

Abstract
Cre recombinase catalyzes site-specific recombination between two 34-bp loxP sites in a variety of DNA substrates. At the start of the recombination pathway, the loxP sites are each bound by two recombinase molecules, and synapsis of the sites is mediated by Cre–Cre interactions. We describe the structures of synaptic complexes formed between a symmetrized loxP site and two Cre mutants that are defective in strand cleavage. The DNA in these complexes is bent sharply at a single base pair step at one end of the crossover region in a manner that is atypical of protein-induced DNA bends. A large negative roll (−49°) and a positive tilt (16°) open the major groove toward the center of the synapse and compress the minor groove toward the protein–DNA interface. The bend direction of the site appears to determine which of the two DNA substrate strands will be cleaved and exchanged in the initial stages of the recombination pathway. These results provide a structural basis for the observation that exchange of DNA strands proceeds in a defined order in some tyrosine recombinase systems. The Cre-loxS synaptic complex structure supports a model in which synapsis of the loxP sites results in formation of a Holliday junction-like DNA architecture that is maintained through the initial cleavage and strand exchange steps in the site-specific recombination pathway.
Most site-specific recombination systems in bacteria and yeast fall into one of two large families that carry out a similar repertoire of genetic rearrangements, including integration and excision of bacteriophage genomes into and out of host chromosomes, resolution of transposition intermediates, regulation of gene expression and plasmid copy number by inversion of DNA segments, and resolution of multimeric circular replicons generated by homologous recombination events (reviewed in refs. 1 and 2). The integrase family, also known as the “tyrosine recombinases” (3), includes over 100 members that share a conserved catalytic domain responsible for cleavage and ligation of the DNA substrates (4). The second large family of site-specific recombinases includes the resolvase enzymes from the γδ- and Tn3 transposons and the Gin and Hin invertases (1).
The integrase family proteins carry out site-specific recombination by stepwise cleavage and exchange of each strand in the DNA substrates. Conserved tyrosine residues first cleave the scissile phosphates in sites recognized specifically by the recombinase proteins to form transient 3′-phosphotyrosine linkages and free 5′-hydroxyl groups (Fig. 1a). Intermolecular attack of the resulting phosphotyrosine linkages by 5′-hydroxyl groups results in the exchange of the first pair of DNA strands and the formation of a Holliday junction intermediate. The Holliday intermediate is then a substrate for a second round of cleavage and strand exchange steps that results in recombinant products. The active site chemistry of the cleavage and ligation steps is similar to that described recently for the eukaryotic topoisomerase Ib enzymes (3, 5, 6). In contrast to the tyrosine recombinases, the resolvase/invertase enzymes cleave all four DNA strands with conserved serine residues in a concerted manner to form 5′-phosphoserine linkages. Intermolecular attacks by the free 3′-hydroxyl groups give recombinant products without formation of a Holliday junction intermediate.
Figure 1.

Cre-loxP site-specific recombination. (a) Schematic representation of the initial steps of the site-specific recombination pathway (11, 12). The Holliday junction intermediate (iii) is a substrate for a second round of cleavage and strand exchange steps, which result in recombinant products. (b) The loxP and loxS DNA sequences. The inverted repeat regions recognized specifically by Cre are underlined. Cleavage sites are indicated by arrows, and the three base pairs changed in loxP to create the fully symmetric loxS site are boxed. The additional 5′-T overhangs in loxS were found to facilitate crystallization.
Cre recombinase is one of the simplest members of the integrase family. The 38-kDa recombinase protein is a monomer in solution (7) and binds cooperatively to 34-bp loxP sites (Fig. 1b) with subnanomolar affinity (7, 8). Cre catalyzes the complete recombination reaction between loxP-containing DNA substrates in vitro in the absence of accessory proteins and in the absence of accessory DNA sequences (7). The simplicity of this system has led to its widespread use as a tool for both in vitro manipulations of DNA and in vivo DNA rearrangements in transgenic plants, insects, and mammals (9, 10). We have been using the Cre-loxP system as a model for studying the structures of reaction intermediates in the site-specific recombination pathway with the goal of developing a three-dimensional mechanistic framework for this multistep process. Although structural models for the covalent intermediate (ii in Figs. 1a and 2) and for the Holliday junction intermediate (iii in Figs. 1a and 2) in the Cre-loxP pathway have recently been reported (11, 12), there is no high-resolution structural information available for the synapsed DNA sites (i in Figs. 1a and 2) for any member of the tyrosine recombinase family.
