

Abstract
Myocardial infarction is linked to atherosclerosis, yet the sequence leading from silent coronary atherosclerosis to acute myocardial infarction has remained unclear. Here we show that hypercholesterolemic apolipoprotein E−/− low density lipoprotein receptor−/− mice develop not only coronary atherosclerosis but also myocardial infarction. Exposure of mice to mental stress or hypoxia led to acute ischemia, which, in a large proportion of the mice, was followed by electrocardiographic changes, leakage of troponin T, and loss of dehydrogenase from the myocardium, all indicative of acute myocardial infarction. Apoptotic death of cardiomyocytes was followed by inflammation and fibrosis in the heart. All these pathological changes could be prevented by a blocker of the endothelin type A receptor. Thus, stress elicits myocardial infarction through endothelin receptor signaling in coronary atherosclerosis caused by hypercholesterolemia.
Keywords: atherosclerosis, cholesterol, gene deletion, hypoxia, stress
Myocardial infarction remains the most common cause of death in the Western world and is increasing also in the developing world (1). It is almost invariably associated with coronary atherosclerosis, but the mechanisms leading from atherosclerosis to infarction have remained obscure (2, 3). Murine models of atherosclerosis recently have been developed by targeted deletion of genes regulating cholesterol metabolism (4). Deletions of the genes for apolipoprotein E (apoE; refs. 5 and 6) and the low density lipoprotein (LDL) receptor (7) result in severe atherosclerosis, with the formation of lesions strikingly similar to those found in humans (8–10). Studies using these models show that atherosclerosis is initiated by hypercholesterolemia, which leads to cholesterol accumulation in the artery wall, vascular inflammation with local activation of macrophages and T cells, and a fibrotic response involving smooth muscle cells (8–12).
Lack of suitable experimental methods have precluded a similar dissection of the sequence of events leading from atherosclerosis to myocardial infarction. Histopathological studies of culprit lesions in humans have identified fissures and thrombi at sites of intense plaque inflammation (13, 14), suggesting a process initiated by inflammation and leading via local proteolytic and procoagulant activity to occluding thrombosis (3). However, myocardial infarction has not been detected in atherosclerotic rabbits or monkeys, and induction of myocardial infarction in experimental animals has required ligation of coronary arteries (15). Such surgical intervention obviously is not suitable for dissecting the pathogenesis of the myocardial infarction that occurs as a consequence of atherosclerosis.
We recently have set up methods for analyzing hemodynamics and cardiac electrophysiology in the laboratory mouse (16, 17) and applied them to study coronary atherosclerotic disease in gene-targeted mice. Using this approach, we now report that hypercholesterolemic apoE−/− × LDL receptor−/− (E0 × LDLR0) mice develop coronary atherosclerosis and myocardial infarction.
MATERIALS AND METHODS
Mice.
Male E0 × LDLR0 mice (7) on the C57BL/6J background were put on a “Western diet” at 4–7 weeks of age and sacrificed at 4, 7, 9, and 12 months of age. The diet was based on corn starch, glucose, sucrose, cocoa butter, cellulose, minerals, cholesterol, and a vitamin mix and contained 0.15% cholesterol and 21% (wt/wt) total fat (18). Male, age-matched wild-type C57BL/6J mice were used as controls, and male E0 single-knockout mice (5) at 4–12 months of age were studied in pilot experiments. All experiments complied with national guidelines and were approved by the regional ethical committee.
ECG.
For the mental stress experiments, continuous telemetric ECG monitoring from freely moving mice was obtained as described (16, 19). Briefly, we used a telemetry system (Datasciences, Minneapolis) comprising a transmitter implanted in the peritoneal cavity and a receiver (RA1010) placed underneath the home cage. The signal from the transmitter was acquired digitally, and the electrical activity was computer-analyzed. During hypoxic stress, ECG was recorded by using s.c. bipolar limb leads in anesthetized mice (isofluorane, 1.2 ± 0.2%). Instant ECG was acquired and analyzed digitally (17). The ischemic burden was calculated as the STU area, i.e., the area covered by the STU (repolarization) segment shift from the isoelectric line. The latter was calculated automatically as the mean level in the interval 5–20 msec preceding the Q wave.
