

Abstract
Microbial populations under nonlethal selection can give rise to mutations that relieve the selective pressure, a phenomenon that has come to be called “adaptive mutation.” One explanation for adaptive mutation is that a small proportion of the cells experience a period of transient hypermutation, and that these hypermutators account for the mutations that appear. The experiments reported here investigated the contribution that hypermutators make to the mutations occurring in a Lac− strain of Escherichia coli during selection for lactose utilization. A broad mutational screen, loss of motility, was used to compare the frequency of nonselected mutations in starved Lac− cells, in Lac+ revertants, and in Lac+ revertants carrying yet another nonselected mutation. These frequencies allowed us to calculate that the hypermutating subpopulation makes up ≈0.06% of the population and that its mutation rate is elevated ≈200-fold. From these numbers we conclude that the hypermutators are responsible for nearly all multiple mutations but produce only ≈10% of the adaptive Lac+ mutations.
Although spontaneous mutations occur at random while cells are proliferating, apparently static microbial populations under nonlethal selective pressure also accumulate mutations. In some cases, these mutations appear to be confined to genes that give an adaptive phenotype, and for this reason the phenomenon has come to be called directed or adaptive mutation (1). Most models for adaptive mutation invoke a genomewide process that produces genetic variants at random but postulate that these variants are transitory unless the cell achieves a mutation that relieves the selective pressure (2). The most extreme of these “trial-and-error” models is the “hypermutable state” proposed by Hall (3). According to this model, most cells in a population under selection do not mutate, but a minority transiently experience a high mutation rate and soon die unless a useful mutation occurs.
Escherichia coli strain FC40 is unable to catabolize lactose (Lac−) but reverts to lactose utilization (Lac+) when lactose is its sole carbon and energy source. Although this reversion has been considered an example of adaptive mutation, during lactose selection nonselected mutations in a nearby gene accumulate in the Lac− population at about the same rate as do Lac+ mutations (4). Thus, the mutational process in FC40 does not meet the original definition of adaptive mutation, which specified that only adaptive mutations should appear. However, we continue here to call the Lac+ mutations appearing during lactose selection “adaptive” simply to distinguish them from the Lac+ mutations occurring during nonselective growth and from nonselected mutations occurring during lactose selection.
A specific prediction of the hypermutable-state model is that nonselected mutations occur at a higher frequency among cells that bear adaptive mutations than among cells that do not (3). In four cases in which this has been tested, the prediction has been confirmed (3–6). However, it is not possible to determine from those results what proportion of the mutations that occur during selection arise from hypermutating cells—that will depend on the proportion of cells that are in the hypermutable state and the degree to which their mutation rate is elevated (7, 8). Two independent measurements are needed to solve for these two unknowns.
In the experiments reported here, we used a broad mutational screen, loss of motility, to compare the frequency of nonselected mutations in starved Lac− cells, in selected Lac+ revertants, and in those few Lac+ revertants that carried an additional mutation. This procedure allowed us to solve the equations given in the Appendix and thereby to estimate both the size of the hypermutating subpopulation, p, and the magnitude of its increase in mutation rate, M. Our results show that, even though the hypermutating minority is responsible for nearly all of the multiple mutations, it makes only a small contribution to the adaptive Lac+ mutations occurring during lactose selection in E. coli strain FC40.
MATERIALS AND METHODS
Bacterial Strains.
E. coli strain FC722 is a rifampicin-resistant (RifR) derivative of P90C [F− ara Δ(lac proB)X111 thi (9)] that carries the F′128 episome with a mutant lac allele, Φ(lacI33-lacZ) (10), and a tetracycline-sensitive (TetS) defective Tn10 element (4). FC722 is isogenic to strain FC40 (11) except for the episomal TetS element. FC1259 is FC722 but mutL∷Kan [allele from strain GM4250 (12)] and was constructed by P1vir transduction. Strain FC691 is F− but carries the Φ(lacI33-lacZ) allele on its chromosome. FC691 is a derivative of GM4270 (obtained from M. G. Marinus, University of Massachusetts Medical School) created by curing its episome with acridine orange (see ref. 13). FC29 is rifampicin-sensitive (RifS) and carries a nonrevertible Lac− allele on its episome (11).
Media.
