

Abstract
Signal transducer and activator of transcription 3 (STAT3) is a DNA-binding transcription factor activated by multiple cytokines and interferons. High expression of STAT3 has also been implicated in cancer and lymphoma. Here, we show a case of B cell lymphoma in which a defective human immunodeficiency virus 1 (HIV-1) integrated upstream of the first STAT3 coding exon. The lymphoma cells with anaplastic large cell morphology formed multiple nodular lesions in the lung of an acquired immunodeficiency syndrome (AIDS) patient with Kaposi’s sarcoma. The provirus had a 5’ long terminal repeat (LTR) deletion, but the 3’ LTR had stronger promoter activity than the STAT3 promoter in reporter assays. Immunohistochemistry showed increased expression of STAT3 in the nuclei of lymphoma cells. Transfection of STAT3 resulted in transient cell proliferation in primary B cells in vitro. Although this is a very rare case of HIV-1-integrated lymphoma, these data suggest that up-regulation of STAT3 caused by HIV-1 integration resulted in the development of B cell lymphoma in this special case.
Keywords: HIV-1, Integration, AIDS-related lymphoma, STAT3
1. Introduction
Malignant lymphoma is an important complication of patients with acquired immunodeficiency syndrome (AIDS). A large part of AIDS-related lymphomas are of B cell linage, and positive for Epstein–Barr virus (EBV) or Kaposi’s sarcoma-associated herpesvirus (KSHV)[1-4]. Since human immunodeficiency virus 1 (HIV-1) is not usually detected in AIDS-related lymphoma cells, HIV-1 infection plays an indirect role in lymphomagenesis by impairing host immune surveillance. However, proviral DNA can either disrupt expression of tumor suppressor genes or enhance expression of cellular oncogenes. Alternatively, retroviral promoters can integrate into the host genome in such a manner that expression of a nearby oncogene is enhanced by a strong promoter within the proviral 3’-long terminal repeat (3’LTR). In humans, abnormal T cell proliferation following gene therapy for severe combined immunodeficiency resulted from retroviral integration into the intron of the LMO2 proto-oncogene [5]. In AIDS patients, some cases of lymphomas had HIV-1 integration within the fur gene, just upstream from the c-fes/fps proto-oncogene [6]. That report, however, did not investigate the functional effect of this integration event. These observations suggest that HIV-1 may contribute directly to lymphomagenesis by inserting an active promoter into a cellular oncogene [6]. In the present study, we report a case of AIDS-related lymphoma in which HIV-1 integrated upstream of the STAT3 gene. The association of HIV-integration and lymphomagenesis was investigated.
2. Materials and methods
2.1 Samples
Lymphoma tissues in the lung of a patient with HIV-1 infection were obtained at autopsy. Formalin-fixed pathological samples of lymphoma, including nine unrelated cases of AIDS-related lymphoma and 15 cases of non-Hodgkin lymphoma in HIV-1-uninfected individuals, were studied. All samples were obtained with informed consent according to the Declaration of Helsinki. The study protocol was approved by the institutional review board of National Institute of Infectious Diseases (Approval No. 93).
2.2 Immunohistochemistry and in situ hybridization
Immunohistochemistry was performed as described before [7, 8]. Primary antibodies were: anti-CD3 (Dako, Copenhagen, Denmark), CD20 (Dako), CD30 (Dako), CD45 (Dako), CD45RO (Dako), CD79a (Dako), CD138 (Serotec, Oxford, UK), and p80NPM/ALK (Nichirei, Tokyo, Japan), STAT3 (sc8019, Santa Cruz Biotechnology, Santa Cruz, CA), pSTAT3 (sc8059, Santa Cruz), KSHV-encoded LANA [8], and vIL-6 [7] antibodies. In situ hybridization for EBERs was performed as described before [9].
2.3 PCR and DNA sequences
PCR detection for KSHV-encoded open reading frame (ORF) 26, EBV W region, HIV-1 V3, and β-globin gene was performed as described previously [9, 10]. For PCR amplification of HIV-1 3’LTR and STAT3 junction, HIV3LTR-F (5’-TCTGAGCCTGGGAGCTCTCT-3’, 9561-9580 in GenBank K03455) and Stat3intron-R (5’-AGTGCATGGCACATAACAGA-3’, 41131-41150 in GenBank AY572796) were used. For amplification of HIV-1 5’LTR and STAT3 junction, 6 reverse primers of 5’LTR (55R 5’-TCAGGGAAGTAGCCTTGTGTGTGGT-3’, 78R 5’-GCCCTGGTGTGTAGTTCTGTCAATC-3’, 348R 5’-GAAAGTCCCCAGTGGAAAGTCCCTT-3’, 495R 5’-GCAGTGGGTTCCCTAGTTAGCC-3’, 563R 5’-TTACCAGAGTCACACAACAGACGGG-3’, and 612R 5’-CACTGCTAGAGATTTTCCACACTGAC-3’), and a reverse primer positioning between 5’LTR and gag (676R 5’-CGAGTCCTGCGTCGAGAGATCTCCT-3’) were used with a forward primer of Stat3-intronF2 (5’-CATTTTTCTTTCCTTCTCTGTTGTC-3’, 40881-40905 in GenBank AY572796). These primers for HIV-1 were designed based on the sequence of HIV-1 IIIB (GenBank K03455).
2.4 Cloning of HIV-1 integration sites
The methods used were essentially as described for the Gene Walker Kit (BD Clontech, Palo Alto, CA). Lung tumor DNA was cleaved with four different blunt cutting enzymes (DraI, EcoRV, PvuII and SspI). Gene specific primers for HIV-1 LTR were 5’-ACCACACACAAGGCTACTTCCCTGA-3’ (GSP-1) and 5’-AAGGGACTTTCCACTGGGGACTTTC-3’ (GSP-2).
2.5 Real-time PCR
Copy numbers of HIV-1 integration site and STAT3 gene were measured with real time PCR as described previously[11]. Two probe and primer sets were used (Set 1: forward primer: 5’-CTAGAGATCCCTCAGACCATTTTAGTC-3’, reverse: 5’-AAAAGTATAAATGAGGATCCAGGAAGAT-3’, probe: 5’-6FAM-TGTGGAAAATCTCTAGCAGAATCTCAGG-TAMRA-3’; Set 2: forward primer: 5’-GCAGCTTGACACACGGTACCT-3’, reverse: 5’-AAACTGCCGCAGCTCCATT-3’, probe: 5’-6FAM-AGCAGCTCCATCAGCTCTACAGTGACAGC-TAMRA-3’).