Stress Tests.
Mental stress. A method developed previously for rats (20) was adopted for mice (17). Mice were implanted 5 days ahead with the telemetric transmitters, and continuous ECG monitoring (ECG averaging and recording every 30 min) lasted 10 days. On days 3 and 9, air was forced into the cage for 30 min (mental stress, ECG averaging and recording every 2 min).
Hypoxic stress.
Under isofluorane anesthesia, the mixture of O2 and N2 in the ventilatory mask was modulated and monitored continuously with the help of a spirometer. The percentage of oxygen was modified manually starting at 21% and reducing it to 16, 14, 12, and 10% (2 min each step). Recovery was achieved by reestablishing O2 at 21%, and ECG was monitored continuously during the acute hypoxia and continued for 20 min after O2 restoration. A subgroup of mice received the endothelin type A (ETA) receptor blocker LU135252 (21) (kindly provided by M. Raschack, Knoll, Ludwigshafen, Germany). A stock solution was dissolved in 1 M NaOH/150 mM NaCl, and the pH was adjusted to 7.5 with 100 mM HCl. The stock solution was diluted further in 150 mM NaCl and injected i.p. at 1 mg/kg of body weight 15 min before hypoxic stress.
Echography.
During hypoxic stress, anesthetized mice also were monitored by an Echo-Doppler probe (PW, 9 MHz) fixed on the right sternal border. Two-dimensional mode was used to localize the aortic root, whereas PW-Doppler mode was used to measure the aortic pulse peak as an index of cardiac pump function.
Serum Analyses.
Peripheral blood was obtained at sacrifice. Serum was harvested and analyzed for troponin T by using a standard immunochemical assay (Boehringer Mannheim). Serum cholesterol was determined by using a Cobas Mira (Roche Diagnostics) analyzer. Lipoproteins were fractionated by fast protein liquid chromatography through Superose-6 (Amersham) (22).
Perfusion and Angiography.
Under anesthesia, animals were sacrificed by exsanguination. The heart was perfused from the apex of the left ventricle with ice-cold PBS for 1 min at a constant pressure of 100 mmHg (1 mmHg = 133 Pa) followed by triphenyl tetrazolium chloride (TTC) (23) perfusion for 1 min. In a subset of mice, barium was injected into the coronary system at constant pressure through a cannula placed in the thoracic aorta and the heart was fixed in PBS/4% formaldehyde. Coronary angiograms were taken with a microangiographic technique by using a highly focused x-ray source and a digital radiologic acquisition system (24).
Microscopic Analysis.
One-millimeter sections of TTC-stained, formaldehyde-fixed hearts were checked for loss of staining (TTC stains dehydrogenase activity in living tissue red). The fixed hearts were embedded in paraffin, and 5-μm cross sections were stained with Masson’s trichrome stain. Fluorescein-dUTP and terminal transferase (In Situ Cell Death Kit; Boehringer Mannheim) were used for terminal deoxynucleotidyltransferase-mediated UTP end labeling (TUNEL) analysis of apoptosis.
RESULTS
Coronary Atherosclerosis and Spontaneous Myocardial Infarction in Hypercholesterolemic Mice.
Atherosclerosis-prone E0 × LDLR0 male mice (7) were fed a “Western” diet containing 0.15% cholesterol (18). This led to severe hypercholesterolemia with elevated very low density lipoprotein (VLDL) and LDL fractions. Serum cholesterol levels at 8–9 months of age were 26.3 ± 5.2 mmol/liter in E0 × LDLR0 mice, which was not significantly different from those in E0 single-knockout mice (29.3 ± 5.0 mmol/liter). As expected (7, 25), both E0 and E0 × LDLR0 but not wild-type C57BL/6J mice showed prominent lipoprotein peaks in the VLDL range, and E0 × LDLR0 mice but not E0 single-knockout mice had additional peaks in the LDL range (data not shown). Both E0 and E0 × LDLR0 mice showed advanced aortic atherosclerosis (7, 8, 26), and increased mortality was observed in E0 × LDLR0 mice older than 7 months. To evaluate whether the coronary tree was affected by disease, we performed postmortem coronary angiography on E0 × LDLR0 mice (Fig. 1 a and b). It showed severe reduction in the lumen diameter of all three main coronary arterial branches in all mice and total occlusion in 28 of 36 mice. Typically, stenoses were most severe in the proximal segments of the coronary arteries (Fig. 1b).
