

SUMMARY
BMP Receptors determine the intensity of BMP signals via Smad1 C-terminal phosphorylations. Here we show that a finely controlled cell biological pathway terminates this activity. The duration of the activated pSmad1Cter signal was regulated by sequential Smad1 linker region phosphorylations at conserved MAPK and GSK3 sites required for its polyubiquitinylation and transport to the centrosome. Proteasomal degradation of activated Smad1 and total polyubiquitinated proteins took place in the centrosome. Inhibitors of the Erk, p38 and JNK MAPKs, as well as GSK3 inhibitors, prolonged the duration of a pulse of BMP7. Wnt signaling decreased pSmad1GSK3 antigen levels and redistributed it from the centrosome to cytoplasmic LRP6-signalosomes. In Xenopus embryos, it was found that Wnts induce epidermis, and that this required an active BMP-Smad pathway. Epistatic experiments suggested that the dorso-ventral (BMP) and antero-posterior (Wnt/GSK3) patterning gradients are integrated at the level of Smad1 phosphorylations during embryonic pattern formation.
INTRODUCTION
Understanding how cells integrate multiple signaling pathways to achieve specific cell differentiations is one of the major challenges in cell and developmental biology. Embryonic patterning in Xenopus is regulated by gradients of growth factors and their antagonists, with Bone Morphogenetic Proteins (BMPs) controlling dorsal-ventral (D-V) and Wnt signals anterior-posterior (A-P) cell fates (Niehrs, 2004). This positional information must be seamlessly integrated, for when a blastula is cut in half the embryo can self-regulate forming perfect identical twins (De Robertis, 2006). In the ectoderm, the main cell differentiation decision is between neural and epidermal tissues, for which excellent molecular markers exist. Neural tissue differentiates when BMP signaling is inhibited by BMP antagonists or depletion by anti-BMP morpholino (MO) oligos, whereas epidermis is formed at high BMP signaling levels (Harland, 2000; Reversade and De Robertis, 2005). BMP receptors (BMPR) are Serine/Threonine protein kinases that signal by phosphorylating the transcription factors Smad1/5/8 at C-terminal sites (SS[PO3]VS[PO3]), causing their activation and nuclear translocation (Shi and Massagué, 2003; Feng and Derynck, 2005).
Neural tissue can also be induced by receptor tyrosine kinases (RTKs) such as FGF and IGF receptors via the activation of Mitogen Activated Protein Kinase (MAPK) (reviewed in Wilson and Edlund, 2001; De Robertis and Kuroda, 2004; Stern, 2005). This neural-inducing activity can be explained in part by an inhibitory phosphorylation in the linker (middle) region of Smad1 at four conserved MAPK (PXS[PO3]P) sites (Pera et al., 2003; Kuroda et al., 2005). This linker region MAPK phosphorylation was first discovered in cultured cells treated with EGF (Kretzschmar et al., 1997) and recently reported to promote polyubiqutinylation of Smad1 by the Smurf1 E3-ubiquitin ligase (Zhu et al., 1999; Sapkota et al., 2007), a finding independently confirmed here. FGF/MAPK signals are known to oppose BMP/Smad1 in many developing organs (De Robertis and Kuroda, 2004). Remarkably, mouse phosphorylation-resistant mutations in the MAPK sites of Smad1, introduced by homologous knock-in, generated embryonic fibroblasts in which the transcriptional activation of a reporter gene by BMP becomes resistant to repression by addition of FGF (Aubin et al., 2004; Sapkota et al., 2007). Thus, the role of Smad1 as an interface for integrating RTK and BMP signals is firmly established.
Although less generally recognized, the Wnt signaling pathway also influences neural induction. Wnts play multiple roles during development: at the early blastula stage canonical Wnt signaling provides a dorsalizing signal via activation of xTcf3 (Harland, 2000; Heasman, 2006) and at the neurula stage it regulates neuronal differentiation via inhibition of NeuroD (Marcus et al., 1998). At the gastrula stage, overexpression of Wnt8 causes anti-neural effects in Xenopus (Christian and Moon, 1993). Wnt antagonists such as Dickkopf-1 (Dkk1) and secreted Frizzled-related proteins (sFRPs), promote neural differentiation in Xenopus, chick and mouse ES cells (Glinka et al., 1998; Wilson and Edlund, 2001; Aubert et al., 2002). The canonical Wnt pathway signals through the inhibition of Glycogen Synthase Kinase 3 activity (Logan and Nusse, 2004). GSK3 is a protein kinase that usually requires pre-phosphorylated substrates, phosphorylating Ser/Thr residues located four amino acids upstream of sites primed by other kinases (Cohen and Frame, 2001). Frequently, as in the case of β-Catenin, such coupled phosphorylations by two protein kinases are followed by polyubiquitination and degradation in the proteasome (Liu et al., 2002; Cohen and Frame, 2001). This project was initiated when we noticed conserved GSK3 sites in vertebrate Smad1/5/8 proteins that could be phosphorylated by GSK3 after priming by MAPK. This was exciting, because a pathway in which
could potentially explain the pro-neural effects of anti-Wnts.
In the present study we demonstrate that phosphorylation at the GSK3 sites represses the transcriptional activity of Smad1 by enhancing proteasomal degradation of pSmad1Cter. We found that GSK3 phosphorylation is an essential requirement for polyubiquitination. Pulse-chase experiments with BMP7 revealed a novel cellular pathway that regulates the duration of the BMP signal. First, BMPR causes C-terminal phosphorylation (pSmad1Cter) and nuclear translocation. Next, the pSmad1MAPK phosphorylation is mediated by MAPK enzymes (Erk, p38 and JNK) in the nucleus. Subsequently, GSK3 recognizes the pre-phosphorylated linker region, generating pSmad1GSK3 at an unknown cellular location. The triply phosphorylated protein is transported along microtubule-like structures to the centrosome. The centrosome is the cellular location at which proteasomes normally accumulate in many cell types (Badano et al., 2005). The GSK3 activity that phosphorylates Smad1 was regulated by Wnt3a protein addition in cultured cells, leading to the accumulation of pSmad1Cter. A constitutively-active low-density lipoprotein receptor-related protein 6 (CA-LRP6) caused redistribution of pSmad1GSK3 from the centrosome to cytoplasmic particles resembling the recently described LRP6-signalosomes (Bilic et al., 2007). In vivo epistatic experiments indicated that Smad1 phosphorylations by GSK3 play a key role in mediating the effects of Wnt signaling on neural development at the gastrula stage. In ectodermal cells, overexpression of Wnt8 and its coreceptor LRP6 induced epidermal differentiation in a Smad1/5/8-dependent manner. The results suggest that a branch of the canonical Wnt pathway signals through the stabilization of BMP-Smad signals.
RESULTS
GSK3 Phosphorylation Inhibits Smad1 Function
Inspection of the Smad1 sequence revealed four GSK3 sites located four amino acids upstream of the PXSP Erk/MAPK sites in the linker region (Figure 1A). These potential GSK3 sites were conserved in BMP-responsive Smad1/5/8 across vertebrates and in Drosophila Mad (Figure S1). Phosphorylation-resistant mutations (Ser/Thr to Ala) were introduced into a human Smad1 expression construct (Kretzschmar et al., 1997) previously characterized in Xenopus embryos (Pera et al., 2003; Kuroda et al., 2005). These sites were mutated individually or in combination; strongest effects were found when all four GSK3 sites were mutated (data not shown) in a construct designated SGM (Figure 1B). The phenotypic effects of SGM were compared to those of Smad1 wild-type (SWT) and Smad1 mutated at the MAPK sites (designated SMM). Overexpression of mRNA encoding GSK3 or MAPK phosphorylation-resistant mutants in early Xenopus embryos resulted in hyperactive Smad1 proteins that caused strongly ventralized phenotypes, as indicated by transcript accumulation of the BMP-inducible marker Sizzled (Figure 1C–1F). A constitutively-active phospho-mimetic form of Smad1, in which the C-terminal SVS phosphorylation sites were mutated into EVE (designated SEVE, Figure 1G) provided a novel reagent independent of BMPR signaling (compare Figure 1D to 1H). GSK3 and MAPK phosphorylation-resistant mutants of SEVE strongly induced Sizzled, not only ventrally but also ectopically in the floor plate (Figure 1I and 1J, insets). We conclude that the function of the putative GSK3 phosphorylations in vivo, like those of MAPK, is to downregulate Smad1 activity.
Figure 1. Smad1 Is Phosphorylated and Inhibited by GSK3.