2.6 Plasmids
For the promoter assay, genes of the HIV-1 3’LTR, STAT3-intron (40951–41959 of GenBank AY572796), and STAT3-promoter (1–1998 of GenBank AY572796) were amplified from DNA of the HIV-1-integrated lymphoma using the LTR-MluI-F, 5’-GAGACGCGTTGGAAGGGCTAATTCACTCCC-3’ and LTR-XhoI-R, 5’-GTGCTCGAGTGCTAGAGATTTTCCACACT-3’, the Intron-MluI-F, 5’-GAGACGCGTGAATCTCAGGCAGATCTTCC-3’ and Intron-XhoI-R, 5’-CACCTCGAGCCTGCTAAAATCAGGGGTCCC-3’, and, the Stat3prom-MluF1, 5’-GAGACGCGTACCCATAGTCGCAGAGGTAGA-3’ and Stat3prom-XhoR1, 5’-GAGCTCGAGCGCTGAATTACAGCCCCTTCA-3’, respectively. Enzyme sites are indicated in italics. A fragment of the HIV-1 3’LTR was amplified also from HIV-1 pNL4-3 (GenBank AF324493). The PCR product was subcloned into MluI-XhoI site of pGL3-basic vector (Promega, Madison, WI). For the STAT3-expression plasmid, STAT3 cDNA was amplified from the mammalian gene collection-human (MGC-1607, American type culture collection, Manassas, VA) using forward primer (STAT3-HpaI-F10 5’-CACCGTTAACGGATCCTGGACAGGCACCC-3’) and reverse primer (STAT3-R24 5’-CATGTCAAAGGTGAGGGACTCAAA-3’). The PCR product was TA cloned using pcDNA 3.1 Directional TOPO Expression kit (Invitrogen, Carlsbad, CA). For cell proliferation experiment, the STAT3 expression vector was digested with Hind III and EcoRV and ligated into BsmBI and EcoRV sites of pMACS 4-IRES.II vector, which is a bicistronic expression vector containing multiple cloning site followed by an internal ribosome entry site (IRES) element from encephalomyocarditis virus and the truncated (non-functional) CD4 cDNA (Miltenyl Biotec, Auburn CA).
2.7 Promoter assay
Plasmids were transiently transfected into HeLa cells with a renilla reporter gene construct using Lipofectamine Plus (Invitrogen). Luciferase activity was measured with a dual luciferase assay system (Promega). In the HIV-1-Tat (+) group, an HIV-1-Tat expression vector, kindly provided by Dr. Kenzo Tokunaga, National Institute of Infectious Diseases, Tokyo, Japan, was cotransfected.
2.8 DNA methylation analysis
Methylation of the cytosine residue of the CpG site was analyzed by the bisulfite genomic sequencing method, as described previously [12]. The primer pair for selective analysis was as follows: sense primer, 5’-TATAAACCAGCATGGGATGGATGA -3’; antisense primer, 5’-CCCAGGCTCGGATCTGGTCTAACC-3’.
2.9 Cell proliferation assay for primary lymphocytes
Primary B cells were negatively selected from whole blood of healthy volunteers using RosetteSep B cell enrichment (StemCell Technology, Vancouver, BC, Canada) [13]. Cell proliferation assay was performed using BrdU Cell proliferation ELISA kit (Roche Molecular Biochemicals, Indianapolis, IN).
3. Results
3.1 HIV-1 was concentrated in lymphoma cells in a case of AIDS-related lymphoma
A 59-year-old, homosexual, HIV-1-positive male with a CD4 cell count of 6/mm3 showed high fever and multiple KS skin lesions. Computed tomography scanning revealed multiple nodules in the lung (Fig. 1A). Despite treatments with antibiotics and combined chemotherapy, with intensive care, he died 30 days after admission. The clinical course of the patient was also reported previously [14]. At autopsy, multiple nodules were present in the lung (Fig. 1B). Histologically, these nodules were composed of large atypical cells with anaplastic large cell morphology infiltrating into interstitial and alveolar areas in the lung tissue (Fig. 1C). Immunohistochemistry demonstrated that the tumor cells were CD3−, CD20−, CD30+, CD45+, CD45RO+, CD79a−, CD138−, and p80NPM/ALK−, suggesting that the lung tumor was composed of lymphoma cells (Fig. 1D and data not shown) [14]. Southern blot hybridization of DNA extracted from the lung tumor with an immunoglobulin junction hinge (JH) probe demonstrated immunoglobulin gene rearrangement, confirming a B cell linage (data not shown). Since KS lesions were found in the oral cavity, stomach, sole and some lymph nodes at autopsy, we examined KSHV positivity in the lymphoma (lung tumor). KSHV-encoded ORF26 was amplified in both gastric KS lesions and lung tumor by PCR (data not shown). However, immunohistochemistry demonstrated that expression of KSHV LANA was very weak or absent in the lymphoma cells, whereas KS cells in the stomach strongly expressed LANA (Fig. 1E). Immunohistochemistry also demonstrated that the lung tumor cells were negative for KSHV-encoded vIL-6 (data not shown). The lymphoma cells were positive for EBV by PCR (data not shown), but in situ hybridization failed to detect EBERs (Fig. 1F). Thus, these data suggest that KSHV and EBV were present in the lymphoma at low copy numbers. Surprisingly, HIV-1 DNA was detected in the lymphoma cells by PCR, but not in other organs besides the lymph nodes (Fig. 1G). Semi-quantitative PCR revealed that there was a 100-fold higher copy number of HIV-1 DNA from the lymphoma than from surrounding lung tissue (Fig. 1H). PCR products of HIV-1 V3 region were TA-cloned and each 10 clones were sequenced. Although two (clones L2 and T3) and three (clones T1, T3, and T6) kinds of sequences were obtained from the lymph nodes and lymphoma, respectively, all sequences coded the same amino acid sequence in the V3 loop (net charge=+7). Basic amino acids at positions 11 and 25 of the gp120 V3 loop and a high positive net charge strongly suggest that fusogenic X4 viruses were detected in the lymphoma cells and lymph nodes (Fig. 1I) [15].