M9 minimal medium (14) was supplemented with 0.1% lactose (Lac), maltose (Mal) (both from Difco), glycerol (Gly), or d-galacturonic acid (GA) (both from Sigma). When required, antibiotics were added at 100 μg/ml rifampicin (Rif), 10 μg/ml tetracycline (Tet), 22.5 μg/ml kanamycin (Kan), or 10 μg/ml spectinomycin (Spc) (all from Sigma). Resistances to 5-fluorocytosine (5FC) and 5-fluorouracil (5FU) were screened on M9-Gly medium supplemented with 20 μg/ml 5FC or 10 μg/ml 5FU (both from Sigma). Mutators were screened on MacConkey Base Agar (Difco) supplemented with 1.0% arabinose (Mac-Ara) or salicin (Mac-Sal). Motility was screened on LB medium (14) solidified with 0.35% Bacto Agar (Difco).
Experimental Techniques.
Lac+ revertants were selected on M9-Lac plates as described (11). About 107 FC722 cells, 106 FC1259 cells (each with 109 FC29 scavengers), or 109 FC691 cells (without scavengers) were plated on each of 60, 48, and 80 M9-Lac plates, respectively. Each plate received cells from an independent culture grown to saturation in M9-Gly medium. Each day for 5 days after plating, new Lac+ colonies were counted, and their positions were marked. The plates were incubated at 37°C for an additional day to allow the colonies that appeared on day 5 to grow to the same size as earlier arising colonies. Plates were then stored at 4°C. Each colony was assigned an identification number specifying its position and day of appearance, and then some of its cells were transferred (patched) with a sterile wooden toothpick to a rectangular Nunc Omni tray containing 50 ml of solid M9-Lac + Rif medium for strains FC722 and FC1259 or M9-Lac medium for strain FC691. The cells were patched in a pattern corresponding to the array of a standard 96-well microplate. After transfer, the Omni trays were incubated at 37°C for 48 hours.
Starved Lac− cells were obtained by taking plugs from the lactose plates each day (11) and plating an appropriate dilution on M9-Gly plates (plus Rif for strains FC722 and FC1259), which were then incubated at 37°C for the duration of the experiment. These colonies were also assigned identification numbers and then patched to Omni trays containing M9-Gly + Rif medium for strains FC722 and FC1259 or M9-Gly medium for strain FC691.
Cells were transferred from the first Omni trays to screening media by using a 96-pin replicator (Boekel Scientific, Feasterville, PA). When transferring to motility agar, the cells were embedded in the agar by piercing the surface with the prongs of the replicator. Motility (Mot) was scored after 16–19 hr of incubation at 37°C. Depending on the area of cell-swimming from the center, the motility of each clone was scored as wild-type, increased, slightly reduced, reduced, very reduced, or minus. To confirm these phenotypes, roughly one-third of the clones were retested. The entire original colony was resuspended, dilutions were plated on minimal medium, and at least five single colonies for each original colony were patched to Omni trays containing LB + Rif or LB medium. These were replicated to LB motility agar and scored as above. The various Lac+ and motility phenotypes differed in their reproducibility, and the numbers reported below have been reduced by the appropriate correction factor (see Table and Fig. legends).
A possible motility gene has been found near 5.3 minutes of the E. coli chromosome (15) and may be carried on F′128. To test whether any Mot phenotypes were caused by mutations in this gene conferring a dominant-negative phenotype, the episomes from 83 Lac+ clones of FC722 that had motility defects were mated into a Mot+ Pro− Lac− StrepR recipient by selecting for Pro+ StrepR. All exconjugates were Lac+ Mot+.
Cells were also replicated to M9-Mal, M9-GA, M9-Gly + 5FC, M9-Gly + 5FU, and LB + Tet media. Because these selections picked up few potential mutants, we rechecked every one of them as described above. Mutants that were 5FCR or 5FUR were also tested for their ability to transfer their resistances by conjugation. Cells were screened for being heritable mutators on Mac-Ara, Mac-Sal, and LB + Spc media; these screens were confirmed to identify mutators by testing them with derivatives of FC40 carrying the known mutator alleles mutL and dnaQ49. Starved cells were confirmed to be Lac− by replicating them onto M9-Gly + X-Gal (5-bromo-4-chloro-3-indolyl β-d-galactoside), on which medium Lac+ cells are blue.
RESULTS
During lactose selection, Lac+ mutants accumulate at a nearly constant rate whether the Φ(lacI33-lacZ) allele is located on the episome (strain FC722) or on the chromosome (strain FC691) (Fig. 1). However, when the Lac− allele is on the chromosome, adaptive mutation to Lac+ has different genetic requirements and occurs at an >100-fold lower rate (ref. 13, and unpublished results; Fig. 1). In contrast, loss of methyl-directed mismatch repair (MMR) (16) increases the rate of adaptive mutation to Lac+ 100-fold (17). Strain FC1259 is isogenic to FC722 but defective in mutL, which encodes a component of the MMR system (18). We tested for nonselected mutations in Lac+ revertants and starved Lac− cells of each of these three strains.
