

Abstract
We have identified a pair of related yeast proteins, Sro7p and Sro77p, based on their ability to bind to the plasma membrane SNARE (SNARE) protein, Sec9p. These proteins show significant similarity to the Drosophila tumor suppressor, lethal giant larvae and to the neuronal syntaxin–binding protein, tomosyn. SRO7 and SRO77 have redundant functions as loss of both gene products leads to a severe cold-sensitive growth defect that correlates with a severe defect in exocytosis. We show that similar to Sec9, Sro7/77 functions in the docking and fusion of post-Golgi vesicles with the plasma membrane. In contrast to a previous report, we see no defect in actin polarity under conditions where we see a dramatic effect on secretion. This demonstrates that the primary function of Sro7/77, and likely all members of the lethal giant larvae family, is in exocytosis rather than in regulating the actin cytoskeleton. Analysis of the association of Sro7p and Sec9p demonstrates that Sro7p directly interacts with Sec9p both in the cytosol and in the plasma membrane and can associate with Sec9p in the context of a SNAP receptor complex. Genetic analysis suggests that Sro7 and Sec9 function together in a pathway downstream of the Rho3 GTPase. Taken together, our studies suggest that members of the lethal giant larvae/tomosyn/Sro7 family play an important role in polarized exocytosis by regulating SNARE function on the plasma membrane.
Keywords: exocytosis, SNARE complex, cell polarity, tumor suppressor, tomosyn
Yeast cells grow asymmetrically by delivering proteins and lipids to specific sites on the plasma membrane. The site of exocytic delivery that corresponds to a small region at the growing bud tip for much of the cell cycle becomes delocalized within the bud as it enlarges and, finally, relocalizes to the mother–bud junction before cell division (Tkacz and Lampen 1972, Tkacz and Lampen 1973; Field and Schekman 1980). This process requires a continual coordination between the exocytic machinery and the actin cytoskeleton. In particular, the integrity and polarity of the actin cytoskeleton (Novick and Botstein 1985; Pruyne et al. 1998) as well as the function of an unconventional type V myosin, Myo2 (Lillie and Brown 1994), have been shown to be required for the recruitment of some of the spatial landmarks for the targeting and fusion of post-Golgi vesicles to specific sites on the plasma membrane (TerBush et al. 1996; Ayscough et al. 1997; Finger et al. 1998). In addition, the Rab GTPase Sec4 (Walch-Solimena et al. 1997; Guo et al. 1999) that is required for transport from the Golgi apparatus to the plasma membrane in yeast was found to be functionally intertwined with the Rho GTPase Rho3, previously identified for its maintenance of actin polarity in yeast (Imai et al. 1996). More recently, Rho3 has been suggested to have a direct role in exocytosis. Two-hybrid analysis has suggested that it has the capacity to interact with elements of the exocytic machinery (Robinson et al. 1999), and we have shown a direct involvement of Rho3 both in exocytic fusion and in the polarized delivery of vesicles from the mother to the bud (Adamo, J., G. Rossi, and P. Brennwald, manuscript submitted for publication).
Once the post-Golgi vesicle has been delivered to the target membrane, formation of a tightly packed complex between the vesicle SNAP receptor (SNARE)1 proteins Snc1/2p (Protopopov et al. 1993) and the plasma membrane SNARE proteins Sec9p (Brennwald et al. 1994) and Sso1/2p (Aalto et al. 1993) finally determines a late event in the fusion of the vesicle with the target membrane. The SNARE complex structure is composed of four helical domains whose COOH termini must converge to bring about membrane fusion (Katz et al. 1998). The SNARE proteins have been structurally reclassified into arginine-containing and glutamine-containing (Q-) SNAREs and fusion competent SNARE complexes generally consist of four helix bundles of three Q-SNAREs to one arginine-containing SNARE (Fasshauer et al. 1998). Since the post-Golgi Q-SNAREs (Sec9p and Sso1/2p) in yeast are evenly distributed around the perimeter of the cell (Brennwald et al. 1994), additional modulators of such proteins are required to activate the target Q-SNAREs at the site of polarized exocytosis.
One such protein was identified recently in neurons, named tomosyn (Fujita et al. 1998). Tomosyn is part of a larger family including the yeast proteins Sro7 and Sro77 (Kagami et al. 1998) and the Drosophila tumor suppressor, lethal giant larvae (Mechler et al. 1985). These proteins have also been implicated in cytoskeletal functions. The lethal giant larvae protein was shown to be physically associated with the nonmuscle myosin II and cofractionates with cytoskeletal components (Strand et al. 1994a). SRO7 was identified as a high copy suppressor of rho3Δ and has been shown to have strong genetic interactions with two yeast myosins, Myo1 and Myo2 (Kagami et al. 1998). Tomosyn was identified based on its ability to bind syntaxin and appears to act as a modulator of neuronal SNARE function by relieving the Q-SNARE syntaxin of its inhibitor Sec1 (Fujita et al. 1998).
In this paper, we describe the identification and characterization of the role of Sro7 and Sro77 in post-Golgi transport in yeast. We find that these proteins bind to the target SNARE (t-SNARE) Sec9p in vivo and in vitro, and that like Sec9, loss of their function results in a pronounced defect in Golgi-to-cell surface transport. We postulate that like its neuronal homologue tomosyn, Sro7 may act to regulate SNARE assembly on the plasma membrane, and that by acting downstream of Rho3 it may contribute to the polarity of this process.
Materials and Methods
Media and Reagents
All strains were grown either in YP medium (1% bacto-yeast extract and 2% bacto-peptone; Difco Laboratories Inc.) with 2% glucose or in S minimal media (0.67% yeast nitrogen base; Difco Laboratories Inc.) with 2% glucose supplemented with the required amino acid. For whole cell labeling, strains were cultured in S minimal media with 2% glucose and without methionine. The absorbance of cell suspensions was measured at 599 nm in an Ultrospec Plus UV/visible spectrophotometer (Pharmacia). Sorbitol, sodium azide, N-ethylmaleimide, β-mercaptoethanol, O-dianasidine, glucose oxidase, peroxidase, Triton X-100, TRITC-phalloidin, 4′,6-diamidino-2-phenylindole (DAPI), imidazole GDP, acid molybdate, Fiske-Soubbarow reducer, cytochrome c oxidase, rotenone, NADPH polylysine, and protease inhibitors were obtained from Sigma Chemical Co. Cacodylate, glutaraldehyde, osmium oxide, uranyl acetate, Spurr resin, and 37% formaldehyde were obtained from EM Sciences. [35S]Methionine, 35S-Express label (a mixture of [35S]methionine and [35S]cysteine), and 125I–protein A were purchased from NEN Life Science Products Inc. Protein A–Sepharose CL-4B was purchased from Pharmacia Biotech. Rhodamine-X–conjugated affinity-purified goat α-rabbit IgG was purchased from Jackson ImmunoResearch Laboratories. Molecular weight markers and Tween 20 were obtained from Bio-Rad Laboratories.
Yeast Genetic Techniques
Yeast transformation was performed using the lithium acetate method (Becker and Guarente 1991) and transformants were selected on minimal medium supplemented with the appropriate amino acid at 25°C. Crosses of strains, sporulation of diploids, and tetrad dissections were performed as described (Sherman et al. 1986).
Two-Hybrid Screening
The two-hybrid screening and assay protocols were similar to those described previously (Durfee et al. 1993). SEC9 and the COOH-terminal SNAP-25–like domain of SEC9 were amplified by PCR and inserted into the GAL4-binding domain vector, pAS1-CYH2. A yeast cDNA library, prepared in the GAL4 activation domain vector pACT, was coexpressed with the GAL4BD-SEC9 fusion vector in the yeast strain Y190. Interaction of the GAL4 BD fusion and GAL4 AD fusion proteins allows for expression of the HIS3 and lacZ reporter genes, thereby enabling cells to grow in the absence of histidine and to exhibit β-Gal activity. To assay β-Gal activity, a filter lift assay was used (Bartel et al. 1993). From 106 transformants, 250 clones could grow in the absence of histidine. 147 of these clones exhibited β-Gal activity. These clones were replated on +His plates and retested for β-Gal activity. 77 clones exhibiting the strongest β-Gal activity were grown on plates containing both histidine and tryptophan with 2.5 μg/ml cycloheximide. 63 clones lost β-Gal activity after treatment with cycloheximide. Plasmids from these clones were recovered and reexamined for their ability to give rise to growth on −His plates in combination with the GAL4 BD fused to SEC9 or the GAL4 BD fused to a control construct. Two clones were found to give rise to growth on −His plates containing 50 mM 3-aminotriazol when in combination with the GAL4 BD fused to SEC9 construct but not with the GAL4 BD fused to the control plasmid.
In Vitro Binding Reactions
The Sro7 sequence corresponding to its COOH-terminal 510 amino acids and the Sro77 sequence corresponding to its COOH-terminal 521 amino acids were placed under control of a T7 promoter in the pCITE-4c vector (Novagen Inc.). The resulting plasmids were added to a reticulocyte lysate–coupled in vitro transcription–translation system (TnT; Promega) in the presence of [35S]methionine. For binding of the radiolabeled COOH-terminal domains of Sro7 and Sro77 to glutathione-Sepharose–bound fusion proteins, the [35S]methionine-labeled in vitro transcription–translation reaction mixture was preincubated with glutathione–Sepharose for 30 min on ice followed by centrifugation. The resulting supernatant was used for binding reactions with glutathione-S-transferase (GST) fusion proteins bound to glutathione-Sepharose as described previously (Rossi et al. 1997). The recombinant GST-Sec9p fusion contained the NH2-terminal 150 residues of SEC9 fused in frame to the synthetic SEC9 SNAP-25 domain described previously (Rossi et al. 1997). This protein corresponds to the minimal fully functional form of the protein.
Construction of Deletion Strains
The SRO7 disruption construct was created by subcloning 750 bp immediately upstream and 770 bp immediately downstream of the SRO7 ORF (YPR032w) into the LEU2 integration vector pRS305. The vector was linearized and transformed into the yeast strain BY24 (MAT a/α, ura3-52/ura3-52; leu2-3,112/leu2-3,112; his3-Δ200/his3-Δ200). Transformants were sporulated and subjected to meiotic analysis. Tetrads exhibited a 2:2 segregation pattern for leucine prototrophy but showed no defect in growth at any temperature. The SRO77 disruption construct was prepared by subcloning 560 bp upstream and 860 bp immediately downstream of the SRO77 ORF (YBL106c) into the URA3 integration vector pRS306. The vector was linearized and transformed into the yeast strain BY24. Transformants were sporulated and subjected to meiotic analysis. Tetrads exhibited a 2:2 segregation pattern for uracil prototrophy and showed no defect in growth at any temperature. The sro7Δ, sro77Δ double-disruptant strain was created by crossing the sro7Δ disruptant strain with the sro77Δ disruptant strain. Diploids selected on −leu,−ura media were sporulated and subjected to meiotic analysis. The RHO3 disruption construct was created by subcloning 1,031 bp immediately upstream and 941 bp immediately downstream of the RHO3 ORF into the LEU2 integration vector pRS305. The vector was linearized between the two flanking DNA fragments and transformed into the yeast strain BY23 (MAT a/α, ura3-52/ura3-52, leu2-3,112/leu2-3,112). Disruption was confirmed by PCR analysis on genomic DNA from two independent transformants. Transformants were sporulated and subjected to meiotic analysis.