Figure 1. Pathological findings of tumors in the lung of a patient with AIDS.
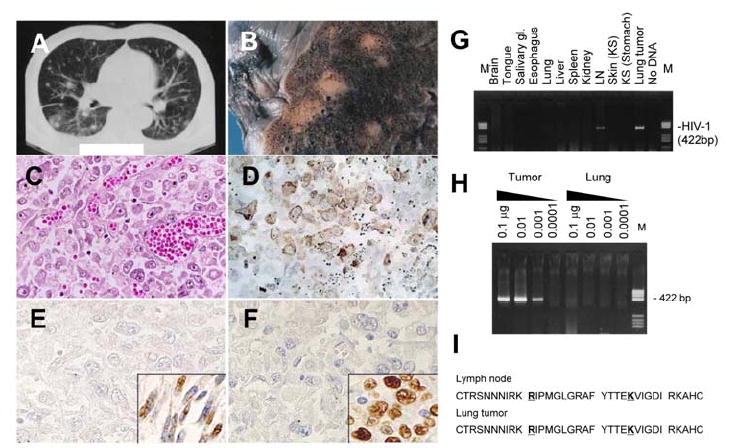
CT scan (A), macroscopic view (B) and Hematoxylin and eosin staining (C) of the lung tumor. (D) Immunohistochemistry of CD45RO. (E) Immunohistochemistry for KSHV-LANA in the lung tumor cells. Inset shows gastric KS cells from the patient. (F) In situ hybridization for EBV-EBER in the lung tumor cells. Inset shows a positive control of EBV-positive lymphoma from an unrelated patient. (G) PCR detection for HIV-1 V3 region in various organs of the patient. LN, lymph node; M, DNA molecular weight marker (pBR322/HaeIII). (H) Semi-quantitative PCR for HIV-1. DNA quantities are indicated at the top of the panel. DNA extracted from the lung tumor and surrounding lung tissues was tested. (I) Predicted amino acid sequence of HIV-1 gp120 V3 loop of HIV-1 amplified from the lymph node and lung tumor by PCR. Positions 11 and 25 are indicated by bold letters with underlines. DNA sequences are deposited in GenBank under accession numbers DQ116951 to DQ116954 (HIV-1 envelope from LN and lung tumor).
3.2 HIV-1 integration in the STAT3 gene
A high copy number of HIV-1 in the lymphoma suggested integration of HIV-1 into the genome of lymphoma cells. Genome walking PCR produced a 400 bp fragment which contained a 300 bp fragment with >99% identity with the HIV-1 IIIB 3’LTR sequence (GenBank K03455) and A 40 bp genomic segment just before the first coding exon of STAT3 (Fig. 2A). PCR using primers in HIV-1 3’LTR and STAT3-intron yielded an independent amplicon with HIV-1 3’LTR and the predicted STAT3 genomic sequences from DNA of the lymphoma cells (Fig. 2B). These data confirmed that HIV-1 had integrated into the intervening sequence just before the first coding exon of STAT3. PCR using a primer pair binding to the STAT3 intron and upstream of HIV-1 gag demonstrates that the 5’LTR of the integrated HIV-1 was truncated (Fig. 2C). The sequence analysis revealed that the integrated HIV-1 lacked a fragment at the position of 1-587 in the 5’LTR (Figs. 2A and 2D, GenBank AF538307). Compared with the sequence of the 3’ integration site, HIV-1 integration resulted in duplication of the cellular 5 bp (GAATC) and addition of a dinucleotide at the integration site by HIV-1 integrase, which is commonly seen among retrovirus integrases [16, 17]. Consequently, the integration event was produced by a defective virus (Figs. 2A and 2D). The absence of p24-staining of the tumor is consistent with this conclusion (data not shown).
Figure 2. Identification of HIV-1 integration site in the lymphoma cells with genome walking.
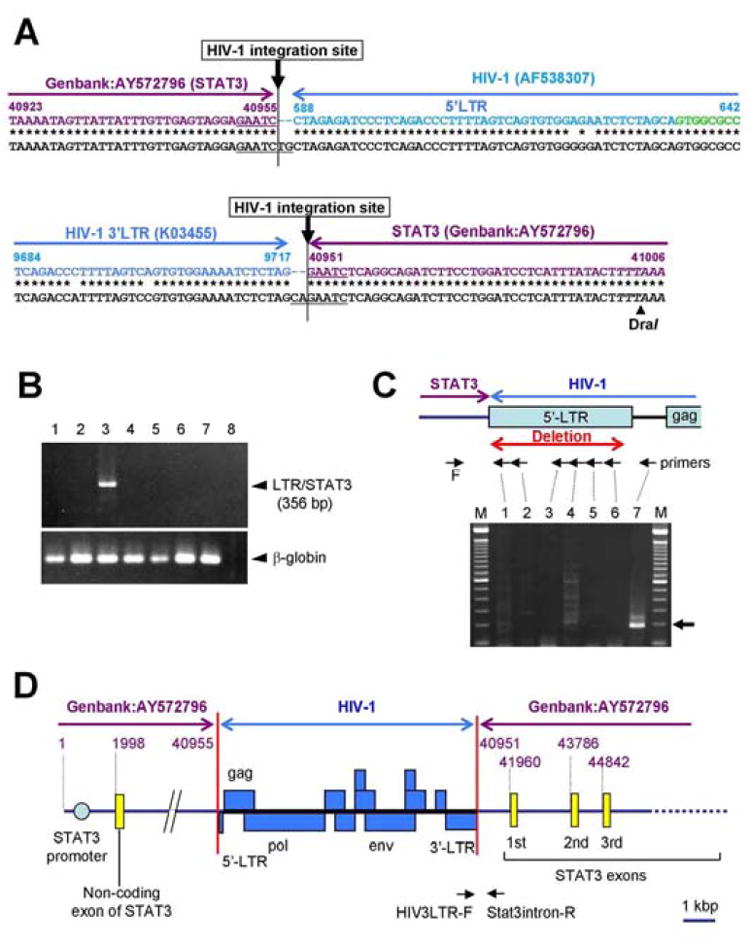
(A) Sequence of the HIV-1 5’-LTR (upper panel) and 3’-LTR (lower panel) insertion site in the lymphoma genome. Whole sequences of PCR products are registered as GenBank DQ355432 (5’-LTR, 190 bp) and DQ117603 (3’-LTR, 1.5 kbp), respectively. The sequence of the lymphoma genome is shown in the lower line in black letters. The upper colored line indicates the HIV-1 LTR sequence (blue, GenBank K03455 or AF538307) and STAT3 genomic sequence (violet, GenBank AY572796). HIV-1 intervening sequence between 5’LTR and gag is indicated by green. Duplication of the cellular 5 bp (GAATC) and additional dinucleotides (TG in 5’-LTR and CA in 3’-LTR) by HIV-1 integrase are underlined. DraI site is indicated by italics. (B) PCR for the junction region of 3’LTR and STAT3 gene using HIV3LTR-F and Stat3intron-R primers (see Fig. 3D). 1, PBMCs from a healthy donor; 2, HIV-1-positive Molt4 cell line; 3, lymphoma cells with HIV-1integration; 4, KS lesion from the patient; 5, AIDS-related lymphoma from an unrelated patient; 6, lymphoma from a non-HIV-1infected patient; 7, BCBL-1 (KSHV-positive B cell line); 8, No DNA. The lower panel shows the results of an internal control (β-globin gene). (C) PCR of genomic DNA with a STAT3-intron forward primer (F in this figure, Stat3-intronF2) in combination with 5’ LTR reverse primers (lanes 1–6, 55R, 78R, 348R, 495R, 563R and 612R), and a reverse primer positioning between 5’LTR and gag (lane 7, 676R). The upper panel shows the positions of these primers. A 188 bp product was identified when the 676R primer was used with the STAT3 intron primer (lane 7). If the 5’LTR was intact, the predicted size of this amplicon would have been 777 bp. (D) Map of the defective HIV-1 insertion site in the STAT3 gene. Violet numbers indicate the number in GenBank AY572796 (STAT3). Blue boxes are HIV-1 genomes.