Figure 1.

The accumulation during lactose selection of Lac+ revertants of the Φ(lacI33-lacZ) allele on the episome and on the chromosome. ■ (left axis), strain FC722 (episomal lac allele); ○ (right axis), strain FC691 (chromosomal lac allele). Values given are the means of 60 FC722 and 40 FC691 cultures. (The Lac+ counts from jackpots in two cultures of FC722 and five cultures of FC691 were eliminated from the results.) Error bars are SEMs, some of which are smaller than the symbols.
Mutations in as many as 55 genes, ≈1% of the E. coli genome, could affect motility (15, 19). Because motility is a novel mutational screen, we tested our assay for reproducibility (see Materials and Methods). Of the five mutant motility phenotypes identified (here collectively called Mot*), Mot− (no swimming) was the most reproducible, and so it is the only phenotype given in Tables 1 and 3 and Fig. 2. Eighty-five percent of the Lac+ clones grew vigorously on lactose; of these strongly Lac+ clones, 2% had an apparent Mot− phenotype in the initial screen, and 70% of these were confirmed to be composed entirely of motility-defective cells on retesting (see below for the characteristics of the remaining clones). If the cells were only weakly Lac+, this value dropped to 15%; poor growth on the lactose medium used for the initial gridding results in fewer cells being transferred to the motility-screening medium, giving an apparent motility deficiency.
Table 1.
The frequencies of Mot− clones
| Strain | mutL genotype | Location of the lac allele | Number of Mot− clones/total tested
|
Ratio of the frequency of Mot−Lac+ to Mot−Lac− | |
|---|---|---|---|---|---|
| Lac+ revertants (%) | Lac− cells (%) | ||||
| FC722 | + | Episome | 48/3,168 (1.5) | 2/2,878 (0.07) | 22 |
| FC691 | + | Chromosome | 12/583 (2.1) | 0/956 (<0.1) | >20 |
| FC1259 | − | Episome | 12/480 (2.5) | 12/437 (2.8) | 0.9 |
To correct for efficiency of detection, the numbers of Mot− clones were reduced by 30% in strongly Lac+ clones, by 85% in the few weakly Lac+ clones, and by 0% in Lac− clones (see Materials and Methods).
Table 3.
Calculation of the size of the hypermutating subpopulation and its increase in mutation rate
| Phenotype | Cells, no. | Cell-days | Mot−, no. | Mot− per cell, % | Mot− per cell-day, % |
|---|---|---|---|---|---|
| Lac− zeros | 2,878 | 6,990 | 2 | 0.07 | 0.029 |
| Lac+ singles | 3,168 | 7,006 | 48 | 1.5 | 0.68 |
| Lac+ doubles | 13 | 34 | 2 | 15 | 5.9 |
p and M calculations are as follows.
R1/0 = 0.68/0.029 = 24.
R2/0 = 5.9/0.029 = 203.
p = R1/0/(R2/0)2 = 24/4.2 × 104 = 5.7 × 10−4.
M = R2/0[1 − (pR2/0)] = 203/(1 − 0.12) = 231.
Proportion of single mutants that arise from the hypermutating subpopulation,
pM/[pM + (1 − p)] = 0.13/[0.130 + (1 − 5.7 × 10−4)] = 0.12.
Proportion of double mutants that arise from the hypermutating subpopulation,
pM2/[pM2 + (1 − p)] = 30/[30 + (1 − 5.7 × 10−4)] = 0.97.
Figure 2.

The accumulation of Mot− mutations among Lac+ and Lac− cells of FC722 during lactose selection. ■, Lac+ revertants; □, Lac− cells. Because it takes 2 days for a Lac+ colony to become visible, the curve for the Lac+ cells has been displaced 2 days to the left. To correct for efficiency of detection, the numbers of Mot− clones were reduced by 30% in strongly Lac+ clones, by 85% in the few weakly Lac+ clones, and by 0% in Lac− clones (see Materials and Methods).
