mediate signals converging into or emanating from the cytoskeleton. Should the protein substrates that are directly phosphorylated by DAP-kinase be identified, then the detailed mechanisms coupling the kinase to downstream targets may be deciphered. In any case, the multidomain structure of this enzyme predicts the formation of multiprotein complex around DAP-kinase. Taken together with its broad involvement in cell death induced by several different triggers, DAP-kinase appears to be a major player in apoptotic pathways.
Acknowledgments
We thank D. Wallach (Weizmann Institute of Science), M. Tewari (Temple University, Philadelphia, PA), R. Stein (Tel-Aviv University), C. Kahana (Weizmann Institute of Science), and S.J. Korsmeyer (Washington University, Seattle, WA) for providing the different plasmids and P. Krammer (DKFZ) for anti–APO-1 agonistic antibodies.
This work was supported by the Israel Foundation, which is administered by the Israel Academy of Science and Humanities, and by QBI Enterprises. A. Kimchi is the incumbent of the Helena Rubinstein Chair of Cancer Research.
Footnotes
1.used in this paper: ΔCAM, deletion of calmodulin regulatory domain; CARD, caspase-recruiting domain; DAP, death-associated protein; DD, death domain; DISC, death-inducing signaling complex; DN-MORT/FADD, dominant negative mutant of MORT/FADD; GFP, green fluorescent protein; ICE, interleukin 1β–converting enzyme; IFN-γ, interferon-γ; PARP,poly (ADP–ribose) polymerase; TNF, tumor necrosis factor; TNF-R, TNF-α receptor
References
- Beidler D.R., Tewari M., Friesen P.D., Poirier G., Dixit V.M. The baculovirus p35 protein inhibits Fas- and tumor necrosis factor–induced apoptosis. J. Biol. Chem. 1995;270:16526–16528. doi: 10.1074/jbc.270.28.16526. [DOI] [PubMed] [Google Scholar]
- Boldin M.P., Varfolomeev E.E., Pancer Z., Mett I.L., Camonis J.H., Wallach D. A novel protein that interacts with the death domain of Fas/APO1 contains a sequence motif related to the death domain. J. Biol. Chem. 1995;270:7795–7798. doi: 10.1074/jbc.270.14.7795. [DOI] [PubMed] [Google Scholar]
- Boldin M.P., Goncharov T.M., Goltsev Y.V., Wallach D. Involvement of MACH, a novel Mort1/FADD–interacting protease, in Fas/APO-1 and TNF receptor-induced cell death. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- Brancolini C., Benedetti M., Schneider C. Microfilament reorganization during apoptosisthe role of Gas2, a possible substrate for ICE-like proteases. EMBO (Eur. Mol. Biol. Organ.) J. 1995;14:5179–5190. doi: 10.1002/j.1460-2075.1995.tb00202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnaiyan A.M., O'Rourke K., Tewari M., Dixit V.M. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan A.M., Tepper C.G., Seldin M.F., O'Rourke K., Kischkel F.C., Hellbardt S., Krammer P.H., Peter M.E., Dixit V.M. FADD/MORT1 is a common mediator of CD95 (Fas/APO-1) and tumor necrosis factor receptor-induced apoptosis. J. Biol. Chem. 1996;271:4961–4965. doi: 10.1074/jbc.271.9.4961. [DOI] [PubMed] [Google Scholar]
- Cohen, O., E. Feinstein, and A. Kimchi. 1997. EMBO (Eur. Mol. Biol. Organ.) J. DAP-kinase is a Ca2+/calmodulin-dependent, cytoskeletal-associated protein kinase, with cell death-inducing functions that depend on its catalytic activity. 16:998–1008. [DOI] [PMC free article] [PubMed]
- Cryns V., Yuan J. Proteases to die for. Genes Dev. 1998;12:1551–1570. doi: 10.1101/gad.12.11.1551. [DOI] [PubMed] [Google Scholar]
- Deiss L.P., Kimchi A. A genetic tool used to identify thioredoxin as a mediator of a growth inhibitory signal. Science. 1991;252:117–120. doi: 10.1126/science.1901424. [DOI] [PubMed] [Google Scholar]
- Deiss L.P., Feinstein E., Berissi H., Cohen O., Kimchi A. Identification of a novel serine/threonine kinase and a novel 15-kD protein as potential mediators of the gamma interferon-induced cell death. Genes Dev. 1995;9:15–30. doi: 10.1101/gad.9.1.15. [DOI] [PubMed] [Google Scholar]