Figure 1.

Coronary atherosclerosis and myocardial postinfarction scars in hypercholesterolemic mice. (a and b) Coronary angiograms from a wild-type mouse (a) and a 7-month-old E0 × LDLR0 mouse (b). Red arrows indicate severe coronary stenosis, and black arrows indicate total occlusion. (c–e) Histological analysis of coronary arteries from wild-type (c) and E0 × LDLR0 mice (d and e). Atherosclerotic plaques are evident in the latter. In e, a myocardial scar surrounds the occluded atherosclerotic artery. [Masson’s trichrome stain, original magnification = ×200 (c and d) and ×100 (e).]
Histologic analysis of heart cross sections (Fig. 1c) confirmed the presence of advanced atherosclerotic plaques in the coronary arteries of E0 × LDLR0 mice (Fig. 1d) as well as E0 mice (data not shown). Myocardial scars were detected around occluded coronary arteries, suggesting that they were caused by myocardial infarction (Fig. 1e). These findings suggest that myocardial infarction can occur as a consequence of coronary atherosclerosis in genetically hypercholesterolemic mice.
Mental Stress Elicits Myocardial Infarction in Mice with Coronary Atherosclerosis.
To test whether myocardial infarction could be induced experimentally in freely moving atherosclerotic mice, we subjected awake mice to a mental stress test. Air was forced into the cage, causing a mental stress that leads to increased heart workload in mice (17). The ECG was recorded through a s.c. transmitter implanted 1 week in advance (19). E0 × LDLR0 mice older than 7 months showed small deviations of the repolarization ECG segment that were suggestive of myocardial ischemia (27) already at rest (Fig. 2). These deviations were increased substantially during the stress test (Fig. 2). Pathological ECG patterns occurred within 1 min after initiation of the stressful stimulus and remained for 12 ± 7 min. They were never found in younger E0 × LDLR0 mice or age-matched wild-type controls.
Figure 2.

Mental stress induces ECG changes and myocardial infarction in E0 × LDLR0 mice. ECG tracings (Left), serum troponin T levels (Center), and TTC staining for dehydrogenase activity in the myocardium (Right) are shown. Wild-type C57BL/6J (Top), a resting E0 × LDLR0 mouse (Middle), and an E0 × LDLR0 mouse exposed to the mental stress test (Bottom). ECG: STU segment deviations (arrows) are apparent in resting E0 × LDLR0 mice and exacerbated by stress. Troponin T concentrations are significantly elevated in E0 × LDLR0 mice exposed to mental stress. Concentrations >1 μg/liter are diagnostic for myocardial infarction in humans. Box-and-whisker plot is shown, in which the line in the box corresponds to the median and the top and bottom of the box correspond to the 75th and 25th percentile, respectively. The whiskers extend from the 10th to the 90th percentile. ∗, Significantly different from the other two groups (P < 0.05). TTC staining of heart segments is shown, where red staining indicates viable myocardium with dehydrogenase activity. There is a slight reduction in TTC staining in the myocardium of the resting E0 × LDLR0 mouse and a drastic loss of TTC dehydrogenase activity in the myocardium of the E0 × LDLR0 mouse exposed to mental stress.