Of the strongly Lac+ clones that had Mot* phenotypes (including Mot−), about 16% proved to be composed of mixtures of wild-type and Mot* cells. Note that pure Lac+ Mot* colonies would be produced only if the two mutations occurred simultaneously or if the Mot* mutation occurred first. Lac+ colonies with a large number of Mot* cells could indicate that the hypermutational state lasts for several cell divisions after a Lac+ revertant begins to grow. Alternatively, the Mot* mutation could have persisted as uncorrected heteroduplex DNA, which would produce a 50:50 mixture of Lac+ Mot* and Lac+ Mot+ cells. Although potentially interesting, mixed clones have been excluded from the results presented here. The reasons for nonreproducibility of the motility defects among the remaining strongly Lac+ clones are currently unknown but probably include broth-sensitive mutants, temporary metabolic problems caused by nutritional shift, irregularities in the screening plates, and gridding and picking errors.
The proportions of selected Lac+ and starved Lac− cells that had a Mot− phenotype are given in Table 1. There was little difference among the three strains in the proportion of Lac+ revertants that were Mot− (χ2 = 2.9, P = 0.24). However, whether the lac allele was on the episome (strain FC722) or the chromosome (strain FC691), the frequency of Mot− mutations among Lac+ revertants was significantly elevated relative to starved Lac− cells (χ2 = 37 and 17, respectively, P ≪ 0.01). Thus, in these MMR-competent cells, selection for Lac+ increased the frequency of nonselected Mot− mutations at least 20-fold. In contrast, loss of MMR (strain FC1259 = mutL, episomal lac allele) had little effect on the frequency of Mot− mutations among the Lac+ cells but increased the frequency of Mot− mutations among the Lac− cells 40-fold so that the frequency of Mot− mutations was the same in both populations (χ2 = 0.5, P = 0.82). This result suggests that MMR (or some other MutL-dependent pathway) is wholly or partially deficient in the hypermutators (see Discussion).
Fig. 2 shows the percentage of Lac+ and Lac− cells of FC722 (= MutL+, episomal lac allele) that were Mot− as a function of time after plating on lactose medium. Among the Lac+ cells, the frequency of mutations increased linearly. The number of Mot− mutants among the nonselected Lac− cells was too small to allow us to determine whether their frequency was changing with time.
To test whether any of the Lac+ or Lac− clones were stable mutators, cells were replicated onto Mac-Ara and Mac-Sal to score for increased papillation and onto LB-Spc to score for an increase in the number of resistant colonies. Among the 3,168 Lac+ revertants of FC722 (= MutL+, episomal lac allele), 11 (0.35%) were stable mutators. Four of these also had motility defects (two being Mot−), but none had any of the other screened phenotypes (see below). No stable mutators were found among starved Lac− FC722 cells, nor among Lac+ or Lac− cells of FC691 (= MutL+, chromosomal lac allele). The 0.35% genotypic mutators among a selected population is in good agreement with the 0.5% found in a previous study (20) of the enrichment of mutators achieved simply by selecting for new mutations.
In addition to motility, we screened Lac+ and starved Lac− cells for five other phenotypes (Table 2).¶ Three of these were also included in a previous study by Torkelson et al. (6). The 0.06% Mal− mutants and 0.25% 5FCR mutants among the Lac+ FC722 cells we obtained is in good agreement with their results (0.07% and 0.27%, respectively). The major difference between the two studies is that we found a 10-fold lower frequency of 5FUR mutants among Lac+ cells (0.03% versus 0.3%). A possible explanation is that the replica-plating method used by Torkelson et al. overestimated the number of 5FUR clones by allowing a density-dependent protection of some of the cells transferred onto the 5FU medium. Mutations in the episomal codBA operon give a 5FCR phenotype, whereas mutations in the chromosomal upp gene give both 5FUR and 5FCR phenotypes (21) (although our 5FUR mutants were only weakly 5FCR). Thus, our 8-fold difference in the frequencies of 5FCR and 5FUR mutants is consistent with the behavior of the Φ(lacI33-lacZ) allele itself, which reverts at a lower rate when on the chromosome (Fig. 1).
Table 2.
Additional phenotypes found among 3,168 Lac+ clones of FC722
| Phenotype | Number Found | Mot Phenotype of the Double Mutants
|
||
|---|---|---|---|---|
| Mot+ | Mot− | Mot* | ||
| TetR | 1 | 0 | 0 | 1 |
| Mal− | 2 | 0 | 1 | 2 |
| 5FCR | 8 | 3 | 1 | 5 |
| 5FUR | 1 | 1 | 0 | 0 |
| GA− | 1 | 1 | 0 | 0 |
| Total | 13 | 5 | 2 | 8 |
Mot* are cells with any motility phenotype, including Mot−.