Secretion Assays
Invertase assays were performed on several tetratype tetrads resulting from a cross between the sro7Δ and sro77Δ single-disruptant strains. Cultures were grown overnight to mid-log phase in YP with 2% glucose (YPD) medium at the permissive temperature of 37°C. Strains were simultaneously shifted to 0.1% glucose media and to the restrictive temperature of 19°C for 3 h. Internal and external invertase activity was measured at the beginning and end of the shift as described previously (Nair et al. 1990). The percentage of total invertase secretion was calculated by the following formula:  . For analysis of the glycosylation state of invertase and carboxypeptidase (CPY), cells were grown overnight to mid-log phase at 37°C in YPD. The strains were shifted to YP medium containing 0.1% glucose and incubated at 19°C for an additional 3 h. The control strains sec18-1 and sec4-8 strains were grown in YPD at 25°C to mid-log phase and shifted to 37°C for 2 h. The strains were transferred to YP medium containing 0.1% glucose and incubated at 37°C for an additional 3 h. Whole cell glass bead lysates were prepared. Samples were subjected to 12.5% SDS-PAGE, transferred to nitrocellulose, and probed with affinity-purified polyclonal α-invertase antibody or polyclonal α-CPY antibody followed by 125I–protein A as secondary.
. For analysis of the glycosylation state of invertase and carboxypeptidase (CPY), cells were grown overnight to mid-log phase at 37°C in YPD. The strains were shifted to YP medium containing 0.1% glucose and incubated at 19°C for an additional 3 h. The control strains sec18-1 and sec4-8 strains were grown in YPD at 25°C to mid-log phase and shifted to 37°C for 2 h. The strains were transferred to YP medium containing 0.1% glucose and incubated at 37°C for an additional 3 h. Whole cell glass bead lysates were prepared. Samples were subjected to 12.5% SDS-PAGE, transferred to nitrocellulose, and probed with affinity-purified polyclonal α-invertase antibody or polyclonal α-CPY antibody followed by 125I–protein A as secondary.
Electron Microscopy
Transmission electron microscopy was performed essentially as described previously (Walworth et al. 1992). Wild-type and sro7Δ, sro77Δ double disruptions were grown overnight in YPD to early log at the permissive temperature of 37°C. Cells were incubated in YP with 0.1% glucose at 19°C for 3 h. Approximately 10 OD599 units were filtered onto a 0.45-μm nitrocellulose filter (Nalgene), washed once with 0.1 M cacodylate buffer, pH 6.8, resuspended in 0.1 M cacodylate buffer with 3% glutaraldehyde, incubated for 1 h at 25°C, and then overnight at 4°C. Samples were washed one time with 5 ml of 50 mM potassium phosphate, pH 7.5, and spheroplasted in 1 ml of KPi with 0.3 mg/ml Zymolyase 100T for every 1 OD599 unit of cells for 40 min at 37°C. Spheroplasts were pelleted and washed two times with 1 ml of ice-cold cacodylate buffer and incubated with 2% osmium tetraoxide (diluted in cacodylate buffer) for 1 h on ice in a hood. The cell pellet was rinsed three times with water and incubated with 1.5 ml of 2% uranyl acetate at 25°C for 1 h. Samples were dehydrated with 5-min successive washes of 50, 70, 90, and 100% ethanol. After a final wash with 100% acetone, the samples were incubated with 50% acetone/50% SPURR medium for 3 h before incubating with the final 100% SPURR solution overnight at room temperature. After baking for 48 h at 80°C, samples were sectioned and layered onto an uncoated copper grid and poststained with lead citrate and uranyl acetate before viewing. Cells were viewed on a JEOL 100CXII electron microscope.
Fluorescence Microscopy and Imaging
For actin staining, cells were grown to mid-log in YPD media and either shifted to the restrictive temperature of 19°C for 3 h or kept at the permissive temperature before fixation. Formaldehyde was added to 3.7% directly to the media and incubated for 10 min at room temperature and centrifuged for 5 min at 3,000 g. A second round of fixation was performed by resuspending the cell pellet in with 3.7% formaldehyde/0.1 M KPO4 buffer, pH 6.5, incubated for 30 min at room temperature, and then pelleted as above. The cell pellets were transferred to a sorbitol buffer (1.2 M sorbitol, 0.1 M KPO4, pH 7.5) for overnight storage at 4°C. The next day, cells were permeabilized for 10 min in 0.1% Triton X-100 and washed two times with PBS. Cells were resuspended in 100 μl of PBS and stained in the dark for 25 min with 35 μl of 3.3 μM TRITC-phalloidin dissolved in methanol. Cells were washed six times with PBS, and then resuspended in the appropriate volume of mounting media (90% glycerol with DAPI to visualize DNA and O-phenylenediamine to retard photobleaching). Stained cells was viewed on a Nikon Eclipse Z600 microscope, images were captured with a Princeton Instruments CCD camera and Metamorph imaging software.
For immunofluorescence staining with affinity-purified α-Sro7 antibodies, yeast strains containing high copy vector only (pB23) or high copy SRO7 (pB497) were grown overnight in selective media to early log phase. 0.1 vol of 1 M KPO4 buffer, pH 6.5, and 0.1 vol of 37% formaldehyde were added directly to the media and incubated for 30 min at room temperature. Cells were centrifuged for 5 min at 3,000 g. A second round of fixation was performed by resuspending the cell pellet in with 3.7% formaldehyde/0.1 M KPO4 buffer, pH 6.5, incubated for 90 min at room temperature, and then pelleted as above. The cell pellets were transferred to a sorbitol buffer (1.2 M sorbitol, 0.1 M KPO4, pH 7.5) for overnight storage at 4°C. Fixed cells were processed for immunofluorescence after permeabilization with 0.5% SDS as described (Redding et al. 1991). Affinity-purified rabbit α-Sro7 was used at a 1:900 dilution. The Sro7 antibody was incubated for 30 min at 25°C with permeabilized cells from a strain containing the sro7 disruption before use. Rhodamine-X–conjugated affinity-purified goat α–rabbit IgG (Jackson ImmunoResearch Laboratories, Inc.) was used at a 1:50 dilution. Stained cells were viewed on a Zeiss LSM 510 confocal microscope and the images captured with LSM 510 software.
Antisera Production and Purification
Rabbit antisera was raised to the carboxy terminus of Sro7 by generating a GST fusion protein with the COOH-terminal 219–amino acids of the Sro7 open reading frame. IgG was first purified from the crude rabbit serum on a DEAE–AffiGel blue column (Bio-Rad Laboratories) according to the manufacturer's directions. The IgG fraction was affinity-purified by binding to the GST-Sro7 fusion protein that had been chemically cross-linked to glutathione–Sepharose beads (Lane and Harlow 1988), and then eluted with 0.2 M glycine, pH 2.8. The peak of IgG was collected, dialyzed against TS buffer (10 mM Tris, pH 7.5, 150 mM NaCl), and then absorbed with GST protein that had been cross-linked to glutathione–Sepharose. The remaining IgG was dialyzed against 50% glycerol (vol/vol) in 1× PBS buffer and stored at –20°C. The concentration of IgG was determined by absorbance at 280 nm and confirmed by SDS-PAGE and Coomassie staining. Affinity-purified antibodies to Sec9p, Sso1/2p, and Snc1/2p were prepared in a similar fashion. Analysis of the α-Sso1/2p and α-Snc1/2 antibodies has demonstrated that they each react well against both isoforms of their respective duplicated gene families. Preimmune IgG for each rabbit was purified on protein A–agarose columns, dialyzed into 50% glycerol/PBS, and quantitated by absorbance at 280 nm.
Subcellular Fractionation
Cells containing vector only (pB23) or SRO7 on high copy (pB497) were grown overnight in selective media, harvested, and grown in rich media for 2 h. Approximately 108 OD599 were washed with 10 mM Tris, pH 7.5, plus 10 mM azide and spheroplasted in 7.2 ml of spheroplast buffer (0.1 M Tris, 10 mM azide, 1.2 M sorbitol, and 21 mM β-mercaptoethanol with 0.1 mg/ml Zymolyase 100T) for 30 min at 37°C. The spheroplasts were lysed in 3 ml of ice-cold triethanolamide-EDTA (TEAE)/sorbitol (10 mM TEA, 1 mM EDTA, pH 7.2, and 0.8 M sorbitol) with 1× protease inhibitor cocktail (protease inhibitor cocktail [PIC], 2 mM 4-(2-aminoethyl) benzenesulfonyl fluoride, 0.5 mM PMSF, 20 μM pepstatin A, and 1 μg/ml each of leupeptin, antipain, and aprotinin) and spun at 450 g for 3 min in a cold centrifuge to get rid of unbroken cells. The lysate was divided into two aliquots and diluted 1:1 with TEAE/sorbitol or TEAE/sorbitol/2% Triton X-100. The lysates were spun at 30,000 g for 15 min in a Sorvall and the supernatant and pellet fraction separated. The supernatant was ultracentrifuged at 100,000 g for 1 h at 4°C. All pellets were normalized to the volume of the supernatant fractions. Samples were boiled in SDS sample buffer, run on a 7% or 12.5% SDS–polyacrylamide gel, and blotted with affinity-purified α-Sro7p antibody (1:200) and polyclonal α-Sso1/2p antibody (1:1,000), respectively. All gels were put on the PhosphorImager screen and radiolabeled bands were quantitated with the STORM using ImageQuant software (both from Molecular Dynamics).
Sucrose Gradient Fractionation
Wild-type cells were grown in rich media, harvested, washed with 10 mM Tris, pH 7.5, plus 10 mM azide and converted to spheroplasts as described above. Spheroplasts prepared from ∼300 OD599 units of cells were resuspended in 6 ml of ice-cold TEAE/sorbitol (see above) and spun at 450 g for 3 min at 4°C to pellet unbroken cells. The cell-free lysate was spun at 36,000 g for 10 min and the supernatant fraction discarded. The pellet fraction containing most of the cell membranes was resuspended in 2 ml of ice-cold 15% sucrose (wt/wt) in TEA and 1 ml corresponding to ∼100 OD599 units was loaded on a sucrose gradient as described (Brennwald et al. 1994). Enzyme assays were carried out as previously described (Walworth and Novick 1987). The GDPase activity is expressed as nanomoles of liberated phosphate/fraction/minute. The cytochrome c reductase is expressed as the rate of increase of the A550 of the reaction using 25 μl of each fraction. Sro7p and the plasma membrane t-SNARE Sso1/2p were detected by immunoblotting. All gels were put on the PhosphorImager screen and radiolabeled bands were quantitated as described above.