Figure 2.

Common architectures of Cre-DNA intermediates in the initial steps of the site-specific recombination pathway as shown schematically in Fig. 1a. Structure i is the Cre/loxS synaptic complex described in this work. Structures ii and iii are described in refs. 11 and 12, respectively. Subunits activated for cleavage of the DNA substrate are colored green and the view is from the catalytic domain side of the synapse.
Here we describe two crystal structures representing the synaptic complex formed between Cre-bound loxP sites at the start of the recombination reaction. When a symmetric loxP site (loxS; Fig. 1b) and active site recombinase mutants R173K or Y324F were used, a stable synaptic complex was formed and crystallized in the absence of cleaved DNA intermediates. Both the 2.2-Å structure of the Cre R173K/loxS complex and the 2.7-Å structure of the Cre Y324F/loxS complex reveal a twofold-symmetric synapse containing four recombinase subunits and two DNA duplex substrates. The loxS sites are sharply and asymmetrically bent within the crossover region in a manner that is atypical of protein-induced DNA bends. The structure of this initial complex in the Cre-loxP site-specific recombination pathway indicates that the location of the kink within the crossover region is coupled to the direction of bending in the loxP site, and both dictate which DNA strand is cleaved first in the recombination pathway.
MATERIALS AND METHODS
Crystallization, Data Collection, and Refinement of Cre-loxS Complexes.
Expression plasmids for Cre R173K and Cre Y324F were constructed and proteins were purified as described previously for Cre R173K (12). Plate-shaped crystals (200 × 200 × 50 μm) of the Cre R173K/loxS complex were grown over a period of 6 weeks from 6-μl hanging drops containing 13 μM protein, 10 μM loxS site, 20 mM sodium acetate at pH 5.6, 20 mM CaCl2, 1 mM dithiothreitol (DTT), and 25% (vol/vol) 2-methyl-2,4-pentanediol (MPD) that were equilibrated against reservoirs of 20 mM sodium acetate at pH 5.0, 20 mM CaCl2, 35% MPD at 18°C. Smaller plates of Cre Y324F/loxS (150 × 150 × 40 μm) were grown from 3-μl hanging drops after centrifugation of 100 μM protein, 75 μM loxS site, 50 mM sodium acetate at pH 5.6, 24% MPD, 20 mM CaCl2, 40 mM NaCl, and 2 mM DTT and equilibration against reservoirs containing 100 mM sodium acetate at pH 5.0, 25% MPD, and 20 mM CaCl2 at 18°C.
Crystals were flash frozen in liquid propane and stored in liquid nitrogen prior to data collection as described (13). Cre R173K/loxS data were measured at 100 K at the Cornell High Energy Synchrotron Source (CHESS) F1 beamline using a QUANTUM4 charge-coupled device detector and processed by using mosflm (14). The crystals are orthorhombic, space group C2221, a = 108.8, b = 122.1, c = 181.0 Å, with two Cre R173K recombinase molecules and one loxS site in the asymmetric unit. Cre Y324F/loxS diffraction data were measured at the National Synchrotron Light Source (NSLS) X25 beamline, using a MAR Research image plate detector. The crystals belong to the same space group, with a = 107.1, b = 121.1, c = 180.0 Å.
Both structures were determined by rigid-body refinement of the Cre-loxA protein domains and 13-bp DNA arms as starting models (11). Iterative cycles of positional refinement in x-plor (15) and model building in o (16) were performed at 2.8-, 2.5-, and 2.2-Å resolution for Cre R173K/loxS and at 3.0- and 2.7-Å resolution for Cre Y324F/loxS. The C-terminal residues (320–341) of the recombinases and the central eight base pairs of loxS were excluded from all refinements until they could be unambiguously fit into unbiased electron density maps. A summary of data collection and refinement statistics is given in Table 1 and representative electron density is shown in Fig. 4.
Table 1.