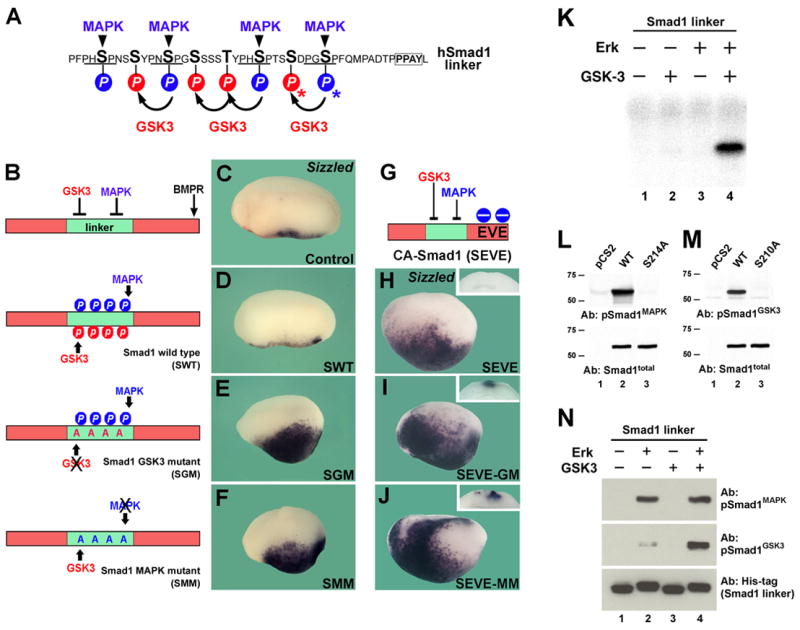
(A) Smad1 contains MAPK (blue) and GSK3 (red) phosphorylation sites in its linker region. The PPAY binding site of Smurf1 is boxed and serines 210 and 214 used to raise antibodies indicated by asterisks.
(B) Smad1 constructs encoding Smad1 wild-type (SWT) or phosphorylation-resistant mutants for GSK3 (SGM) and MAPK (SMM) sites.
(C–F) Injection of SGM or SMM, but not of SWT mRNA, increased expression of the ventral marker sizzled in Xenopus embryos.
(G) A BMP-independent phospho-mimetic activated Smad1 (SEVE) in which the SVS terminus was mutated into EVE.
(H–J) Activity of SEVE is increased by phosphorylation-resistant linker mutations.
(K) GSK3 radioactively phosphorylates Smad1 in vitro, but only when primed by MAPK. The recombinant Smad1 linker substrate was about 90% pure in polyacrylamide gels (data not shown).
(L and M) Phospho-specific antibodies for hSmad1 Ser 214 (pSmad1MAPK) and Ser 210 (pSmad1GSK3 antibody B was used).
(N) Phospho-specific antibodies (pSmad1MAPK and pSmad1GSK3-A) demonstrate that GSK3 phosphorylation of recombinant Smad1 requires MAPK priming, in non-radioactive western blots. Recombinant Smad1 substrate was used in the same amount as in panel K.
We next tested whether Smad1 was a substrate for GSK3 that required priming by MAPK (Cohen and Frame, 2001). The entire linker region of human Smad1 was tagged with 6 histidines, expressed in E. coli, and purified. In vitro phosphorylations were performed in two stages, and it was found that GSK3β caused the incorporation of γ32P-ATP by Smad1 linker protein, but only after pre-phosphorylation by Erk/MAPK (Figure 1K, compare lanes 2 and 4).
Antibody reagents specific for human phospho-Smad1GSK3 (p-Serine 210) and phospho-Smad1MAPK (p-Serine 214) were developed. A synthetic peptide (SS[PO3]DPGS[PO3]PFQMPADT) proved extremely antigenic. After affinity-purification on a column of the same peptide phosphorylated only in Ser 214, one rabbit produced a very high titer antibody for pSmad1MAPK, such that it detected overexpressed hSmad1-wt, but not the phosphorylation-resistant mutant S214A, at dilutions of 1:200,000 (Figure 1L). A second rabbit produced a phospho-specific antibody for pSmad1GSK3 Ser 210 active at dilutions of 1:30,000 after affinity-depletion with phospho-Ser 214 peptide and positive purification with phospho-Ser 210 peptide (antibody A). An additional anti-pSmad1GSK3 Ser 210 (antibody B) derived using peptide PHSPTSS[PO3]DPGSPFQ as antigen, detected overexpressed hSmad1-wt but not the phosphorylation-resistant mutant S210A (Figure 1M). Immunoblots of non-radioactive biochemical reactions confirmed that Ser 214 in recombinant Smad1 protein was indeed phosphorylated by Erk/MAPK and that the phosphorylation of Ser 210 by GSK3 was strongly dependent on pre-phosphorylation by MAPK, and underscored the phospho-specificity of our antibodies (see Figure 1N, lanes 2–4). We conclude that Smad1 is phosphorylated by GSK3 in vitro in a MAPK-dependent fashion.
GSK3 phosphorylations were required for the polyubiquitinylation of Smad1 (Figure 2A). The Smad ubiquitination regulatory factor 1 (Smurf1) E3 ubiquitin ligase binds to a PPXY sequence (Zhu et al., 1999) located near the Smad1 linker MAPK phosphorylation sites (Figure 1A). We used the assay of Zhu et al. (1999) in 293T cells, in which flag-tagged Smad1-WT or its GSK3 or MAPK mutants were co-transfected with HA-tagged ubiquitin and Smurf1, and Smad1 immunoprecipitated via its flag-tag. Polyubiquitinylation of GSK3 or MAPK phosphorylation-resistant Smad1 proteins (SGM and SMM) was greatly reduced when compared to their wild-type counterpart (Figure 2A, lanes 2–4). The mono- or di-ubiquitinylated forms were less affected. In addition, polyubiquitinylation of SWT in 293T cells was inhibited by the GSK3 inhibitors SB415286 or Lithium chloride (LiCl) (Figure 2B). We conclude that phosphorylation of Smad1 by GSK3 is essential for its polyubiquitinylation in cultured mammalian cells.
Figure 2. GSK3 and MAPK Phosphorylation Sites Regulate Polyubiquitinylation of Smad1 and its Transcriptional Activity.

(A) Polyubiquitinylation of Smad1 requires GSK3 and MAPK phosphorylation sites. Cells were co-transfected with Smad1-flag, Smurf1 and Ubiquitin-HA.
(B) GSK3 inhibitors decrease polyubiquitinylation of human Smad1. SB415286 (SB4, Biomol) was used at 40 μM, and LiCl at 30 mM, for 24 hours.
(C) SGM and SMM are more active than SWT mRNA in a BRE-luciferase transcriptional reporter assay in ectodermal Xenopus explants.
(D) The GSK3 inhibitor LiCl increases BMP4-induced transcriptional activation of BRE-luciferase in 293T cells 8 hrs after co-transfection. Data are represented as mean ± standard deviation, three independent experiments.
We next tested whether the transcriptional activity of Smad1 was regulated by GSK3 phosphorylations. Experiments were performed in Xenopus ectodermal explants, which have physiological endogenous BMP signaling levels sufficient for reporter gene studies (Kuroda et al., 2005). It was found that the phosphorylation-resistant mutants SGM and SMM caused significantly stronger induction (3–4 fold) of a BMP reporter (BREx2-Id1-luciferase; Korchynskyi and ten Dijke, 2002) than SWT (Figure 2C). In addition, the effect of the GSK inhibitor LiCl on BMP-dependent transcription was tested. Co-transfection of BMP4 and BREx2-Id1-luciferase DNAs into 293T cells showed that LiCl treatment significantly increased the transcriptional activity of endogenous BMP-Smads (Figure 2D). These results were congruent with the microinjection of SGM and SEVE-GM mRNAs in Xenopus embryos (Figure 1E and 1I), and suggested that GSK3 activity normally represses Smad1 signaling by promoting proteasomal degradation.
GSK3 and MAPKs Regulate the Duration of BMP Signals
We next investigated whether the coupled inhibitory phosphorylations by MAPK and GSK3 regulated the duration of the BMP7 signal in pulse-chase experiments (Figure 3). The intensity of the Smad1 signal is determined by the levels of C-terminal phosphorylation (pSmad1Cter) by BMPR (Feng and Derynck, 2005). The phosphorylations of endogenous Smad were analyzed in L-cells, which respond particularly well to BMP in serum-free medium (serum complicates analyses because it contains BMP and FGFs). Note that in these and all subsequent cultured cell experiments endogenous Smad1 was analyzed. Using this assay, we found that the MAPK and GSK3 phosphorylations were triggered by BMPR activity.
Figure 3. The Duration of the pSmad1Cter Signal Is Regulated by MAPK and GSK3.
