

Abstract
A simple and rapid high performance liquid chromatographic method for the determination of plasma amino acids was developed. The method uses minimal sample volume and automated online precolumn derivitization of amino acids with o- phthalaldehyde and fluorescent detection. Amino acids are separated by a simplified gradient without column heating. The assay is linear from 5 to 1000μmol/L for all amino acids. Recovery of amino acids was between 91 and 108%, intra-assay coefficient of variation (CV) was 1–7%, and inter-assay CV was 2–12%. The simple sample preparation and minimal sample volume make the method useful for the quantitation of amino acids in both patient and experimental animal samples.
Keywords: chromatography, amino acids, HPLC, reverse phase, fluorescence
1. INTRODUCTION
The determination of amino acids is important in assessing nutritional requirements and status in health and during disease. [1–6] For over thirty years most amino acid analyses have been performed by ion-exchange chromatography with post- column derivatization with ninhydrin. [7] Alternatively, amino acids can be analyzed by precolumn fluorescent derivatization and then separated by reverse-phase high- performance liquid chromatography (HPLC). [8] The most common agent used for the fluorescent derivatization of amino acids is o-phthalaldehyde (OPA). OPA is a highly effective derivatization agent for amino acids because it readily reacts with amino groups to form highly fluorescent products when in the presence of excess thiols. However, these isoindole derivatives are unstable. [9] Previous studies have demonstrated that the formation of OPA derived fluorescent amino compounds are more stable in the presence of the thiol 3-mercaptopropionic acid (MPA) instead of the more commonly used 2-mercaptoethanol. [10–12] In this manuscript we describe a simple and rapid automatic online precolumn derivatization HPLC method for the quantitation of plasma amino acids. The separation of the amino acid derivatives is by reversed-phase chromatography and quantitation by fluorescent detection.
2. EXPERIMENTAL
2.1 Chemicals
All amino acids including norvaline as an internal standard, o-phthalaldehyde (OPA), 3-mercaptopropionic acid (3-MPA), tetrahydrofuran, and potassium tetraborate were obtained from Sigma (St. Louis, MO). Perchloric acid, potassium dihydrogen phosphate, HPLC grade methanol and acetonitrile were obtained from Fisher scientific (Pittsburgh, PA). deionized water was processed through a Milli-Q purification system (Millipore, USA)
2.2 Samples
EDTA plasma samples were collected after an overnight fast from ten healthy female volunteers recruited as approved by the institutional review board and informed consent was obtained from all subjects. Samples were stored at -70°C until assayed.
2.3 Sample preparation
Plasma samples (20 μL) were prepared by adding an equal volume (20 μL) of an internal standard (norvaline 62.5 μmol/L, Sigma, St. Louis, MO) and HPLC grade water (160 μL) for a final volume of 200 μL. The plasma proteins were precipitated by adding 200 μL of 0.5 mol/L perchloric acid. After protein precipitation, the samples were vortexed and centrifuged at 15,000xg for 5 minutes at room temperature. After the samples were centrifuged, 150 μL of the supernatant was collected and filtered in a Spin-X 0.2-μm micro-centrifuge filter (Fisher Scientific, Pittsburgh, PA) by centrifugation at 15,000xg for 1 minute. One hundred microliters (100 μL) of the filtered sample was collected and split between 2 sample vials for HPLC analysis. Samples were split between 2 vials to minimize the time between the derivatization and injection on to the HPLC system. The technique limits any loss in fluorescent signal intensity.
2.4 Standard preparation
All amino acid standards, except glutamine, were prepared in 100 mmol/L HCl and stored at –70°C. A separate glutamine standard was prepared in water, stored at –70°C and added to the standard stock before use. [13] Amino acid standards were prepared similar to samples and set to cover the reported physiologic range. The linearity of the response for each individual standard across five different concentrations ranging from 5 to 1000 μmol/L by plotting the peak area for each amino acid divided by the area of the internal standard vs. concentration. The correlation coefficients for all curves were >0.99.