We also analyzed serum levels of cardiac troponin T, a contractile protein that leaks out of injured cardiomyocytes (28) and is used commonly as a sensitive assay to detect myocardial infarction in humans (29). Troponin T levels were elevated significantly after mental stress in atherosclerotic mice and not in controls, suggesting that the transient ECG changes observed in the atherosclerotic mice during the stress test were due to myocardial infarction (Fig. 2). A subset of mice (14/32, 44%) with more prolonged ECG changes (>20 min) showed large infarcted zones with loss of dehydrogenase activity from the myocardium (Fig. 2). Together, the results of these experiments show that stress can cause myocardial infarction in mice with coronary atherosclerosis.
Hypoxic Stress Induces Myocardial Ischemia Followed by Infarction.
A fully standardized induction of myocardial infarction was deemed necessary to dissect the pathogenetic mechanism, and, therefore, we exposed anesthetized, artificially ventilated mice to global hypoxia by reducing the oxygen supply. Hypoxia causes reflex coronary vasoconstriction (30), which may result in dynamic coronary occlusion in the presence of severe atherosclerotic stenoses. Whereas age-matched wild-type mice did not show ECG changes or echographic signs of reduced myocardial contractility even at very low oxygen levels (down to 8%), atherosclerotic mice showed typical ECG deviations within 2 min after reduction of O2 below 12% (Fig. 3). Echocardiographic analysis revealed reduced (22 ± 7%) peak aortic blood flow velocity during the hypoxic stress, probably reflecting depressed left ventricular function caused by massive ischemia (31).
Figure 3.
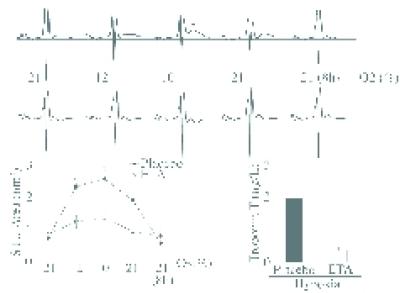
Hypoxic stress induces ECG changes and myocardial infarction, which are prevented by the ETA blocker LU135252. (Top) ECG tracings during hypoxia in 7-month-old E0 × LDLR0 mouse. Arrow indicates STU segment elevation at 10% oxygen, which gradually returns to baseline during normoxia. Below are ECGs from the same mouse subjected to the same test 1 week later. This time, LU135252 was given i.p. 15 min before hypoxia. No STU elevation is seen. (Bottom Left) Average STU deviations during hypoxic stress in E0 × LDLR0 mice with LU135252 (ETA, n = 7) or placebo (n = 9). ∗, Significantly different from ETA group (P < 0.05). (Bottom Right) Serum troponin T elevations 1 week after hypoxic stress in E0 × LDLR0 mice were prevented by LU135252 (ETA). ∗, Significantly different from ETA group (P < 0.05).
In most mice, ECG deviations disappeared upon cessation of hypoxia, and only 33% of them exhibited persistent ECG changes at 8 h after reoxygenation. However, a total of 31 of 57 mice (54%) developed altered QRS complexes in the recovery phase, suggesting that the initial, reversible ischemic phase was followed by irreversible myocardial damage. This was confirmed by analysis of plasma troponin T, which was elevated (>0.2 μg/liter) at 72 h through to 14 days after hypoxia (Fig. 3). At autopsy, 22 of 31 E0 × LDLR0 mice with persistent ECG changes (71%) had morphological signs of infarction, i.e., loss of dehydrogenase activity in the myocardium. The structural development of infarcted hearts was analyzed in mice killed at day 1 (n = 6), 3 (n = 7), 7 (n = 6), 14 (n = 6), and 21 (n = 6) after hypoxia. Histologic analysis revealed an early phase of apoptosis (day 1–3), followed by inflammatory infiltration (day 3), removal of dead myocytes, development of granulation tissue (day 7), and subsequent healing by a dense collagenous scar (day 21) (Fig. 4). Thus, ECG, biochemical, and histologic data show that hypoxic stress elicits myocardial infarction in E0 × LDLR0 mice. Hypoxic stress also induced myocardial infarction in 12-month-old but not in 7-month-old E0 single-knockout mice (data not shown).
Figure 4.