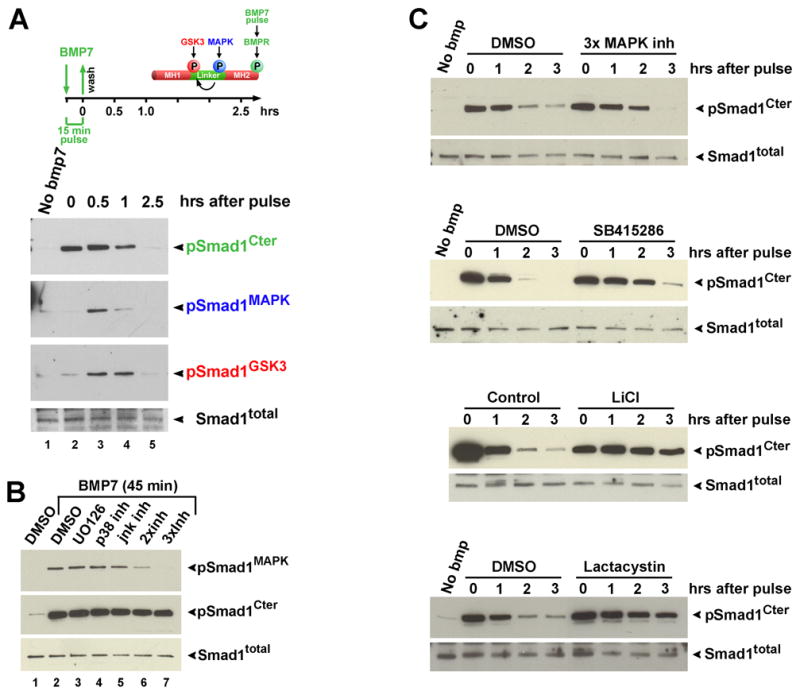
(A) Sequential phosphorylation of Smad1 by BMPR, MAPK and GSK3 after a pulse of BMP7.
(B) Induction of pSmad1MAPK by BMP7 is blocked by triple inhibition of MAPKs (lane 7). Inhibitors used were: MEK/Erk (10 μM U0126), p38 (10 μM Calbiochem CFPD p38 inhibitor), and JNK (25 μM SP600125). Lane 6 contained both p38 and JNK inhibitors.
(C) The duration of pSmad1Cter signal is prolonged by inhibition of MAPKs, GSK3 (40 μM SB415286 or 30 mM LiCl), or proteasome activity (50 μM Lactacystin).
A strong pSmad1Cter signal was elicited at time 0 after a 15 min pulse of 5 nM BMP7 (Figure 3A, compare lanes 1 and 2), which partially decreased after 1 hr, and greatly diminished by 2.5 hrs (lanes 3–5). Interestingly, pSmad1MAPK antibodies also detected a BMP7-induced band, but with a 0.5 hr delay. The pSmad1GSK3 antibody detected BMP-induced phosphorylations 0.5 and 1 hr after treatment, which decreased by 2.5 hrs (Figure 3A, lanes 2–5). Total Smad1 protein levels did not change during this period; this is in agreement with observations that only a small fraction of the total Smad1 is phosphorylated at physiological BMP levels, with a large reservoir of inactive Smad1 persisting at most times (e.g., Kuroda et al., 2005). BMP treatment does not increase activity levels of the three main cellular MAPKs (Erk, p38 and JNK) (Kuroda et al., 2005; Sapkota et al., 2007; data not shown). Experiments using chemical inhibitors (U0126 for MEK/Erk, Calbiochem CFPD inhibitor for p38, SP600125 for JNK) showed that the Smad1 MAPK phosphorylation triggered by BMP7 results from the combined constitutive activity of these three MAPKs in cultured cells (Figure 3B; Figure S2). We have also observed, using a phosphorylation-resistant C-terminal mutant, that MAPK and GSK3 can phosphorylate Smad1 in a BMPR-independent fashion (data not shown); this activity may help explain why neural induction by Chordin requires an intact MAPK pathway (Pera et al., 2003). Recently, Sapkota et al. (2007), reported that BMP induced MAPK phosphorylation, but suggested the involvement of a different and as yet unknown kinase; however, they did not test a cocktail of the three inhibitors as in Figure 3B, lane 7. We conclude from these pulse-chase experiments that BMP treatment triggers three sequential phosphorylations in Smad1: first by BMPR, second by MAPKs, and third by GSK3.
The sequential nature of these events suggested that they might function in the regulation of the duration of the BMP signal, which would be terminated by the coupled activity of MAPK and GSK3 via degradation in the proteasome. To test this hypothesis, we performed BMP7 pulse-chase experiments in the presence of the MAPK inhibitor cocktail, the GSK3 inhibitors LiCl or SB415286, or the proteasome inhibitor Lactacystin. As shown in Figure 3C, all these treatments significantly increased the duration of the pSmad1Cter signal. These results suggest a molecular pathway in which, following phosphorylation by BMPR, the duration of the pSmad1Cter signal is controlled by coupled MAPK and GSK3 phosphorylations and protein degradation in the proteasome.
pSmad1MAPK/GSK3 Is Targeted to the Centrosome
Unexpectedly, when the subcellular localization of the various forms of phosphorylated Smad1 in cultured cells was examined, it was found that Smad1 targeted for degradation accumulated in the centrosomal region (Figure 4). The pSmad1Cter antigen accumulates in the nucleus of Cos7 cells after BMP4 treatment in serum-free conditions, but we noted that in cells treated with the proteasome inhibitor Lactacystin, pSmad1Cter was also found in a cytoplasmic region (Figure 4B and 4C) that appeared to correspond to the centrosome (when a kidney-shaped nucleus is present, as in panel 4C, the centrosome is almost invariably located in its concavity; Wilson, 1928). The centrosome, the region of cytoplasm that surrounds the centrioles, is a center for ubiquitin-mediated proteolysis in which the proteasomal machinery becomes concentrated (Badano et al., 2005). We corroborated this by staining Cos7 cells with proteasomal S20α5 subunit antibodies. Proteasomes were present both in nucleus and cytoplasm, but were more concentrated in the centrosome (Figure 4D, arrows).
Figure 4. MAPK and GSK3 Phosphorylations Target Smad1 and Total Polyubiquitinated Proteins to the Centrosome in Cos7 Cells.
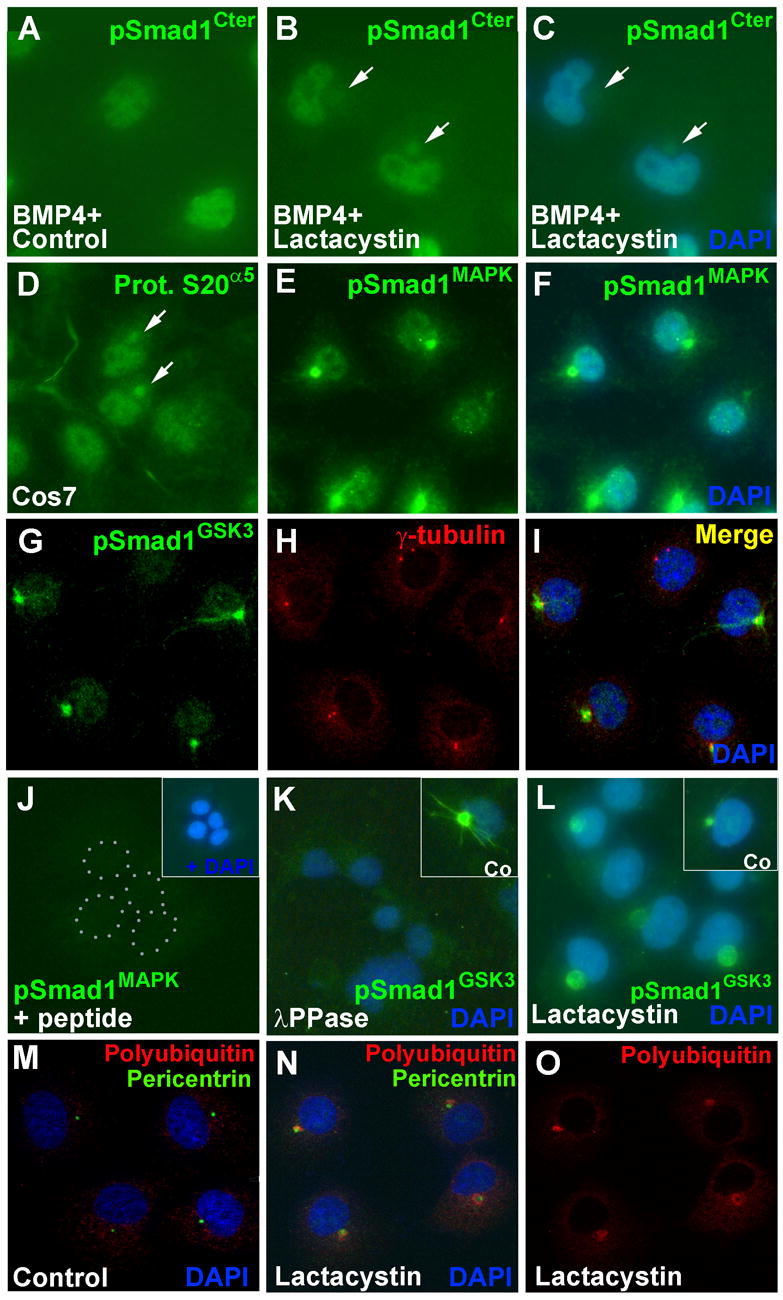
(A–C) pSmad1Cter accumulates in the centrosome (arrows) in response to proteasomal inhibition by Lactacystin (24%, n=323).