Of 583 Lac+ revertants of FC691(= MutL+, chromosomal lac allele), none had any of the third phenotypes given in Table 2; however, lacking the dTn10 element, this strain could not have become TetR and was, for some unknown reason, weakly 5FCR. Of 480 Lac+ revertants of FC1259 (= mutL, episomal lac allele), one was Mal− and three were 5FCR. None of Lac− starved cells screened in any background had any of these third phenotypes.
Cairns (8) has modeled the contribution that a hypermutating minority would make to the frequency of selected mutations among the population under selection. In this simplest of possible models, the population is assumed to contain only two cell types: the majority plus a small proportion (p) with a mutation rate (for every class of mutation) M times higher than the majority. In this model, there are two unknowns (p and M), and they can be determined with the following two independent measurements (see Appendix): (i) R1/0, the frequency of Mot− mutations among the Lac+ revertants (singles) divided by the frequency of Mot− mutations among the Lac− cells (zeros); and (ii) R2/0, the frequency of Mot− mutations among Lac+ revertants that bear another detected mutation (doubles) again divided by the frequency of Mot− mutations among the Lac− cells (zeros). Our data in Tables 1 and 2 for FC722 (= MutL+, episomal lac allele) are sufficient to give values of R1/0 and R2/0 (see below).
The least reliable numbers entering into these calculations are the number of Mot− mutations in the Lac− zeros, 2/2,878, and in the Lac+ doubles, 2/13 (Table 2). Although each frequency is significantly different from 48/3,168, the frequency of Mot− mutations among Lac+ cells in general (χ2 = 36.7, P ≪ 0.001, and χ2 = 8.4, P = 0.004, respectively), 2 is a very small number. However, we can increase our confidence in the estimates of p and M by including the cells that had other Mot phenotypes. These phenotypes were not always reproducible, but extrapolating from the clones that were retested (about 30% of the total), the frequency of all classes of Mot mutations (Mot*) among the Lac+ cells (the singles) was 210/3,168 (6.6%). Because their numbers were far fewer, all of the apparent Mot* mutants found in the Lac− zeros and the Lac+ doubles were retested, and the actual frequency of Mot* mutations was 10/2,878 (0.35%) and 8/13 (61.5%) among the zeros and doubles, respectively. Each of these three frequencies is significantly different from the others (χ2 > 60, P ≪ 0.001 in each case).
The calculations of p and M based on the Mot− data for FC722 (= MutL+, episomal lac allele) are given in Table 3. Because the frequency of multiple mutations increases linearly with time (Fig. 2) but different numbers of cells were screened each day, we corrected for this by calculating the frequencies per cell-day (the number of cells screened multiplied by the number of days they were on the lactose plates before starting to form a colony). From these calculations, the proportion of hypermutators, p, is 6 × 10−4, and their mutation rate, M, is 230-fold higher than that of the main population. If the more frequent Mot* mutations are used, p = 8 × 10−4 and M = 190.
DISCUSSION
Recent experiments (4, 6) have shown that nonselected mutations in strain FC40 arise under adaptive-mutation conditions. Because nonselected mutations occur at a higher frequency among the selected Lac+ cells than among the nonselected Lac− cells, some or all of the mutations must be arising in a hypermutating subpopulation. To determine what proportion of cells are hypermutators and by how much their mutation rate is elevated, we measured the frequency of a common nonselected mutation, loss of motility (Mot−), in nonselected Lac− cells (zeros), in selected Lac+ cells (singles), and in selected Lac+ cells that also carried an additional nonselected mutation (doubles).
If all mutation were confined to a hypermutating minority, every Lac+ mutation would have arisen in a hypermutator. The frequency of Mot− mutations would then be much higher in Lac+ singles than in the population as a whole, but it would not be still higher in Lac+ doubles. Because we find that the frequency of Mot− mutations is far higher in doubles than in singles, it follows that most Lac+ revertants are arising in the general population rather than in the hypermutating minority.
The simplest model postulates that a certain proportion (p) of all cells are hypermutators, and in them the mutation rate is M-fold higher than in the rest of the population. Our experiments have given us two independent measurements: the factor R1/0, by which Mot mutations are more common in Lac+ singles than in the general Lac− population, and the factor R2/0, by which Mot mutations are more common in Lac+ doubles than in the general Lac− population. These two measurements allow us to calculate the values of the two independent parameters, p and M (see Appendix). This and other experiments then lead to certain conclusions about the hypermutating minority.
The Frequency and Mutation Rate of the Hypermutators.
