

Abstract
The hemolytic uremic syndrome (HUS) is a triad of microangiopathic hemolytic anemia, thrombocytopenia, and renal impairment. Genetic studies demonstrate that heterozygous mutations of membrane cofactor protein (MCP;CD46) predispose to atypical HUS (aHUS), which is not associated with exposure to Shiga toxin (Stx). Among the initial 25 MCP mutations in patients with aHUS were 2, R69W and A304V, that were expressed normally and for which no dysfunction was found. The R69W mutation is in complement control protein module 2, while A304V is in the hydrophobic transmembrane domain. In addition to 3 patients with aHUS, the A304V mutation was identified in 1 patient each with fatal Stx-HUS, the HELLP (hemolysis, elevated liver enzymes, and low platelets) syndrome, and glomerulonephritis with C3 deposits. A major goal was to assess if these putative mutations lead to defective complement regulation. Permanent cell lines expressing the mutated proteins were complement “challenged,” and membrane control of C3 fragment deposition was monitored. Both the R69W and A304V MCP mutations were deficient in their ability to control the alternative pathway of complement activation on a cell surface, illustrating the importance of modeling transmembrane proteins in situ.
Introduction
The hemolytic uremic syndrome (HUS) is a triad of microangiopathic hemolytic anemia, thrombocytopenia and renal impairment. Atypical HUS (aHUS) accounts for about 5% to 10% of all cases and has a poor prognosis compared with HUS associated with Shiga toxin (Stx) producing Escherichia coli.1 Genetic studies indicate that heterozygous mutations of complement factor H (CFH),2–6 membrane cofactor protein (MCP;CD46),7–11 and factor I (CFI)9,10,12,13 predispose to aHUS.
MCP mutations are present in approximately 15% of patients with aHUS.14 It is a widely expressed type I transmembrane glycoprotein that binds to C3b and C4b deposited on the cell surface on which it is expressed.15,16 MCP then serves as a cofactor for factor I (FI), a serine protease in plasma, to inactivate C3b and C4b by limited cleavage.17 The fragments remaining attached to the target membrane, iC3b and C4d, do not participate in C3 and C5 convertase formation. Through this process, known as cofactor activity, MCP is particularly important in controlling the amplification loop of the alternative pathway (AP).18–22
The initial 25 mutations in MCP identified in aHUS have been reviewed.14 About 50% of these mutations lead to proteins that are not expressed, and thus the individual is haploinsufficient. In the other major group, there is normal surface expression, but the protein has a functional defect. We previously reported 2 MCP mutations in patients with aHUS, R69W and A304V, in which no apparent dysfunction was found in binding and cofactor studies.10,14 The R69W mutation is in complement control protein 2 (CCP 2), and A304V is in the transmembrane domain. The A304V mutation has also been described in a form of glomerulonephritis featuring almost exclusively deposits of C3 fragments.23 In this report, we identified the A304V mutation in 2 other syndromes, fatal Stx-HUS and the HELLP (hemolysis, elevated liver enzymes, and low platelets) syndrome.
To further evaluate the function of R69W and A304V on the cell surface, permanent cell lines expressing the mutated proteins were established. Ligand binding and fluid phase cofactor activity assays over a range of salt concentrations of solubilized cell extracts demonstrated no defect. The cell lines were also challenged using a complement-activating protocol in which the membrane control by MCP of this process could be quantitatively assessed. In situ, both proteins were deficient in their ability to control the AP of complement activation.
Methods
Approval for these studies was obtained from the Human Research Protection Office (HRPO) of Washington University in St Louis, MO; the Departement de la Recherche Clinique et du Developpement, Delegation Regionale a la Recherche Clinique Ile de France, France; the Bioethics Committee of province of Bergamo Azienda Sanitaria Locale della Provincia di Bergamo, Italy; and the Northern and Yorkshire Multi-Centre Research Ethics Committee, United Kingdom. Informed consent was obtained in accordance with the Declaration of Helsinki.
MCP mutations
Previously reported patients with R69W and A304V MCP mutations are noted (Tables 1,2). The unreported descriptions of the patients' clinical courses have been gathered through personal communication as indicated. The mutants are numbered beginning with the first amino acid of the mature protein. R69W was originally reported as R103T,9 but was subsequently corrected to R103W.24 Their numbering system includes the 34-amino acid leader sequence.
Table 1.
Summary of patients with R69W MCP mutations
| Patient | Disease | Sex | Age of onset, y | Relapses, no. | Renal sequelae, no. | Inheritance | Complement protein tested† | Other mutations | Source |
|---|---|---|---|---|---|---|---|---|---|
| 1 | aHUS | M | NA | NA | NA | Het | NA | Het N151S CFI, hom MCPggaac risk haplotype | Esparza-Gordillo et al,9 Rodríguez de Córdoba, written communication, December 2006 |
| 2 | aHUS | F | NA | NA | NA | Het | NA | Het c.905–925del21n MCP, het N151S CFI, het MCPggaac risk haplotype | Rodríguez de Córdoba, written communication, December 2006 |
| 3 | aHUS | M | Childhood | 0 | None | Hom | C3, FB, FH, FI, MCP | None | V.F.B., December 2006 |
| 4 | aHUS | F | 2 | 2 | None | Het | C3, FB, FH, FI, MCP | Het P32A CFI | V.F.B., December 2006 |
NA indicates not available; Het, heterozygous; and Hom, homozygous.
*Patients 1 and 2 and patients 3 and 4 are father and daughter, respectively.
Components listed were measured and were normal; MCP was measured by FACS.
Table 2.
Summary of patients with A304V MCP mutations
| Patient | Disease | Sex | Age of onset, y | Relapses, no. | Renal sequelae | Inheritance | Complement protein tested* | Other mutations | Source |
|---|---|---|---|---|---|---|---|---|---|
| 5 | aHUS | F | 6 | 4 | proteinuria | Het | FH, FI, CR1 | None | Caprioli et al10 |
| 6 | aHUS | M | 0.9 | Multiple | CRF | Het | C3, C4, CH50, FH, FI | None | C. Belsha, written communication, February 2007 |
| 7 | aHUS/TTP | M | 63 | 2 | None | NA | C3, C4, FH, FI | P553S CFI | T.H.J.G., March 2007 |
| 8 | Stx-HUS | F | 4 | NA | NA | Het | FH, FI, CR1 | None | C.J.F., J.P.A., April 2007 M.N., October 2006 |
| 9 | HELLP | F | 30 | NA | CRF | NA | C3, C4, FB, FH, FI, MCP | None | V.F.B., December 2006 |
| 10 | GN with C3 deposits | M | 22 | NA | proteinuria, hematuria | Het | C3, C4, FB, FH, FI, MCP | Het V181M MCP† | Servais et al23 |
NA indicates not available; Het, heterozygous; GN, glomerulonephritis; and CRF, chronic renal failure.