- Enari M., Hug H., Nagata S. Involvement of an ICE-like protease in Fas-mediated apoptosis. Nature. 1995;375:78–81. doi: 10.1038/375078a0. [DOI] [PubMed] [Google Scholar]
- Feinstein E., Wallach D., Boldin M., Varfolomeev E., Kimchi A. The death domaina module shared by proteins with diverse cellular functions. Trends Biochem. Sci. 1995;20:342–344. doi: 10.1016/s0968-0004(00)89070-2. [DOI] [PubMed] [Google Scholar]
- Green D., Kroemer G. The central executioners of apoptosiscaspases or mitochondria? Trends Cell Biol. 1998;8:267–271. doi: 10.1016/s0962-8924(98)01273-2. [DOI] [PubMed] [Google Scholar]
- Gossen M., Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inbal B., Cohen O., Polak-Charcon S., Kopolovic Y., Vadai E., Eisenbach L., Kimchi A. DAP kinase links the control of apoptosis to metastasis. Nature. 1997;390:180–184. doi: 10.1038/36599. [DOI] [PubMed] [Google Scholar]
- Kimchi A. DAP genesnovel apoptotic genes isolated by a functional approach to gene cloning. Biochim. Biophys. Acta. 1998;1377:F13–F33. doi: 10.1016/s0304-419x(98)00002-x. [DOI] [PubMed] [Google Scholar]
- Kischkel F.C., Hellbardt S., Behrmann I., Germer M., Pawlita M., Krammer P.H., Peter M.E. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO (Eur. Mol. Biol. Organ.) J. 1995;14:5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissil J., Feinstein E., Cohen O., Jones P.A., Tsai Y.C., Knowles M.A., Eydmann M.E., Kimchi A. DAP-kinase loss of expression in various carcinoma and B-cell lymphoma cell linespossible implications for role as tumor suppressor gene. Oncogene. 1997;15:403–407. doi: 10.1038/sj.onc.1201172. [DOI] [PubMed] [Google Scholar]
- Kothakota S., Azuma T., Reinhard C., Klippel A., Tang J., Chu K., McGarry T.J., Kirschner M.W., Koths K., Kwiatkowski D.J., Williams L.T. Caspase-3-generated fragment of gelsolineffector of morphological change in apoptosis. Science. 1997;278:294–298. doi: 10.1126/science.278.5336.294. [DOI] [PubMed] [Google Scholar]
- Lazebnik Y.A., Takahashi A., Moir R.D., Goldman R.D., Poirier G.G., Kaufmann S.H., Earnshaw W.C. Studies of lamin proteinase reveal multiple parallel biochemical pathways during apoptotic execution. Proc. Natl. Acad. Sci. USA. 1995;92:9042–9046. doi: 10.1073/pnas.92.20.9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema J.P., Scaffidi C., Kischkel F.C., Shevchenko A., Mann M., Krammer P.H., Peter M.E. FLICE is activated by association with the CD95 death-inducing signaling complex (DISC) EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:2794–2804. doi: 10.1093/emboj/16.10.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvesen G.S., Dixit V.M. Caspasesintracellular signaling by proteolysis. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- Scaffidi C., Fulda S., Srinivasan A., Friesen C., Li F., Tomaselli K.J., Debatin K.M., Krammer P.H., Peter M.E. Two CD95 (APO-1/Fas) signaling pathways. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:1675–1687. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari M., Dixit V.M. Fas- and tumor necrosis factor-induced apoptosis is inhibited by the pox virus crmA gene product. J. Biol. Chem. 1995;270:3255–3260. doi: 10.1074/jbc.270.7.3255. [DOI] [PubMed] [Google Scholar]
- Villa P., Kaufmann S.H., Earnshaw W.C. Caspases and caspase inhibitors. Trends Biochem. Sci. 1997;22:388–393. doi: 10.1016/s0968-0004(97)01107-9. [DOI] [PubMed] [Google Scholar]
- Wallach D., Boldin M.P., Kovalenko A.V., Malinin N.L., Mett I.L., Camonis J.H. The yeast two-hybrid screening technique and its use in the study of protein–protein interactions in apoptosis. Curr. Opin. Immunol. 1998;10:131–136. doi: 10.1016/s0952-7915(98)80240-9. [DOI] [PubMed] [Google Scholar]