(D) The proteasomal subunit S20α5 normally accumulates in the centrosome in Cos7 cells (arrows).
(E and F) Nuclear and centrosomal localization of pSmad1MAPK.
(G–I) pSmad1GSK3- A (green) colocalizes with the centrosome marker γ-Tubulin (red). Note that in panels E through I BMP4 was not added, but the medium contained 10% fetal calf serum, which provides growth factors such as BMP and FGF.
(J and K) Specificity of pSmad1MAPK and pSmad1GSK3-A staining: phospho-peptide competition and λ-phosphatase sensitivity.
(L) pSmad1GSK3 accumulates in the centrosome 8 hrs after Lactacystin (50 μM) treatment (a 25-fold increase in volume).
(M–O) Treatment of Cos7 cells with Lactacystin induces accumulation of total cellular polyubiquitinylated proteins in the centrosomal region marked by Pericentrin in red. In one cell a ring of pericentrosomal staining is seen.
Phospho-Smad1MAPK was observed both in the nucleus and in a bright centrosome-like body in the cytoplasm (Figure 4E and 4F). The pSmad1GSK3 antibody stained the nucleus weakly and the cytoplasm strongly, along microtubule-like filaments that converged on the centrosome, which was unequivocally identified by co-localization with the established centrosomal marker γ-Tubulin (Figure 4G to 4I). Specificity controls showed that centrosomal phospho-Smad1 staining was eliminated or reduced by competition with the corresponding phosphorylated peptide (Figure 4J), by λ-phosphatase treatment (Figure 4K), and by the MAPK inhibitor cocktail or the GSK3 inhibitor SB415286 (Figure S2). Centrosomal localization was greatly enhanced by treatment of Cos7 cells with the proteasomal inhibitor Lactacystin (Figure 4N), supporting the view that Smad1 is degraded by proteasomes located in the centrosome.
Finally, we investigated whether this cellular mechanism represented a wider phenomenon that might apply to other proteins targeted for degradation. An antibody against total polyubiquitin chains stained diffusely both nucleus and cytoplasm (Figure 4M), but after Lactacystin treatment polyubiquitin antigen became clearly concentrated in the pericentrosomal region overlapping, and in some cases surrounding, the Pericentrin centrosomal marker (Figure 4N and 4O). We conclude that the cellular degradation pathway we have identified probably is used by many other proteins marked for destruction in addition to Smad1, since total polyubiquitinylated proteins also became concentrated in centrosomes when proteasome activity was inhibited.
Smad Degradation Is Regulated by Wnt
As shown in the model in Figure 5A, the experiments presented so far suggest that an elaborate cellular regulatory pathway is involved in the termination of BMP signaling. Nuclear C-terminal phosphorylated Smad1 is subjected first to MAPK phosphorylation in the linker region and then to GSK3 phosphorylation, triggering the polyubiquitinylation of Smad1. Once targeted for degradation, Smad1 is transported, probably along microtubules, to the centrosome, where the triply phosphorylated and poly-ubiquitinylated Smad1 is degraded by proteasomes. A key question, with important implications for the integration of patterning signals, is whether GSK3 phosphorylation of Smad1 is regulated by canonical Wnt signaling.
Figure 5. Wnt3a Regulates Smad1 Phosphorylation.
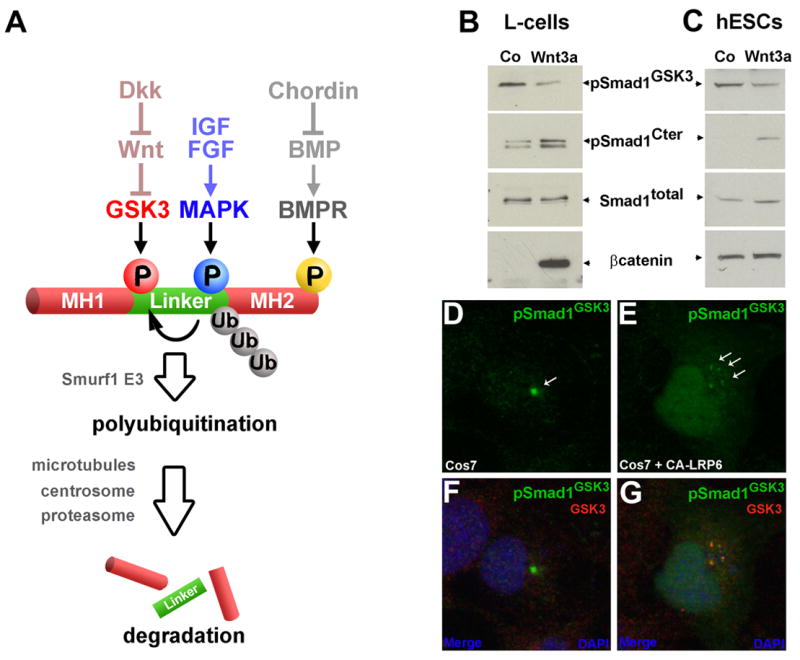
(A) Model of biochemical and cellular signaling pathway integration at the level of Smad1 phosphorylations.
(B) Wnt3a protein (300 ng/ml, R&D Systems) inhibits Smad1 phosphorylation by GSK3 and stabilizes pSmad1Cter. L-cells cultured in the absence of serum were treated with purified Wnt3a protein for 1 hour; 0.3 nM BMP7 and 40 ng/ml FGF2 were added after 20 minutes of Wnt treatment.
(C) Wnt3a (300 ng/ml) stabilizes pSmad1Cter and β-Catenin in hESCs after two hours.
(D–E) Activation of the Wnt pathway using transfected CA-LRP6 (Tamai et al., 2004) disperses pSmad1GSK3 from the centrosome into small cytoplasmic puncta. Note that in CA-LRP6 transfected cells the levels of pSmad1GSK3 in the nucleus are elevated.
(F–G) The cytoplasmic puncta containing pSmad1GSK3 most likely correspond to LRP6-signalosomes because they co-stain with anti-GSK3 antibodies. Transfected cells were identified by co-transfection of CFP, not shown here.
To investigate this, the effects of treating cultured L-cells (in the absence of serum and in the presence of BMP7 and FGF2) with purified Wnt3a protein were examined. The levels of pSmad1GSK3 were decreased, but not eliminated, one hour after Wnt3a addition, while the levels of pSmad1Cter increased (Figure 5B). The increase in pSmad1Cter was due to stabilization, rather than to secondary induction of BMP ligands, because transfection of β-catenin DNA (both in wild-type and stabilized mutant forms) did not significantly affect pSmad1Cter phosphorylation levels in L-cells (data not shown). Wnt3a protein also affected Smad1 phosphorylations in human embryonic stem cells (hESCs), causing pSmad1GSK3 to decrease moderately and pSmad1Cter to accumulate (Figure 5C). We conclude that the levels of pSmad1Cter are stabilized by Wnt3a treatment, in agreement with the pathway proposed in Figure 5A.
We next investigated whether the activation of Wnt signaling caused a change in the cellular localization of the pSmad1GSK3 antigen. A recent study has demonstrated that Wnt signals cause the co-localization of components of the β-Catenin degradation complex, such as GSK3 and Axin, with intracellular membrane vesicles containing the Wnt receptor LRP6 (Bilic et al., 2007). Transfection of CA-LRP6 DNA (Tamai et al., 2004) into Cos7 cells caused a dispersion of the centrosomal pSmad1GSK3 antigen into small cytoplasmic particles when compared to untransfected cells (Figure 5D and 5E). These cytoplasmic puncta most likely correspond to LRP6-signalosomes (Bilic et al., 2007) because they co-localized with endogenous GSK3 protein (Figure 5G). In addition to dispersing the centrosomal material into smaller particles, CA-LRP6 caused the pSmad1GSK3 antigen to accumulate in the cell nucleus (Figure 5D to 5G). Perhaps transport to the centrosome is required for nuclear export of Smad1 marked for destruction. We conclude that activation of the Wnt pathway triggers a major redistribution of Smad1 forms targeted for degradation within the cell.
We next turned to the Xenopus embryo. An old enigma is why overexpression of Wnt antagonists, such as Dkk1, cause dorsalized phenotypes (expanded neural plate at the expense of epidermis) almost indistinguishable to those of embryos microinjected with BMP antagonists, such as Chordin protein (Figure 6A to 6C). This suggested the possibility that Wnt inhibitors might regulate BMP signaling at gastrula through the pathway indicated in Figure 6D. This hypothesis was tested by co-injection of Dkk1 and Xenopus BMP4 mRNA (Figure 6E to 6H). BMP4 overexpression inhibited expression of the forebrain/eye marker Rx2a, and Dkk1 mRNA blocked this inhibition (Figure 6, compare panels G and H). Dkk1 is one of the strongest and best characterized inhibitors of canonical Wnt signaling, which functions by removing the LRP6 Wnt coreceptor from the cell surface (Glinka et al., 1998; Niehrs, 2004). Therefore, in principle, Dkk1 should not have inhibited BMP4 signaling.