3.3 Copy number of the integrated HIV-1 in the lymphoma tissue
Generally, pathological tissues obtained from lymphoma lesions contain not only lymphoma cells, but also surrounding CD4-positive T cells or alveolar macrophages. Although immunohistochemistry demonstrated no or rare CD4-positive cells in the lymphoma tissue, we tried to determine a copy number of the integrated HIV-1 in the lymphoma tissue by a real time PCR targeting genes near the integration site to deny the possibility that HIV-1 integration was originated in the contaminated CD4-positive cells (Fig. 3). A fragment of HIV-1-integration site was amplified at 12,570 copies/ 100 ng of DNA by the real time PCR, whereas exon 1 of STAT3 gene was amplified at 121,597 copies/ 100 ng. Since each cell has two copies of STAT3 gene on two alleles, these data suggest that HIV-1 integration occurred about 20% of the population that the DNA was extracted from. As shown in Fig. 1C, the lymphoma tissue contained many cells other than lymphoma cells, such as alveolar epithelial cells, macrophages, and endothelial cells. However, CD4-positive T cells were rare in the tissue, and the HIV-1 was X4 virus. Therefore, these data suggest that the HIV-1 might be detected from lymphoma cells, not from contaminated T cells or macrophages, and integrate into more than 20% of the lymphoma cells.
Figure 3. Quantitative analysis of genes for HIV-1 integration site.
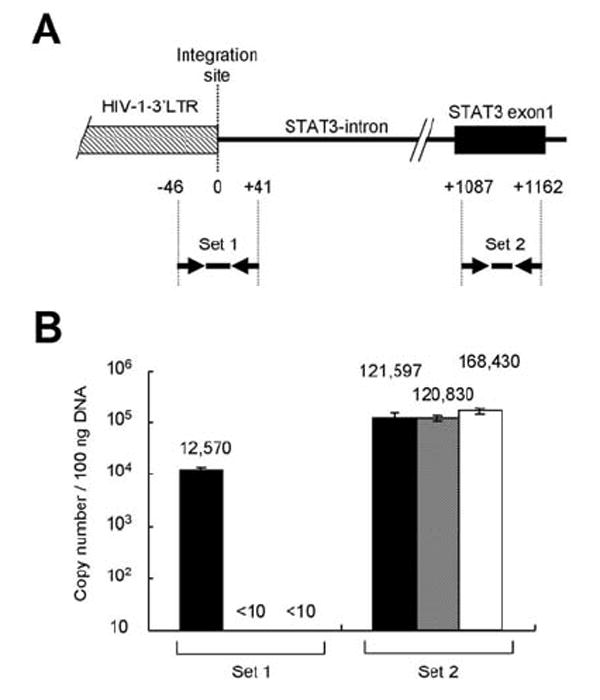
(A) Probe-primer sets for real time PCR. The top line with boxes is a genome map around HIV-1 integration site of HIV-1 3’LTR. Numbers with plus and minus under the genome map indicate distances (bp) from the integration site. Arrows and heavy lines are probe-primer sets of real time PCR. (B) Copy numbers of HIV-1 integration site and STAT3 gene. Black, gray and white bars indicate mean copy number per 100 ng DNA of this case, HIV-1-positive Molt4 cell line, and TY-1 (HIV-1-negative, KSHV-positive B cell line), respectively. Copy numbers per 100 ng DNA are indicated on the top of each bar. Error bars indicate standard errors of triplicate samples.
3.4 Promoter activity and methylation of HIV-1 3’LTR
LTRs of HIV-1 usually have a promoter activity in HIV-1-infected T cells and macrophages [18]. To investigate if the HIV-1 3’LTR contained a functional promoter, we constructed a plasmid containing the patient’s HIV-1 3’LTR or upstream intron sequence of STAT3 before a luciferase reporter gene. Transfection of the plasmid to HeLa cells revealed that the sequence of 3’LTR derived from the patient had significant promoter activity at a similar level to that of 3’LTR in HIV-1 NL4-3, but the upstream intron sequence of STAT3 did not (Fig. 4A). 3’LTR was a stronger promoter than the STAT3 promoter derived from the patient. Cotransfection with a plasmid expressing HIV-1-Tat enhanced the activity of the patient’s 3’LTR 31-fold, whereas the activity of the STAT3 promoter was not enhanced. These data suggest that the HIV-1 3’LTR contains promoter activity. It is known that DNA CpG methylation inactivates retroviral promoter including HIV-1 LTR [12, 19]. However, a bisulfite genomic sequence revealed that the fragment of HIV-1 3’LTR did not have any CpG or non-CpG methylation in the DNA extracted from the lymphoma (Fig. 4B and 4C). These data suggest that methylation might not reduce or inhibit the transcriptional activity of HIV-1 3’LTR in the HIV-1-integrated lymphoma cells.