Components listed were measured and were normal; MCP and CR1 were measured by FACS.
V181M MCP mutant is normally expressed and has no abnormalities in C3b and C4b binding and cofactor activity (unpublished data).
Mutagenesis, expression, and functional assessment
For mutagenesis and expression experiments, MCP isoform BC1 was used as a template, and stable transfections were performed in Chinese hamster ovary (CHO) K1 cells, as previously reported.7,11 CHO cells transfected with MCP cDNA in a reverse orientation served as a control (CHO). MCP expression was evaluated by enzyme-linked immunosorbent assay (ELISA), Western blotting, and fluorescence-activated cell sorter (FACS).25 Ligand binding, ELISA, and cofactor assays were performed on cell lysates as described previously.25,26 In short, cells were lysed in phosphate-buffered saline (PBS) containing 1% Nonidet P-40 (NP-40), 0.05% SDS, and 2 mM phenylmethylsulfonyl fluoride. MCP has proven remarkably stable using 1% NP-40 while providing high yields15,17 (unpublished data, J.P.A., January 1985). Purified C3b and C4b (both from Complement Technology, Tyler, TX) were coated on microtiter wells, and binding of MCP in the cell lysates to these substrates were evaluated. MCP was detected with rabbit anti-MCP antiserum (gift of Millennium Pharmaceuticals, Cambridge, MA). For analysis of cofactor activity, biotinylated ligands were treated with samples that contained MCP and purified FI (Quidel, San Diego, CA) over a range of ionic strengths.25 The generation of cleavage fragments was monitored by SDS-PAGE and then quantitated with a laser densitometer using ChemiImager software (Alpha Innotech, San Leandro, CA).
Protein purification by affinity chromatography
A monoclonal Ab to MCP, TRA-2–10, was coupled to CNBr-activated Sepharose 4B (GE Healthcare, Chalfont St Giles, United Kingdom). Wild-type and mutant cell lysates (400 μL each) were incubated on a rotator with 100 μL TRA-2–10–Sepharose for 30 minutes at 4°C. More than 90% of MCP in the lysates bound to TRA-2–10–Sepharose. The beads were washed twice with PBS (centrifuged at 420g for 3 minutes). MCP was eluted with 50 mM ethylamine (pH 11.5). The eluates were neutralized with 75 μL/mL of 250 mM sodium acetate buffer (pH 4.6) before being quantitated and functionally assessed.
Challenge assays
As previously described,21,22 cell lines stably expressing mutant and wild-type MCP were sensitized with anti-CHO antibody (purified IgG) that was prepared by a commercial vendor (Harlan, Indianapolis, IN) in rabbits injected with CHO cells. After washing, cells were exposed to a source of complement (usually human C7–deficient [C7d] serum) for the indicated time points. To assess the magnitude of activation, mAbs to complement fragments deposited after “challenge” were assessed by FACS.
Initiation of complement activation
Standard procedure for these complement challenge assays has been described.21,22,27 Briefly, CHO cells were grown to approximately 80% confluency and trypsinized (0.5 g trypsin and 0.2 g EDTA), washed in PBS, and resuspended in 1% heat-inactivated fetal calf serum (FCS) in PBS (FCS-PBS) at 4°C. Rabbit anti-CHO IgG was added to the cells with mixing for 30 minutes at 4°C. After 2 washes with 1% FCS-PBS, C7d serum (donated by P. Densen, University of Iowa, Iowa City) diluted in gelatin veronal–buffered saline was added, and the mixture was incubated at 37°C for up to 60 minutes. The C7d serum serves as a source of complement components, but cell lysis is avoided. Untreated, Ab-sensitized and serum-treated CHO cells served as controls.
To block the CP and allow for AP activation, gelatin veronal–buffered saline containing 10 mM EGTA and 7 mM magnesium chloride (Mg2+-EGTA) was used. EGTA preferentially chelates calcium, which is required for C1 function and thereby abrogates the classical pathway. Cells were harvested at indicated time points, and C4 and C3 fragment deposition was monitored by FACS.
FACS analysis of complement fragment deposition
As previously reported,21 murine mAbs to the human complement component fragments C4c, C4d, and C3d (Quidel) were added for 30 minutes at 4°C to the cell lines used in the challenge experiments. Next, the cells were washed twice with 1% FCS-PBS followed by incubation with FITC-conjugated goat anti-mouse IgG that had been preadsorbed with rabbit serum-agarose and rabbit IgG-agarose (both from Sigma-Aldrich, St Louis, MO) to remove cross-reactive material. Finally, cells were washed with 1% FCS-PBS, fixed in 0.5% paraformaldehyde, and analyzed using a BD Biosciences FACSCalibur system (Franklin Lakes, NJ).
Case reports
Selected parameters of these patients are presented in Tables 1 and 2.
R69W
Patients 1 and 2: R69W mutation in a family with MCP and CFI mutations and the MCPggaac risk haplotype.
Patient 1, previously reported,9 is heterozygous for the R69W MCP mutation and N151S CFI mutation. He is also homozygous for the MCPggaac haplotype which increases the risk of developing aHUS approximately 2-fold. Patient 2, the daughter of patient 1, also carries the CFI mutation and the MCPggaac risk haplotype. In addition, she carries a second MCP mutation, c. 905-925del21n, which causes a 7–amino acid deletion in CCP 4 on a separate allele inherited from her mother. She has approximately 50% of the normal expression level of MCP. The healthy mother of patient 2 carries the c905-925del21n MCP mutation, and she also has approximately 50% of the normal expression level of MCP. Since the R69W mutant protein is normally expressed, the reduced expression in the patient is secondary to the allele carrying the deletion.