Figure 6. The Dorsalizing Effects of Wnt Inhibition Are Rescued by SGM.
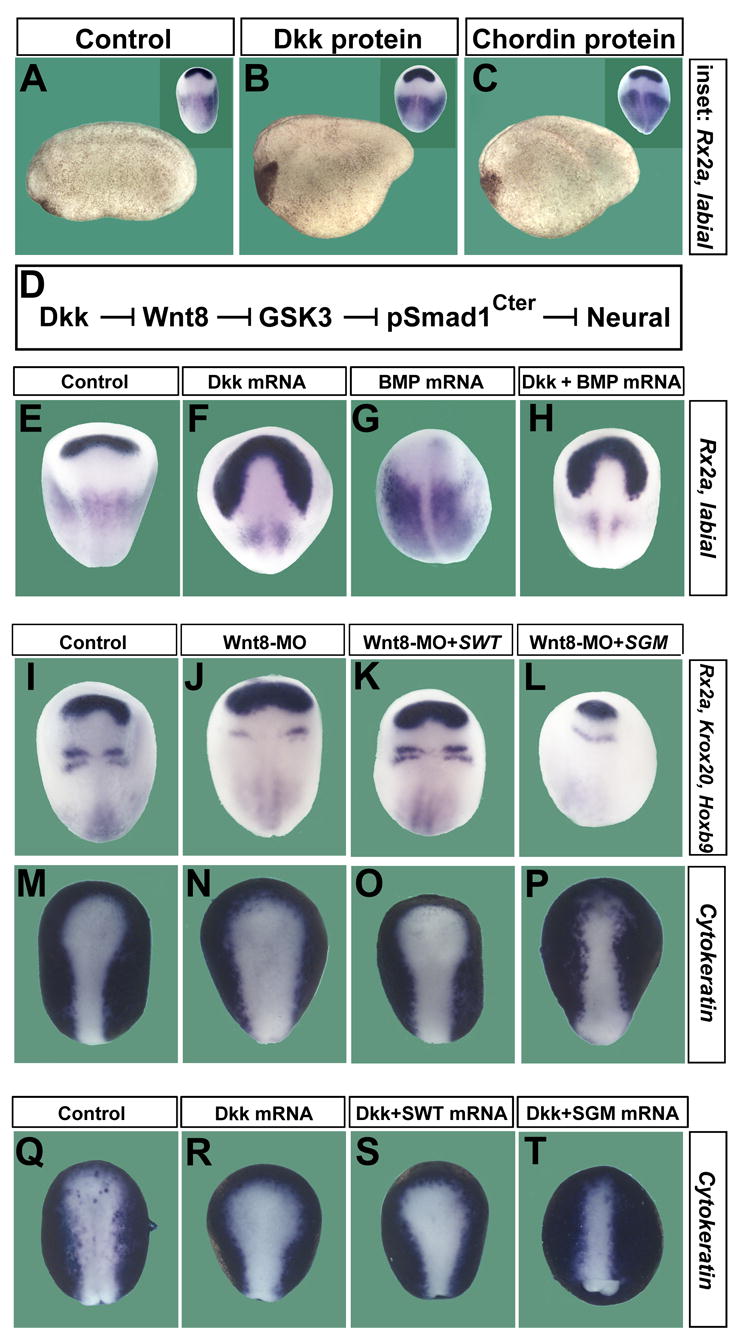
(A–C) Dkk1 (n=28) and Chordin (n=45) protein (R&D) injections into the blastula cavity (60 nl of 2.5 μM and 0.5 μM, respectively) present considerable phenotypical similarities.
(D) Proposed molecular pathway.
(E–H) Dkk1 mRNA suppresses the anti-neural phenotype of BMP4 overexpression (n for each sample was 18, 13, 23 and 18, respectively).
(I–P) Wnt8 MO causes dorsalization, which is blocked by GSK3-resistant SGM, but not by SWT mRNA (n for each sample was 22, 17, 23 and 26, respectively).
(Q–T) The expansion of the neural plate by Dkk1 mRNA is rescued by GSK3-resistant SGM, but not SWT mRNA (n=30, 22, 21 and 27, respectively).
To explore this in vivo in a Wnt loss-of-function situation, we used a previously characterized Xenopus laevis Wnt8 MO (Lee et al., 2006). xWnt8 is the main Wnt expressed posteriorly during gastrulation (Christian and Moon, 1993) and its knockdown causes dorsalization with expansion of brain at the expense of epidermis (marked by Cytokeratin) (Figure 6J and 6N). Microinjection of wild-type Smad1 mRNA had little effect on the xWnt8 depletion phenotype (Figure 6K and 6O), yet injection of the same amount of SGM mRNA was able to abrogate the dorsalizing effects of Wnt8 MO (Figure 6L and 6P). This indicates that the GSK3 phosphorylation sites in microinjected Smad1 play a crucial role in the dorsalizing effects caused by Wnt8 depletion. The phosphorylation of Smad1 GSK3 sites requires MAPK priming and, in agreement with this, phosphorylation-resistant SMM, but not SWT mRNA, also inhibited the effects of Wnt8 MO in the embryo (Figure S3).
Dkk1 overexpression is expected to block all canonical Wnt signaling at gastrula. Dkk1 mRNA microinjection expanded the neural plate and overexpressed wild-type Smad1 was ineffective at counteracting this (Figure 6Q to 6S), whereas the GSK3 phosphorylation-resistant mutant SGM was able to reverse the enlargement of the neural plate produced by Dkk1 mRNA (Figure 6, compare panels S and T). These epistatic experiments support the hypothesis that the GSK3 phosphorylation sites in Smad1 are critical targets of Dkk1 and Wnt8 signaling during Xenopus development.
The Anti-neural Effects of Wnt/GSK3 Require Smad1 but not Tcf3
We next investigated the molecular pathway by which Wnt/GSK3 regulates the decision between epidermal and neural cell fates in vivo. In Xenopus, it is sufficient to dissociate ectodermal (animal cap) cells to switch from an epidermal to a neural fate (Wilson and Hemmati-Brivanlou, 1995). This default neural differentiation is caused by sustained activation of MAPK/Erk (Kuroda et al., 2005). Ectodermal explants normally differentiate into epidermis, while dissociated animal cap cells form neural tissue (Figure 7A, compare lanes 2 and 3). LiCl, an inhibitor of GSK3 inhibits neural tissue and causes epidermal differentiation (Figure 7A, lanes 3–6).
Figure 7. Wnt Signaling Induces Epidermis in a Smad1/5/8 and β–Catenin Dependent, but Tcf3 Independent, Manner.
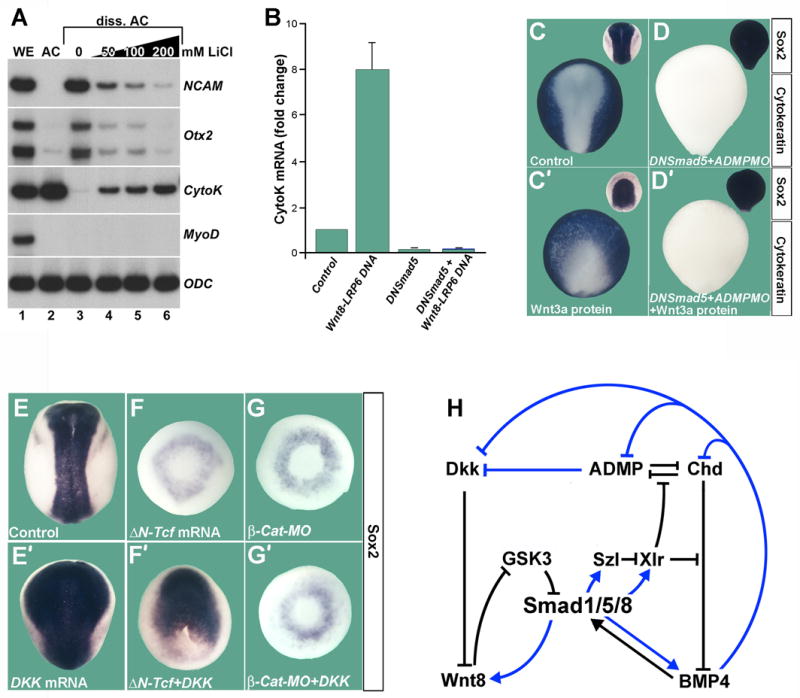
(A) LiCl induces epidermis (Cytokeratin) and inhibits neural differentiation (NCAM, Otx2). Radioactive RT-PCR analysis of whole embryos (WE), animal cap (AC) explants and dissociated animal cap cells at stage 13. MyoD indicates lack of mesoderm induction and Ornithine decarboxylase (ODC) equal loading.