Data collection and refinement statistics
| R173K/loxS | Y324F/loxS | |
|---|---|---|
| Data collection | ||
| Resolution, Å | 32.3–2.2 | 48.2–2.7 |
| Unique reflections | 55,807 | 32,242 |
| Redundancy | 2.4 | 4.7 |
| Completeness,* % | 92/94 | 97/94 |
| Rsym* | 0.062/0.261 | 0.066/0.230 |
| Refinement | ||
| R/Rfree† | 0.213/0.252 | 0.222/0.295 |
| No. solvent atoms | 473 | 314 |
| rms bonds, Å | 0.007 | 0.009 |
| rms angles, deg | 1.3 | 1.2 |
Overall value/highest resolution shell.
Rfree was calculated for 5% of the data not included in the refinement. Definitions of R, Rfree, and Rsym are given in ref. 13.
Figure 4.

Stereo representation of electron density (weighted 2Fo − Fc; contoured at 1.2σ) for the loxS kink. Coloring of the DNA strands is the same as in Fig. 3 b and c. Well-defined solvent molecules are drawn as red spheres, and Lys-86 from the inactive subunit is indicated.
Geometric Calculations and Illustrations.
Base pair tilt (+16°) and roll (−49°) parameters reported in the text and figures are the vector-based definitions used by Dickerson in the freehelix program (17). Estimates of base pair tilt and roll in the loxS bend depend on the computational method used. freehelix and the program curves (18) both calculate an inter-base pair tilt of +22° with respect to a local helical axis and curves computes a +16° tilt with respect to a global axis. Base pair roll values are −49°, −45°, and −38°, obtained by using the freehelix and curves local helical axis and the curves global axis methods, respectively. Figs. 2 and 3 were produced with ribbons (19), and Fig. 4 was generated with molscript (20).
Figure 3.

The Cre R173K/loxS complex and bending of the loxS DNA. (a) A “cleaving” Cre subunit bound to a loxS half-site. Active-site side chains and side chains contacting the continuous DNA strand are drawn in red. The scissile phosphate in the active site is represented by a blue sphere. The view is from the center of the synapse. (b) Synapsed loxS sites, with scissile phosphates circled and the bound recombinases omitted. Phosphates activated for cleavage are indicated. Angles are between the 13-bp inverted repeat segments of the DNA arms (see text). (c) Close-up stereo representation of the loxS bend shown in b. The view directions of b and c are the same as in Fig. 2.
RESULTS AND DISCUSSION
Design and Construction of Synaptic Cre-DNA Complexes.
To avoid cleavage of DNA substrates during crystallization experiments, two active-site mutants of Cre recombinase (Y324F and R173K) were used in these studies. Tyr-324 and Arg-173 are strictly conserved amino acids in the integrase family and are essential for catalytic activity (21). Mutation of Tyr-324 removes the attacking nucleophile and results in a recombinase that is defective in strand cleavage. Changing Arg-173 to lysine results in an enzyme with undetected in vitro activity in the complete recombination reaction (22) or in cleavage assays with suicide DNA substrates (data not shown). For these studies, we used a symmetric version of the loxP site (loxS; Fig. 1b), which differs from the wild-type sequence by three base pair changes. The loxS site contains two identical wild-type half-sites and is competent in in vitro recombination assays with linear DNA substrates (data not shown). This symmetric sequence is the same as that used to form a covalent Cre-DNA intermediate with a nicked suicide substrate (loxA; ref. 11)) and is the same as that used to form a nicked Cre–Holliday junction complex (HJ2; ref. 12).
Overall Structure of the Synaptic Complex.
For both the Cre R173K/loxS and Cre Y324F/loxS complexes, synchrotron radiation was used to optimize the resolution of the diffraction data, and a combination of rigid-body optimization and torsion-angle dynamics was used to determine and refine the structures. Details of the refinements are outlined in Materials and Methods, and the results are summarized in Table 1. As expected from the preliminary crystallographic data indicating nearly isomorphous structures, the overall architecture of the Cre-loxS synapse is closely related to that seen in the covalent Cre-DNA intermediate (11) and in the Cre–Holliday junction complexes (12) (Fig. 2). The two Cre/loxS halves of the synapse are related by exact twofold symmetry by virtue of a crystallographic dyad that passes through the center of the overall complex. One recombinase subunit binds to each of the four loxS inverted repeats in the synaptic complex (Fig. 3a), resulting in a pseudo-fourfold symmetric arrangement of Cre-bound loxS half-sites. These Cre-half-site complexes become the “arms” of the Holliday junction intermediate after the first strand exchange step. The DNA arms are sharply bent within the crossover region of each loxS site (Figs. 3 b and c and 4), and the four arms in the synapse lie nearly in the same plane. The N-terminal domains of the recombinases form a network of intersubunit contacts on one side of the substrate plane and the C-terminal domains interact primarily through a cyclic exchange of helices between subunits on the opposite side.