DSP Cross-linking and Coimmunoprecipitation
Strains containing high copy myc-tagged SEC9 (pB37) and either full-length SRO7 or SRO7-CT Sro7 in a CEN vector expressed under the regulation of the inducible GAL1 promoter (pB363 and pB367) were grown overnight in synthetic media with raffinose. The cells were shifted to synthetic media with 2% galactose and grown for 1 h at 30°C. Approximately 6 OD599 were labeled with [35S]methionine and cysteine in 3 ml for 1 h at 30°C. Cross-linking and immunoprecipitation were carried out as described (Brennwald et al. 1994). After the labeling, cells were washed in Tris-azide, spheroplasted, and lysed in 300 μl of PBS with PIC. The lysate from each strain was divided into two pools each of 100 μl and treated with or without the chemical cross-linker dithiosuccinnimidylpropionate (DSP). After boiling and dilution with immunoprecipitation buffer, the samples were treated with mAb 9E10. The samples were subjected to a second round of denaturing immunoprecipitations by resuspending the beads from the first immunoprecipitation in reducing boiling buffer.
Second immunoprecipitations were carried out with mAb 9E10 and pAb α-Sro7. Samples were boiled in sample buffer and loaded on a 7 and 10% gel. The gels were treated with stain, destain, dried, and exposed to film. Immunoprecipitations on detergent extracts were done as follows: wild-type yeast strains were grown to mid-log phase overnight in rich media at 25°C. Cells corresponding to ∼200 OD599 were washed in ice-cold 10 mM Tris with 10 mM azide and spheroplasted in 13.3 ml of spheroplast buffer (50 mM KPi, 1.2 M sorbitol, 21 mM β-mercaptoethanol with 0.1 mg/ml Zymolyase 100T) for 30 min at 37°C. Cells were lysed in 6.6 ml of ice-cold extraction buffer (20 mM Hepes-KOH, 150 mM KCl, 0.5% NP-40) with PIC, left to equilibrate on ice for 5 min, and then spun in a cold microfuge to pellet the insoluble material. The supernatant fractions were pooled and used to set up six 1-ml immunoprecipitations on ice. Affinity-purified antibodies to Sro7p, Sec9p, Snc1/2p, and Sso1/2p were added in saturating amounts and equivalent amounts of preimmune sera purified from each respective rabbit was used as a control immunoprecipitation. After 1 h on ice, 60 μl of 1:1 protein A–Sepharose was added for 1 h at 4°C on a nutator. Samples were washed four times in extraction buffer and the beads boiled in 100 μl of sample buffer. Samples were run on SDS–polyacrylamide gels and blotted onto nitrocellulose. Sro7p, Sec9p, Snc1/2p, and Sso1/2p were detected by immunoblot analysis with the respective antibodies. All gels were quantitated as described above.
Native Immunoprecipitations of Supernatant and Pellet Fractions
Wild-type cells corresponding to ∼200 OD599 were washed in ice-cold 10 mM Tris, 10 mM azide, and spheroplasted in 13.3 ml of spheroplast buffer (100 mM Tris, 10 mM azide, 1.2 M sorbitol, 21 mM β-mercaptoethanol, and 0.1 mg/ml of Zymolyase 100T). Spheroplasts were lysed in 5 ml of ice-cold TEAE/sorbitol (see above) with PIC and spun at 450 g for 3 min at 4°C to get rid of unbroken cells. The lysate was spun at 36,000 g to separate supernatant and pellet fractions, and the pellets were normalized to the volume of the supernatants. Both fractions were treated with 5× extraction buffer (100 mM Hepes-KOH, 750 mM KCl, 2.5% NP-40) with PIC to a final concentration of 1×, equilibrated on ice for 15 min and spun in a cold microfuge to pellet insoluble material. Native immunoprecipitations with saturating amounts of affinity-purified α-Sec9p, α-Sro7p, α-Sso1/2p, and α-Snc1/2p polyclonal antibodies and preimmune antibodies were carried out and analyzed as described above.
Results
Identification of Sro7 and Sro77 as Sec9-binding Proteins
Using a yeast two-hybrid system (Durfree et al., 1993) to isolate proteins that interact with Sec9p, we identified two distinct cDNAs that yielded a strong and specific interaction (as measured by histidine prototrophy and β-galactosidase activity) when present in reporter cells with the GAL4-SEC9 fusion (Fields and Song 1989). Sequence analysis of one of the interacting cDNAs revealed that it encoded the COOH-terminal half of a recently described gene known as SRO7 (Kagami et al. 1998). The other cDNA encoded an unrelated protein and will be described elsewhere. The interaction of Sec9p with the corresponding region of Sro7p was specific since it did not yield a positive interaction when present with the GAL4 DNA–binding domain alone or the GAL4 DNA–binding domain fused to several control proteins. Likewise, Sec9p did not yield a positive interaction with the GAL4 activation domain alone or the GAL4 activation domain fused with several control proteins (data not shown).
To corroborate the two-hybrid interaction between Sec9 and Sro7, we in vitro translated the region of Sro7 identified above in a rabbit reticulocyte lysate and examined its ability to interact with the immobilized GST-Sec9p fusion construct (see Materials and Methods for details about recombinant Sec9p construct). This COOH-terminal region of Sro7p specifically associated only with the GST-Sec9p construct (Fig. 1 A, left). Furthermore, Sro7p did not interact with GST alone, GST-Sso1p, or GST-Snc1p. Yeast contain a second gene, previously designated as SRO77, with 55% identity to SRO7 (Fig. 1 B), and has been shown to have overlapping function with SRO7 (Kagami et al. 1998). In a parallel experiment, we in vitro translated the corresponding domain of Sro77p and examined its ability to interact with GST fusion constructs. Sro77p associated with the GST construct containing Sec9p, but not significantly with GST-Sso1p and GST-Snc1p (Fig. 1 A, right).
Figure 1.

Identification of Sro7p and Sro77p as Sec9p-binding proteins. (A) Interaction of in vitro translated COOH-terminal domains of Sro7 and Sro77p with Sec9p. The COOH-terminal domain of Sro7p (left) identified in the two-hybrid screen and the homologous region of Sro77p were radiolabeled by coupled transcription/translation in rabbit reticulocyte lysates. In vitro translation reactions were incubated with various recombinant SNARE proteins immobilized on GST beads (each at ∼1 μM in the binding reaction), washed, and the bound material was analyzed by SDS-PAGE and autoradiography. The positions of molecular mass markers (expressed in kD) are indicated on the left of the gels. The bands corresponding to primary translation products of Sro7-CTp and Sro77-CTp are indicated by arrows. The lower molecular mass band presumably represents breakdown products. (B) Alignment of Sro7p and Sro77p with three lethal giant larvae family members and tomosyn: mouse LGL (MgCl-1, GenBank accession number D16141), human LGL (Hugl, GenBank accession number X86371), and Drosophila LGL (lg(2)l, Swiss protein accession number P08111) and tomosyn (accession number U92072). Each rectangle is drawn proportional to the length of each coding sequence. The positions of the predicted WD-40 repeats and the VAMP-like domain are indicated. The arrows indicate the region in Sro7p recovered by two-hybrid and used for the in vitro binding assays. The BLAST analysis shows the smallest sum probability (in parentheses) and highest scoring region (scores <10−3 are considered potentially significant).
Sro7 is predicted to encode a 110-kD protein containing two β-transducin–like WD-40 repeats that are contained in the NH2-terminal half of the protein (Neer et al. 1994). Only the COOH-terminal half of SRO7 is present in the original isolate, suggesting that the WD-40 repeats are not required for Sec9p interaction. Sro7 and Sro77 are the only yeast members of a family of related proteins first identified in Drosophila by the tumor suppressor protein, lethal giant larvae (Mechler et al. 1985). Like Sro7 and Sro77, the metazoan lethal giant larvae proteins are of similar size and contain two predicted WD-40 repeats in similar regions of the protein (Fig. 1 B). While the two regions of highest conservation between the yeast and metazoan proteins are centered around the two WD-40 repeats, three regions of conservation are also found in the COOH-terminal halves of both Sro7 and Sro77 (corresponding to residues 549–576, 675–719, and 813–849 in the SRO7 coding sequence).
Recently, a neuronal-specific protein named tomosyn was identified as a syntaxin-binding protein that shows an extensive similarity to both lethal giant larvae and Sro7/77 (Fig. 1 B). This group suggested that tomosyn may act as an activator of SNARE formation in neuronal cells (Fujita et al. 1998). However, neither Sro7 nor any of the other members of this family, described to date, contain a VAMP-like domain, demonstrating that this is not a conserved feature of this family (Fig. 1 B).
Phenotypic Characterization of sro7Δ, sro77Δ mutants
To examine the effect of loss of Sro7 and Sro77 function in vivo, we disrupted each of the chromosomal copies of these genes individually in a diploid yeast strain. After sporulation and tetrad dissection, there was no observable effect on growth of the resulting haploid disruptants containing either the sro7Δ or sro77Δ allele. However, when sro7 disruptants were crossed with sro77 disruptants, a pronounced growth defect was observed at 25°C in haploid segregants containing both null alleles. Therefore, these genes represent a functionally redundant gene family (Fig. 2 A). Growth of these strains at lower temperatures, such as 14 or 19°C (not shown), further enhances this growth defect, whereas growth at 37°C reduces the growth defect, demonstrating that the sro7Δ and sro77Δ double mutants have a severe cold-sensitive growth defect.
Figure 2.

SRO7 and SRO77 form a functionally redundant gene family and the sro7Δ, sro77Δ mutant exhibits a severe cold-sensitive phenotype. (A) Tetrad dissection of a cross between sro7Δ and sro77Δ single disruptants. Tetrads were dissected and grown on YPD plates at 25°C. Pinpoint colonies correspond to haploid segregants containing disruptions of both SRO7 and SRO77. Large colonies correspond to haploid segregants containing wild-type copies of both SRO7 and SRO77 or individual disruptions in either gene. T refers to a tetratype and NPD refers to nonparental ditype segregation patterns observed after scoring for auxotrophic markers for each disruption. (B) Cold sensitivity of the sro7Δ, sro77Δ double-disruptant strain. Each strain of a tetratype tetrad was struck out onto YPD plates and grown at either 37, 25, or 14°C. The genotype of the four strains is as indicated.
This conditional growth defect allowed us to assess directly Sro7 and Sro77 function in the yeast secretory pathway. We examined the secretory capacity of sro7Δ, sro77Δ double-disruptant cells after a shift from the permissive temperature of 37°C to the nonpermissive temperature of 19°C. In particular, we determined the ability to secrete invertase (Esmon et al. 1981), a protein that follows the classical secretory pathway from the ER to the extracellular periplasmic space. We performed invertase secretion assays on wild-type (SRO7, SRO77), each of the two single-disruptants (sro7Δ) and (sro77Δ), and the double-disruptant (sro7Δ, sro77Δ) strains. After a 3-h shift to low glucose media (to derepress expression of invertase) at 19°C, the wild-type strain and each of the single disruption strains secreted 88–98% of the total invertase produced in this period. In contrast, the sro7Δ, sro77Δ double mutant strain secreted only 53% of the total invertase produced during the shift, with the remaining 47% being intracellular (Fig. 3 A). These results demonstrate a clear defect in the secretory pathway in response to loss of both Sro7 and Sro77.
Figure 3.