(B) Quantitative PCR of dissociated animal caps injected with pCSKA-Wnt8 and pCS2-LRP6 DNA. DN-Smad5 mRNA was co-injected to block Smad1/5/8 activity. Cytokeratin mRNA levels at stage 13 were normalized for ODC mRNA and the standard deviation from three independent experiments is indicated.
(C,C′) Wnt3a protein (60 nl of 16 ng/μl) microinjected into the blastula cavity at stage 9 inhibits anterior neural plate and expands epidermis (n=40 and 42, respectively).
(D,D′) DN-Smad5 converts the entire ectoderm into neural tissue and is epistatic to Wnt3a protein injection (n=27 and 35). ADMP MO was co-injected to eliminate all traces of epidermis. (E,E′) Dkk1 mRNA expands the neural plate (n=100 and 53).
(F) ΔN-Tcf3 mRNA eliminates the neural plate; only a ring of Sox2 expression in ventral mesoderm remained (90%, n=30).
(F′) Dkk1 mRNA rescues neural plate in the presence of ΔN-Tcf3 in 60% of embryos (n=70).
(G,G′) The induction of neural plate by Dkk1 mRNA has a complete requirement for β-Catenin (100%, n=17 and 55, respectively).
(H) Model in which the BMP (D–V) and Wnt (A–P) patterning pathways are integrated at the level of Smad1/5/8 phosphorylations. Black arrows indicate direct protein-protein interactions and blue arrows transcriptional regulation by Smad1/5/8; all interactions are supported by overexpression or morpholino studies in Xenopus (Lee et al., 2006, and data not shown).
Microinjection of pCSKA-Wnt8 and pCS2-LRP6 DNA, which are expressed at gastrula stage (Christian and Moon, 1993), induced epidermis 8-fold in dissociated animal cap cells (Figure 7B). Importantly, epidermal induction by Wnt was blocked by co-injection of mRNA encoding a dominant-negative Smad5 mutant mimicking the zebrafish somitabun mutant (mouse DN-Smad5; Beck et al., 2001) that blocks all BMP/Smad1/5/8 signaling (Figure 7B). The epidermal-inducing effect of Wnt8/LRP6 DNA, which was in itself a novel observation, was not caused by an increase of BMP4 mRNA levels, which did not change significantly when compared to uninjected cells (mean 0.9 + 0.2 standard deviation) in the same samples (Figure 7B). This is in agreement with previous work in Xenopus showing that xWnt8 is unable to induce BMP4 (Hoppler and Moon, 1998). In whole embryos, DN-Smad5 eliminates epidermal differentiation, causing ubiquitous neural differentiation of the entire ectoderm (particularly in the presence of MO for ADMP, a BMP expressed at low Smad1/5/8 signaling levels, Reversade and De Robertis, 2005) (Figure 7C and 7D). Microinjection of Wnt3a protein at late blastula expanded the embryonic epidermis, but was without effect in a DN-Smad5 background (Figure 7C′ and 7D′). Similarly, GSK3 MO, which expands epidermis and inhibits neural tissue in the whole embryo, was unable to induce epidermis in DN-Smad5 embryos (Figure S4). We conclude that Wnt/GSK3 signals induce epidermis in Xenopus embryos and that this requires an active BMP/Smad1/5/8 pathway.
The canonical Wnt pathway signals by preventing the degradation of β-Catenin, an adaptor protein containing armadillo repeats, which accumulates in the nucleus, binds to the repressor T-cell factor 3 (Tcf3) and activates the transcription of Tcf target genes (Molenaar et al., 1996; Logan and Nusse, 2004). This pathway controls patterning in the Xenopus embryo during cleavage stages, causing the formation of the early embryonic dorsal axis (Harland, 2000; Heasman, 2006). At the gastrula stage, Wnts (and LiCl) have the opposite effect, promoting the formation of epidermis and ventral tissues (Figure S5B). Since late Wnt signaling appeared to be mediated in part through the regulation of Smad1 activity, we investigated whether the late effects of Wnt signaling in Xenopus were dependent on Tcf3 and β-Catenin. Dkk1 mRNA provides an excellent reagent to explore this since it specifically inhibits all gastrula stage canonical Wnt signaling. Dkk1 greatly expands the anterior neural plate (Figure 7E′). ΔN-Tcf encodes a 31 amino acid deletion lacking the β-Catenin binding site (Molenaar et al., 1996), which once bound to DNA blocks early Wnt signaling, resulting in embryos devoid of neural plate (Figure 7E and 7F). Remarkably, it was found that Dkk1 mRNA was able to restore neural plate development in 60% of these embryos (Figure 7F′). We also investigated whether the loss of neural plate caused by depletion of β-Catenin could be rescued by Dkk1 and found, to our surprise, a complete requirement for β-Catenin (figure 7G and 7G′). We conclude from these epistatic experiments that Wnt signaling promotes epidermal development via a Smad-dependent pathway, and that Dkk1 promotes neural development in a Tcf-independent, yet β-Catenin-dependent, manner.
DISCUSSION
We investigated the function of the four conserved GSK3 phosphorylation sites in Smad1. During the course of the work we generated new phospho-specific antibody reagents for pSmad1MAPK and pSmad1GSK3 that revealed a cell biological pathway by which the duration of the BMP signal is exquisitely modulated. Three novel observations were made. First, we found that BMP signaling triggers three sequential Smad1 phosphorylations by BMPR, MAPK and GSK3. Second, we found that the GSK3 and MAPK phosphorylations specifically regulate the duration of the BMP signal. Finally, experiments in cultured mammalian cells and in Xenopus embryos indicated that Smad1 phosphorylation by GSK3 serves to integrate the BMP and Wnt signaling pathways.
Sequential Smad1 Phosphorylations
GSK3 phosphorylation required priming by phosphorylated MAPK sites located in the linker region of Smad1. GSK3 phosphorylation-resistant mutations generated hyperactive forms of Smad1, as in the case of MAPK-resistant mutations (Kretzschmar et al., 1997; Pera et al., 2003; Aubin et al., 2004; Sapkota et al., 2007). High-titer phospho-specific antibody reagents for GSK3 (Ser 210) and MAPK (Ser 214) revealed the biochemical pathway of sequential phosphorylations summarized in Figure 5A. C-terminal phosphorylation by BMPR triggered nuclear translocation (Shi and Massagué, 2003), and was followed about 30 minutes later by phosphorylation at MAPK sites, presumably in the nucleus. This served as priming for phosphorylation by GSK3. The intracellular site of the GSK3 phosphorylation is not known since GSK3 is found both in nucleus and cytoplasm (Huang et al., 2007). The combination of both MAPK and GSK3 phosphorylations is essential for the polyubiquitinylation of Smad1 by the Smurf1 E3 ubiquitin ligase (Zhu et al., 1999). Recently, an independent study showed that MAPK phosphorylation is essential for Smad1 polyubiquitinylation (Sapkota et al., 2007), a discovery fully confirmed here. One difference between the two studies is that here we report an essential requirement for GSK3 for polyubiquitinylation in vivo, whereas Sapkota et al. (2007) found only a partial impairment in vitro studies. The broad theme that emerges from these complementary studies is that linker phosphorylations are required for Smad1 polyubiquitinylation in a negative feedback loop (Sapkota et al., 2007; this work).
Triply phosphorylated Smad1 is polyubiquitinylated, binds to cytoplasmic microtubule-like filaments and is transported to the centrosome (Figure 4). The centrosome has been described as the proteolytic center of the cell (Badano et al., 2005). Inhibition of the proteolytic activity of proteasomes with Lactacystin caused a marked accumulation of phospho-Smad1 destined for degradation specifically in the centrosome. Since, as demonstrated here, total polyubiquitin chains also accumulate in the centrosome after lactacystin treatment, many other proteins targeted for degradation must also use this pathway. The study of Smad1 has led to the identification of an elaborate cellular pathway involved in the degradation of this transcription factor.
The Duration of the Smad1 Signal Is Regulated by a Cellular Proteolytic Pathway
Smad1 is a transcriptional activator that controls the activity of hundreds of downstream target genes (Shi and Massagué, 2003; Feng and Derynck, 2005). During signaling it is important to control both the intensity and the duration of the signal. The classic example is provided by PC12 cells, in which a transient stimulus of MAPK/Erk by EGF causes cell proliferation, whereas the more sustained activation of Erk caused by NGF induces neuronal differentiation and neurite outgrowth (Marshall, 1995). While the intensity of the Smad1 signal is determined by the dose of BMP, its duration is regulated by the coupled phosphorylations mediated by MAPK and GSK3. Thus, the half-life of phospho-Smad1Cter was prolonged in pulse-chase experiments by inhibiting the three main endogenous MAPKs (Erk, p38 and JNK), GSK3, or proteasomal degradation (Figure 3). This novel biochemical pathway offers rich possibilities for the differential regulation of target genes by the Smad1 signal.