Development of myocardial infarction after hypoxic stress in E0 × LDLR0 mice. (Left) Apoptotic cardiomyocytes (TUNEL stain, fluorescent microscopy). (Center and Right) Masson’s trichrome stain (MT). At day 1 after hypoxic stress, dead cardiomyocytes and a thrombosed coronary artery with extravasation of erythrocytes [MT (×400)] are evident. At day 3, scattered inflammatory cells are present between dead cardiomyocytes [TUNEL; MT (×400)]; atherosclerotic and collapsed intramural coronary arteries can be seen [MT (×200)]. At day 7, granulation tissue and collagen deposits are formed [blue, MT (×200)]; detail of the neovessels in the granulation tissue is shown at MT (×400). At day 21, dead cardiomyocyte fibers have been replaced by a dense collagenous scar (blue). A few remaining cardiac muscle cells are present (red).
ETA Receptor Mediates Hypoxia-Induced Myocardial Infarction.
The sudden but transient development of ECG changes and signs of depressed pump function during hypoxic stress in atherosclerotic mice suggested that myocardial ischemia might develop through a vasoconstrictor reflex response to hypoxia (30) in the atherosclerotic coronary artery (32, 33). Hyperlipidemia and hypoxia increase endothelium-dependent vasoconstriction (34). Among vasoactive molecules, endothelin plays a pivotal role as a vasoconstrictive peptide that is present in endothelial cells and macrophages of active coronary atherosclerotic plaques (35). It is released upon hypoxic stress (36) and elevated in patients with myocardial infarction because of increased secretion and/or reduced clearance (37, 38).
Because the vasoconstrictor effects of endothelin are mediated by the ETA receptor, which is expressed by vascular smooth muscle cells (39), we exposed atherosclerotic mice to hypoxic stress before and after administration of an ETA receptor blocker, LU135252 (1 mg/kg of body weight). Such treatment dramatically attenuated the ECG signs of myocardial ischemia and also reduced the extent of subsequent myocardial infarction as assessed by troponin T leakage (Fig. 3). To rule out that the first exposure to hypoxia preconditioned the heart to make it resistant to a second hypoxic stress, we repeated the experiment but exposed the mice to LU135252 during the first rather than the second hypoxic episode. Again, ETA receptor blockade prevented the ECG signs of myocardial ischemia. One week later, when hypoxia was performed without the ETA blocker, the ECG alterations reappeared.
DISCUSSION
These studies demonstrate, through combined use of electrophysiological, biochemical, radiological, and morphological methods, that (i) hypercholesterolemia induces coronary atherosclerosis that leads to myocardial infarction, (ii) infarction can be precipitated by mental or hypoxic stress in mice with coronary atherosclerosis, and (iii) ETA receptor signaling mediates the ischemic response to hypoxia in the atherosclerotic heart.
Although the correlation between hypercholesterolemia and myocardial infarction is well established by a large series of epidemiological studies, no previous experimental data have demonstrated that hypercholesterolemia causes myocardial infarction. Mice deficient in apoE develop severe atherosclerosis involving the coronary arteries (8), and the occurrence of occasional fibrotic scars in their myocardium (8) suggests that they may develop ischemic heart disease. In the present study, we extend these observations by showing that E0 × LDLR0 mice develop angiographic stenosis and occlusions, which are indicative of severe coronary heart disease, ECG changes characteristic for myocardial damage, and elevated plasma troponin T levels, which are diagnostic for myocardial infarction. Thus, hypercholesterolemia induces coronary atherosclerosis, which, in turn, elicits myocardial infarction.
Both E0 and E0 × LDLR0 mice developed coronary atherosclerosis, myocardial scars, and myocardial infarction upon hypoxic stress. However, the atherosclerotic process was accelerated in the latter model, which also became sensitive to stress at an earlier stage. This is in line with the observation that E0 × LDLR0 mice develop more extensive aortic atherosclerosis than E0 mice (25). The two strains differ significantly in lipoprotein profiles but not in serum cholesterol levels (refs. 7 and 25; this study), suggesting that LDL-like lipoproteins and/or the lack of LDL receptors add to the atherogenicity of hypercholesterolemia and increased chylomicrons and remnant particles, which are common to both models. The finding that myocardial infarction could be elicited only when coronary atherosclerosis was present indicates that atherosclerosis rather than hyperlipoproteinemia per se was needed for the development of ischemic heart disease.