Our data imply that ≈0.06% of all cells are hypermutators and that their mutation rate is ≈200-fold higher than that of the rest of the population. These estimates are not very precise because they depend on some rather small numbers (see Tables 1–3), but they are not significantly different from the values suggested by Ninio (7) on theoretical grounds.
We have assumed that there are only two populations of cells, high and low mutators. However, there may be many populations of different sizes and with different mutation rates, which together give our values for p and M. To determine whether another population exists, a third independent variable, e.g., R3/0, would have to be determined.
The Proportion of Mutants That Arise in Hypermutators.
If P = 0.06% and M = 200, ≈10% of all single mutations and >95% of all double mutations will occur in hypermutators (see Eqs. 2 and 4 in the Appendix). Our preliminary results indicate that the sequence changes that revert the episomal lac allele (strain FC722) do not differ between singles and doubles (unpublished results). This suggests, but does not prove, that the mechanism that generates Lac+ revertants of the episomal lac allele is the same in normal and hypermutating cells (see below).
The Source of the Hypermutating Minority.
Both Ninio (7) and Boe (22) suggested that hypermutators could be created as the result of occasional errors in translation or transcription leading to defects in the proteins used for DNA replication or repair. Deficiencies in MMR might be particularly likely because the levels of certain MMR proteins have been found to decline in stationary-phase cells (23, 24). Our data support the hypothesis that hypermutators are MMR-deficient. We find that loss of MutL does not have a great effect on the frequency of Mot− mutations in Lac+ singles but raises the frequency of Mot− mutations in Lac− zeros to the level found in singles (Table 1). Thus, loss of MutL has a far smaller effect on the hypermutators than on the population at large, implying that hypermutators owe their high mutation rate to a complete or partial deficiency in MMR (or some other repair pathway that requires MutL). Although the majority of Lac+ adaptive mutations arise in cells that are not hypermutators, the spectra of mutations that revert the episomal lac allele do not appear to differ between the two populations (as mentioned above). Therefore, the simplest hypothesis is that the mechanism that generates Lac+ mutations on the episome is the same in all the cells in the population but that a deficiency in MMR allows more of these to be retained in the hypermutators. Other, nonselected mutations on the episome and elsewhere would also necessarily be retained, although these may occur by different mechanisms.
The Kinetics of Mutation in Hypermutators.
Hall (3) was the first to propose that mutation in populations under selective pressure depends on a hypermutating minority. He suggested that the mutations appear to be specific to the selection because every hypermutator rapidly dies unless it produces a mutation that allows growth to resume. This hypothesis predicts that, although nonselected mutations may occasionally be picked up as fortuitous hitchhikers, their frequency in the population of selected mutants should not increase with time. In fact, we find that the frequency of Mot− mutations among selected Lac+ revertants increases linearly with time (Fig. 2). The simplest interpretation is that the hypermutators are not continually turning over to a significant degree, at least during the course of our experiments. Nevertheless, few cells would be expected to survive for long with a mutation rate so high that 2 of every 13 of them suffered a null mutation in a group of genes comprising only 1% of the genome (Table 2). Thus, it will be interesting to determine what allows the hypermutating minority to survive its ever-increasing number of mutations.
The Mutational Targets for the Hypermutating Minority.
The hypermutating minority can be identified whether the allele undergoing selection, Φ(lacI33-lacZ), is on the episome or on the chromosome (Table 1), although the frequency and genetic requirements for adaptive Lac+ mutation are different in the two cases. Thus, the pathways for the two classes of mutation can each be divided into two components, one that partially overlaps (so that hypermutators are hypermutators for both) and one that is specific to the position of the lac allele (because different gene products are involved). This is consistent with the hypothesis that all mutations, no matter how generated, are more likely to be retained in hypermutators. Our data also indicate that a conjugal plasmid is not required to induce the hypermutable state.
The Frequency of Transient and of Genetically Stable Hypermutators.
Continuous selection for phenotypes that increase competitive fitness (25, 26) or repeated selection for a succession of novel phenotypes (20) selects for cells that are stable mutators. But in the real world, cells may more often be confronted with some particular barrier to growth rather than a succession of hurdles. To meet these complex challenges, which may require several mutations, transient mutators may be more important than genetically stable mutators. (Indeed, of the population enriched for hypermutators, the Lac+ Mot− doubles, we found only 2% to be genetically stable.) In addition, cells that succeed because of a transient increase in mutation rate would not continue to be burdened with a potentially deleterious mutation rate.
Implications for the Mechanism of Adaptive Mutation.