Figure 4. Promoter activity of HIV-1 3’LTR and STAT3 expression in the lymphoma.
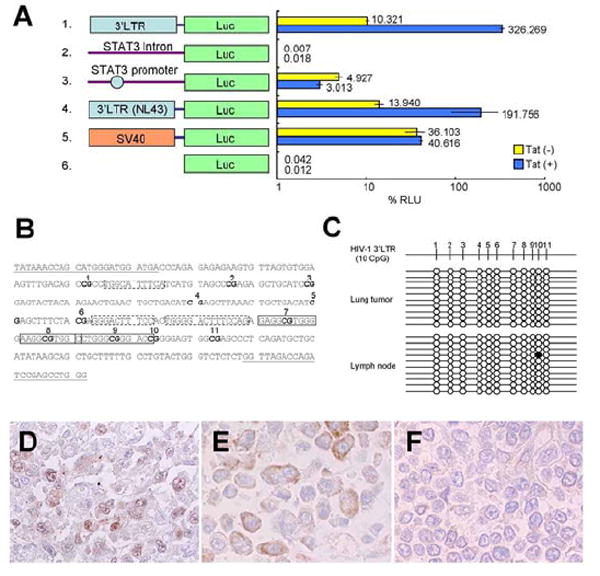
(A) Promoter activity of HIV-1 3’LTR by reporter assay. Schematic representation of promoter constructs used in transient transfection assays is shown on the left. Forty-eight hours after transfection, cells were collected and the luciferase activity was measured. The percentage relative luminescence units (RLU) were calculated by dividing firefly activity by renilla activity. Horizontal bars indicate standard deviations of three independent experiments. (B and C) No methylation in a promoter enhancer region of HIV-1 3’LTR in the HIV-1-integrated lymphoma. (B) CpG sites in the promoter enhancer region of 3’LTR of the HIV-1 provirus in the patient with HIV-1-integrated lymphoma (218-529 in GenBank DQ117603). CpG sites are in boldface and numbered from the 5’ end of the LTR (1–11). Nuclear factor-κB and Sp1 sites identified with Motif Search (Kyoto University Bioinfomatics center, Kyoto, Japan, http://motif.genome.jp/) at a 75% cut-off value are indicated by boxes with broken and solid lines, respectively. Sequences used for primers are indicated by underlining. (C) Levels of CpG methylation of the promoter enhancer region of HIV-1 3’LTR in the HIV-1-integrated lymphoma and lymph nodes in the patient. Results of bisulfite genomic sequencing coupled with TA cloning are shown. The methylation status of 10 clones for each sample is presented; methylation of each CpG site is expressed as a filled circle, and unmethylated sites are shown as open circles. Top, schematic description of CpG sites in the 3’LTR of (B). (D-F) Immunohistochemistry of STAT3. The HIV-1-integrated lymphoma cells expressed STAT3 predominantly in the nucleus (D), however, signals of STAT3 were weak and localized in the cytoplasm in the other case of KSHV-positive, AIDS-related lymphoma (E), and were very weak in a case of EBV-positive, AIDS-related lymphoma (F). Original magnification is x400.
3.5 Expression of STAT3 in the HIV-1-integrated lymphoma
We investigated expression of STAT3 in the case of HIV-1-integrated lymphoma. Immunohistochemistry demonstrated a high level of STAT3 expression predominantly in the nuclei of the HIV-1-integrated lymphoma cells (Fig. 4D). To know the phospholyration status of STAT3, we immunostained the slide using an anti-pSTAT3 (Tyr-705) antibody as a primary antibody. However, any signal was not found in the lymphoma cells (data not shown). We also examined STAT3 expression in 24 cases of lymphoma, including nine cases of AIDS-related lymphoma and 15 of non-AIDS-related lymphoma, normal tonsilar tissues and lymph nodes derived from unrelated patients. The nine cases of AIDS-related lymphoma contained seven of EBV-positive diffuse large B cell lymphoma (DLBCL), and two cases of Hodgkin’s disease. The 15 cases of non-AIDS-related lymphoma contained 12 EBV-positive or EBV-negative DLBCL and three cases of Hodgkin’s disease. Immunohistochemistry revealed that several cases of AIDS-related lymphoma and one of HIV-unrelated lymphoma expressed STAT3 predominantly in the cytoplasm (Fig. 4E); however, no case expressed STAT3 predominantly in the nucleus (Table 1). STAT3 expression was not found, or was weak, in other cases examined (Fig. 4F). These data suggest that the integration of HIV-1 induced high expression of STAT3 in the lymphoma cells of the patient.
Table 1.
STAT3 expression in AIDS-related and unrelated lymphoma.
| STAT3 expression | Nucleus | Cytoplasm | No expression | Total |
|---|---|---|---|---|
| AIDS-related lymphoma | 1* | 7 | 2 | 10 |
| Non-AIDS-related lymphoma | 0 | 1 | 14 | 15 |
HIV-integrated lymphoma reported in the present study.
3.6 Transfection of STAT3 expression plasmid to primary B cells in vitro
To investigate if expression of STAT3 induces cell growth, we constructed an expression plasmid for STAT3 and transfected the plasmid to B cells. At first, to confirm expression of STAT3 by Nucleofector transfection, His-tagged STAT3 was expressed in TY-1, a KSHV-positive B cell line. Immunofluorescence assay using anti-STAT3 and anti-6x His antibodies revealed that transfection efficiency to lymphocytes was 30-40% in this experiment (Fig. 5A). Addition of IL-6 to culture medium of transfected TY-1 altered the localization of STAT3 from the cytoplasm to the nucleus, suggesting that the transfected STAT3 reacted with IL-6 stimulation (Fig. 5B). Then, we investigated the proliferation of STAT3-transfected primary B cells. Cell proliferation assay after 48 hours transfection showed that the proliferation of STAT3-transfected primary B cells were slightly higher than that of vector-transfected primary B cells (Fig. 5C, Man-Whitney test, p<0.01). However, 4 days after transfection, the difference was not statistically significant (data not shown). The transfection of STAT3 to B cells was repeated 4 times with similar results. These data suggested that transfection of STAT3 might induce a transient proliferation in the primary B cells in vitro.