The role of stress in ischemic heart disease has been controversial. Although clinicians have observed that patients suffering from acute coronary syndromes often have experienced stressful stimuli in their recent past, experimental studies have failed to clarify the role of stress in coronary disease. We exposed atherosclerotic mice to two different types of stressful stimuli: a mental stress that may increase heart workload and a hypoxic stress that reduces oxygen delivery to the heart muscle. Both stimuli led to acute myocardial infarction in atherosclerotic mice but did not cause disease in wild-type mice. This experimental evidence shows that stress causes myocardial infarction in hyperlipidemia-induced atherosclerosis. It confirms the importance of an imbalance between oxygen demand and the supply through an atherosclerotic coronary artery for development of acute ischemia. That endothelin receptor blockade completely prevented myocardial infarction during hypoxic stress shows the importance of vasodynamic responses in the sequence of events leading from silent coronary atherosclerosis to acute myocardial infarction.
Clinical studies previously have suggested that endothelin may be important in myocardial infarction (37), and ETA receptor blockade can restore endothelial function in E0 mice (40) and protect the myocardium from neutrophil injury during ischemia/reperfusion (41). We now provide direct experimental evidence that coronary vasoconstriction through ETA receptor signaling plays a pivotal role in the development of stress-induced myocardial infarction in the atherosclerotic mouse. It is likely that endothelin, increased rapidly upon hypoxia (34, 39), induces smooth muscle contraction, which, in turn, causes vasospasm of the atherosclerotic coronary arteries (32, 33). This may explain the sudden but reversible ECG deviations observed at the time of provocative tests in the E0 × LDLR0 mice. Stress may, by inducing endothelin-dependent vasoconstriction in the atherosclerotic coronary artery, initiate a process that results in irreversible myocardial injury, which is detectable as apoptosis, loss of dehydrogenase from cardiomyocytes, leakage of troponin T, and, eventually, inflammation and formation of a fibrotic scar in the myocardium. It should now be possible to dissect all the critical steps involved in this sequence of events by using specific blockers and compound mutant mice.
Acknowledgments
We thank Xinghua Zhou for helpful discussions, Inger Bodin for excellent technical assistance, Anders Hamsten and Knut Pettersson for cholesterol and lipoprotein analyses, and Manfred Raschack for providing LU135252. This work was supported by grants from the Swedish Medical Research Council (projects 6816 and 4764), Heart–Lung Foundation, Hedlund, Johnson, and Osterman Funds, and King Gustav V 80th Anniversary Fund. G.C. is the recipient of fellowship grants from the European Society of Cardiology and the Swedish Heart and Lung Foundation.