The sro7Δ, sro77Δ mutant has a pronounced defect in Golgi-to-cell surface transport. (A) Invertase secretion is deficient in the sro7Δ, sro77Δ mutant strain. Invertase secretion assays were performed after a temperature shift on several tetratype tetrads and a representative example is shown. The left frame shows a comparison of the percentage of invertase secreted into the periplasm after a 3-h shift to the restrictive temperature (19°C). The right frame shows the partitioning of invertase activity in the internal (intracellular) and external (periplasmic) fractions of these cells. All samples were measured in duplicate and the SD determined from two separate experiments. (B) The invertase that accumulates in sro7Δ, sro77Δ cells is fully glycosylated and transport of CPY to the vacuole is unaffected. Wild-type, single-disruptant, and double-disruptant strains, in addition to sec4-8 and sec18-1 strains, were grown and shifted as in A, except that sec4-8 and sec18-1 strains were grown for 2 h at 37°C before lysis. Equivalent amounts of each strain were lysed with glass beads, boiled in SDS sample buffer, subjected to SDS-PAGE, transferred to nitrocellulose, and probed with antibodies raised against invertase (top) or CPY (bottom). Mutants that block secretion after exit from the Golgi apparatus, such as sec4-8, accumulate internal pools of fully glycosylated invertase, whereas mutants that block at earlier stages, such as sec18-1, accumulate the core-glycosylated or ER-modified form of invertase. Likewise post-Golgi sec mutants do not affect CPY transport and maturation (and therefore show only the mature, mCPY form), unlike mutants that block earlier in the pathway such as sec18-1, which accumulates the core-glycosylated, p1 form of CPY. The forms of invertase and CPY found in the sro7Δ, sro77Δ cells suggest it is primarily affecting Golgi-to-cell surface transport.
To determine the stage of the secretory pathway that was blocked in the sro7Δ, sro77Δ cells, we examined the glycosylation state of invertase and the vacuolar enzyme CPY (Valls et al. 1987) after the temperature shift described above. Whole cell lysates (including the secreted periplasmic fraction) were prepared from each of the sro mutant strains and subjected to SDS-PAGE and immunoblot analysis. As a control for defects in ER-to-Golgi and post-Golgi transport, lysates from sec18-1 and sec4-8 (Novick et al. 1980) strains were prepared after a 2-h shift to 37°C. The results, shown in Fig. 3 B, demonstrate that under conditions in which nearly half of the invertase is accumulated internally in sro7Δ, sro77Δ cells, virtually all of the invertase is present in the fully glycosylated state. This is similar to that seen with sec4-8, a late sec mutant blocked in Golgi-to-cell surface transport (Fig. 3 B). In contrast, the sec18-1 mutant shows a dramatic accumulation of core-glycosylated invertase, indicative of a block in ER-to-Golgi transport. To confirm that the secretory defect observed in the sro7Δ, sro77Δ cells is at a post-Golgi step, we examined the maturation of CPY in these strains. CPY is a vacuolar enzyme that follows the same secretory pathway as invertase until a late Golgi compartment, where it is sorted away from secreted proteins into a distinct pathway that brings it to the vacuole to be cleaved into its mature form (Valls et al. 1987). A diagnostic feature of post-Golgi–specific secretory mutants is the lack of an effect on CPY transport. The results, shown in Fig. 3 B, demonstrate that, as expected, the ER-blocked, core-glycosylated (p1) form of CPY is the major form present in a sec18-1 strain. However, only mature CPY (mCPY) is detected in both the sec4-8 and the sro7Δ, sro77Δ mutants as well as the single mutants and wild-type strains. Therefore, the pronounced secretion defect observed in the sro7Δ, sro77Δ mutant strain appears to be primarily a post-Golgi–specific defect.
Another feature common to all known mutants blocked in Golgi-to-cell surface transport in yeast is the accumulation of 80–100 nm secretory vesicles (Novick et al. 1980). To examine the morphological effects of loss of Sro7 and Sro77, we performed thin section electron microscopy (Walworth et al. 1992) on the double-disruptant strain following the same cold temperature shift protocol used in the invertase secretion assays. Representative mutant and wild-type cells are shown in Fig. 4 A. While the mutant cells generally appeared somewhat larger on average than the wild-type cells, the most striking feature was that virtually all of the budded cells observed in the sro7Δ, sro77Δ mutant strain showed a dramatic accumulation of 80–100 nm vesicles. Furthermore, in most of the budded cells the vesicles appeared to be significantly more abundant in the bud than in the mother cell as is the case with most of the late sec mutants (Walch-Solimena et al. 1997). As expected, because of the normally rapid rate of exocytosis in yeast, vesicles were quite rare in wild-type (SRO7, SRO77) cells analyzed in parallel with the mutant strain.
Figure 4.
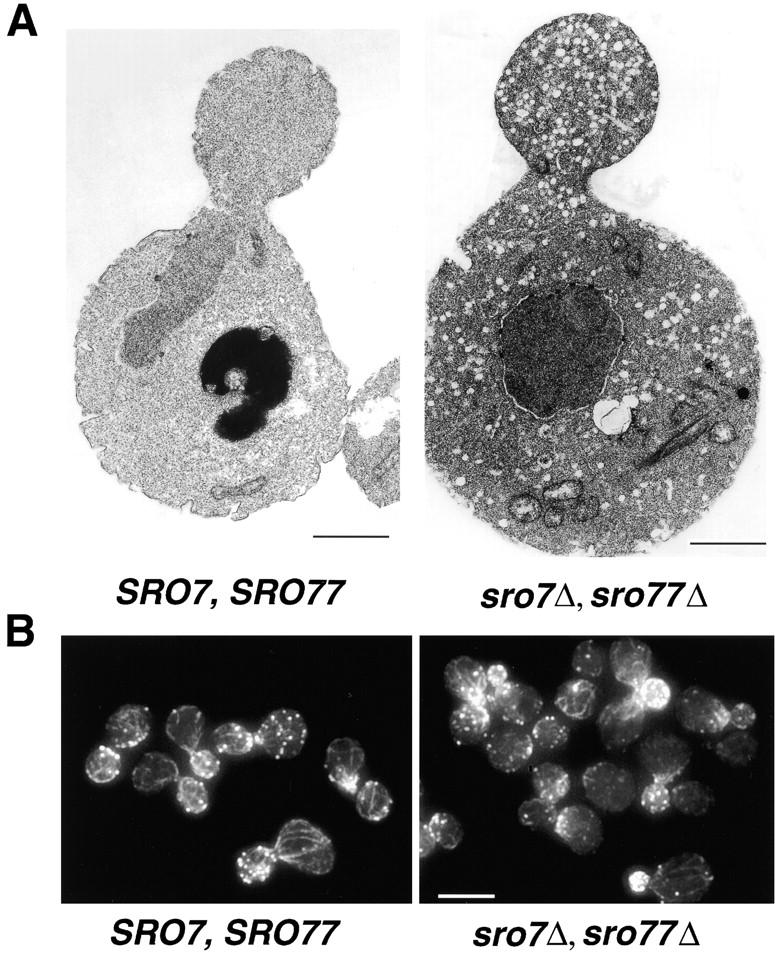
(A) Loss of Sro7 and Sro77 leads to the accumulation of a large number of post-Golgi secretory vesicles at the nonpermissive temperature. The wild-type (SRO7, SRO77) and double-disruptant (sro7Δ, sro77Δ) strains were grown to mid-log phase at 37°C, shifted to the restrictive temperature of 19°C for 3 h, and then processed for thin section electron microscopy. High magnification micrographs of the accumulated vesicles demonstrated that >90% of the vesicles had diameters between 80 and 100 nm, identical to that reported for other post-Golgi sec mutants (Novick et al. 1980). (B) Actin localization in wild-type (SRO7, SRO77) and double-disruptant (sro7Δ, sro77Δ) strains. Haploid cells were grown overnight at the permissive temperature of 37°C to log phase, and then shifted to the restrictive temperature of 19°C for 3 h before fixing, permeabilizing, and staining with TRITC-phalloidin. Bars: A, 1 μm; B, 5 μm.
sro7Δ, sro77Δ Mutants Do Not Exhibit Defects in Actin Polarity
Previously, Kagami et al. 1998 reported defects in the polarity and integrity of the actin cytoskeleton in sro7Δ, sro77Δ mutants after an 18-h shift to 19°C. However, we observed pronounced secretory defects and accumulation of secretory vesicles after only 3 h at 19°C. Therefore, we examined the status of the actin cytoskeleton by rhodamine-phalloidin staining after a 3-h shift from 37 to 19°C. The results, shown in Fig. 4 B, demonstrate that both the polarized distribution of the cortical actin patches and the integrity of the actin cables are normal in the sro7Δ, sro77Δ mutant. Therefore, the secretory defect appears to be the primary phenotype in this mutant and the cytoskeletal defects are likely to result from the loss of vesicular transport to the cell surface.
Subcellular Localization of Sro7
Since Sro7p/77p both bind to Sec9p and function at the same stage of exocytosis, we reasoned that the localization of these proteins within the cell might overlap with that of Sec9p. To examine the Sro7 protein function within yeast cells and determine its subcellular localization, we raised antibodies to the COOH-terminal 219 residues of the protein (see Materials and Methods). The affinity-purified antibodies recognize a protein of ∼105 kD that corresponds to the Sro7 protein, as it is absent in a strain that lacks Sro7 and is greatly enhanced in cells carrying a plasmid overexpressing the Sro7 protein (Fig. 5 A).
Figure 5.