Figure 5. Transfection of STAT3 into B cells in vitro.
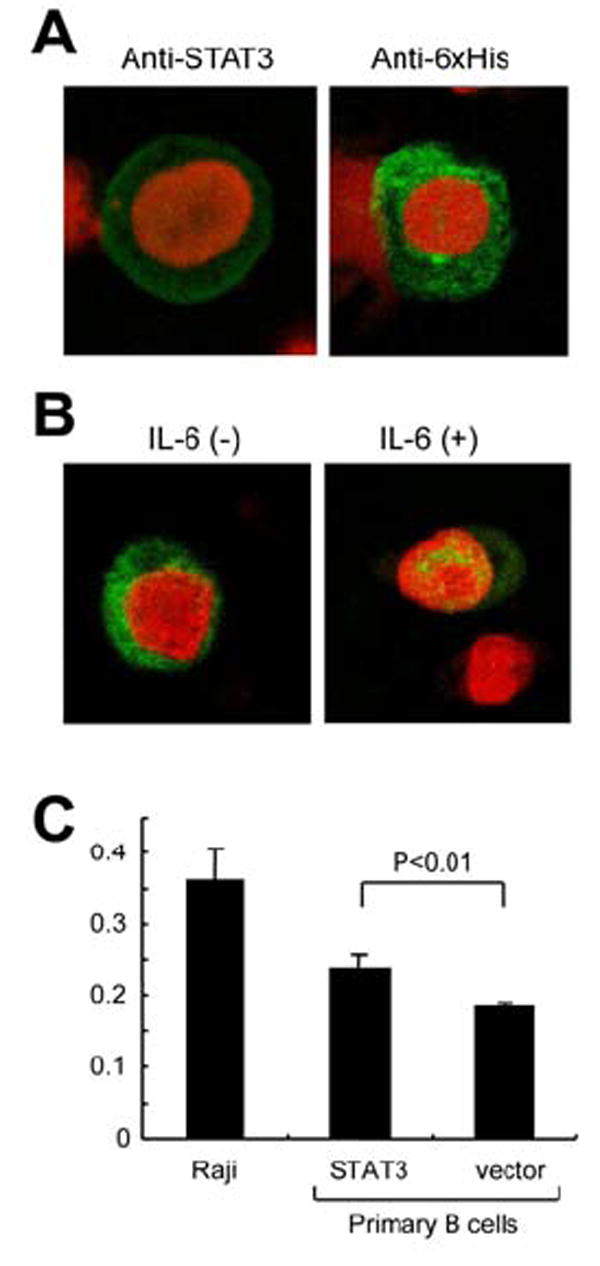
(A) STAT3 expression in the STAT3-transfected TY-1, a KSHV-positive B cell line. The cells were transfected with STAT3 expression vector by Nucleofector (Amaxa, Cologne, Germany) using O-06 program. STAT3 expression was detected by anti-STAT3 mouse monoclonal antibody (green in left panel) and anti-6xHis antibody, followed by Alexa 488-conjugated anti-mouse IgG antibody (Molecular probe, green in right panel). Red color indicates nuclear counterstaining of propidium iodide. (B) Localization of transfected STAT3 in TY-1. His-tagged STAT3 was detected by anti-6x His antibody in the cytoplasm of B cells (left panel). In the presence of IL-6 (Peprotech, Rocky Hill, NJ, 0.1 ng/ml), transfected STAT3 localizes in the nucleus predominantly (right panel). (C) Cell proliferation assay for STAT3-transfected primary B lymphocytes. Primary B cells were isolated from PBMC. The purity of B cell (CD19+) was >95%. The cells were transfected with STAT3 expression vector expressing STAT3 and CD4 by Nucleofector using U-15 program. Transfection efficiency to primary B cells was around 20%. To increase the proportion of transfected cells, the transfected B cells were separated with CD4 microbeads after 16 hours of the transfection (Miltenyl Biotec, Auburn CA). 48 hours after transfection of STAT3 or vector to primary B cells, the proliferation rate was measured with BrdU ELISA (Roche). Raji is an EBV-positive Burkitt lymphoma cell line (untransfected). Numbers in Y-axis indicates absorbance in ELISA. Error bars indicate standard errors of 8 independent experiments.
4. Discussion
In the present study, we present a case of AIDS-related B cell lymphoma with HIV-1 integration. HIV-1 with defective 5’LTR integrated into the upstream region of the first STAT3 coding exon. The 3’ LTR had strong promoter activity, resulting in increased expression of STAT3 in the nuclei of lymphoma cells. This is the first case report describing dysregulation of STAT3 by HIV-1 integration, resulting in B cell lymphoma development.
STAT3 is an important molecule for IL-6-type cytokines that signal and stimulate proliferation and terminal differentiation of B cells [20]. STAT3 also plays some oncogenic roles. Activated and phosphorylated STAT3 has been observed in a variety of experimental and numerous human malignancies [21-23]. A recent study reveals that high expression of unphosphorylated STAT3 results in up-regulation of oncogenes, suggesting that overexpression of either form of STAT3, phosphorylated and unphosphorylated, might induce cancer [24]. Although we failed to detect phosphorylated STAT3, high expression of STAT3 in the nucleus implies that activated STAT3 may bind to DNA and activate some genes constitutively. Alternatively, it implies that overexpression of unphosphorylated STAT3 in the nucleus might induce various oncogenes such as cdc2, cyclin B1 and mras [24]. However, our transfection study of STAT3 resulted in transient cell proliferation in the primary B cells (Fig. 5), suggesting that additional factors other than STAT3 expression might be required for complete transformation of primary B cells. HIV-1 integrated into c-fes/fps in other reported cases of AIDS-related lymphoma [6], and it has been demonstrated that c-fes activates STAT3 [25]. Thus, STAT3 may play some roles in the lymphomagenesis in the cases of HIV-1-integrated lymphoma.