2.5 Chromatographic system
The HPLC system consisted of a Waters 600E dual piston pump system controller, a Waters 474 fluorescence detector and a Waters 717 plus autosampler all controlled by a Waters Empower software system (Milford, MA) for chromatographic analysis. The HPLC separation of the derivatized amino acids required two mobile phases. Mobile phase A consists of 30 mmol/L potassium dihydrogen phosphate buffer with 0.4% tetrahydrofuran adjusted to pH 7.0 with 4 mol/L KOH, and mobile phase B consists of 50% HPLC grade acetonitrile mixed with HPLC grade water. All buffers were filtered through a 0.2-μm filter and degassed with ultra pure helium. The derivatized amino acids were separated on a Waters (Milford, MA) XTerra RP18 column (100mm x 4.6mm i.d., 5μm particle size) and a Waters XTerra MS C18 guard column (3.9mm x 20 mm, 5μm particle size). The chromatographic separation was performed at room temperature (22°C). The detection was performed fluorometrically with an excitation wavelength of 340 nm and an emission wavelength of 455 nm, and gain at 100. The flow rate of the mobile phase was 1 ml/minute throughout the analysis. All gradient changes were linear. The gradient conditions were as follows: initial conditions are 100% mobile phase A; from time 0 to 22 minutes the gradient changes to 52% mobile phase A and 48% mobile phase B; from 22 to 34 minutes the gradient changes to 40% mobile phase A and 60% mobile phase B; from 34 to 35 minutes the gradient changes to 100% mobile phase A and remains at this condition until the next injection. The total HPLC run time for the separation of the derivatized amino acids in a single sample or standard is 35 minutes. The column is equilibrated in 100% mobile phase A for 10 minutes before the next sample injection.
The amino acid derivatization reagent was prepared fresh each day by dissolving10 mg of OPA in 0.2 mL of methanol and 1.8 mL of 200 mmol/L potassium tetraborate buffer (pH9.5), and 10 μL of 3-MPA was added. This derivatization reagent was further diluted with 8 mL of 200 mmol/L potassium tetraborate buffer (pH9.5) to obtain the final working solution. The final concentrations of OPA and 3-MPA were 7.45 and 11.4 mmol/L, respectively. The derivatization of the amino acids in the samples and standards was performed in an automated fashion using a Waters 717plus autosampler kept at 4°C. The derivatization reaction was started by transferring 50 μL of the derivatization reagent to 50 μL of the prepared sample spiked with the internal standard. The sample and derivatization reagent were mixed three times by the autosampler and incubated for 30 minutes before injection. The final pH of the derivatized samples and standards was 7.8. The Waters XTerra column is stable at higher pH and therefore it was not necessary to further neutralize samples or standards after derivatization. After this incubation 10 μL of the derivatized sample was injected onto the equilibrated HPLC column. The needle of the autosampler was rinsed with 50% methanol/water between injections. During chromatography of the sample, the derivatization of the next sample is started in order that there is no delay in the injection of the next sample.
2.6 Recovery and variability
The recovery of the plasma amino acids was between 91 and 108% as determined by mixing each amino acid standard separately to a final concentration of 62.5μM in a pooled plasma sample, and recovery was calculated as the difference between spiked and unspiked plasma samples. The intra-assay coefficient of variation (CV) was between 1 and 7% as determined by replicate analysis of a standard solution (n=10) and a quality control plasma sample (n=10) in a single run. The inter-assay CV was between 2 and 12% as determined by replicate analysis of the same quality control plasma sample stored at –70°C and analyzed ten separate times over a period of 6 months.
3. RESULTS AND DISCUSSION
The chromatographic separation of an amino acid standard and a plasma sample are shown in the figure (A and B respectively). The method demonstrated good chromatographic separation of 22 amino acids including the internal standard norvaline.
Figure.
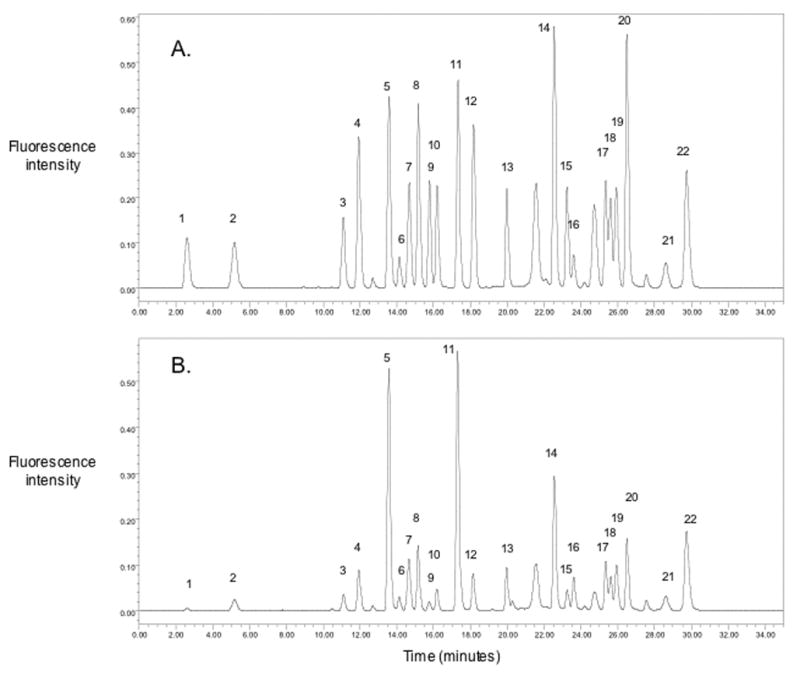
Typical chromatograms of amino acids in a standard (A) and in a human plasma sample (B). 1. aspartic acid, 2. glutamic acid, 3. asparagine, 4. serine, 5. glutamine, 6. histidine, 7. glycine, 8. threonine, 9. citrulline, 10. arginine, 11. alanine, 12. taurine, 13. tyrosine, 14. valine, 15. methionine, 16. norvaline, 17. isoleucine, 18. tryptophan, 19. phenylalanine, 20. leucine, 21. ornithine, 22. lysine.