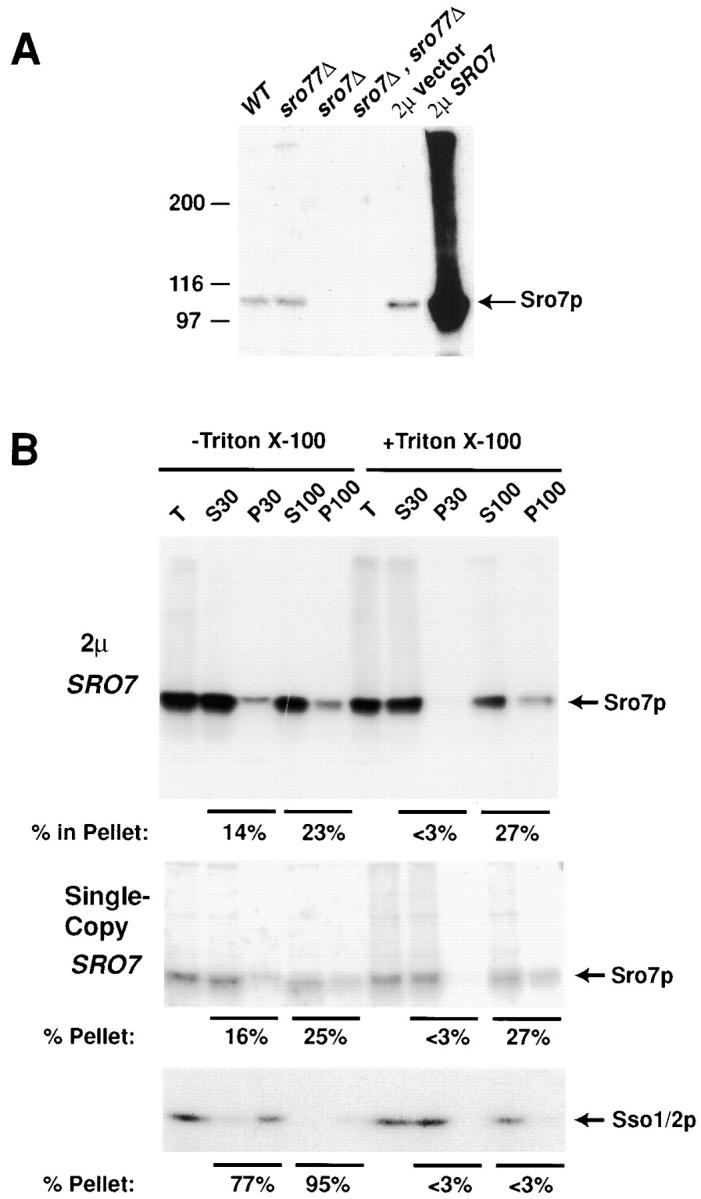
Sro7p is found in both cytosolic- and membrane-bound pools. (A) Affinity-purified antibodies to Sro7p recognize a protein of ∼105 kD on SDS-PAGE. Cells from wild-type (SRO7, SRO77), single disruptants (sro77Δ or sro7Δ), double disruptants (sro77Δ, sro7Δ), and strains containing high copy SRO7 (2μ SRO7) were spheroplasted, lysed, and boiled in SDS sample buffer. The samples were examined by 7% SDS-PAGE gels followed by immunoblotting with affinity-purified α-Sro7p antibodies. The higher molecular mass forms present in the SRO7 high copy lane probably represent denatured aggregates since they are much less apparent when samples are diluted before boiling and do not change appreciably during pulse-chase experiments (data not shown). (B) Sro7p is found in the soluble and membrane fractions of the cell. Cells containing vector only (pB23) or SRO7 on high copy (pB497) were grown to logarithmic phase, spheroplasted, lysed, and spun to remove unbroken cells. The lysate was treated with detergent or a mock control and subjected to two successive centrifugations: 30,000 g for 15 min, and then the S30 supernatant was centrifuged at 100,000 g for 1 h. Pellet fractions were resuspended in the same volumes as the supernatants to normalize. Samples from each fraction were boiled, analyzed by 7% SDS-PAGE, and immunoblotted with affinity-purified α-Sro7p antibody. Samples were also run on a 12.5% gel and immunoblotted with α-Sso1/2p polyclonal antibody as an internal control of the fractionation procedure.
Fractionation (Fig. 5 B) of cells carrying Sro7 on single copy (pB23) or on a high copy expression plasmid (pB497) showed that ∼14% of Sro7 pellets were at a 30,000 g spin, whereas ∼23% pellets were at 100,000 g. The 30,000 g pellet is presumably due to Sro7 association with a membrane fraction as it is affected by the addition of 1% Triton X-100 to the cell lysate. The 100,000 g pellet is probably not a membrane-associated fraction, as it is unperturbed by the addition of detergent. This fraction could be either a large complex of Sro7 in the cell or could be Sro7p that is associating with the cytoskeleton. Treatment with 1 M NaCl and 100 mM sodium bicarbonate also solubilized the protein indicating that Sro7 is a peripheral membrane protein, although the stability of the protein largely diminished under these conditions (data not shown).
To determine the subcellular compartment with which Sro7 is associated, fractionation was performed by velocity sedimentation through sucrose gradients (Fig. 6 A). A lysate from a wild-type strain was layered directly onto a 20–55% sucrose gradient. After centrifugation, the positions of various membrane compartments within the gradient were determined by enzymatic assays or by quantitative immunoblotting of marker proteins. Sro7 protein was found in two fractions of the gradient. Both peaks were clearly distinct from the Golgi marker, GDPase and the ER marker, and cytochrome c reductase. One peak cofractionated with the plasma membrane marker Sso1/2p, consistent with the localization of Sro7p to the plasma membrane. The second peak was found in the top fractions presumably representing a fraction of the membrane-associated pool that has been stripped of the plasma membrane because of high sucrose concentration. These localization data were also confirmed by immunofluorescence (Fig. 6 B) on wild-type cells (pB23) or cells containing high copy SRO7. Like Sec9p, Sro7p is found to localize to the cytoplasm and all along the plasma membrane.
Figure 6.
Sro7p is associated with the plasma membrane in yeast. (A) Sro7p colocalizes with the plasma membrane marker Sso1/2p on a sucrose density gradient. Wild-type cells grown to logarithmic phase were spheroplasted, lysed, and spun at 30,000 g to pellet the membrane-associated pool of Sro7p that was analyzed on a 20–50% sucrose density gradient. The top shows the fractionation profile of Sro7p and the plasma membrane t-SNARE Sso1/2p in the gradient as determined by immunoblotting. The bottom shows the distribution of the GDPase activity (a Golgi marker) and cytochrome c reductase activity (an ER marker) in the gradient. Fraction 1 corresponds to the top of the gradient. Sro7p is found in two pools on the gradient: one that has been released from the membrane and is found at the top of gradient and a second pool that comigrates with the plasma membrane marker Sso1/2p. (B) Indirect immunofluorescence localization of Sro7p to the plasma membrane and soluble pool of the cell. Wild-type cells containing vector only (pB23) or SRO7 on high copy (pB497) were grown in selective media overnight and shifted in YPD for 2 h before fixing with formaldehyde. Cells were permeabilized using 0.05% SDS and stained with affinity-purified α-Sro7p antibody and detected using a rhodamine-conjugated α-rabbit secondary antibody. Stained cells were observed on a Zeiss LSM 510 confocal microscope and individual Z-slices were captured with LSM 510 software. Plasma membrane and cytosolic staining is evident in cells containing the high copy SRO7 but no specific staining was observed in the empty vector control cells containing a single copy of SRO7.


To determine whether Sro7p is associated with post-Golgi secretory vesicles, we isolated vesicles accumulated in either of two late acting sec mutants, sec1-1 (Fig. 7) or sec6-4 (not shown). Mutants were shifted to 37°C for 2 h to accumulate vesicles, the cells were lysed, and the vesicle-enriched P3 membrane fraction (Walworth and Novick 1987) was layered onto a velocity gradient. After centrifugation, the gradients were divided into 16 fractions and each fraction was assayed for invertase activity and for the vesicle marker Snc1/2. As expected, both vesicle markers migrated as a major peak in the center of the gradient but no detectable peak of Sro7 was found in this region of the gradient. Instead, the majority of Sro7 in the gradient was found at the top of the gradient, whereas a smaller fraction pelleted at the bottom with the plasma membrane markers (Fig. 7). Identical results were found using vesicles isolated from a sec6-4 strain, with the profile of Sro7p demonstrating no Sro7p in the region of the gradient containing the vesicle peak (not shown). Therefore, like Sec9p, Sro7p does not associate with post-Golgi secretory vesicles.
Figure 7.
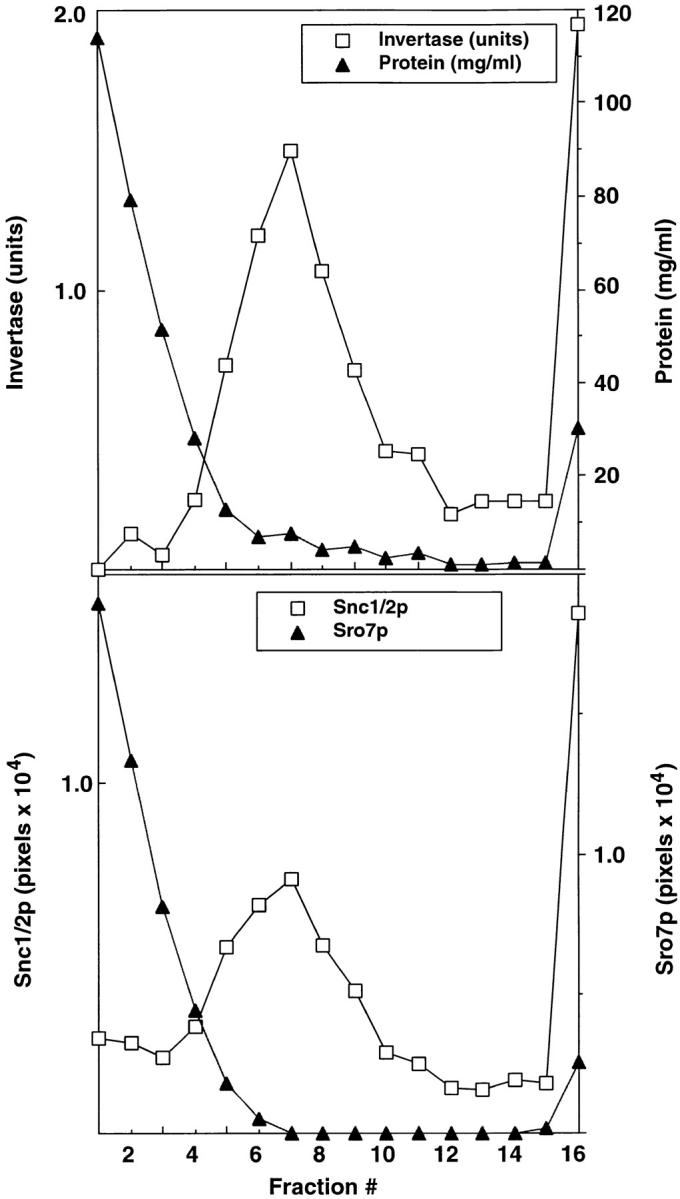
Sro7p is not present on post-Golgi secretory vesicles. Cells from a BY29 (sec1-1, ura3-52) strain were shifted to 37°C in YP medium with 0.1% glucose for 2 h to accumulate post-Golgi vesicles and induce expression of invertase. Spheroplasts were prepared from these cells that were lysed osmotically by gentle resuspension in lysis buffer containing 0.8 M sorbitol. A 100,000 g membrane fraction (P3) enriched in post-Golgi vesicles was prepared from a 10,000 g supernatant fraction by differential centrifugation as described previously (Walworth and Novick 1987). The P3 vesicle fraction was resuspended in lysis buffer and layered onto a 20–40% sorbitol velocity gradient as described previously (Brennwald et al. 1994). The top shows the fractionation profile of latent invertase (a marker for secretory vesicles) compared with the distribution of total protein in the gradient. Invertase activity is expressed as micromoles of glucose per minute per fraction. Protein concentrations were determined according to Bradford 1976. The bottom shows the fractionation of Sro7p compared with Snc1/2, a marker for post-Golgi vesicles (Protopopov et al. 1993). The presence of Sro7p and Snc1/2 in the gradients was determined by immunoblotting followed by quantitation on a STORM PhosphorImager using ImageQuant software. Similar results were obtained using a P3 fraction prepared from a sec6-4 strain.