This case was B cell lymphoma. HIV-1 usually infects and integrates into T cells or macrophages, and it is uncommon for HIV-1 to infect B cells. In the report by other group, HIV-1 provirus was frequently detected in macrophages infiltrating lymphomas, not in lymphoma cells [6]. However, in our case, we concluded that the HIV-1 integration occurred in the lymphoma cells, not in T cells or macrophages infiltrating in the lymphoma, because of following reasons; (1) there were few T cells in the lymphoma tissue by immunohistochemistry for CD3 (data not shown), (2) HIV-1 DNA was detected in the lymphoma at a high copy number, that is very rare or none in AIDS-related lymphoma [26], (3) HIV-1 sequences suggested the possibility of X4 viruses, which leads the integrated HIV-1 sequences are usually not found in the macrophages, (4) some different HIV-1 V3 sequences were identified between the lymphoma and lymph node, and (5) the titer of HIV-1 DNA in the lymphoma were higher than that in the lymph node (Fig. 1G). Then, how did HIV-1 infect B cells in the patient? Although detail mechanism of HIV-1 infection to B cells in this case was still unknown, we presume that KSHV played an important role in HIV-1 infection to B cells. This case of lymphoma was positive for KSHV and EBV by PCR, however, KSHV and EBV did not play a direct role in the oncogenesis of the lymphoma because of the low or absent expression of LANA and EBERs. It is possible that KSHV infection might increase susceptibility of B cells expressing CD4 and CXCR4 to infection with the X4 genotype of the HIV-1 [27]. Moreover, it is demonstrated that KSHV-encoded ORF50 protein increases susceptibility of B cells to infection with HIV-1 [28]. Although ORF50, CD4 and CXCR4 were not detected in the lymphoma cells by immunohistochemistry (data not shown), it is possible that KSHV-infected B cells might be infected and integrated by HIV-1 in the early stage of lymphoma development.
Although an intensive study revealed that there were many hot spots of HIV-1 integration [29], the STAT3 gene was not included in the list of hot spots. Thus, the STAT3 gene is a novel target of HIV-1 integration. Since HIV-1 DNA has not been detectable in DNAs extracted from AIDS-related lymphoma cases by Southern blot hybridization, so far [26], HIV-1 integration should be rare in AIDS-related lymphoma. A recent study demonstrates a decrease in EBV-positive lymphoma among patients with AIDS because of introduction of highly active antiretroviral therapy (HAART) [30]. Therefore, novel mechanisms other than oncogenesis by EBV or KSHV may have been involved in the lymphomagenesis of AIDS-related lymphoma recently. There is no report describing a frequency of HIV-1 integration among AIDS related lymphoma. HIV-1 usually infects T cells or macrophages in AIDS patients, however, T cell lymphoma is still rare among AIDS-related lymphoma in the HAART era [30]. In addition, HIV-1 infection to B cells would occur in a very special condition, such as under KSHV infection. Taken together, although the case we described in the present study contained an important scientific phenomenon on STAT3, HIV-1-integrated lymphoma should be very rare among AIDS-related lymphoma.
Acknowledgments
This study was supported by Health and Labor Sciences Research Grants on HIV/AIDS from the Ministry of Health, Labor and Welfare (grants H15-AIDS-005 to H.K.), a Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (grant 19590485 to H.K.), a grant for Research on Health Sciences focusing on Drug Innovation from Japan Health Sciences Foundation (grant SA14831 to H.K.), NIH MO1 RR00096 (to W.N.R.), NIH HL57879 (to M.D.W.), NIH HL 59832 (to M.D.W.), NIH DA022162 (to Y.H.), American Lung Association (to M.D.W.), Japanese Foundation for AIDS Prevention (to Y.H.), Uehara Memorial Foundation (to Y.H.) and the New York University Center for AIDS Research (to Y.H.). The authors declare that they have no competing financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Carbone A. Emerging pathways in the development of AIDS-related lymphomas. Lancet Oncol. 2003;4:22–29. doi: 10.1016/s1470-2045(03)00957-4. [DOI] [PubMed] [Google Scholar]
- 2.Boshoff C, Weiss R. AIDS-related malignancies. Nat Rev Cancer. 2002:373–382. doi: 10.1038/nrc797. [DOI] [PubMed] [Google Scholar]
- 3.Moore PS, Chang Y. Kaposi’s sarcoma-associated herpesvirus. Lippincott Williams & Wilkins; Philadelphia: 2001. [Google Scholar]
- 4.Chadburn A, Hyjek E, Mathew S, Cesarman E, Said J, Knowles DM. KSHV-positive solid lymphomas represent an extra-cavitary variant of primary effusion lymphoma. Am J Surg Pathol. 2004;28:1401–1416. doi: 10.1097/01.pas.0000138177.10829.5c. [DOI] [PubMed] [Google Scholar]
- 5.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, Lim A, Osborne CS, Pawliuk R, Morillon E, Sorensen R, Forster A, Fraser P, Cohen JI, de Saint Basile G, Alexander I, Wintergerst U, Frebourg T, Aurias A, Stoppa-Lyonnet D, Romana S, Radford-Weiss I, Gross F, Valensi F, Delabesse E, Macintyre E, Sigaux F, Soulier J, Leiva LE, Wissler M, Prinz C, Rabbitts TH, Le Deist F, Fischer A, Cavazzana-Calvo M. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 6.Shiramizu B, Herndier BG, McGrath MS. Identification of a common clonal human immunodeficiency virus integration site in human immunodeficiency virus-associated lymphomas. Cancer Res. 1994;54:2069–2072. [PubMed] [Google Scholar]
- 7.Katano H, Sato Y, Kurata T, Mori S, Sata T. Expression and localization of human herpesvirus 8-encoded proteins in primary effusion lymphoma, Kaposi’s sarcoma, and multicentric Castleman’s disease. Virology. 2000;269:335–344. doi: 10.1006/viro.2000.0196. [DOI] [PubMed] [Google Scholar]