To compare the calculated plasma amino acid concentrations to other published methods, we analyzed plasma samples collected after an overnight fast from 10 healthy female volunteers. Plasma amino acid concentrations were similar to those reported previously (table). [11,14] The separation of isoleucine, tryptophan and phenylalanine were the most difficult. However, their inter-assay CV was still <10% over a period of 6 months and their calculated values agreed with those reported by others. In addition, we observed that the isoindole derivatives for taurine, ornithine and lysine were more susceptible to degradation over time. However, this was overcome by the online derivitization procedure and standardization of the timing between derivitization and injection of the samples. In this analysis we also observed that perchloric acid protein precipitation was superior to 5-sulfosalicylic acid (SSA) as reported in other methods because SSA interfered with the chromatography of the aspartic acid and glutamic acid peaks. In addition, in this analysis perchloric acid precipitation was not found to adversely affect the quantitative results for the amino acids (table), consistent with a previous report by Qureshi and Qureshi [15] in which they reported no significant difference in amino acid recoveries between SSA and perchloric acid protein precipitation. While this is a reasonably rapid and simple method for the analysis of many plasma amino acids, there are still several important amino acids that are not identified by this method including proline, cysteine, homocysteine, and α-amino isobutyric acid. In addition, we were only able to investigate the plasma amino acid concentration in a small number of healthy nonpregnant female subjects. However, despite the small sample size we were still able to determine that this method was reliable, accurate and consistent with previously reported values.
Table.
Performance characteristics of HPLC-fluorescent detection for plasma amino acids.
| Amino Acid | Elution time (min) [%RSD] | Intra- assay CV (%) | Inter- assay CV (%) | Recovery (%) | Plasma concentration (μmol/L) [range] |
|---|---|---|---|---|---|
| Aspartic Acid | 2.6 [0.9] | 7.03 | 12.01 | 96 | 2.3±0.7 [1.4–3.0] |
| Glutamic Acid | 5.0 [0.9] | 2.03 | 5.90 | 91 | 29.7±7.7 [21.9–44.4] |
| Asparagine | 10.9 [0.4] | 1.42 | 3.21 | 92 | 46.7±8.2 [37.1–63.8] |
| Serine | 11.7 [0.5] | 2.05 | 4.85 | 91 | 85.9±11.4 [73.6–102.8] |
| Glutamine | 13.4 [0.4] | 1.60 | 5.44 | 98 | 533.3±60.6 [423.1–632.2] |
| Histidine | 14.0 [0.4] | 2.58 | 7.21 | 108 | 85.7±9.2 [68.9–100.4] |
| Glycine | 14.5 [0.4] | 3.26 | 4.80 | 103 | 227.8±60.9 [134.5–336.2] |
| Threonine | 15.0 [0.4] | 1.52 | 2.08 | 90 | 146.9±38.4 [98.3–227.9] |
| Citrulline | 15.6 [0.3] | 1.63 | 2.39 | 102 | 34.5±6.2 [23.2–40.6] |
| Arginine | 16.0 [0.4] | 1.46 | 3.01 | 99 | 55.6±15.6 [23.6–79.1] |
| Alanine | 17.2 [0.4] | 1.49 | 2.40 | 93 | 347.8±69.9 [294.7–481.9] |
| Taurine | 18.0 [0.3] | 2.07 | 12.49 | 103 | 47.6±18.2 [32.7–94.2] |
| Tyrosine | 19.9 [0.2] | 1.65 | 5.54 | 102 | 51.1±15.3 [34.6–88.1] |
| Valine | 22.5 [0.2] | 1.53 | 2.32 | 92 | 196.1±20.1 [159.5–225.6] |
| Methionine | 23.2 [0.2] | 3.07 | 6.44 | 103 | 24.4±3.1 [20.3–29.4] |
| Norvaline | 24.0 [0.4] | na | na | na | Internal Standard |
| Isoleucine | 25.3 [0.4] | 1.84 | 2.98 | 93 | 51.4±7.9 [37.6–65.9] |
| Tryptophan | 25.5 [0.2] | 7.42 | 9.49 | 99 | 50.4±8.1 [34.0–64.0] |
| Phenylalanine | 25.9 [0.2] | 2.78 | 3.23 | 101 | 50.5±8.7 [39.6–65.9] |
| Leucine | 26.5 [0.4] | 1.70 | 2.96 | 93 | 105.2±17.2 [85.1–137.2] |
| Ornithine | 28.7 [0.3] | 2.79 | 7.47 | 102 | 52.9±17.7 [32.8–83.8] |
| Lysine | 29.9 [0.3] | 5.18 | 5.37 | 98 | 154.1±11.5 [129.8–168.8] |
Data are mean±SD. (na= not applicable)
In summary, this method demonstrates good precision and reproducibility for all the amino acids analyzed. The required sample volumes are small and no special processing for the samples is needed. The chromatographic method allows for the separation and quantification of amino acids using a simplified gradient and without the need for a column heater. The method shows good precision and reproducibility, it is relatively simple and allows the determination of most plasma amino acids.