Patients 3 and 4: R69W mutation in a family with MCP and CFI mutations.
In this French family with R69W MCP and P32A CFI mutations, patient 3 is homozygous for the R69W mutation and has had 1 episode of aHUS during childhood. Patient 4, the daughter of patient 3, is heterozygous for both the MCP and CFI mutation. This 8-year-old girl developed aHUS at age 2 years, with recurrences at ages 5 and 7 years. After each episode, she has had complete recovery of renal function.
A304V
Patient 5: A304V MCP mutation in a patient with aHUS.
This 13-year-old girl, beginning at age 6 years, has had 5 episodes of aHUS that appear to be triggered by upper airway infections.10 Each resolved without plasma exchange. Renal function is normal, but she has persistent proteinuria.
Patient 6: A304V MCP mutations in a family with aHUS.
This 7-year-old boy developed aHUS at age 11 months of age that was managed with plasma treatment. Multiple recurrences were similarly treated but the patient progressed to ESRD by age 7 years. No mutations were identified in CFH or in CFI, but a heterozygous A304V mutation was detected in MCP. His healthy father shares this abnormality.
Patient 7: A304V MCP mutation in a patient with aHUS/TTP.
A 63-year-old man presented with the acute onset of dysphagia, confusion, and tunnel vision. At 2 weeks prior, he had paresthesias in both arms. On admission, he was anemic (hemoglobin, 97 g/L), thrombocytopenic (platelet count, 29 × 109/L) and in acute renal failure (creatinine, 167.96 μM). Lactate dehydrogenase was increased to 1084 U/L (< 460 U/L), reticulocyte count was 8.9%, and haptoglobin was less than 0.4 g/L (.1-.3 g/L). Peripheral blood smear showed fragmented erythrocytes. A diagnosis of aHUS or thrombotic thrombocytopenic purpura (TTP) was suspected, and he was treated with fresh frozen plasma (FFP) for 21 days, and dexamethasone, azathioprine, and 1 dose of rituximab. He relapsed 4 days after stopping FFP, and had plasma exchange daily for another 8 days. At 8 days later, he again relapsed and was treated with FFP 5 U daily for 5 more days. The abnormal hematologic parameters resolved. The patient was treated with azathioprine for 6 months. He currently has normal renal function. This patient also carries a P553S mutation in CFI. This patient could be classified as having TTP. Antibodies to ADAMST13 were not measured, and the presence of mutations in MCP and CFI point more toward aHUS.
Patient 8: A304V MCP mutation in a fatal case of Stx-HUS.
A 4-year-old previously healthy girl was admitted to the hospital because of hemorrhagic colitis, seizures, and anuria. One week earlier, the brother had had nonbloody diarrhea without fever. There was no family history of HUS, and the patient's parents were unrelated. On admission, she was anemic (hemoglobin, 50 g/L [5 g/dL]; hematocrit, 14.8%), thrombocytopenic (platelet count, 52 × 109/L), and in acute renal failure (creatinine, 424.32 μM [4.8 mg/dL]). Serum lactate dehydrogenase was increased at 4198 U/L (< 460 U/L). A diagnosis of HUS was suspected, and hemodialysis was initiated. She also received multiple blood and plasma exchanges and was treated with diuretics and antiepileptics. The patient's condition rapidly deteriorated. She became comatose and developed respiratory insufficiency, requiring mechanical ventilation. Anasarca, bilateral pleural effusions, and ascites necessitated pulmonary and abdominal drainage procedures and hemodialysis. The patient died 4 days after admission with a multiorgan failure syndrome. At autopsy, she had a thrombotic microangiopathy with ischemic-hemorrhagic necrosis of the kidneys, intestine, lungs, and myocardium. There was an acute enteritis and cerebral edema with prominent perivascular microhemorrhages. In the patient's stool specimen, a Stx-producing E coli (STEC) strain of serotype O55 was isolated. The patient and her parents were screened for CFH, MCP, and CFI gene mutations. No mutations were identified in CFH or in CFI, but a heterozygous A304V mutation was detected in MCP. The father is a carrier but has not had aHUS. Serum complement profile demonstrated a low C3 of .65 g/L (.66-1.25 g/L) and a borderline low C4 of .14 g/L (.093-.38 g/L).
Patient 9: A304V MCP mutation in a patient with chronic renal failure following HELLP.
A 30-year-old female had HELLP syndrome during a second pregnancy after an uncomplicated first pregnancy. She developed chronic renal failure 1 year later.
Patient 10: A304V MCP mutation in a patient with glomerulonephritis featuring isolated C3 deposits.
This individual (patient 7 in Servais et al23) is a 22-year-old male with hypertension, hematuria, and mild proteinuria (300-400 mg of total protein/24 hours). A kidney biopsy showed glomerulonephritis with isolated C3 deposits but not HUS. He is also heterozygous for V181M mutation in MCP.
Results
A major goal of this investigation was to assess the complement regulatory capability of the R69W and A304V mutations in MCP. R69W is in CCP 2 and is solvent exposed (Figure 1A,B). CCPs 2, 3, and 4 are required for C3b and C4b binding and cofactor activity. A304V is found in the transmembrane domain (Figure 1A-C). We had previously shown, using a transient transfection system, that both mutants were expressed normally and migrated identically to wild type on SDS-PAGE.10,14 We also demonstrated, using our standard screening low-ionic-strength conditions, that R69W and A304V bound C3b and C4b in an ELISA-based system comparable with wild type.10,14 In addition, in fluid phase experiments, the cofactor activity of both R69W and A304V was similar to that of wild type.14
Figure 1.

MCP structure. (A) Schematic diagram of protein structure. R69W mutation is in CCP 2. The A304V mutation is in the transmembrane domain. (B) Crystal structure of CCP 2 highlighting the location of R69W. (C) Helical wheel diagram of predicted segments and transmembrane representation by SOSUI (http://bp.nuap.nagoya-u.ac.jp/sosui) of A304V MCP mutation versus that of wild type. Yellow arrows indicate mutation of MCP at position 304 from alanine (left) to valine (right).