Integrating the A-P and D-V Axes via Smad1 Phosphorylations
In Xenopus, the D-V axis is regulated by a gradient of BMP maximal in the ventral and lowest in the dorsal, and the A-P axis by a gradient of Wnt lowest in the head and highest in the posterior blastopore where xWnt8 is expressed (Niehrs, 2004). When normal patterning is challenged by transplanting a new organizer or cutting the embryo in half, the new pattern is seamlessly integrated (De Robertis, 2006). The model shown in Figure 7H indicates how a self-regulating patterning mechanism may integrate the A-P and D-V axes. This model is based on recent studies on BMP signals and their antagonists (Reversade and De Robertis, 2005; Lee et al., 2006). Smad1/5/8 are presented as key players that can transcriptionally activate ventral genes (BMP4, the protease Xolloid-related, and its inhibitor Sizzled) as well as xWnt8. Previous work has shown that BMP4 is required for the expression of xWnt8, while xWnt8 does not affect BMP4 levels in the Xenopus gastrula (Hoppler and Moon, 1998). On the dorsal side, BMP signals inhibit the transcription of genes such as ADMP (a dorsal BMP-like molecule), Chordin and Dkk1. The additional layer of regulation of Smad1/5/8 by MAPKs activated by RTKs such as FGFR, IGFR and EGFR was not included for simplicity. The key new node of interaction in this modified patterning network is that Wnt8 inhibits the phosphorylation of Smad1 by GSK3, resulting in a longer BMP signal which, in turn, is inhibited by Dkk1. In this view, the D-V axis would be specified by the intensity of the BMP signal and the A-P positional information by its duration.
Our model proposes that Wnt enhances the BMP/Smad1 signal. Such a molecular pathway could have implications for the pathogenesis of hereditary bone diseases. BMPs were isolated as potent inducers of bone morphogenesis, yet recent genetic studies in humans and mice have shown that the Wnt coreceptor LRP5 plays a central role in bone mineralization. Loss-of-function mutations in LRP5 cause osteoporosis and gain-of-function mutations osteopetrosis (reviewed in Koay and Brown, 2005). Conversely, heterozygosity of Dkk1, an LRP5/6 inhibitor, causes excessive bone mass formation (Morvan et al., 2006). Like Dkk1, Sclerostin (SOST) is a Wnt antagonist that binds to LRP5/6, and leads to increased bone mass in loss-of-function mutations (Semenov et al, 2005). It would be interesting to determine whether these osteoblast differentiation syndromes are caused by misregulation of the duration of the Smad1/5/8 signal through the Wnt/GSK3 pathway.
The canonical Wnt pathway can inhibit GSK3 activity causing β-Catenin stabilization (Logan and Nusse, 2004). Wnt3a protein moderately decreased pSmad1GSK3 levels while increasing the activated pSmad1Cter form (Figure 5). In cytological analyses, activation of the Wnt pathway by a CA-LRP6 caused a striking change. The pSmad1GSK3 antigen lost its centrosomal localization and was detected in intracellular puncta corresponding to the recently discovered LRP6-signalosomes that contain GSK3 and other components of the Wnt-regulated protein degradation complex (Bilic et al., 2007). In addition, pSmad1GSK3 levels in the nucleus increased in cells transfected with CA-LRP6. This redistribution from the centrosome into putative signalosomes explains the residual levels of pSmad1GSK3 found in Wnt-treated cells. Relocation of GSK3 away from the cellular proteolytic center located in the centrosome may disrupt the flow of Smad1 targeted for degradation and suffice to explain the stabilization of pSmad1Cter caused by Wnt3a protein, as well as the increase of pSmad1GSK3 levels in the nucleus. Perhaps other features of the canonical Wnt/GSK3 pathway, such as the stabilization of β-Catenin, are mediated by the relocation of Wnt-regulated protein destruction complexes away from the centrosomal proteasomes (Bilic et al., 2007).
The new Wnt signaling branch identified here requires Smad1 and is independent of Tcf3, yet requires β-Catenin (Figure 7). This requirement suggests that both β-Catenin and Smad1 might be degraded by a common Wnt-regulated destruction complex containing GSK3, Axin and other components (Logan and Nusse, 2004). In this regard, we note that β-Catenin has a primary structure related to armadillo domain adaptor proteins and may help carry Smad1, and perhaps other proteins, to their destruction. A direct interaction between Smad4 and β-Catenin has been reported (Nishita et al., 2000). In addition, β-Catenin binds to the microtubular motor Dynein (Ligon et al., 2001) and β-Catenin can localize to microtubules (Huang et al., 2007). An attractive working hypothesis is that the destruction of β-Catenin could serve to drive the flow of Smad1 targeted for degradation to the centrosome. In Drosophila, work currently under completion supports the view that Wingless can stabilize the Mad transcription factor (E.E., L.C.F., J. Clemens and E.M.D.R., manuscript in preparation). With 1500 transcription factors in the human genome, one might predict that in future the degradation of other proteins may also be found to be stabilized by Wnt/GSK3 signaling.
EXPERIMENTAL PROCEDURES
Mammalian Cell Culture Experiments
Mouse L fibroblasts, Cos7 and HEK293T cells were cultured in DMEM supplemented with 10% fetal bovine serum (Gibco) and cultured at 37°C in 5% CO2. For pulse-chase experiments, L-cells were incubated in serum-free medium (⅓ Iscove’s, ⅔ F12 DMEM, Gibco) for 4 hrs, and 5 nM BMP7 added for 15 min. Chemical inhibitors were added 1 hr prior to the BMP7 pulse. Wnt3a protein (R&D Systems) was added at 300 ng/ml in the absence of serum. FGF-2 (Invitrogen) and BMP4 (R&D Systems) were added at 40 ng/ml and 0.3 nM, respectively. CA-LRP6 (LRP6–ΔN, Tamai et al., 2004), was transfected into Cos7 cells using Fugene HD (Roche). We note that the pericentrosomal region is best visualized in sparse cell cultures well below confluence at the G1 phase. Ubiquitination assays were performed as described by Zhu et al. (1999).
Western Blots and Immunostainings
Western blots of endogenous proteins were as described (Kuroda et al., 2005), using polyclonal rabbit antibodies against pSmad1MAPK (1:30,000); pSmad1GSK3-A (1:15,000); pSmad1Cter (1:1000; Cell Signaling) and total Smad1 (1:1000; Zymed). β-catenin antibody (Sigma, 1:8000), GSK3β antibody (BD Transduction Labs, 1:1000), anti-His antibody (Santa Cruz, 1:1000) and anti-flag-HRP conjugate (Sigma, 1:1500) were also utilized. Synthetic peptide was used to immunize two rabbits (Covance). A second pSmad1GSK3 antibody phosphorylated at site 210 (designated B) that works on western blots was generated. For cell immunostaining, primary antibodies were used as follows: pSmad1MAPK (1:2500), pSmad1GSK3 (1:1500), total polyubiquitin (1:500, Biomol), proteasome 20Sα5 subunit (1:200, Abcam), γ-Tubulin (1:500, Sigma) and Pericentrin (1:1000. Abcam). For additional immunostaining methods, see supplementary material S2.
In Vitro Phosphorylations
His-tagged linker region (residues 146-264) of Smad1 was cloned in the periplasmic expression vector pET26b, expressed in E. coli BL21 cells and purified using His Bind Resin (Novagen). The priming reaction was performed for 40 min at 30°C in kinase buffer (8 mM MOPS, pH 7.3, 0.2 mM EDTA, 10 mM MgCaH3PO4 and 20 mM MgCl2) containing 1 mM cold ATP and 5 U/μl recombinant p42/Erk2 (New England Biolabs). After heat inactivation of Erk (65°C for 20 min), a second reaction containing [γ-32P]ATP (Amersham) and recombinant GSK3β (Upstate) was performed for 1 hr at 30°C.
Xenopus Microinjections
All microinjections were four times marginal (or in the animal pole for ectodermal explants) at the 4-cell stage. Amounts injected per blastomere were as follows: Xwnt8 MO (10 ng; Lee et al., 2006), ADMP-MO (8.5 ng), β-catenin-MO (11 ng at 2-cell stage), pCSKA-Wnt8 DNA (30 pg, Christian and Moon, 1993), pCS2-LRP6 DNA (10 pg, Tamai et al., 2004), ΔN-xTCF3 mRNA (200 pg, Molenaar et al., 1996), DN-Smad5 mRNA (100 pg, Beck et al., 2001), Dkk1 mRNA (50 pg per embryo, Glinka et al., 1998), xBMP4 mRNA (50 pg), hSmad1 mRNA or its linker mutants (375 ng), and the constitutively active phospho-mimetic hSmad1 SEVE or its linker mutants (200 pg). Luciferase assays were as described in Kuroda et al. (2005). For quantitative PCR, RNA was isolated using Absolutely RNA preparation kit (Stratagene), DNAse treated, and reverse transcribed with random primers. SYBR Green real time quantitative PCR was performed in a MX3000 Cycler (Stratagene). Methods for mRNA synthesis, whole-mount in situ hybridizations and PCR primers are available at http://www.hhmi.ucla.edu/derobertis/index.html.