Sro7p and Sec9p Are Directly Associated In Vivo
The two-hybrid interaction, post-Golgi secretion defect, and subcellular localization data strongly suggest that Sro7p function is likely to involve it binding to Sec9p in vivo. To test this directly, we examined whether we could coprecipitate Sec9 and Sro7 proteins after treatment with the chemical cross-linker, DSP. Yeast strains were generated that expressed either full-length Sro7p or the COOH-terminal domain of Sro7p (Fig. 1) behind the GAL1 promoter along with either a myc-tagged Sec9p construct or an untagged Sec9p as a control. Cells were radiolabeled with [35S]methionine/cysteine in selective galactose-containing media (to induce the expression of the full-length or COOH-terminal domain of Sro7p), spheroplasted, and lysed. Lysates were either treated with the cleavable cross-linker, DSP, dissolved in DMSO (+DSP) or mock-treated with DMSO alone (−DSP), boiled in 1% SDS (so that only covalent associations with myc-Sec9p are retained), diluted in buffer, and subjected to immunoprecipitation with the α-myc mAb 9E10. These immunoprecipitates were boiled in buffer containing 1% SDS/0.1 M DTT to cleave the cross-linker, and then subjected to immunoprecipitation with either α-Sro7p or α-Sec9p antibodies to monitor the results of the cross-linking. Fig. 8 shows that full-length Sro7p can be coprecipitated with myc-Sec9p in a manner that is dependent both on the presence of the DSP cross-linker and the epitope tag on Sec9p. Interestingly, this interaction was not observed in the strains expressing only the COOH-terminal half of the Sro7 protein, suggesting that whereas this domain is sufficient for binding in vitro, in vivo the interaction is much more efficient with the full-length Sro7 protein.
Figure 8.

Full-length Sro7p, but not the COOH-terminal half of Sro7p, can be directly cross-linked to Sec9p in yeast lysates. Coprecipitation analysis was performed on radiolabeled yeast strains transformed with either myc-tagged YEpSEC9-myc plasmid (pB37) or, as a control, an otherwise identical untagged YEpSEC9 plasmid (pB37) in strains which contained a construct overexpressing either full-length Sro7 (pB363) or CT Sro7 (pB367) under control of the inducible GAL1 promoter. All strains were induced in galactose-containing media for 2 h before 1 h labeling of cells with 35S-Express label in galactose-containing media. The cells were spheroplasted and lysed osmotically in PBS. Lysates were immediately subjected to treatment with the protein cleavable cross-linking agent (DSP) dissolved in DMSO or a DMSO control for 20 min on ice and subsequently boiled in 1% SDS buffer before dilution with IP buffer. Association because of cross-linking was monitored by a two-step immunoprecipitation protocol with the first immunoprecipitation being with the α-myc or directly with α-Sro7p antibodies (α-Sro7 IP). After washing, samples were boiled in 1% SDS/0.1 M DTT (to cleave the cross-linker), diluted with IP buffer, and subjected to a second round of immunoprecipitation with either α-Sro7p (top) or α-Sec9p (bottom) polyclonal antibodies. Samples were boiled in sample buffer, resolved by SDS-PAGE, dried, and exposed to film. The expression from the GAL1-SRO7 and GAL1-SRO7-CT constructs were similar (compare first and last lanes, marked α-Sro7 IP). A cross-linker and myc tag–dependent interaction is apparent between full-length Sro7p and Sec9p, strongly suggesting that these proteins are directly associated with each other in vivo. In contrast, the COOH-terminal domain does not show a detectable cross-linking in this assay.
To further investigate the interaction between Sro7p and Sec9p, we performed immunoprecipitations from whole cell detergent extracts of wild-type yeast cells expressing only the endogenous Sro7 and Sec9 proteins (Fig. 9 A). We used saturating amounts of affinity-purified IgG (or an equivalent amount of preimmune IgG as a control) directed against either Sro7p or each of the three yeast post-Golgi SNARE proteins, Sec9p, Sso1/2p, and Snc1/2p. Fig. 9 B shows that in total yeast extracts, α-Sro7p IgG can coimmunoprecipitate ∼6% of Sec9 and 4% of the post-Golgi SNAREs Sso1/2p and Snc1/2p present in these lysates. Under the same conditions, however, only the Sec9p immunoprecipitation showed detectable amounts of Sro7p (0.5%).
Figure 9.
Sro7p interacts with the post-Golgi SNARE proteins in detergent extracts. (A) Wild-type cells, grown to logarithmic phase were spheroplasted and lysed in buffer containing 0.5% NP-40. Detergent extracts were subjected to immunoprecipitations with either saturating amounts (3–10 μg of IgG/1 ml immunoprecipitation) of affinity-purified antibodies against either Snc1/2p, Sso1/2p, Sec9p, Sro7p, or an equivalent amount of IgG purified from a preimmune bleed of the respective rabbit. After recovery of immune complexes on protein A–Sepharose beads, beads were washed four times with the same buffer, and then boiled in SDS sample buffer. The samples were analyzed by SDS-PAGE, and then blotted with α-Sro7p, α-Sec9p, α-Snc1/2p, and α-Sso1/2p antibodies, respectively. To quantitate the samples that contained the same precipitating and immunoblotting antibody, we found it necessary to dilute these samples with SDS sample buffer before electrophoresis as follows: in the Snc1/2 blot, the Snc1/2 immunoprecipitates (and the control) were diluted 1:4; in the Sso1/2 blot, the Sso1/2 IPs were diluted 1:8; for the Sec9 blot, the Sec9 IPs were diluted 1:2; and for the Sro7 blot, the Sro7 IPs were diluted 1:2. A fraction of the total detergent lysate was immunoblotted in parallel to determine the total protein present in the immunoprecipitation reaction. The efficiency of the immunoprecipitation with saturating amounts of affinity-purified antibodies was calculated as follows: the amount of target protein immunoprecipitated by a given antibody was quantitated after immunoblotting and (after adjusting for dilution and amount loaded) divided by the amount of protein present in the total sample (after adjusting for the amount loaded). From this, the efficiency of precipitation by each antibody was determined to be as follows: α-Snc1/2p, 93%; α-Sso1/2p, 63%; α-Sec9p, 76%; and α-Sro7p, 98%. (B) Quantitation of the coimmunoprecipitations. The values are expressed as the amount of coimmunoprecipitating proteins present as a percentage of the total protein present (see above). 125I–protein A signals on the immunoblots were quantitated on a Storm PhosphorImager using ImageQuant software. Note that the numbers that are expressed as a percentage of the total protein are higher for Sec9p than for the other three proteins. This is likely due to the fact that the levels of Sec9 protein found in wild-type yeast are 5–10-fold less than that of Snc1/2p and Sso1/2p, and at least threefold less than Sro7p. Thus, Sec9p is the limiting factor in these associations.


Since both Sec9p and Sro7p have significant pools of protein on both the plasma membrane and in the cytosol, we examined whether the interaction seen between the two proteins occurred in one or both pools. For these experiments, it was necessary to use yeast cells expressing Sec9p on a multicopy plasmid (which results in about a fivefold increase in Sec9p levels) to readily detect the interaction after the separation of the cytosol and membrane fractions. Lysates were centrifuged at 30,000 g to separate the soluble and membrane fractions, and then each was diluted 1:1 with lysis buffer containing 1% NP-40 to solubilize the membranes. The samples were subjected to immunoprecipitation exactly as for the whole cell lysates. The results, shown in Fig. 10 A, show that Sro7p and Sec9p can coprecipitate each other in both the membrane and supernatant fractions of the cell. As expected the transmembrane SNARE proteins Sso1/2 and Snc1/2 are predominantly found in association with each other in the 30,000 g membrane fraction. However, a significant amount of association between Sro7p and Sec9p was seen in both the supernatant and membrane fractions.
Figure 10.

Sro7p interacts specifically with Sec9p in the cytoplasm and at the plasma membrane. (A) Cells containing high copy SEC9 (pB35) were grown to logarithmic phase, spheroplasted, and lysed. The lysate was centrifuged at 30,000 g to separate cytosol and membrane fractions, the samples were adjusted to 0.5% NP-40, centrifuged at 13,000 g for 10 min to eliminate insoluble material, and subjected to coimmunoprecipitation analysis as in Fig. 8. Samples were analyzed by SDS-PAGE and immunoblotted with α-Sec9p, α-Sro7p, α-Sso1/2p, and α-Snc1/2p antibodies. (B) Serial coprecipitation analysis demonstrates that Sro7p interacts with Sso1/2p and Snc1/2p only through the interaction with Sec9p. A 30,000 g membrane fraction was solubilized with detergent as above and was precleared of Sec9 protein by immunoprecipitation with saturating amounts of affinity-purified α-Sec9p antibody or equivalent amounts of preimmune IgG as a control. Fractions were subjected to a second round of immunoprecipitations with α-Sec9p, α-Sro7p, α-Sso1/2p, and α-Snc1/2p polyclonal antibodies and analyzed as above.
Interestingly, as observed for the whole cell immunoprecipitates described above, the α-Sro7p antibodies were found to coimmunoprecipitate significant amounts of the two other post-Golgi SNARE proteins, Sso1/2p and Snc1/2p, from the membrane fraction. To determine if this was likely to be a direct interaction between Sro7p and these two SNARE proteins or an indirect interaction through Sec9p, we examined the effect of the preclearing of Sec9 protein on coprecipitation. Immunoprecipitations of the 30,000 g membrane pool were set up as described above, and then treated with either saturating amounts of α-Sec9p IgG or an equivalent amount of preimmune IgG. The supernatant of these two immunoprecipitations was subjected to precipitation with either α-Sro7p or α-Sec9p IgG (to monitor the effectiveness of the preclearing), and the effect on the coprecipitation of Sso1/2p and Snc1/2p was determined. The results of this experiment, shown in Fig. 10 B, demonstrate the dramatic loss of Sso1/2p and Snc1/2p coprecipitation by Sro7p when Sec9 protein is removed from the extracts. This strongly suggests that Sro7p does not directly associate with these other SNARE proteins but that it can bind to Sec9p, not only on its own, but also within the context of a SNARE complex.
SRO7 and SEC9 Suppress Both rho3 and Post-Golgi sec Mutants
SRO7 was independently isolated as a multicopy suppressor of a mutant in the Rho GTPase, RHO3, which is thought to play a role in regulating actin polarity of the yeast cell. rho3 mutants appear to have severe defects in cytoskeletal organization. Specifically, cortical actin patches, normally present almost exclusively in the bud, are found randomly distributed throughout the mother and bud of the rho3 mutant cells (Imai et al. 1996). Rho3 has also been implicated in polarized exocytosis. This observation stems from the recent identification, by our laboratory, of the RHO3 gene as a high copy suppressor of sec4-P48, a cold-sensitive Sec4 effector domain mutant. Furthermore, we have demonstrated that rho3 mutant cells have a defect in polarity and accumulate post-Golgi secretory vesicles in the mother cell at the restrictive temperature (Adamo, J., G. Rossi, and P. Brennwald, manuscript submitted for publication). Genetic interactions involving RHO3 and SEC4 prompted us to determine if both SRO7 and SEC9 can act as suppressors of a rho3 deletion mutant. Sporulation of a diploid heterozygous for disruption of the chromosomal RHO3 gene results in a 2:2 segregation pattern for growth at 25°C that is linked to the rho3 disruption. Transformation of the high copy vector, pRS426, into the heterozygous diploid did not alter this segregation pattern after sporulation (Fig. 11, top). Expression of high copy SRO7 strongly suppressed the growth defect associated with disruption of rho3 (Fig. 11, middle). SEC9, when expressed on high copy, was even more potent in its suppression of the rho3 deletion (Fig. 11, bottom).