Preparation of stable CHO cell lines
Stable CHO lines were prepared expressing wild type and the 2 mutant proteins at approximately 100 000 copies/cell, as determined by ELISA. R69W and A304V mutants were also shown to be expressed in equal copy number by FACS and to migrate in SDS-PAGE comparable with wild type (Figure 2A,B). These FACS and Western blot results are consistent with those using a transient transfection system.10,14
Figure 2.

Expression profile and electrophoretic mobility of R69W and A304V mutants. (A) Flow cytometry of MCP expression on CHO cell lines. Stable transfectants R69W and A304V express similar copy number of MCP/cell (approximately 100 000) compared with wild type. (B) Western blot of these same CHO cell lysates probed with a rabbit polyclonal Ab to MCP. R69W and A304V have similar Mr to wild type. Representative experiment of 3.
C3b and C4b binding
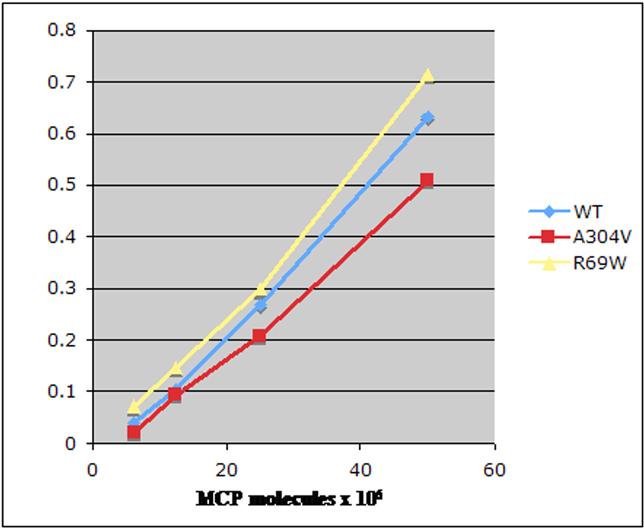
Transient and permanent CHO cell lines were lysed, and the quantity of MCP expressed was quantitated. Next, the activity of the mutant MCPs was compared with wild type in ELISA-based C3b- and C4b-binding assays over a range of ionic strengths (25, 50, and 100 mM NaCl). At 100 mM or higher concentrations of NaCl, neither the mutants nor wild-type MCP bound sufficient C3b or C4b for a reliable comparison (data not shown). The R69W mutant protein had similar C3b- and C4b-binding activity to wild type at 25 mM (Figure S1, available on the Blood website; see the Supplemental Materials link at the top of the online article) and 50 mM (Figure 3) salt concentration. Although in prior published studies, there was a modest reduction in A304V mutant binding (84% ± 6% for C3b and 82% ± 6% for C4b, with wild type being set at 100%),10 repeat studies at 25 mM (Figure S1) and 50 mM (Figure 3) salt concentration showed similar binding of A304V compared with wild type for C3b and C4b. Studies using wild-type and mutant R69W and A304V proteins obtained following purification also showed no detectable differences in ligand-binding activity (Figure S2).
Figure 3.

Analysis of MCP binding to C3b and C4b. C3b and C4b binding assays were performed at 25 mM (Figure S1) and 50 mM (shown here) salt concentration. CHO cell lysates (5 × 106 to 25 × 106 MCP molecules as measured by ELISA) of R69W, A304V, and wild type from both transient transfections and stable cell lines were used. Data represent means (± SD) for 4 experiments at 50 mM.
C3b and C4b cofactor activity
Cofactor activity for C3b and C4b in fluid phase assays was tested at increasing ionic strengths in kinetic assays. At 5-, 10-, 15-, 30-, 45-, and 90-minute time points for each of 3 salt concentrations (25, 50, and 100 mM), no reproducible difference was demonstrable in C3b or C4b cofactor activity between the 2 mutants and wild type. For example, for the C3b cofactor assay at 90 minutes, the loss of α′ chain was evaluated relative to the β chain, and activity was designated as a percentage of wild type. Both mutants had similar cofactor to wild type (98% ± 7.5% and 108% ± 10.9%, respectively). The electrophoretic mobility of bands produced by the mutants and wild type was also the same. In some cases, as the representative results at 50 mM NaCl for 90 minutes show in Figure 4, 1 of the mutants appears to be more efficient, but this is not reproducibly observed in kinetic analyses.
Figure 4.

Analysis of C3b and C4b cofactor activity. Lysates of CHO cells expressing R69W, A304V, or wild-type MCP were incubated at 50 mM NaCl for 90 minutes with C3b or C4b and factor I. Cleavage of the α′ chain and generation of the α1 and α2 by the mutants are comparable with that of wild type. For the Western blot C3b cofactor activity (A), α′ and β represent the 2 chains of C3b. α1 corresponds to the larger proteolytic product of the α′ chain of C3b. The bottom portion of panel A is a 3-fold longer exposure of the same gel to show the smaller α2. SDS-PAGE, 10% gel, reducing conditions. Representative experiment of 3. For the Western blot of C4b cofactor activity (B), cleavage fragments C4d and α4 are similar between the mutants and wild type. The α3 is poorly biotinylated and not seen. The light band at approximately 60 kDa is most likely C4d (45 kDa) + α4 (15 kDa) and again is similar for the mutants in comparison with wild type. SDS-PAGE, 4% to 10% gel, reducing conditions. Representative experiment of 3.
Complement challenge assays
To further assess the role of the 2 mutants versus wild-type MCP, AP of complement was activated on CHO cell lines. The quantity of sensitizing antibody and the serum concentration were varied. To monitor the magnitude of complement activation, the deposited C3b and its cleavage fragments were assessed by FACS. In the absence of a sensitizing antibody, there is no spontaneous activation of AP, and thus no C3d is deposited (Figure S3).
Under routinely used conditions to activate the AP (1.0 mg/mL anti-CHO antibody and 10% serum21,22; Figure 5; Table 3), the R69W mutant did not inhibit C3d deposition as efficiently as wild-type MCP. For example, at 1, 4, and 8 mg/mL of anti-CHO Ab, mean fluorescence intensity (MFI) for C3 fragment deposition was 30, 49, and 59 for R69W compared with 14, 25, and 32 for wild type, respectively. Increasing the serum concentration to 30% further pointed out the regulatory defect of R69W, as the differences between the R69W MCP mutant and wild type (Figure 5; Table 3) were magnified. Increasing the serum concentration to 30% and the sensitizing antibody level to 4 mg/mL (or greater), the protection against C3b deposition by R69W MCP mutant was similar to control CHO cells lacking MCP. Under the same conditions, wild-type MCP offered substantial protection (reduced C3 binding by approximately 52%). We conclude that R69W is defective in its capacity to control complement activation on the cell surface.