Supplementary Material
Acknowledgments
We thank, Drs. G. Thomsen, J. Massagué, D. Bohmann, X. He, R. Moon, J. Slack, H. Clevers, P. ten Dijke, and C. Niehrs for reagents, Y. Sun for advice, and members of our laboratory for comments on the manuscript. This work was supported by the NIH (HD21502-21). C.H. was a recipient of a Heart & Stroke Foundation of Canada Fellowship. E.M.D.R. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aubert J, Dunstan H, Chambers I, Smith A. Functional gene screening in embryonic stem cells implicates Wnt antagonism in neural differentiation. Nat Biotechnol. 2002;20:1240–1245. doi: 10.1038/nbt763. [DOI] [PubMed] [Google Scholar]
- Aubin J, Davy A, Soriano P. In vivo convergence of BMP and MAPK signaling pathways: impact of differential Smad1 phosphorylation on development and homeostasis. Genes Dev. 2004;18:1482–1494. doi: 10.1101/gad.1202604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badano JL, Teslovich TM, Katsanis N. The centrosome in human genetic disease. Nat Rev Genet. 2005;6:194–205. doi: 10.1038/nrg1557. [DOI] [PubMed] [Google Scholar]
- Beck CW, Whitman M, Slack JM. The role of BMP signaling in the outgrowth of the Xenopus tail bud. Dev Biol. 2001;238:303–314. doi: 10.1006/dbio.2001.0407. [DOI] [PubMed] [Google Scholar]
- Bilic J, Huang YL, Davidson G, Zimmermann T, Cruciat CM, Bienz M, Niehrs C. Wnt induces LRP6 Signalosomes and promotes Dishevelled-dependent LRP6 phosphorylation. Science. 2007;316:1619–1622. doi: 10.1126/science.1137065. [DOI] [PubMed] [Google Scholar]
- Christian JL, Moon RT. Interactions between Xwnt-8 and Spemann organizer signaling pathways generate dorsoventral pattern in the embryonic mesoderm of Xenopus. Genes Dev. 1993;7:13–28. doi: 10.1101/gad.7.1.13. [DOI] [PubMed] [Google Scholar]
- Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- De Robertis EM. Spemann’s organizer and self-regulation in amphibian embryos. Nat Rev Mol Cell Bio. 2006;7:296–302. doi: 10.1038/nrm1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Robertis EM, Kuroda H. Dorsal-ventral patterning and neural induction in Xenopus embryos. Ann Rev Cell Dev Biol. 2004;20:285–308. doi: 10.1146/annurev.cellbio.20.011403.154124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng XH, Derynck R. Specificity and versatility in TGF-β signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- Glinka A, Wu W, Delius H, Monaghan AP, Blumenstock C, Niehrs C. Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nature. 1998;391:357–362. doi: 10.1038/34848. [DOI] [PubMed] [Google Scholar]
- Harland R. Neural induction. Curr Opin Genet Dev. 2000;10:357–362. doi: 10.1016/s0959-437x(00)00096-4. [DOI] [PubMed] [Google Scholar]
- Heasman J. Patterning the early Xenopus embryo. Development. 2006;133:1205–1217. doi: 10.1242/dev.02304. [DOI] [PubMed] [Google Scholar]
- Hoppler S, Moon RT. BMP-2/-4 and Wnt-8 cooperatively pattern the Xenopus mesoderm. Mech Dev. 1998;71:119–129. doi: 10.1016/s0925-4773(98)00004-5. [DOI] [PubMed] [Google Scholar]
- Huang P, Senga T, Hamaguchi M. A novel role of phospho-β-catenin in microtubule regrowth at centrosome. Oncogene. 2007;26:4357–4371. doi: 10.1038/sj.onc.1210217. [DOI] [PubMed] [Google Scholar]
- Koay MA, Brown MA. Genetic disorders of the LRP5-Wnt signalling pathway affecting the skeleton. Trends Mol Med. 2005;11:129–137. doi: 10.1016/j.molmed.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Korchynskyi O, ten Dijke P. Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J Biol Chem. 2002;277:4883–4891. doi: 10.1074/jbc.M111023200. [DOI] [PubMed] [Google Scholar]
- Kretzschmar M, Doody J, Massagué J. Opposing BMP and EGF signaling pathways converge on the TGF-β family mediator Smad1. Nature. 1997;389:618–622. doi: 10.1038/39348. [DOI] [PubMed] [Google Scholar]
- Kuroda H, Fuentealba L, Ikeda A, Reversade B, De Robertis EM. Default neural induction: neuralization of dissociated Xenopus cells is mediated by Ras/MAPK activation. Genes Dev. 2005;19:1022–1027. doi: 10.1101/gad.1306605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HX, Ambrosio AL, Reversade B, De Robertis EM. Embryonic dorsal-ventral signaling: secreted frizzled-related proteins as inhibitors of tolloid proteinases. Cell. 2006;124:147–159. doi: 10.1016/j.cell.2005.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon LA, Karki S, Tokito M, Holzbaur EL. Dynein binds to β-Catenin and may tether microtubules at adherens junctions. Nat Cell Biol. 2001;3:913–917. doi: 10.1038/ncb1001-913. [DOI] [PubMed] [Google Scholar]
- Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. Control of β-Catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837–847. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Ann Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- Marcus EA, Kintner C, Harris W. The role of GSK3β in regulating neuronal differentiation in Xenopus laevis. Mol Cell Neurosci. 1998;12:269–280. doi: 10.1006/mcne.1998.0713. [DOI] [PubMed] [Google Scholar]
- Marshall CJ. Specificity of receptor tyrosine kinase signaling: treatment versus sustained extracellular signal regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destrée O, Clevers H. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell. 1996;86:391–399. doi: 10.1016/s0092-8674(00)80112-9. [DOI] [PubMed] [Google Scholar]
- Morvan F, et al. Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J Bone Min Res. 2006;21:934–945. doi: 10.1359/jbmr.060311. [DOI] [PubMed] [Google Scholar]
- Niehrs C. Regionally specific induction by the Spemann-Mangold organizer. Nat Rev Genet. 2004;6:425–434. doi: 10.1038/nrg1347. [DOI] [PubMed] [Google Scholar]
- Nishita M, Hashimoto SO, Ogata S, Laurent MN, Ueno N, Shibuya H, Cho KW. Interaction between Wnt and TGF-β signalling pathways during formation of Spemann’s Organizer. Nature. 2000;403:781–785. doi: 10.1038/35001602. [DOI] [PubMed] [Google Scholar]
- Pera EM, Ikeda A, Eivers E, De Robertis EM. Integration of IGF, FGF, and anti-BMP signals via Smad1 phosphorylation in neural induction. Genes Dev. 2003;17:3023–3028. doi: 10.1101/gad.1153603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reversade B, De Robertis EM. Regulation of ADMP and BMP2/4/7 at opposite embryonic poles generates a self-regulating morphogenetic field. Cell. 2005;123:1147–1160. doi: 10.1016/j.cell.2005.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapkota G, Alarcon C, Spagnoli FM, Brivanlou AH, Massagué J. Balancing BMP signaling through integrated outputs into the Smad linker. Mol Cell. 2007;25:441–454. doi: 10.1016/j.molcel.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Semenov M, Tamai K, He X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem. 2005;280:26770–26775. doi: 10.1074/jbc.M504308200. [DOI] [PubMed] [Google Scholar]
- Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Stern CD. Neural induction: old problem, new findings, yet more questions. Development. 2005;132:2007–2021. doi: 10.1242/dev.01794. [DOI] [PubMed] [Google Scholar]
- Tamai K, Zeng X, Liu C, Zhang X, Harada Y, Chang Z, He X. A mechanism for Wnt coreceptor activation. Mol Cell. 2004;13:149–156. doi: 10.1016/s1097-2765(03)00484-2. [DOI] [PubMed] [Google Scholar]
- Wilson E. The Cell in Development and Heredity. New York: The MacMillan Company; 1928. pp. 24–31. [Google Scholar]
- Wilson S, Edlund T. Neural induction: toward a unifying mechanism. Nature Neuroscience. 2001;4:1161–1168. doi: 10.1038/nn747. [DOI] [PubMed] [Google Scholar]
- Wilson PA, Hemmati-Brivanlou A. Induction of epidermis and inhibition of neural fate by BMP4. Nature. 1995;376:331–333. doi: 10.1038/376331a0. [DOI] [PubMed] [Google Scholar]
- Zhu H, Kavsak P, Abdollah S, Wrana JL, Thomsen GH. A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature. 1999;400:687–693. doi: 10.1038/23293. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.