Although the architectural features of the Cre R173K/loxS and Cre Y324F/loxS complexes are the same, the quality of the electron density in key regions of the structures is quite different. The R173K complex is very well ordered nearly everywhere, with a clear image of the crossover region DNA (see Fig. 4), the active sites of the recombinases, and the solvent-mediated Cre-DNA interface. In contrast, the Y324F complex is less well defined overall, as indicated by the lower resolution of the diffraction data obtained. This structure has quite weak density in the region of active site residue Phe-324, presumably because the wild-type Tyr-324 forms important hydrogen bonds (shown schematically in Fig. 5) that dock helix M (residues 319–325) into the active site. Electron density for bases T1′, A1, and T2 in the crossing strand is also poorly defined in this complex. The descriptions of the loxS geometry will therefore refer to the Cre R173K/loxS structure in the discussions that follow, with similar results obtained for the Y324F/loxS structure.
Figure 5.

(a) Model for bending and cleavage of the loxP site during site-specific recombination. Bending in the “left” half of the crossover region stimulates cleavage in the “right” half (indicated by arrow). Bending in the right half of the crossover region stimulates cleavage in the left half. The two alternative bend locations shown require opposite bending directions of the loxP site. (b) Schematic of direct and solvent-mediated Cre–DNA interactions in the central eight base pairs of the loxS site. The contacts shown have distances between non-hydrogen atoms less than 3.5 Å. Water molecules and the scissile phosphates are drawn as filled circles.
DNA-Bending in the Cre-loxS Synapse.
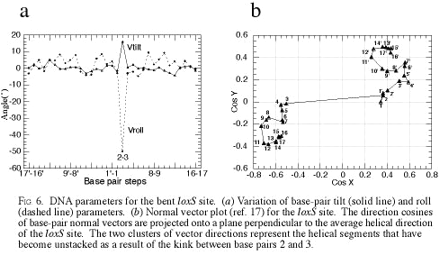
It was anticipated, on the basis of the structures of other Cre-DNA reaction intermediates, characterization of synaptic Cre-loxP complexes in solution (23), and DNA-bending studies in the Flp system (24), that the loxP sites would be bent in the synaptic complex. The location and type of bend, however, were not easily predicted from previous studies. As shown in Figs. 2–4, the loxS sites are bent sharply and asymmetrically within the crossover region. The resulting angle between DNA arms (defined here as the angle between inertial axes of the 13-bp inverted repeats) is 102°, which is identical to the angle observed between DNA arms in the Cre–Holliday junction intermediate (12). The 78° deviation from a straight duplex comes primarily from a kink between the second and third base pairs (T2⋅A2-G3⋅C3) from one end of the central crossover region. A large negative roll (−49°) and a positive tilt (+16°; see Materials and Methods) produce a single kink in the duplex loxS site that results in an opening of the major groove toward the center of the synapse and a compression of the minor groove in the direction of the bend. The kink also causes a 22° buckle between the T2 and A2 bases. On both sides of the kink, the loxS DNA adopts nearly canonical B-DNA helical parameters, with only modest curvature observed within the two inverted repeat segments. The localized distortion is most evident in Fig. 6, which is published as supplemental data on the PNAS web site, www.pnas.org. Fig. 6 shows the variation in tilt and roll parameters and a base pair normal vector distribution for the synapsed loxS site.
The most striking consequence of the loxS bend is the exposure of T⋅A and G⋅C base pairs to solvent (Fig. 4). These base pairs are entirely unstacked, with no intercalation of protein side chains. A total of 300 Å2 of solvent-accessible surface is exposed by the two base pairs, compared with ≈135 Å2 expected in standard B-DNA. There is electron density present for at least one ordered solvent molecule in this gap that does not form hydrogen bonds with the base heteroatoms (see Fig. 4). A second feature of the loxS bend is the unusually large tilt angle (16°–22°; see Materials and Methods) between T2⋅A2 and G3⋅C3 base pairs. This type of deformation from standard B-DNA helicoidal parameters has rarely been observed in structural studies of protein–DNA complexes (17). To achieve a large tilt angle, the deoxyribose-phosphate backbone must adopt a partially extended conformation in one of the two strands. In the loxS kink, the T-G linkage is the most distorted, as shown in Figs. 3c and 4.