Figure 5.

Comparison of R69W mutant versus wild-type MCP in inhibiting the alternative complement pathway. FACS analysis of C3d deposition using 1, 4, or 8.3 mg/mL of sensitizing Ab with 10% or 30% C7d serum for 60 minutes in Mg2+-EGTA. Representative experiment of 3. See Table 3 for MFIs.
Table 3.
MFI of FACS analysis in Figure 5 at 10% or 30% serum
| 10% serum |
30% serum |
|||||
|---|---|---|---|---|---|---|
| 1 mg/mL | 4 mg/mL | 8.3 mg/mL | 1 mg/mL | 4 mg/mL | 8.3 mg/mL | |
| WT | 14 | 25 | 32 | 45 | 65 | 84 |
| R69W | 30 | 49 | 59 | 109 | 141 | 151 |
| CHO | 165 | 228 | 234 | 181 | 150 | 209 |
We next evaluated the A304V mutant. The A304V MCP mutant was comparable with wild type in protecting against C3b deposition if challenged with 1.0 mg/mL anti-CHO antibody and 10% serum (Figure 6; Table 4) However, a functional defect in A304V was observed with increasing serum and antibody concentrations. The A304V MCP mutant allowed more C3b deposition than wild type at anti-CHO antibody concentration of 4 mg/mL (MFI of anti-C3d Ab was 26 for A304V and 19 for wild type) or if the serum concentration was increased to 30% (41 for A304V and 20 for wild type; Figure 6; Table 4). We conclude that A304V also showed a reduced ability to regulate complement activation in situ.
Figure 6.

Comparison of A304V mutant versus wild-type MCP in inhibiting the alternative pathway. FACS analysis of C3d deposition using 1, 4, or 8.3 mg/mL of sensitizing Ab with 10% or 30% C7d serum for 60 minutes in Mg2+-EGTA. Representative experiment of 3. See Table 4 for MFIs.
Table 4.
MFI of FACS analysis in Figure 6 at 10% and 30% serum
| 10% serum |
30% serum |
|||||
|---|---|---|---|---|---|---|
| 1 mg/mL | 4 mg/mL | 8.3 mg/mL | 1 mg/mL | 4 mg/mL | 8.3 mg/mL | |
| WT | 10 | 19 | 49 | 20 | 50 | 122 |
| A304V | 9 | 26 | 54 | 41 | 71 | 207 |
| CHO | 120 | 177 | 466 | 162 | 302 | 441 |
Kinetic study of C4b cleavage
In our challenge assay system, MCP serves as the cofactor for the cleavage of C4b deposited on CHO cells.21 C4b cleavage was not detected on CHO cells lacking MCP, despite the presence of C4 binding protein in the serum. Over approximately 40 minutes, the majority of C4b was progressively cleaved to C4c and C4d by MCP and FI.21 In order for C4b cofactor activity to occur on the cell surface, endogenous MCP must locate and bind this cell-surface–bound ligand and then, in conjunction with FI, cleave the C4b.
Comparison of the A304V MCP mutant and wild type at 5, 10, 15, 20, and 25 minutes demonstrated no difference in the rate of cleavage of C4b (Figure 7). In 3 experiments at 60 minutes, the percentage of C4b cleaved by A304V versus wild type was 70% (± 1%) and 60% (± 9%), respectively (data not shown). Similar data were obtained for the R69W mutant (data not shown). In contrast to membrane-bound C4b being degraded by MCP, factor H (and not MCP) cleaves bound C3b.21 These data establish that the ability of A304V and R69W proteins embedded in the membrane to locate C4b on the cell membrane and serve as a cofactor for its degradation are comparable with that of wild type.
Figure 7.

Kinetic analysis of cofactor activity for C4b of wild type versus the A304V mutant MCP. Control profile was nearly identical for cells exposed to sensitizing antibody but not to serum or to serum alone. Antibody, 1.0 mg/mL; serum concentration, 10%. Representative experiment of 3.
Discussion
In the initial set of expression and functional assessments,10,14 a deficiency of complement regulatory activity was not observed for the R69W and A304V MCP mutant proteins derived from transient transfections. These assays were performed at low salt concentrations using C3b or C4b on plates (ELISA) or in the fluid phase. In this report, stable cell lines with equivalent expression levels of these 2 MCP mutants and wild type were created. Using higher salt concentrations in the same assay systems, the R69W and A304V mutants still had no detectable differences in ligand binding or cofactor activity versus wild type. MCP, however, is expressed as a transmembrane protein. In this location, it prevents amplification of the AP feedback loop on the same cell on which it is expressed (intrinsic cofactor activation).16 MCP is thought to “scan” the cell membrane in order to find, bind, and then serve as a cofactor for FI to cleave C3b. To mimic this situation, these cell lines were “challenged” with an AP activating system. Both the R69W and the A304V MCP mutants allowed substantial more C3b deposition than did wild type. Thus, in situ, both mutants are defective in their ability to control the alternative complement pathway.
R69W mutation
Whether or not the R69W mutant would have a functional consequence was also unclear, because the original 2 families carrying this mutation had a second complement regulatory protein defect that could have accounted for the aHUS phenotype. CFI mutations are found in approximately 10% of patients with aHUS.9,10,12,13 The N151S (patients 1 and 2) and P32A (patient 4) mutations, however, have not been characterized as to their functional consequences. Further, patients 1 and 2 have additional potential predisposing alterations in their complement regulatory genes: the MCPggaac SNP haplotype and another MCP mutation, c.905-925del21n. The MCPggaac SNP haplotype may reduce transcription activity of the MCP promoter.9 The MCP mutation c.905-925del21n produces a 7-amino acid deletion in CCP 4, likely interfering with folding and transport of MCP to the cell membrane. The 50% reduction in MCP levels of patient 2 and her healthy mother is probably due to the c.905-925del21n mutation, as the R69W mutation does not alter MCP expression9,14 (also this report).