The mutations that revert the episomal lac allele during lactose selection are caused by DNA polymerase errors (27). Currently, there are two theories for the origin of this de novo DNA synthesis—amplification of the Lac− allele (17, 28) and DNA replication initiated during recombinational repair of double-strand breaks (18, 29). The results presented here do not distinguish between these two hypotheses, but do limit them in two ways. First, as previously reported (11, 30), the rate at which Lac+ mutations appear during lactose selection is constant with time for our E. coli strains. Here we show this is true both for the episomal and the chromosomal lac allele (Fig. 1) and is also true for the accumulation of Mot− mutations in the Lac+ population (Fig. 2). Second, no fewer than 5% of the cells must be mutating. This is because the proportion of cells that are mutating cannot be smaller than 1/R1/0 and R1/0 is about 20 (Table 3). The simplest hypothesis consistent with these results is that during lactose selection all cells are capable of producing mutations, and that, for the majority of cells, their probability of doing so is constant with time.
Acknowledgments
We thank J. Cairns for invaluable aid in analyzing our data, preparing this paper, and for the mathematical analysis given in the Appendix. We also thank M. Marinus and J. Miller for strains and helpful discussions. This work was supported by National Science Foundation Grant MCB 97838315.
ABBREVIATIONS
- Lac
lactose
- Rif
rifampicin
- Tet
tetracycline
- Mal
maltose
- Gly
glycerol
- 5FC
5-fluorocytosine
- 5FU
5-fluorouracil
- MMR
mismatch repair
- Mac
MacConkey agar
Appendix
If a small proportion of all cells are high mutators and have a mutation rate that is significantly greater than that of the rest of the population, any given class of mutation will be more frequent in cells that are mutant for some other trait (singles) than in the population as a whole (zeros). R1/0, the frequency of mutation B in A-mutants divided by the frequency of mutation B in all cells, will be simply a function of the proportion (p) of cells that are high mutators and the factor, M, by which their mutation rate is raised.
The relation between R1/0, p, and M has been shown elsewhere (8) to be
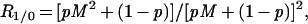 |
1 |
and the proportion of any class of mutant cells that arise in high mutators is
 |
2 |
R2/0, the frequency of mutation C in double-mutant AB cells (doubles) divided by the frequency of mutation C in all cells (zeros), is similarly simply a function of p and M and can be shown to be
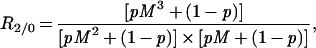 |
3 |
and the proportion of any class of doubly mutant cells that arise in high mutators is
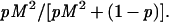 |
4 |
R1/0 and R2/0 can be shown to reach their maximum values when p is 1/(M + 1) and 1/(M3/2 + 1), respectively.
p and M are the two unknowns, and it is possible to calculate their values by measuring R1/0 and R2/0. If p ≪ 1, then R1/0 ≈ (pM2 + 1)/(pM + 1)2, and if pM2 ≫ 1, then R2/0 ≈ M/(pM + 1).
It follows that
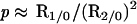 |
5 |
and
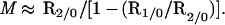 |
6 |
In other words, by measuring the frequency of some class of mutation in the population as a whole, in cells mutant for one other trait, and in cells mutant for two other traits (i.e., measure R1/0 and R2/0), it should be possible to determine p and M. Once p and M are known, it becomes possible to determine the magnitude of the contribution of high mutators to single-mutant and double-mutant cells.
The above analysis is based on the assumption that there are only two classes of cell, the high-mutators and the low-mutators and therefore only two independent variables (p and M). To establish that there are indeed only two classes, it would be necessary to measure the frequency of fourth mutations in cells bearing three mutations (i.e., to determine R3/0). Until that has been done, the possibility remains that the population contains many classes of cell and many values for both p and M.
Footnotes
One Lac+ colony of FC722 was composed entirely of TetR cells, which is a lower frequency of Lac+ TetR double mutants than previously reported for this strain (4). However, we found five apparent Lac+ TetR double mutants that, when retested, segregated the two phenotypes. Four of these were stable mutators. If these five are included, the number of apparent Lac+ TetR doubles is not significantly different between the two studies (χ2 = 2.13, P = 0.14), suggesting that many of the Lac+ TetR double mutants obtained in the previous study were due to stable mutators.