Many proteins that induce severe DNA bends within a specific protein–DNA interface use intercalation of side chains to create sharp kinks (25). In the Cre/loxS complex, the specific protein–DNA interfaces are formed with the 13-bp inverted repeats of the loxP site and involve only modest DNA bending. The kink in the loxS site is produced in the central “nonspecific” strand exchange region, where there are few DNA contacts made by the bound recombinases. Protein–protein interactions between subunits bound to the inverted repeats that flank the kink site are apparently responsible for stabilizing the bent conformation, with further stabilization coming from the additional protein–protein interface generated by synapsis of the sites.
Asymmetric Cre–DNA Interactions in the Central Crossover Region.
The overall asymmetry of the Cre-bound loxS site allows us to clearly distinguish the two unique recombinase active sites and the two unique DNA strands in each half of the synaptic complex. By comparison with the structure of the covalent Cre-DNA intermediate where cleavage has taken place in one, but not the other, active site (11), we can make the following assignments. The recombinase subunits colored green in structure i of Fig. 2 adopt a cleaving configuration (but cannot cleave the substrate because the active sites have been mutated), whereas the subunits colored magenta adopt a noncleaving configuration. Similarly, we refer to the DNA strand containing the activated scissile phosphate as “crossing” and the strand containing the phosphate prevented from cleavage at the initial stage of the reaction as “continuous” (Fig. 3b), in anticipation of strand exchange and of the structural roles of these strands upon formation of the Holliday junction intermediate (12, 26, 27).
The 2.2-Å structure of the Cre R173K/loxS complex provides a more detailed model for the Cre–DNA interface than was available from the structures of other reaction intermediates in this system, particularly with respect to the role of tightly bound solvent. Here we will describe only the interactions that involve the crossover region (summarized in Fig. 5b). The details of the specific interface within the 13-bp inverted repeat segments of loxP will be reported elsewhere. Consistent with the requirement of the DNA strands to move during recombination is the observation that the bases in the central region are primarily contacted by solvent and not by the recombinases. The single exception involves Lys-86, which hydrogen bonds to O6 of G4 and G4′ at the extreme ends of the central region (Fig. 4). In the wild-type loxP spacer this interaction would presumably be found only at G4, because an adenine base would occupy the equivalent position in the opposite half-site (see Fig. 1b).
The scissile phosphates (G4-C3 and G4′-C3′) are contacted in a similar manner by active-site residues from the two different Cre subunits, but the remainder of the phosphate contacts are quite different for the crossing vs. continuous DNA strands. The crossing strand is free from direct Cre contacts throughout the center of the spacer, with water-mediated contacts made to the flanking C3′-A2′, T2-G3, and G3-C4 phosphates. The A2′-T1′, T1′-A1, and A1-T2 phosphates of this strand are directed toward the center of the synapse, away from the recombinase binding surface (Figs. 2 and 3). In contrast, the continuous strand phosphate backbone faces the recombinase dimer bound to the loxS site and is contacted both directly and via water molecules (Figs. 3a and 5b). The pattern of contacts between Cre and the backbone atoms of the DNA crossover region is therefore consistent with one of the two strands being positioned for transfer after cleavage, with the other more tightly bound in the Cre–DNA interface.
Implications of DNA Bending in Strand Exchange.
The crossing strands that are to be cleaved and exchanged between opposite halves of the synapse have helical trajectories that place them in the central strand exchange region, where the phosphate backbones from the two substrates approach one another very closely at the central T1′-A1 step (Fig. 2c). Although the buffers used to crystallize the Cre/loxS complexes contained Ca2+ and Na+, we do not observe electron density consistent with a unique binding site for ions bridging these phosphates. A weak torus of electron density around the phosphate-phosphate vector instead suggests that an ensemble of such interactions might be made. Both the sharp bend in the loxS site and electrostatic repulsion between phosphates in the crossing strands are formed during synapsis with an energetic cost that is most likely provided by formation of the synaptic protein–protein interface (described in ref. 11). An intriguing possibility is that the stereochemical energy stored in DNA bending and in the close phosphate–phosphate interaction is used to promote the strand exchange step after cleavage.