A concurrence of different susceptibility alleles affecting complement regulator expression has been previously shown to influence predisposition to aHUS.28 Likewise, in the first family described in this paper, patient 1 had both the R69W MCP mutation and the N151S CFI mutation. The daughter of patient 1 (patient 2) also has aHUS, as she inherited her father's mutations as well as an additional risk factor from her unaffected mother. No member of this family had the R69W mutation alone.
The second family has 1 member (patient 3) with the R69W mutation, without other defects. He is, though, homozygous for the R69W mutation, and manifested aHUS as a child but with no recurrences. The daughter of patient 3 (patient 4) is heterozygous for R69W but also has the P32A CFI mutation, possibly explaining why she has recurrent aHUS. These data further illustrate that multiple mutations of complement regulatory genes may be present and suggest that they are additive in predisposing to aHUS. They also may help to explain the incomplete penetrance of aHUS in carriers.
Unlike the majority of MCP mutations associated with aHUS, evaluation of R69W uncovered no expression profile defect and no reduction in binding or cofactor activity assays. Only when testing the mutant protein in situ was the defect in regulation of the AP elicited. This illustrates the importance of modeling transmembrane proteins on a cell surface to demonstrate the dysfunctional regulatory activity. Residue 69 is solvent exposed, so a mutation from a polar to a hydrophobic amino acid is likely to alter its interaction with ligands. That the R69 amino acid is an important residue is also supported by it being part of the measles virus binding site29 and a contact-forming residue for adenovirus type 11.30 A viral pathogen would more likely choose as its binding site a region of the protein that is functionally important and therefore conserved.
A304V mutation
Relative to the A304V mutation, it seemed a priori unlikely that such a conservative substitution in the transmembrane domain would affect its binding in solution to C3b and C4b. This was confirmed, as ligand binding activity was assessed at 3 ionic strengths, and no detectable differences in C3b or C4b binding were identified. In kinetic assays, fluid phase cofactor activity also did not differ between the A304V mutant and wild type. In contrast, the mutant protein was defective in its regulatory activity when embedded in the cell membrane. The mechanism underlying this loss of function remains to be defined.
Delineating the dysfunctional in situ cofactor activity of the A304V mutation may help us better understand MCP's role in the pathophysiology of the HUS syndrome and related conditions. The A304V mutation has now been found in 4 types of patients featuring renal pathology: aHUS, fatal Stx-HUS, HELLP, and glomerulonephritis with C3 deposits. All but the last share pathologic features of endothelial cell injury and thrombosis in the microcirculation. In aHUS, cofactor activity for C3b, provided by FI in conjunction with factor H or MCP, is necessary for endothelial cell protection in the kidney microvasculature. Being haploinsufficient is inadequate to protect against microthrombi formation in the setting of a renal endothelial cell injury.
Fatal Stx-HUS
Most children with Stx-HUS recover with supportive care. Fewer than 5% of patients die during the acute phase of Stx-HUS.31 This child is the first example we are aware of to have an MCP mutation with fatal Stx-HUS. The case further supports a “2-hit” model for disease expression in aHUS; in other words, a mutated complement regulator is predisposing, and a trigger, probably less injurious than the Shiga toxin, combine to initiate the disease. The Stx-HUS is sufficiently damaging to the renal endothelium as to not require a complement regulatory protein deficiency for disease development. HUS related to Shiga toxin is an uncommon disease, usually occurring in epidemic fashion. Mutations in CFH, CFI, or MCP are rare. The unlikely consequence of these conditions coming together, though, was a lethal case of HUS with a multiorgan failure syndrome. If a patient presents with a severe case of Stx-HUS, a search for a deficiency in a complement regulatory protein may be warranted.
HELLP
Pregnancy in patients with mutations in the complement regulatory proteins has been a precipitating factor in aHUS.10,12,13,32 Our report highlights an association with an MCP mutation and the HELLP syndrome. Unfortunately, the clinical details regarding patient 9 are limited. Preeclampsia, eclampsia, the HELLP syndrome, and HUS share clinical manifestations, and their distinction may be difficult.33 Nevertheless, a search for complement regulatory proteins in the HELLP syndrome may be warranted. Pregnancy and its prothrombotic state may provide the predisposing factor that can provoke an acute episode in a susceptible woman, such as one with a deficiency in a complement regulatory protein.
Glomerulonephritis with C3 deposits
The phenotype of patient 10 with glomerulonephritis and C3 deposits could also be explained by the V181M mutation or a combined effect with A304V. The V181M change lies near an area of importance for MCP binding and cofactor activity.26 However, transient transfections of the V181M MCP mutant showed normal expression with no alterations in C3b or C4b binding and cofactor activity compared with wild type (Table 2). This patient had a primary glomerulonephritis characterized by isolated mesangial C3 deposits as well as features of type I membranoproliferative glomerulonephritis.23
Conclusions
The syndromes associated with the A304V mutations are characterized by some common features and possibly represent an overlapping spectrum of disease. They are probably triggered by an environmental factor (Shiga toxin, infections) or internally (pregnancy) that induce complement activation secondary to an endothelial insult. In these situations, defective MCP on glomerular endothelial cells does not adequately protect against complement activation. An endothelial injury state followed by excessive complement AP activation results in a prothrombotic microvascular environment, clot formation, and renal injury. This suboptimal MCP activity, though, appears sufficient to protect the host from complement activation under other conditions.
Future studies to address how these 2 mutants are deficient in their ability to control the AP are under way. Perhaps both of these mutations, 1 solvent exposed and the other in the membrane, affect MCP's interaction with other proteins, including itself. In support of MCP oligomerization is the crystal structure of CCP 1 and CCP 2, which was a trimer.29 MCP and DAF synergize in their complement regulatory activity, preventing C3b deposition on the cell surface.34 How MCP and DAF communicate and interact are unknown. MCP is also known to be associated with integrins35–37 and upon cross-linking to signal.38,39 Observing how these 2 mutants signal, cycle, and inhibit complement activation on human endothelial cells may provide additional insights.
Supplementary Material
Acknowledgments
We thank Santiago Rodríguez de Córdoba, Craig Belsha, and Guiseppe Remuzzi for providing information on patients. We thank Shiping Wang (Department of Biostatistics, Washington University School of Medicine, St Louis, MO) for statistical analyses. We thank Richard Hauhart for his assistance in protein purification.