Figure 11.

Suppression of the rho3 deletion by multicopy SRO7 and SEC9. A diploid heterozygous for a disruption of the chromosomal rho3 gene (a/α, rho3Δ::LEU2/RHO3, leu2-3/leu2-3; ura3-52/ura3-52) was transformed with multicopy plasmids containing (top) empty vector (pB23), (middle) SRO7 (pB427), or (bottom) SEC9 (pB35). Tetrads were dissected on YPD plates and grown for 3 d at 25°C.
The results suggest that these proteins act together to promote vesicle transport at the later stages of post-Golgi transport. While the high copy SRO7 and SEC9 suppress a number of the late acting sec genes and rho3Δ, they do not suppress each other (Table ). High copy SRO7 is unable to suppress sec9-4 or sec9-7 mutants and high copy SEC9 cannot suppress the sro7/sro77 double disruption, suggesting that although they act at the same stage, they do not have an identical function. However, they both are strong suppressors of the sec3, sec8, sec10, and sec15 mutants, suggesting that they function downstream of the exocyst, in mediating the final steps of vesicle fusion at the plasma membrane (Table ).
Table 1.
Summary of Suppression Properties of High Copy SR07 and SEC9
| Mutant | 2μ SRO7 | 2μ SEC9 |
|---|---|---|
| rho3Δ | ++ | +++ |
| sro7Δ, sro77Δ | +++ | − |
| sec4-P48 | + | +++ |
| sec1-1 | − | ++ |
| sec2-41 | − | − |
| sec3-2 | ++ | ++ |
| sec4-8 | − | − |
| sec5-24 | − | − |
| sec6-4 | − | − |
| sec8-9 | ++ | ++ |
| sec9-4 | − | +++ |
| sec10-2 | + | − |
| sec15-1 | +++ | ++ |
Late acting sec and rho3 mutants were transformed with either vector (pRS426), SRO7 (pB497), or SEC9 (pB35). Transformants grown on selective media were picked into microtiter plates and tested on YPD plates for growth at four temperatures: 25°, 30°, 34°, and 37°C. Scoring for temperature-sensitive mutants is as follows: triple plus, wild-type growth at the most restrictive temperature (37°C); double plus, good growth at an intermediate temperature (34°C) at which empty vector controls cannot grow; and single plus, slow growth at an intermediate temperature. Scoring for suppression of cold-sensitive mutants was determined by comparing growth relative at 14°C to an isogenic wild-type yeast (+++) and to the vector control (−).
Discussion
In this paper, we describe the identification of Sro7p/Sro77p as novel components of the yeast exocytotic machinery. Our initial identification of these genes was based on their ability to interact with the plasma membrane SNARE protein Sec9. The biological relevance of this interaction has been supported by a number of observations. First, the analysis of the cold-sensitive sro7Δ, sro77Δ mutants demonstrates a secretion phenotype that is identical to that of sec9 mutants; that is a block in the docking and fusion of post-Golgi secretory vesicles with the plasma membrane. Second, the subcellular localization of Sro7p resembles that of Sec9p as it is present both in the cytosol and peripherally associated with the plasma membrane, and is not found associated with post-Golgi secretory vesicles. Third, we have clearly documented its association with Sec9p both in vitro and in vivo. The in vivo association was demonstrated by two methods: first, by chemical cross-linking and, second, by coprecipitation from detergent extracts. We were also able to show that Sec9p and Sro7p are found associated both in the cytosol and on the plasma membrane. Interestingly, we have evidence that Sro7p is also able to associate with Sec9p when it is present in SNARE complexes with Sso1/2p and Snc1/2p. The fact that it associates with Sec9p alone or in the context of the SNARE complex suggests that it likely interacts with a region of Sec9p not directly involved in the SNARE complex. Recent data from our laboratory support this idea. Mapping of the two-hybrid interaction between Sro7p and Sec9p demonstrates that it interacts with the extreme NH2 terminus of Sec9p, a region important for the ability of Sec9 to suppress an effector mutant in Sec4 (Maksimova, M., L. Katz, and P. Brennwald, unpublished data).
Previous work on yeast Sro7/77 and Drosophila lethal giant larvae function has suggested a role for these proteins in the regulation of the actin/myosin cytoskeleton. This stems from several observations. First, the Drosophila lethal giant larvae gene product has been shown to be physically associated with nonmuscle myosin II and appears to cofractionate with the cytoskeleton after detergent extraction (Strand et al. 1994a,Strand et al. 1994b). Second, the yeast Sro7/77 proteins were initially identified as suppressors of the Rho3 GTPase, a protein required for normal actin polarity (Kagami et al. 1998). The phenotypic analysis of the sro7Δ, sro77Δ mutants by Kagami et al. 1998 appears to support this notion since defects in actin polarity and integrity after shifts to the nonpermissive temperature were observed. Furthermore, they demonstrate physical and genetic interactions between Sro7p and Myo1p, a yeast type II myosin (Kagami et al. 1998).
The data presented here demonstrate that the primary function of Sro7/77 is in exocytosis and not in the regulation of the actin cytoskeleton. In the Kagami et al. 1998 report, the sro7Δ, sro77Δ cells were shifted to 19°C for 18 h before staining for actin. In our experiments, we used a much shorter 3-h shift at 19°C for all of our analyses. After this shorter temperature shift protocol, we found that the actin cytoskeleton was well organized, with polarized cortical actin patches and cables of normal length. Under identical temperature shift conditions, we saw a clear and pronounced defect in Golgi-to-cell surface transport, demonstrating that the exocytic defect is the primary defect associated with loss of Sro7/77. The actin polarity defect observed by Kagami et al. 1998 is likely to be a secondary consequence of loss of membrane transport that is necessary for the delivery of the markers of cell polarity to the cell surface.
Tomosyn, the neuronal homologue of Sro7 and lethal giant larvae, was initially identified based on its interaction with syntaxin. At its COOH terminus, tomosyn contains a domain that is very similar to the helical region of the vesicle SNARE, synaptobrevin/VAMP (Fasshauer et al. 1998; Masuda et al. 1998). This domain, which contributes one of the four helices found at the core of the SNARE complex, may serve as a surrogate vesicle SNARE that is used to occupy or stabilize the other three helices of the SNARE complex until the real vesicle-bound SNARE is presented. Interestingly, we find that this domain is absent in both of the yeast homologues Sro7p and Sro77p, as well as in all of the other members of the lethal giant larvae family (Fig. 1 B). Therefore, this domain appears to be a specialized feature unique to the neuronal homologue. However, since the ability to interact with SNARE proteins and SNARE complexes does appear to be conserved in this family of proteins, it is likely that the conserved portions of the protein are also involved in regulating SNARE function and assembly. Consistent with this notion, Yokoyama et al. 1999 have recently shown that this domain is necessary but not sufficient for the high affinity association with syntaxin. The data presented here and by Fujita et al. 1998 demonstrate that these proteins have a conserved role in the regulation of SNARE assembly at the plasma membrane, even though the precise SNARE protein to which they have the highest affinity interaction appears to be somewhat different in the two systems.
While it is clear that the Sro7 family of proteins plays an important role in exocytosis, it is also evident that this is not an essential function since sro7Δ, sro77Δ strains are able to grow and secrete at levels close to that of wild-type cells when grown at 37°C (Adamo, J., and P. Brennwald, unpublished data; not shown). Therefore, it appears that these proteins act in a regulatory role rather than in a structural role in the process of docking and fusion of post-Golgi vesicles with the plasma membrane. What is the nature of this regulatory function? The genetic analysis presented here demonstrates that loss of Sro7/77 function (i.e., in sro7Δ, sro77Δ mutants) has a negative effect on the exocytic function and that gain of Sro7/77 function (i.e., high copy SRO7) has an overall positive effect on exocytosis (seen as suppression of several late acting sec mutants). Therefore, Sro7/77 act as positive, rather than negative, regulators of exocytosis.
The suppression of the rho3Δ mutant by SRO7 and SEC9 suggests that these three gene products act together in regulating exocytosis. Recent studies by our laboratory (Adamo, J., G. Rossi, and P. Brennwald, manuscript submitted for publication) and others (Imai et al. 1996; Robinson et al. 1999) have suggested that Rho3 plays a direct role both in regulating the actin cytoskeleton and exocytosis. An attractive model for the function of Sro7/77 in such a pathway is that it would act to transmit Rho3 function onto the SNARE complex at the plasma membrane. Whereas, like Sec9p, Sro7p does not display a polarized localization on the plasma membrane, it may function in a polarized fashion in conjunction with an activating signal from Rho3. The end result of this would be the localized assembly of Sec9p/Ssop heterodimers in response to the Rho3-GTP signal. The localized assembly of Q-SNAREs to specific sites on the plasma membrane would serve to restrict vesicle docking and fusion to these sites. Since Rho3 also has a role in regulating actin polarity (Matsui and Toh 1992), it may serve to coordinate the dynamic regulation of the polarity of the actin cytoskeleton with polarized delivery of membrane and protein to the cell surface. It is clear that other factors such as the Rab GTPase Sec4 and the exocyst complex play a role in this process as well (Finger and Novick 1997; Guo et al. 1999). It is worth noting in this regard that Sec9, Sro7, and Rho3 have strong genetic interactions with both Sec4 and multiple subunits of the exocyst complex (Table ; Adamo, J., G. Rossi, and P. Brennwald, manuscript submitted for publication). Unraveling the molecular events by which these numerous gene products function in this process will be a major key to understanding how cell polarity is regulated in eukaryotic cells.
Acknowledgments
We would like to thank Luba Katz and Mayya Maksimova for critical comments on the manuscript, Linda Burg Friedman for help with the electron microscopy, and Geri Kreitzer and Lee Cohen-Gould (all from Weill Medical College of Cornell University) for help with the fluorescence microscopy.
This work was supported by grants from the Mathers Charitable Foundation, the National Institutes of Health (GM54712) and Pew Scholars in Biomedical Sciences program (to P. Brennwald).