The scissile phosphate that is activated for cleavage in the Cre R173K/loxS and Cre Y324F/loxS complexes is at the opposite end of the crossover region with respect to the kink. The inactivated phosphate is consequently located adjacent to the kink. If the wild-type loxP site is more readily bent at one end of the crossover region (e.g., at the T2-G3 step as in loxS) vs. the other end (at the C2′-A3′ step), then a corresponding preference for initially cleaving the top vs. the bottom strand should exist (Fig. 5a). The alternative bend in the loxP site differs in sequence, with Cyt and Ade bases instead adopting the most open and solvent-exposed positions. As shown in Fig. 5a, the two alternative bend sites within the loxP crossover region imply opposite bending directions of the overall site. In the Cre-loxP system, a preference for initial cleavage at G4-C3 has been demonstrated for an intramolecular excision reaction (23, 28), but it is not yet clear whether this result represents an intrinsic bias of the loxP site or a preference imposed by the entire DNA substrates used.
Some tyrosine recombinases display a strong preference for the DNA strand that is initially cleaved, but the selectivity is not determined by a bending preference within the crossover region. The requirement for assembly of a specific nucleoprotein architecture with accessory proteins and accessory DNA sequences has the effect of dictating the bend direction (and therefore which strand is initially cleaved) by providing structural constraints on synapsis. Lambda integrase and the XerC/XerD recombinases acting at plasmid sites both behave in this manner at the initial synapsis and cleavage steps in the site-specific recombination pathway (29–31).
The most well-studied member of the tyrosine recombinase family with respect to DNA bending is the Saccharomyces cerevisiae 2-μm circle Flp recombinase (32). Sadowski and co-workers (24, 33), using cyclic permutation gel-shift assays, have demonstrated that Flp bends its recombination site by >140° in the absence of synapsis and that the bend center is approximately at the center of the crossover region. While this result appears to contradict the location of the bend observed in the Cre/loxS structure, Fig. 5a provides a possible explanation. If Flp bends its recombination site equally in both directions, as is suggested by the lack of a strong preference in the order of strand cleavages in the recombination reaction (34), then the electrophoretic mobilities of Flp-DNA complexes may represent that of two species (the alternatives in Fig. 5a) that differ by ≈4 base pairs in bend centers. Alternatively, there may be differences in bending geometry between synapsed and unsynapsed recombination sites.
Regulation of Cleavage in Cre-loxP Site-Specific Recombination.
The active site of Cre recombinase contains five amino acids that are either strictly conserved (Arg-173, His-289, Arg-292, and Tyr-324) or donate a conserved hydrogen bond (Trp-315) in the Int family (4, 11, 35). A comparison of these catalytic residues indicates that they are nearly superimposable (rms difference < 0.3 Å for Cα atoms) between the active sites of the “cleaving” and “noncleaving” Cre subunits. What, then, is the stereochemical basis for formation of an “active” site on the crossing-strand scissile phosphate and formation of an “inactive” site on the continuous-strand scissile phosphate? Although the Cre/loxS structures described here do not alone provide a clear model for this allosteric regulation, the combined structural models of all three of the primary intermediates in the site-specific recombination pathway do provide a great deal of insight and will be discussed in detail elsewhere. Briefly, the mobility of the tyrosine nucleophile, small rotations of the scissile phosphate, and the proximity of Lys-201, an amino acid only recently identified as conserved in the Int family (3, 6) and required for Cre-loxP catalysis (F.G. and G.D.V.D., unpublished data), may all contribute to the active site switch mechanism.
The Cre/loxS structures described here, combined with the structural models of other intermediates in the Cre-loxP pathway (11, 12), lead to the following model for synapsis and cleavage of DNA substrates. Two loxP sites bent in the same direction can associate to form a single antiparallel synaptic complex. Because the scissile phosphate to be initially cleaved is determined by the bend direction of the site, the antiparallel synapses formed from the alternatively bent substrates shown in Fig. 5a are predicted to proceed through the reaction pathway with an opposite order of strand exchanges. If bending of the loxP site occurs readily in both directions, then sites should form parallel synapses as well. In this case, however, base mismatches encountered during the strand-transfer step of the reaction would be expected to block formation of recombinant products (36–39). Formation of parallel synapses between λ-integrase-bound attL sites has recently been visualized by atomic force microscopy (40).