ABBREVIATIONS
- apoE
apolipoprotein E
- LDL
low density lipoprotein
- LDLR
LDL receptor
- E0 × LDLR0
apoE−/− × LDLR−/− mice
- TTC
triphenyl tetrazolium chloride
- ETA receptor
endothelin type A receptor
References
- 1.Murray C J, Lopez A D. Lancet. 1997;349:1436–1442. doi: 10.1016/S0140-6736(96)07495-8. [DOI] [PubMed] [Google Scholar]
- 2.Ross R. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 3.Libby P. Circulation. 1995;91:2844–2850. doi: 10.1161/01.cir.91.11.2844. [DOI] [PubMed] [Google Scholar]
- 4.Breslow J L. Science. 1996;272:685–688. doi: 10.1126/science.272.5262.685. [DOI] [PubMed] [Google Scholar]
- 5.Plump A S, Smith J D, Hayek T, Aalto-Setälä K, Walsh A, Verstuyft J G, Rubin E M, Breslow J L. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- 6.Piedrahita J A, Zhang S H, Hagaman J R, Oliver P M, Maeda N. Proc Natl Acad Sci USA. 1992;89:4471–4475. doi: 10.1073/pnas.89.10.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ishibashi S, Herz J, Maeda N, Goldstein J L, Brown M S. Proc Natl Acad Sci USA. 1994;91:4431–4435. doi: 10.1073/pnas.91.10.4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakashima Y, Plump A S, Raines E W, Breslow J L, Ross R. Arterioscler Thromb. 1994;14:133–140. doi: 10.1161/01.atv.14.1.133. [DOI] [PubMed] [Google Scholar]
- 9.Smith J D, Trogan E, Ginsberg M, Grigaux C, Tian J, Miyata M. Proc Natl Acad Sci USA. 1995;92:8264–8268. doi: 10.1073/pnas.92.18.8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou X, Stemme S, Hansson G K. Am J Pathol. 1996;149:359–366. [PMC free article] [PubMed] [Google Scholar]
- 11.Nicoletti A, Kaveri S, Caligiuri G, Bariéty J, Hansson G K. J Clin Invest. 1998;102:910–918. doi: 10.1172/JCI119892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mach F, Schönbeck U, Sukhova G K, Atkinson E, Libby P. Nature (London) 1998;394:200–203. doi: 10.1038/28204. [DOI] [PubMed] [Google Scholar]
- 13.van der Wal A C, Becker A E, van der Loos C M, Das P K. Circulation. 1994;89:36–44. doi: 10.1161/01.cir.89.1.36. [DOI] [PubMed] [Google Scholar]
- 14.Falk E, Shah P K, Fuster V. Circulation. 1995;92:657–671. doi: 10.1161/01.cir.92.3.657. [DOI] [PubMed] [Google Scholar]
- 15.Maseri A. Ischemic Heart Disease. New York: Churchill Livingstone; 1995. [Google Scholar]
- 16.Johansson C, Thoren P. Acta Physiol Scand. 1997;160:133–138. doi: 10.1046/j.1365-201X.1997.00134.x. [DOI] [PubMed] [Google Scholar]
- 17.Johansson C, Vennstrom B, Thoren P. Am J Physiol. 1998;275:R640–R646. doi: 10.1152/ajpregu.1998.275.2.R640. [DOI] [PubMed] [Google Scholar]
- 18.Zhou X, Paulsson G, Stemme S, Hansson G K. J Clin Invest. 1998;101:1717–1725. doi: 10.1172/JCI1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kramer K, van Acker S A, Voss H P, Grimbergen J A, van der Vijgh W J, Bast A. J Pharmacol Toxicol Methods. 1993;30:209–215. doi: 10.1016/1056-8719(93)90019-b. [DOI] [PubMed] [Google Scholar]
- 20.Lundin S, Ricksten S-E, Thorén P. Acta Physiol Scand. 1984;120:273–281. doi: 10.1111/j.1748-1716.1984.tb00134.x. [DOI] [PubMed] [Google Scholar]
- 21.Münter K, Hergenröder L, Unger L, Kirchengast M. Pharm Pharmacol Lett. 1996;6:90–92. [Google Scholar]
- 22.De Silva H V, Mas-Oliva J, Taylor J M, Mahley R W. J Lipid Res. 1994;35:1297–1310. [PubMed] [Google Scholar]