- 8.Katano H, Sato Y, Kurata T, Mori S, Sata T. High expression of HHV-8-encoded ORF73 protein in spindle-shaped cells of Kaposi’s sarcoma. Am J Pathol. 1999;155:47–52. doi: 10.1016/S0002-9440(10)65097-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Katano H, Hoshino Y, Morishita Y, Nakamura T, Satoh H, Iwamoto A, Herndier B, Mori S. Establishing and characterizing a CD30-positive cell line harboring HHV- 8 from a primary effusion lymphoma. J Med Virol. 1999;58:394–401. [PubMed] [Google Scholar]
- 10.Katano H, Sato Y, Sata T. Expression of p53 and human herpesvirus 8 (HHV-8)-encoded latency-associated nuclear antigen (LANA) with inhibition of apoptosis in HHV-8-associated malignancies. Cancer. 2001;92:3076–3084. doi: 10.1002/1097-0142(20011215)92:12<3076::aid-cncr10117>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 11.Asahi-Ozaki Y, Sato Y, Kanno T, Sata T, Katano H. Quantitative analysis of Kaposi sarcoma-associated herpesvirus (KSHV) in KSHV-associated diseases. J Infect Dis. 2006;193:773–782. doi: 10.1086/500560. [DOI] [PubMed] [Google Scholar]
- 12.Koiwa T, Hamano-Usami A, Ishida T, Okayama A, Yamaguchi K, Kamihira S, Watanabe T. 5’-long terminal repeat-selective CpG methylation of latent human T-cell leukemia virus type 1 provirus in vitro and in vivo. J Virol. 2002;76:9389–9397. doi: 10.1128/JVI.76.18.9389-9397.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoshino Y, Nakata K, Hoshino S, Honda Y, Tse DB, Shioda T, Rom WN, Weiden M. Maximal HIV-1 replication in alveolar macrophages during tuberculosis requires both lymphocyte contact and cytokines. J Exp Med. 2002;195:495–505. doi: 10.1084/jem.20011614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katano H, Suda T, Morishita Y, Yamamoto K, Hoshino Y, Nakamura K, Tachikawa N, Sata T, Hamaguchi H, Iwamoto A, Mori S. Human herpesvirus 8-associated solid lymphomas that occur in AIDS patients take anaplastic large cell morphology. Mod Pathol. 2000;13:77–85. doi: 10.1038/modpathol.3880012. [DOI] [PubMed] [Google Scholar]
- 15.Hoshino Y, Tse DB, Rochford G, Prabhakar S, Hoshino S, Chitkara N, Kuwabara K, Ching E, Raju B, Gold JA, Borkowsky W, Rom WN, Pine R, Weiden M. Mycobacterium tuberculosis-induced CXCR4 and chemokine expression leads to preferential X4 HIV-1 replication in human macrophages. J Immunol. 2004;172:6251–6258. doi: 10.4049/jimmunol.172.10.6251. [DOI] [PubMed] [Google Scholar]
- 16.Brown PO, Bowerman B, Varmus HE, Bishop JM. Retroviral integration: structure of the initial covalent product and its precursor, and a role for the viral IN protein. Proc Natl Acad Sci U S A. 1989;86:2525–2529. doi: 10.1073/pnas.86.8.2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujiwara T, Mizuuchi K. Retroviral DNA integration: structure of an integration intermediate. Cell. 1988;54:497–504. doi: 10.1016/0092-8674(88)90071-2. [DOI] [PubMed] [Google Scholar]
- 18.Hoshino Y, Hoshino S, Gold JA, Raju B, Prabhakar S, Pine R, Rom WN, Nakata K, Weiden M. Mechanisms of PMN-Mediated Induction of HIV-1 Replication in Macrophages during Pulmonary Tuberculosis. J Infect Dis. 2007 doi: 10.1086/513438. in press. [DOI] [PubMed] [Google Scholar]
- 19.Bednarik DP, Cook JA, Pitha PM. Inactivation of the HIV LTR by DNA CpG methylation: evidence for a role in latency. Embo J. 1990;9:1157–1164. doi: 10.1002/j.1460-2075.1990.tb08222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 1998;334(Pt 2):297–314. doi: 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 22.Catlett-Falcone R, Dalton WS, Jove R. STAT proteins as novel targets for cancer therapy. Signal transducer an activator of transcription. Curr Opin Oncol. 1999;11:490–496. doi: 10.1097/00001622-199911000-00010. [DOI] [PubMed] [Google Scholar]
- 23.Lin TS, Mahajan S, Frank DA. STAT signaling in the pathogenesis and treatment of leukemias. Oncogene. 2000;19:2496–2504. doi: 10.1038/sj.onc.1203486. [DOI] [PubMed] [Google Scholar]
- 24.Yang J, Chatterjee-Kishore M, Staugaitis SM, Nguyen H, Schlessinger K, Levy DE, Stark GR. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005;65:939–947. [PubMed] [Google Scholar]
- 25.Nelson KL, Rogers JA, Bowman TL, Jove R, Smithgall TE. Activation of STAT3 by the c-Fes protein-tyrosine kinase. J Biol Chem. 1998;273:7072–7077. doi: 10.1074/jbc.273.12.7072. [DOI] [PubMed] [Google Scholar]
- 26.Pelicci PG, Knowles DM, 2nd, Arlin ZA, Wieczorek R, Luciw P, Dina D, Basilico C, Dalla-Favera R. Multiple monoclonal B cell expansions and c-myc oncogene rearrangements in acquired immune deficiency syndrome-related lymphoproliferative disorders. Implications for lymphomagenesis. J Exp Med. 1986;164:2049–2060. doi: 10.1084/jem.164.6.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Merat R, Amara A, Lebbe C, de The H, Morel P, Saib A. HIV-1 infection of primary effusion lymphoma cell line triggers Kaposi’s sarcoma-associated herpesvirus (KSHV) reactivation. Int J Cancer. 2002;97:791–795. doi: 10.1002/ijc.10086. [DOI] [PubMed] [Google Scholar]
- 28.Caselli E, Galvan M, Santoni F, Rotola A, Caruso A, Cassai E, Luca DD. Human herpesvirus-8 (Kaposi’s sarcoma-associated virus) ORF50 increases in vitro cell susceptibility to human immunodeficiency virus type 1 infection. J Gen Virol. 2003;84:1123–1131. doi: 10.1099/vir.0.18799-0. [DOI] [PubMed] [Google Scholar]
- 29.Schroder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–529. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- 30.Hishima T, Oyaizu N, Fujii T, Tachikawa N, Ajisawa A, Negishi M, Nakamura T, Iwamoto A, Hayashi Y, Matsubara D, Sasao Y, Kimura S, Kikuchi Y, Teruya K, Yasuoka A, Oka S, Saito K, Mori S, Funata N, Sata T, Katano H. Decrease in Epstein-Barr virus-positive AIDS-related lymphoma in the era of highly active antiretroviral therapy. Microbes Infect. 2006;8:1301–1307. doi: 10.1016/j.micinf.2005.12.012. [DOI] [PubMed] [Google Scholar]