The relationship between strand cleavage and bending of the loxP site discussed here is closely related to other models described for tyrosine recombinases (24, 41). Jayaram and colleagues (34), for example, have used bulged DNA substrates for Flp recombination and have analyzed strand cleavage preference as a function of bulge position. When bulges were placed in the crossover region of either the top or bottom strands of the Flp recombination sequence, cleavage of the scissile phosphate located on the bulged strand was greatly stimulated. The Cre/loxS and Cre/loxA (11) complexes indicate that a bulge could be most readily accommodated in the crossing strand of a synaptic complex, which is the strand whose scissile phosphate is activated for cleavage. Similar experiments using bulged loxP substrates have been performed recently in the Cre recombinase system, with results that also support the bending/cleavage relationship inferred from the Cre/loxS structural models (M. Jayaram, personal communication).
Supplementary Material
Acknowledgments
We thank Dr. M. Jayaram for sharing unpublished results and the National Synchrotron Light Source X25 and Cornell High Energy Synchrotron Source F1 beamline staffs for access and support during synchrotron radiation experiments. This work was supported by Grant GM55041 from the National Institutes of Health.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
A Commentary on this article begins on page 7122.
Data deposition: The atomic coordinates of the Cre R173K/loxS and Cre Y324F/loxS structures have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID codes 4CRX and 5CRX, respectively).
References
- 1.Stark W M, Boocock M R, Sherratt D J. Trends Genet. 1992;8:432–439. [PubMed] [Google Scholar]
- 2.Hallet B, Sherratt D J. FEMS Microbiol Rev. 1997;21:157–178. doi: 10.1111/j.1574-6976.1997.tb00349.x. [DOI] [PubMed] [Google Scholar]
- 3.Sherratt D J, Wigley D B. Cell. 1998;93:149–152. doi: 10.1016/s0092-8674(00)81566-4. [DOI] [PubMed] [Google Scholar]
- 4.Nunes-Düby S E, Kwon H J, Tirumalai R S, Ellenberger T, Landy A. Nucleic Acids Res. 1998;26:391–406. doi: 10.1093/nar/26.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stewart L, Redinbo M R, Qiu X, Hol W G J, Champoux J J. Science. 1998;279:1534–1540. doi: 10.1126/science.279.5356.1534. [DOI] [PubMed] [Google Scholar]
- 6.Cheng C, Kussie P, Pavletich N, Shuman S. Cell. 1998;92:841–850. doi: 10.1016/s0092-8674(00)81411-7. [DOI] [PubMed] [Google Scholar]
- 7.Abremski K, Hoess R. J Biol Chem. 1984;259:1509–1514. [PubMed] [Google Scholar]
- 8.Ringrose L, Lounnas V, Ehrlich L, Buchholz F, Wade R, Stewart A F. J Mol Biol. 1998;284:363–384. doi: 10.1006/jmbi.1998.2149. [DOI] [PubMed] [Google Scholar]
- 9.Kilby N J, Snaith M R, Murray J A. Trends Genet. 1993;9:413–421. doi: 10.1016/0168-9525(93)90104-p. [DOI] [PubMed] [Google Scholar]
- 10.Sauer B. Methods. 1998;14:381–392. doi: 10.1006/meth.1998.0593. [DOI] [PubMed] [Google Scholar]
- 11.Guo F, Gopaul D N, Van Duyne G D. Nature (London) 1997;389:40–46. doi: 10.1038/37925. [DOI] [PubMed] [Google Scholar]
- 12.Gopaul D N, Guo F, Van Duyne G D. EMBO J. 1998;17:4175–4187. doi: 10.1093/emboj/17.14.4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Duyne G D, Ghosh S, Maas W K, Sigler P B. J Mol Biol. 1996;256:377–391. doi: 10.1006/jmbi.1996.0093. [DOI] [PubMed] [Google Scholar]
- 14.Abrahams J P, Leslie A G W. Acta Cryst D. 1996;52:30–42. doi: 10.1107/S0907444995008754. [DOI] [PubMed] [Google Scholar]