Footnotes
1.used in this paper: CPY, carboxypeptidase Y; CT, carboxy terminal; DAPI, 4′,6-diamidino-2-phenylindole; DSP, dithiosuccinnimidylpropionate; GST, glutathione-S-transferase; PIC, protease inhibitor cocktail; Q-SNARE, glutamine-containing SNARE; SNARE, SNAP receptor; TEAE, triethanolamine-EDTA; t-SNARE, target SNARE; VAMP, vesicle-associated membrane protein; YPD, yeast extract/peptone/glucose
K. Lehman and G. Rossi contributed equally to this work.
References
- Aalto M.K., Ronne H., Keranen S. Yeast syntaxins Sso1p and Sso2p belong to a family of related membrane proteins that function in vesicular transport. EMBO (Eur. Mol. Biol. Organ.) J. 1993;12:4095–4104. doi: 10.1002/j.1460-2075.1993.tb06093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayscough K.R., Stryker J., Pokala N., Sanders M., Crews P., Drubin D.G. High rates of actin filament turnover in budding yeast and roles for actin in establishment and maintenance of cell polarity revealed using the actin inhibitor latrunculin-A. J. Cell Biol. 1997;137:399–416. doi: 10.1083/jcb.137.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel P., Chien C.T., Sternglanz R., Fields S. Elimination of false positives that arise in using the two-hybrid system. Biotechniques. 1993;14:920–924. [PubMed] [Google Scholar]
- Becker D.M., Guarente L. High-efficiency transformation of yeast by electroporation. Methods Enzymol. 1991;194:182–187. doi: 10.1016/0076-6879(91)94015-5. [DOI] [PubMed] [Google Scholar]
- Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Brennwald P., Kearns B., Champion K., Keranen S., Bankaitis V., Novick P. Sec9 is a SNAP-25-like component of a yeast SNARE complex that may be the effector of Sec4 function in exocytosis. Cell. 1994;79:245–258. doi: 10.1016/0092-8674(94)90194-5. [DOI] [PubMed] [Google Scholar]
- Durfee T., Becherer K., Chen P.L., Yeh S.H., Yang Y., Kilburn A.E., Lee W.H., Elledge S.J. The retinoblastoma protein associates with the protein phosphatase type 1 catalytic subunit. Genes Dev. 1993;7:555–569. doi: 10.1101/gad.7.4.555. [DOI] [PubMed] [Google Scholar]
- Esmon B., Novick P., Schekman R. Compartmentalized assembly of oligosaccharides on exported glycoproteins in yeast. Cell. 1981;25:451–460. doi: 10.1016/0092-8674(81)90063-5. [DOI] [PubMed] [Google Scholar]
- Fasshauer D., Sutton R.B., Brunger A.T., Jahn R. Conserved structural features of the synaptic fusion complexSNARE proteins reclassified as Q- and R-SNAREs. Proc. Natl. Acad. Sci. USA. 1998;95:15781–15786. doi: 10.1073/pnas.95.26.15781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field C., Schekman R. Localized secretion of acid phosphatase reflects the pattern of cell surface growth in Saccharomyces cerevisiae . J. Cell Biol. 1980;86:123–128. doi: 10.1083/jcb.86.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields S., Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- Finger F.P., Novick P. Sec3p is involved in secretion and morphogenesis in Saccharomyces cerevisiae . Mol. Biol Cell. 1997;8:647–662. doi: 10.1091/mbc.8.4.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger F.P., Hughes T.E., Novick P. Sec3p is a spatial landmark for polarized secretion in budding yeast. Cell. 1998;92:559–571. doi: 10.1016/s0092-8674(00)80948-4. [DOI] [PubMed] [Google Scholar]
- Fujita Y., Shirataki H., Sakisaka T., Asakura T., Ohya T., Kotani H., Yokoyama S., Nishioka H., Matsuura Y., Mizoguchi A., Scheller R.H., Takai Y. Tomosyna syntaxin-1-binding protein that forms a novel complex in the neurotransmitter release process. Neuron. 1998;20:905–915. doi: 10.1016/s0896-6273(00)80472-9. [DOI] [PubMed] [Google Scholar]
- Guo W., Roth D., Walch-Solimena C., Novick P. The exocyst is an effector for sec4p, targeting secretory vesicles to sites of exocytosis. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:1071–1080. doi: 10.1093/emboj/18.4.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai J., Toh-e A., Matsui Y. Genetic analysis of the Saccharomyces cerevisiae RHO3 gene, encoding a rho-type small GTPase, provides evidence for a role in bud formation. Genetics. 1996;142:359–369. doi: 10.1093/genetics/142.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagami M., Toh-e A., Matsui Y. Sro7p, a Saccharomyces cerevisiae counterpart of the tumor suppressor l(2)gl protein, is related to myosins in function. Genetics. 1998;149:1717–1727. doi: 10.1093/genetics/149.4.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz L., Hanson P.I., Heuser J.E., Brennwald P. Genetic and morphological analyses reveal a critical interaction between the C-termini of two SNARE proteins and a parallel four helical arrangement for the exocytic SNARE complex. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:6200–6209. doi: 10.1093/emboj/17.21.6200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane D., Harlow E. Antibodies 1988. A Laboratory Manual. Vol 13; Cold Spring Harbor Laboratory: 726 pp. pp. Cold Spring Harbor, NY [Google Scholar]
- Lillie S.H., Brown S.S. Immunofluorescence localization of the unconventional myosin, Myo2p, and the putative kinesin-related protein, Smy1p, to the same regions of polarized growth in Saccharomyces cerevisiae . J. Cell Biol. 1994;125:825–842. doi: 10.1083/jcb.125.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda E.S., Huang B.C., Fisher J.M., Luo Y., Scheller R.H. Tomosyn binds t-SNARE proteins via a VAMP-like coiled coil. Neuron. 1998;21:479–480. doi: 10.1016/s0896-6273(00)80559-0. [DOI] [PubMed] [Google Scholar]
- Matsui Y., Toh E.A. Yeast RHO3 and RHO4 ras superfamily genes are necessary for bud growth, and their defect is suppressed by a high dose of bud formation genes CDC42 and BEM1. Mol. Cell Biol. 1992;12:5690–5699. doi: 10.1128/mcb.12.12.5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechler B.M., McGinnis W., Gehring W.J. Molecular cloning of lethal(2)giant larvae, a recessive oncogene of Drosophila melanogaster . EMBO (Eur. Mol. Biol. Organ.) J. 1985;4:1551–1557. doi: 10.1002/j.1460-2075.1985.tb03816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair J., Muller H., Peterson M., Novick P. Sec2 protein contains a coiled-coil domain essential for vesicular transport and a dispensable carboxy-terminal domain. J. Cell Biol. 1990;110:1897–1909. doi: 10.1083/jcb.110.6.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neer E.J., Schmidt C.J., Nambudripar R., Smith T.F. The ancient regulatory-protein family of WD-repeat proteins. Nature. 1994;371:297–300. doi: 10.1038/371297a0. [DOI] [PubMed] [Google Scholar]
- Novick P., Botstein D. Phenotypic analysis of temperature-sensitive yeast actin mutants. Cell. 1985;40:405–416. doi: 10.1016/0092-8674(85)90154-0. [DOI] [PubMed] [Google Scholar]
- Novick P., Field C., Schekman R. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell. 1980;21:205–215. doi: 10.1016/0092-8674(80)90128-2. [DOI] [PubMed] [Google Scholar]
- Protopopov V., Govindan B., Novick P., Gerst J.E. Homologs of the synaptobrevin/VAMP family of synaptic vesicle proteins function on the late secretory pathway in S. cerevisiae . Cell. 1993;74:855–861. doi: 10.1016/0092-8674(93)90465-3. [DOI] [PubMed] [Google Scholar]
- Pruyne D.W., Schott D.H., Bretscher A. Tropomyosin-containing actin cables direct the Myo2p–dependent polarized delivery of secretory vesicles in budding yeast. J. Cell Biol. 1998;143:1931–1945. doi: 10.1083/jcb.143.7.1931. [DOI] [PubMed] [Google Scholar]
- Redding K., Holcomb C., Fuller R.S. Immunolocalization of Kex2 protease identifies a putative late Golgi compartment in the yeast Saccharomyces cerevisiae . J. Cell Biol. 1991;113:527–538. doi: 10.1083/jcb.113.3.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson N.G., Guo L., Imai J., Toh-e A., Matsui Y., Tamanoi F. Rho3 of Saccharomyces cerevisiae, which regulates the actin cytoskeleton and exocytosis, is a GTPase which interacts with myo2 and exo70. Mol. Cell Biol. 1999;19:3580–3587. doi: 10.1128/mcb.19.5.3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi G., Salminen A., Rice L.M., Brunger A.T., Brennwald P. Analysis of a yeast SNARE complex reveals remarkable similarity to the neuronal SNARE complex and a novel function for the C terminus of the SNAP-25 homolog, Sec9. J. Biol Chem. 1997;272:16610–16617. doi: 10.1074/jbc.272.26.16610. [DOI] [PubMed] [Google Scholar]
- Sherman F., Fink G.R., Hicks J.B. Laboratory Course Manual for Methods in Yeast Genetics 1986. Cold Spring Harbor Laboratory, ; Cold Spring Harbor, NY: pp. 186 pp [Google Scholar]
- Strand D., Jakobs R., Merdes G., Neumann B., Kalmes A., Heid H.W., Husmann I., Mechler B.M. The Drosophila lethal(2)giant larvae tumor suppressor protein forms homo-oligomers and is associated with nonmuscle myosin II heavy chain J. Cell Biol. 127 1994. 1361 1373a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand D., Raska I., Mechler B.M. The Drosophila lethal(2)giant larvae tumor suppressor protein is a component of the cytoskeleton J. Cell Biol. 127 1994. 1345 1360b [DOI] [PMC free article] [PubMed] [Google Scholar]
- TerBush D.R., Maurice T., Roth D., Novick P. The Exocyst is a multiprotein complex required for exocytosis in Saccharomyces cerevisiae . EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:6483–6494. [PMC free article] [PubMed] [Google Scholar]
- Tkacz J.S., Lampen J.O. Wall replication in Saccharomyces speciesuse of fluorescein-conjugated concanavalin A to reveal the site of mannan insertion. J. Gen. Microbiol. 1972;72:243–247. doi: 10.1099/00221287-72-2-243. [DOI] [PubMed] [Google Scholar]
- Tkacz J.S., Lampen J.O. Surface distribution of invertase on growing Saccharomyces cells. J. Bacteriol. 1973;113:1073–1075. doi: 10.1128/jb.113.2.1073-1075.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valls L.A., Hunter C.P., Rothman J.H., Stevens T.H. Protein sorting in yeastthe localization determinant of yeast vacuolar carboxypeptidase Y resides in the propeptide. Cell. 1987;48:887–897. doi: 10.1016/0092-8674(87)90085-7. [DOI] [PubMed] [Google Scholar]
- Walch-Solimena C., Collins R.N., Novick P.J. Sec2p mediates nucleotide exchange on Sec4p and is involved in polarized delivery of post-Golgi vesicles. J. Cell Biol. 1997;137:1495–1509. doi: 10.1083/jcb.137.7.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walworth N.C., Novick P.J. Purification and characterization of constitutive secretory vesicles from yeast. J. Cell Biol. 1987;105:163–174. doi: 10.1083/jcb.105.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walworth N.C., Brennwald P., Kabcenell A.K., Garrett M., Novick P. Hydrolysis of GTP by Sec4 protein plays an important role in vesicular transport and is stimulated by a GTPase-activating protein in Saccharomyces cerevisiae . Mol. Cell Biol. 1992;12:2017–2028. doi: 10.1128/mcb.12.5.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama S., Shirataki H., Sakisaka T., Takai Y. Three splicing variants of tomosyn and identification of their syntaxin-binding region. Biochem. Biophys. Res. Commun. 1999;252:218–222. doi: 10.1006/bbrc.1999.0300. [DOI] [PubMed] [Google Scholar]