- 23.Holmbom B, Näslund U, Eriksson A, Virtanen I, Thornell L E. Histochemistry. 1993;99:265–275. doi: 10.1007/BF00269099. [DOI] [PubMed] [Google Scholar]
- 24.Griffith T M, Edwards D H, Davies R L, Harrison T J, Evans K T. Br J Pharmacol. 1988;93:654–662. doi: 10.1111/j.1476-5381.1988.tb10323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonthu S, Heistad D D, Chappell D A, Lamping K G, Faraci F M. Arterioscler Thromb Vasc Biol. 1997;17:2333–2340. doi: 10.1161/01.atv.17.11.2333. [DOI] [PubMed] [Google Scholar]
- 26.Reddick R L, Zhang S H, Maeda N. Arterioscler Thromb. 1994;14:141–147. doi: 10.1161/01.atv.14.1.141. [DOI] [PubMed] [Google Scholar]
- 27.Pfeffer M A, Pfeffer J M, Steinberg C, Finn P. Circulation. 1985;72:406–412. doi: 10.1161/01.cir.72.2.406. [DOI] [PubMed] [Google Scholar]
- 28.Yamahara Y, Asayama J, Ohta B, Matsumoto T, Miyazaki H, Tatsumi T, Kobara M, Inoue M, Inoue D, Nakagawa M. Basic Res Cardiol. 1993;88:307–313. doi: 10.1007/BF00800637. [DOI] [PubMed] [Google Scholar]
- 29.Rebuzzi A G, Quaranta G, Liuzzo G, Caligiuri G, Lanza G A, Gallimore J R, Grillo R L, Cianflone D, Biasucci L M, Maseri A. Am J Cardiol. 1998;82:715–719. doi: 10.1016/s0002-9149(98)00458-5. [DOI] [PubMed] [Google Scholar]
- 30.Downey H F, Grice D P, Jones C E. J Cardiovasc Pharmacol. 1991;18:657–664. doi: 10.1097/00005344-199111000-00002. [DOI] [PubMed] [Google Scholar]
- 31.Lew W Y, Nishikawa Y, Su H. Circulation. 1994;90:1942–1950. doi: 10.1161/01.cir.90.4.1942. [DOI] [PubMed] [Google Scholar]
- 32.Zeiher A M, Drexler H, Wollschläger H, Just H. Circulation. 1991;83:391–401. doi: 10.1161/01.cir.83.2.391. [DOI] [PubMed] [Google Scholar]
- 33.Ludmer P L, Selwyn A P, Shook T L, Wayne R R, Mudge G H, Alexander R W, Ganz P. N Engl J Med. 1986;315:1046–1051. doi: 10.1056/NEJM198610233151702. [DOI] [PubMed] [Google Scholar]
- 34.Anderson T J, Meredith I T, Charbonneau F, Yeung A C, Frei B, Selwyn A P, Ganz P. Circulation. 1996;93:1647–1650. doi: 10.1161/01.cir.93.9.1647. [DOI] [PubMed] [Google Scholar]
- 35.Zeiher A M, Goebel H, Schächinger V, Ihling C. Circulation. 1995;91:941–947. doi: 10.1161/01.cir.91.4.941. [DOI] [PubMed] [Google Scholar]
- 36.Aversa C R, Oparil S, Caro J, Li H, Sun S D, Chen Y F, Swerdel M R, Monticello T M, Durham S K, Minchenko A, et al. Am J Physiol. 1997;273:L848–L855. doi: 10.1152/ajplung.1997.273.4.L848. [DOI] [PubMed] [Google Scholar]
- 37.Miyauchi T, Yanagisawa M, Tomizawa T, Sugishita Y, Suzuki N, Fujino M, Ajisaka R, Goto K, Masaki T. Lancet. 1989;ii:53–54. doi: 10.1016/s0140-6736(89)90303-6. [DOI] [PubMed] [Google Scholar]
- 38.Dupuis J, Rouleau J L, Cernacek P. Circulation. 1998;98:1684–1687. doi: 10.1161/01.cir.98.16.1684. [DOI] [PubMed] [Google Scholar]
- 39.Lüscher T F. Am Rev Respir Dis. 1992;146:S56–S60. doi: 10.1164/ajrccm/146.5_Pt_2.S56. [DOI] [PubMed] [Google Scholar]
- 40.Barton M, Haudenschild C C, d′Uscio L V, Shaw S, Münter K, Lüscher T F. Proc Natl Acad Sci USA. 1998;95:14367–14372. doi: 10.1073/pnas.95.24.14367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gonon A T, Wang Q-D, Pernow J. Cardiovasc Res. 1998;39:674–682. doi: 10.1016/s0008-6363(98)00167-9. [DOI] [PubMed] [Google Scholar]