This work was supported by National Institutes of Health (NIH) grant no. T32 AR07279 (C.J.F.); grants from Assistance Publique-Hôpitaux de Paris (Progres Medical 2004 and Program Hospitalier de Recherche Clinique [AOM05130/P051065] 2005 to V.F.B.); NIH grant RO1 AI37618 (M.K.L. and J.P.A.); a fellowship from Association Amitié Sans Frontiers (G.P.); grants from Comitato 30 ore per la vita, from Telethon project GGP02162, from Associazione Ricerca Trapianto (ART), from Instituto Superiore di Sanità, and from the Foundation for Children with Atypical HUS along with the Nando Peretti Foundation (M.N.); and the Foundation for Children with atypical HUS and the Robin Davies Trust (T.H.J.G.). V.F.B., M.N. and T.H.J.G. are members of the European Working Party on the Genetics of aHUS.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: C.J.F., M.K.L, and J.P.A. designed the research and analyzed data. C.J.F. and J.P.A. wrote the paper. V.F.B, G.P., M.N. and T.H.J.G. contributed clinical information. C.J.F. performed the research.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: John P. Atkinson, Washington University School of Medicine, Division of Rheumatology, 660 South Euclid Avenue, Campus Box 8045, St Louis, MO 63110; e-mail: jatkinso@im.wustl.edu.
References
- 1.Moake JL. Thrombotic microangiopathies. N Engl J Med. 2002;347:589–600. doi: 10.1056/NEJMra020528. [DOI] [PubMed] [Google Scholar]
- 2.Warwicker P, Goodship THJ, Donne RL, et al. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int. 1998;53:836–844. doi: 10.1111/j.1523-1755.1998.00824.x. [DOI] [PubMed] [Google Scholar]
- 3.Perez-Caballero D, Gonzalez-Rubio C, Gallardo ME, et al. Clustering of missense mutations in the C-terminal region of factor H in atypical hemolytic uremic syndrome. Am J Hum Genet. 2001;68:478–484. doi: 10.1086/318201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Richards A, Buddles MR, Donne RL, et al. Factor H mutations in hemolytic uremic syndrome cluster in exons 18–20, a domain important for host cell recognition. Am J Hum Genet. 2001;68:485–490. doi: 10.1086/318203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caprioli J, Bettinaglio P, Zipfel PF, et al. The molecular basis of familial hemolytic uremic syndrome: mutation analysis of factor H gene reveals a hot spot in short consensus repeat 20. J Am Soc Nephrol. 2001;12:297–307. doi: 10.1681/ASN.V122297. [DOI] [PubMed] [Google Scholar]
- 6.Dragon-Durey M-A, Fremeaux-Bacchi V, Loirat C, et al. Heterozygous and homozygous factor H deficiencies associated with hemolytic uremic syndrome or membranoproliferative glomerulonephritis: report and genetic analysis of 16 cases. J Am Soc Nephrol. 2004;15:787–795. doi: 10.1097/01.asn.0000115702.28859.a7. [DOI] [PubMed] [Google Scholar]
- 7.Richards A, Kemp EJ, Liszewski MK, et al. Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. Proc Natl Acad Sci U S A. 2003;100:12966–12971. doi: 10.1073/pnas.2135497100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noris M, Brioschi S, Caprioli J, et al. Familial haemolytic uraemic syndrome and an MCP mutation. Lancet. 2003;362:1542–1547. doi: 10.1016/S0140-6736(03)14742-3. [DOI] [PubMed] [Google Scholar]
- 9.Esparza-Gordillo J, Goicoechea de Jorge E, Buil A, et al. Predisposition to atypical hemolytic uremic syndrome involves the concurrence of different susceptibility alleles in the regulators of complement activation gene cluster in 1q32. Hum Mol Genet. 2005;14:703–712. doi: 10.1093/hmg/ddi066. [DOI] [PubMed] [Google Scholar]
- 10.Caprioli J, Noris M, Brioschi S, et al. Genetics of HUS: the impact of MCP, CFH and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108:1267–1279. doi: 10.1182/blood-2005-10-007252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fremeaux-Bacchi V, Moulton EA, Kavanagh D, et al. Genetic and functional analyses of membrane cofactor protein (CD46) mutations in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2006;17:2017–2025. doi: 10.1681/ASN.2005101051. [DOI] [PubMed] [Google Scholar]
- 12.Fremeaux-Bacchi V, Dragon-Durey M-A, Blouin J, et al. Complement factor I: a susceptibility gene for atypical haemolytic uraemic syndrome. J Med Genet. 2004;41:e84. doi: 10.1136/jmg.2004.019083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kavanagh D, Kemp EJ, Mayland E, et al. Mutations in complement factor I predispose to the development of atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16:2150–2155. doi: 10.1681/ASN.2005010103. [DOI] [PubMed] [Google Scholar]
- 14.Richards A, Liszewski MK, Kavanagh D, et al. Implications of the initial mutations in membrane cofactor protein (MCP; CD46) leading to atypical hemolytic uremic syndrome. Mol Immunol. 2007;44:111–122. doi: 10.1016/j.molimm.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 15.Cole JL, Housley GA, Jr, Dykman TR, MacDermott RP, Atkinson JP. Identification of an additional class of C3-binding membrane proteins of human peripheral blood leukocytes and cell lines. Proc Natl Acad Sci U S A. 1985;82:859–863. doi: 10.1073/pnas.82.3.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oglesby TJ, Allen CJ, Liszewski MK, White DJG, Atkinson JP. Membrane cofactor protein (MCP;CD46) protects cells from complement-mediated attack by an intrinsic mechanism. J Exp Med. 1992;175:1547–1551. doi: 10.1084/jem.175.6.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seya T, Turner J, Atkinson JP. Purification and characterization of a membrane protein (gp45–70) that is a cofactor for cleavage of C3b and C4b. J Exp Med. 1986;163:837–855. doi: 10.1084/jem.163.4.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seya T, Atkinson JP. Functional properties of membrane cofactor protein of complement. Biochem J. 1989;264:581–588. doi: 10.1042/bj2640581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seya T, Okada M, Matsumoto M, Hong K, Kinoshita T, Atkinson JP. Preferential inactivation of the C5 convertase of the alternative complement pathway by factor I and membrane cofactor protein (MCP). Mol Immunol. 1991;28:1137–1147. doi: 10.1016/0161-5890(91)90029-j. [DOI] [PubMed] [Google Scholar]