References
- 1.Cairns J, Overbaugh J, Miller S. Nature (London) 1988;335:142–145. doi: 10.1038/335142a0. [DOI] [PubMed] [Google Scholar]
- 2.Foster P L. Annu Rev Microbiol. 1993;47:467–504. doi: 10.1146/annurev.mi.47.100193.002343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hall B G. Genetics. 1990;126:5–16. doi: 10.1093/genetics/126.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foster P L. J Bacteriol. 1997;179:1550–1554. doi: 10.1128/jb.179.5.1550-1554.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boe L. Mol Microbiol. 1990;4:597–601. doi: 10.1111/j.1365-2958.1990.tb00628.x. [DOI] [PubMed] [Google Scholar]
- 6.Torkelson J, Harris R S, Lombardo M-J, Nagendran J, Thulin C, Rosenberg S M. EMBO J. 1997;16:3303–3311. doi: 10.1093/emboj/16.11.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ninio J. Genetics. 1991;129:957–962. doi: 10.1093/genetics/129.3.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cairns J. Genetics. 1998;148:1433–1440. doi: 10.1093/genetics/148.4.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coulondre C, Miller J H. J Mol Biol. 1977;117:525–575. doi: 10.1016/0022-2836(77)90056-0. [DOI] [PubMed] [Google Scholar]
- 10.Calos M P, Miller J H. J Mol Biol. 1981;153:39–66. doi: 10.1016/0022-2836(81)90525-8. [DOI] [PubMed] [Google Scholar]
- 11.Cairns J, Foster P L. Genetics. 1991;128:695–701. doi: 10.1093/genetics/128.4.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aronshtam A, Marinus M G. Nucleic Acids Res. 1996;24:2498–2504. doi: 10.1093/nar/24.13.2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Foster P L, Trimarchi J M. Proc Natl Acad Sci USA. 1995;92:5487–5490. doi: 10.1073/pnas.92.12.5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller J H. Experiments in Molecular Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1972. [Google Scholar]
- 15.Blattner F R, Plunkett G, III, Bloch C A, Perna N T, Burland V, Riley M, Collado-Vides J, Glasner J D, Rode C K, Mayhew G F, et al. Science. 1997;277:1453–1479. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 16.Modrich P, Lahue R. Annu Rev Biochem. 1996;65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- 17.Foster P L, Cairns J. Genetics. 1992;131:783–789. doi: 10.1093/genetics/131.4.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Foster P L, Trimarchi J M, Maurer R A. Genetics. 1996;142:25–37. doi: 10.1093/genetics/142.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berlyn M K B, Low K B, Rudd K E. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. 2nd Ed. Neidhardt F C, Curtiss R III, Ingraham J L, Lin E C C, Low K B, Magasanik B, Reznikoff W S, Riley M, Schaechter M, Umbarger H E, editors. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 1715–1902. [Google Scholar]
- 20.Mao E F, Lane L, Lee J, Miller J H. J Bacteriol. 1997;179:417–422. doi: 10.1128/jb.179.2.417-422.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neuhard J, Kellin R A. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. 2nd Ed. Neidhardt F C, Curtiss R III, Ingraham J L, Lin E C C, Low K B, Magasanik B, Reznikoff W S, Riley M, Schaechter M, Umbarger H E, editors. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 580–599. [Google Scholar]
- 22.Boe L. Mol Gen Genet. 1992;231:469–471. doi: 10.1007/BF00292717. [DOI] [PubMed] [Google Scholar]
- 23.Feng G, Tsui H-C T, Winkler M E. J Bacteriol. 1996;178:2388–2396. doi: 10.1128/jb.178.8.2388-2396.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harris R S, Feng G, Ross K J, Sidhu R, Thulin C, Longerich S, Szigety S K, Winkler M E, Rosenberg S M. Genes Dev. 1997;11:2426–2437. doi: 10.1101/gad.11.18.2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cox E C, Gibson T C. Genetics. 1974;77:169–184. doi: 10.1093/genetics/77.2.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sniegowski P D, Gerrish P J, Lenski R E. Nature (London) 1997;387:703–705. doi: 10.1038/42701. [DOI] [PubMed] [Google Scholar]
- 27.Foster P L, Gudmundsson G, Trimarchi J M, Cai H, Goodman M F. Proc Natl Acad Sci USA. 1995;92:7951–7955. doi: 10.1073/pnas.92.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andersson D I, Slechta E S, Roth J R. Science. 1998;282:1133–1135. doi: 10.1126/science.282.5391.1133. [DOI] [PubMed] [Google Scholar]
- 29.Harris R S, Ross K J, Rosenberg S M. Genetics. 1996;142:681–691. doi: 10.1093/genetics/142.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Foster P L. Genetics. 1994;138:253–261. doi: 10.1093/genetics/138.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]