Acknowledgments
Source of financial support: National Institutes of Health grant number 5 P01 HD30367
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ingenbleek Y, Barclay D, Dirren H. American Journal of Clinical Nutrition. 1986;43:310. doi: 10.1093/ajcn/43.2.310. [DOI] [PubMed] [Google Scholar]
- 2.Young VR. Age & Ageing. 1990;19 doi: 10.1093/ageing/19.suppl_1.s10. [DOI] [PubMed] [Google Scholar]
- 3.Sprung CL, Cerra FB, Freund HR, Schein RM, Konstantinides FN, Marcial EH, Pena M. Critical Care Medicine. 1991;19:753. doi: 10.1097/00003246-199106000-00004. [DOI] [PubMed] [Google Scholar]
- 4.Divino Filho JC, Barany P, Stehle P, Furst P, Bergstrom J. Nephrology Dialysis Transplantation. 1997;12:2339. doi: 10.1093/ndt/12.11.2339. [DOI] [PubMed] [Google Scholar]
- 5.MacLaren DP, Nevill AM, Thake CD, Campbell IT, Cheetham E, Keegan MA, Lane C, Roberts NB. Medicine & Science in Sports & Exercise. 2000;32:1244. doi: 10.1097/00005768-200007000-00010. [DOI] [PubMed] [Google Scholar]
- 6.Cynober LA. Nutrition. 2002;18:761. doi: 10.1016/s0899-9007(02)00780-3. [DOI] [PubMed] [Google Scholar]
- 7.Friedman M. Journal of Agricultural & Food Chemistry. 2004;52:385. doi: 10.1021/jf030490p. [DOI] [PubMed] [Google Scholar]
- 8.Furst P, Pollack L, Graser TA, Godel H, Stehle P. Journal of Chromatography A. 1990;499:557. doi: 10.1016/s0021-9673(00)97000-6. [DOI] [PubMed] [Google Scholar]
- 9.Garcia Alvarez-Coque MC, Medina Hernandez MJ, Villanueva Camanas RM, Mongay Fernandez C. Analytical Biochemistry. 1989;178:1. doi: 10.1016/0003-2697(89)90346-1. [DOI] [PubMed] [Google Scholar]
- 10.Godel H, Graser T, Foldi P, Pfaender P, Furst P. Journal of Chromatography A. 1984;297:49. doi: 10.1016/s0021-9673(01)89028-2. [DOI] [PubMed] [Google Scholar]
- 11.Teerlink T, van Leeuwen P, Houdijk A. Clin Chem. 1994;40:245. [PubMed] [Google Scholar]
- 12.Vasanits A, Kutlan D, Sass P, Molnar-Perl I. Journal of Chromatography A. 2000;870:271. doi: 10.1016/s0021-9673(99)00942-5. [DOI] [PubMed] [Google Scholar]
- 13.Grossie VB, Jr, Yick J, Alpeter M, Welbourne TC, Ota DM. Clinical Chemistry. 1993;39:1059. [PubMed] [Google Scholar]
- 14.Le Boucher J, Charret C, Coudray-Lucas C, Giboudeau J, Cynober L. Clinical Chemistry. 1997;43:1421. [PubMed] [Google Scholar]
- 15.Qureshi GA, Qureshi AR. Journal of Chromatography A. 1989;491:281. doi: 10.1016/s0378-4347(00)82846-7. [DOI] [PubMed] [Google Scholar]