- 15.Brünger A T. x-plor Version 3.1 Manual. New Haven, CT: Yale University; 1993. [Google Scholar]
- 16.Jones T A, Zou J Y, Cowan S W, Kjeldgaard M. Acta Cryst A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 17.Dickerson R E. Nucleic Acids Res. 1998;26:1906–1926. doi: 10.1093/nar/26.8.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lavery R, Sklenar H. J Biomol Struct Dyn. 1988;6:63–91. doi: 10.1080/07391102.1988.10506483. [DOI] [PubMed] [Google Scholar]
- 19.Carson M. J Appl Crystallogr. 1991;24:958–961. [Google Scholar]
- 20.Kraulis P. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 21.Wierzbicki A, Kendall M, Abremski K, Hoess R. J Mol Biol. 1987;195:785–794. doi: 10.1016/0022-2836(87)90484-0. [DOI] [PubMed] [Google Scholar]
- 22.Abremski K E, Hoess R H. Protein Eng. 1992;5:87–91. doi: 10.1093/protein/5.1.87. [DOI] [PubMed] [Google Scholar]
- 23.Hoess R, Wierzbicki A, Abremski K. In: Structure and Methods. Sarma R H, Sarma M H, editors. Vol. 1. New York: Adenine; 1990. pp. 203–213. [Google Scholar]
- 24.Luetke K H, Sadowski P D. J Mol Biol. 1995;251:493–506. doi: 10.1006/jmbi.1995.0451. [DOI] [PubMed] [Google Scholar]
- 25.Allemann R K, Egli M. Chem Biol. 1997;4:643–650. doi: 10.1016/s1074-5521(97)90218-0. [DOI] [PubMed] [Google Scholar]
- 26.Azaro M A, Landy A. EMBO J. 1997;16:3744–3755. doi: 10.1093/emboj/16.12.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arciszewska L K, Grainge I, Sherratt D J. EMBO J. 1997;16:3731–3743. doi: 10.1093/emboj/16.12.3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoess R, Wierzbicki A, Abremski K. Proc Natl Acad Sci USA. 1987;84:6840–6844. doi: 10.1073/pnas.84.19.6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nunes-Düby S, Matsumoto L, Landy A. Cell. 1987;50:779–788. doi: 10.1016/0092-8674(87)90336-9. [DOI] [PubMed] [Google Scholar]
- 30.Kitts P A, Nash H A. J Mol Biol. 1988;204:95–107. doi: 10.1016/0022-2836(88)90602-x. [DOI] [PubMed] [Google Scholar]
- 31.Colloms S D, Bath J, Sherratt D J. Cell. 1997;88:855–864. doi: 10.1016/s0092-8674(00)81931-5. [DOI] [PubMed] [Google Scholar]
- 32.Sadowski P D. Prog Nucleic Acids Res Mol Biol. 1995;51:53–91. [PubMed] [Google Scholar]
- 33.Schwartz C J, Sadowski P D. J Mol Biol. 1989;205:647–658. doi: 10.1016/0022-2836(89)90310-0. [DOI] [PubMed] [Google Scholar]
- 34.Lee J, Tonozuka T, Jayaram M. Genes Dev. 1997;11:3061–3071. doi: 10.1101/gad.11.22.3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Argos P, Landy A, Abremski K, Egan J B, Haggard-Ljungquist E, Hoess R H, Kahn M L, Kalionis B, Narayana S V, Pierson L S, 3d, et al. EMBO J. 1986;5:433–440. doi: 10.1002/j.1460-2075.1986.tb04229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nunes-Düby S, Azaro M A, Landy A. Curr Biol. 1995;5:139–148. doi: 10.1016/s0960-9822(95)00035-2. [DOI] [PubMed] [Google Scholar]
- 37.Burgin A B, Nash H A. Curr Biol. 1995;5:1312–1321. doi: 10.1016/s0960-9822(95)00258-2. [DOI] [PubMed] [Google Scholar]
- 38.Lee J, Jayaram M. J Biol Chem. 1995;270:4042–4052. doi: 10.1074/jbc.270.8.4042. [DOI] [PubMed] [Google Scholar]
- 39.Zhu X D, Pan G, Luetke K, Sadowski P D. J Biol Chem. 1995;270:11646–11653. doi: 10.1074/jbc.270.19.11646. [DOI] [PubMed] [Google Scholar]
- 40.Cassell G, Moision R, Rabani E, Segall A. Nucleic Acids Res. 1999;27:1145–1151. doi: 10.1093/nar/27.4.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Voziyanov Y, Pathania S, Jayaram M. Nucleic Acids Res. 1999;27:930–941. doi: 10.1093/nar/27.4.930. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}