- 20.Devaux P, Christiansen D, Fontaine M, Gerlier D. Control of C3b and C5b deposition by CD46 (membrane cofactor protein) after alternative but not classical complement activation. Eur J Immunol. 1999;29:815–822. doi: 10.1002/(SICI)1521-4141(199903)29:03<815::AID-IMMU815>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 21.Barilla-LaBarca ML, Liszewski MK, Lambris JD, Hourcade D, Atkinson JP. Role of membrane cofactor protein (CD46) in regulation of C4b and C3b deposited on cells. J Immunol. 2002;168:6298–6304. doi: 10.4049/jimmunol.168.12.6298. [DOI] [PubMed] [Google Scholar]
- 22.Liszewski MK, Leung MK, Schraml B, Goodship TH, Atkinson JP. Modeling how CD46 deficiency predisposes to atypical hemolytic uremic syndrome. Mol Immunol. 2007;44:1559–1568. doi: 10.1016/j.molimm.2006.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Servais A, Fremeaux-Bacchi V, Lequintrec M, et al. Primary glomerulonephritis with isolated C3 deposits: a new entity which shares common genetic risk factors with haemolytic uraemic syndrome. J Med Genet. 2007;44:193–199. doi: 10.1136/jmg.2006.045328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Esparza-Gordillo J, Goicoechea de Jorge E, Buil A, et al. Predisposition to atypical hemolytic uremic syndrome involves the concurrence of different susceptibility alleles in the regulators of complement activation gene cluster in 1q32. Hum Mol Genet. 2005;14:1107. doi: 10.1093/hmg/ddi066. [DOI] [PubMed] [Google Scholar]
- 25.Liszewski MK, Leung MK, Atkinson JP. Membrane cofactor protein: importance of N- and O-glycosylation for complement regulatory function. J Immunol. 1998;161:3711–3718. [PubMed] [Google Scholar]
- 26.Liszewski MK, Leung M, Cui W, et al. Dissecting sites important for complement regulatory activity in membrane cofactor protein (MCP; CD46). J Biol Chem. 2000;275:37692–37701. doi: 10.1074/jbc.M004650200. [DOI] [PubMed] [Google Scholar]
- 27.Liszewski MK, Atkinson JP. Membrane cofactor protein (MCP; CD46). Isoforms differ in protection against the classical pathway of complement. J Immunol. 1996;156:4415–4421. [PubMed] [Google Scholar]
- 28.Esparza-Gordillo J, Jorge EG, Garrido CA, et al. Insights into hemolytic uremic syndrome: segregation of three independent predisposition factors in a large, multiple affected pedigree. Mol Immunol. 2006;43:1769–1775. doi: 10.1016/j.molimm.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 29.Casasnovas JM, Larvie M, Stehle T. Crystal structure of two CD46 domains reveals an extended measles virus-binding surface. EMBO J. 1999;18:2911–2922. doi: 10.1093/emboj/18.11.2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Persson BD, Reiter DM, Marttila M, et al. Adenovirus type 11 binding alters the conformation of its receptor CD46. Nat Struct Mol Biol. 2007;14:164–166. doi: 10.1038/nsmb1190. [DOI] [PubMed] [Google Scholar]
- 31.Elliott EJ, Robins-Browne RM, O'Loughlin EV, et al. Nationwide study of haemolytic uraemic syndrome: clinical, microbiological, and epidemiological features. Arch Dis Child. 2001;85:125–131. doi: 10.1136/adc.85.2.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caprioli J, Castelletti F, Bucchioni S, et al. Complement factor H mutations and gene polymorphisms in haemolytic uraemic syndrome: the C-257T, the A2089G and the G2881T polymorphisms are strongly associated with the disease. Hum Mol Genet. 2003;12:3385–3395. doi: 10.1093/hmg/ddg363. [DOI] [PubMed] [Google Scholar]
- 33.O'Brien JM, Barton JR. Controversies with the diagnosis and management of HELLP syndrome. Clin Obstet Gynecol. 2005;48:460–477. doi: 10.1097/01.grf.0000160309.73197.35. [DOI] [PubMed] [Google Scholar]
- 34.Brodbeck WG, Mold C, Atkinson JP, Medof ME. Cooperation between decay-accelerating factor and membrane cofactor protein in protecting cells from autologous complement attack. J Immunol. 2000;165:3999–4006. doi: 10.4049/jimmunol.165.7.3999. [DOI] [PubMed] [Google Scholar]
- 35.Lozahic S, Christiansen D, Manie s, et al. CD46 (membrane cofactor protein) associates with multiple beta 1 integrins and tetraspans. Eur J Immunol. 2000;30:900–907. doi: 10.1002/1521-4141(200003)30:3<900::AID-IMMU900>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 36.Kurita-Taniguchi M, Hazeki K, Murabayashi N, et al. Molecular assembly of CD46 with CD9, alpha 3-beta 1 integrin and protein tyrosine phosphatase SHP-1 in human macrophages through differentiation by GM-CSF. Mol Immunol. 2002;38:689–700. doi: 10.1016/s0161-5890(01)00100-6. [DOI] [PubMed] [Google Scholar]
- 37.McLaughlin BJ, Fan W, Zheng JJ, et al. Novel role for a complement regulatory protein (CD46) in retinal pigment epithelial adhesion. Invest Ophthalmol Vis Sci. 2003;44:3669–3674. doi: 10.1167/iovs.02-0813. [DOI] [PubMed] [Google Scholar]
- 38.Crimeen-Irwin B, Ellis S, Christiansen D, et al. Ligand binding determines whether CD46 is internalized by clathrin-coated pits or macropinocytosis. J Biol Chem. 2003;278:46927–46937. doi: 10.1074/jbc.M308261200. [DOI] [PubMed] [Google Scholar]
- 39.Kemper C, Atkinson JP. T-cell regulation: with complements from innate immunity. Nat Rev Immunol. 2007;7:9–18. doi: 10.1038/nri1994. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}