

Summary
Mastadenoviruses represent one of the four major genera of the Adenoviridae family comprising a variety of mammalian pathogens including human adenovirus (Ad), whose genomes encode a gene for minor core protein V (pV), not found in other genera of Adenoviridae. Deletion of other genus-specific genes (gene IX and E3 genes) from Ad type 5 (Ad5) genome has been experimentally studied in vitro and the results on biological characterization of the mutants support the phylogenetic evidence of those genes being non-essential for Ad viability. On this basis it seemed logical to suggest that a deletion of gene V from the Ad5 genome could also be tolerated. To test this hypothesis we constructed and rescued the first pV-deletion mutant of human Ad5. As compared to Ad5, this mutant formed small plaques, had dramatically reduced thermostability and lower infectivity. A subsequent thermoselection screen of the pV-deleted Ad5 allowed isolation of a suppressor mutant Ad5-dV/TSB with restored biological characteristics. Since replication and viral assembly of Ad5-dV/TSB could still occur in the absence of pV, we conclude that pV is a non-essential component of the virion. The observed rescue of the biological defects appears to be associated with a cluster of point mutations in the gene encoding the precursor for the other core protein, X/Mu. This finding, thus, suggests possible roles of pV and protein X/Mu precursor in viral assembly. It also provides an interesting insight into genetic events that mediate molecular adaptation of viruses to possible changes in the genetic background in the course of their evolutionary divergence. The possible mechanism of the observed genetic suppression is discussed.
Keywords: Mastadenovirus, adenovirus, core proteins, pV deletion, thermoselection
Introduction
The Adenoviridae family is classified into four major genera comprising Atadenovirus, Aviadenovirus, Mastadenovirus and Siadenovirus.1 All members of the Adenoviridae family are non-enveloped, icosahedral viruses containing double-stranded DNA genomes ranging from 26 to 45 kb in size and replicate in the cell nucleus.1 Interestingly, the genome of Mastadenoviruses, which include various serotypes of the human adenovirus (Ad), encodes a few genus-specific genes missing in the other three viral taxons.1 These comprise genes encoding the minor capsid protein IX (pIX), minor core protein V (pV), and a group of seven different non-structural Ad genes encoded by the E3 region of the genome.1–3 The pIX is a structural component of the Ad capsid associated with the group-of-nine (GON) hexons in its homotrimeric form which “cements” them in the planar facets of the icosahedral shell thereby stabilizing the capsid structure.2,4 Each of the capsid’s 20 facets comprises four pIX trimers which make a total of 240 copies of the pIX monomer per virion. In addition to its role as a structural protein, pIX is also implicated in transcriptional trans-activation of the E1A, E4 and the major late (MLP) promoters, as well as the cellular TATA-containing promoters. Protein IX also appears to be involved in the Ad-induced nuclear reorganization.5,6
The functional roles of various proteins encoded by the E3 region of the Ad genome are related to protection of the Ad-infected cells from killing mediated by the host immune defenses, particularly cytotoxic T cells and death-inducing cytokines such as tumor necrosis factor (TNF), Fas ligand, and TNF-related apoptosis-inducing ligand (TRAIL).7,8 Despite the importance of the protein products encoded by the genus-specific genes for Ad biology, deletion of gene IX as well as all of the E3 genes from the Ad5 genome can be tolerated in vitro.9–11 Specifically, deletion of the pIX gene results in thermolability and only a minor reduction of viral growth,12,13 whereas deletion of one of the E3 genes (ADP/11.6 K protein gene) reduces cell lysis and release of the viral progeny from infected cell but does not affect viral replication.9 Deletion of most E3 genes is commonly used in Ad vector construction to reduce vector DNA size and accommodate excessive sequences of large trans-genes 14–17. The non-essential nature of IX and E3 genes is, thus, consistent with their absence from genomes of other Ad genera members revealed by phylogenetic analysis.1
In addition to the pIX and E3 genes mentioned above, Mastadenoviruses have yet another unique gene V. The protein product of gene V along with two other proteins, X/Mu and VII, has been implicated in the formation of the virion nucleocapsid structure (“core”). Protein V is also speculated to function as a bridge between the core and the capsid proteins.18–20 Each Ad virion contains 157 copies of pV,21 non-specifically associated with the double stranded viral genomic DNA.22–24 Protein V intrinsically localizes to both the nucleus and the nucleolus and redistributes nucleolin/C23 and nucleophosmin/B23 from the nucleolus to the cytoplasm.25 Although Ad infection inhibits biosynthesis and processing of ribosomal RNA in the nucleolus, expression of pV per se does not interfere with this important nucleolar function25 and, thus, the role of pV nucleolar localization remains unclear.
Based on the phylogenetic and experimental lines of evidence for dispensability of pIX and E3 genes, it was logical to suggest that deletion of genus-specific gene V from Ad5 genome would be tolerated as well. To validate the hypothesis and to elucidate the possible roles of pV in Ad biology, we engineered the first Ad mutant carrying gene V deletion in its genome. Although we succeeded in rescuing the pV-deleted Ad5, a dramatic reduction in thermostability and infectivity was observed as a result of the pV-deletion. We further employed a modified thermoselection screening approach26,27 to isolate a thermostable variant of this mutant. Sequence analysis of the thermoselected variant revealed a cluster of suppressor mutations in the coding sequence of the protein X/Mu precursor, which is another adenoviral core protein.
Results
Generation of pV-deleted Ad5
In order to generate an Ad5 genome carrying a deletion of the minor core protein V (pV) gene we first removed the entire pV gene sequence from pShuttle-V-EGFP-pA(−) to generate pShuttle-dV-kan by a PCR based approach as described in the Materials and Methods. The above deletion was then introduced into pTG360228 backbone by homologous recombination with the shuttle vector. The resulting rescue vector was subsequently digested with ClaI restriction enzyme and self-ligated to remove the kanamycin resistance (kan) gene. Three restriction sites, BsrGI, ClaI and HindIII, were introduced in place of the deleted pV gene sequence as a result of vector construction (Figure 1(a)). PCR analysis of Ad genomic DNA from CsCl-purified virus and total DNA extracted from virus-infected A549 cells was carried out with primers flanking the pV-deletion site and the fiber gene-specific primers (Figure 1(b)). Amplification of the pV region by PCR using the wild type Ad5 genome as a template produced DNA fragment of approximately 1.2 kb in size comprising the entire gene V sequence (Figure 1(c), left panel, lane 1). In contrast, the genome of the Ad5-dV used for amplification with the same primer set produces an approximately 100-bp DNA fragment (Figure 1(c), left panel, lane 2). On the other hand, PCR amplification of the fiber region using both the wild type and the pV-deleted genomes as templates produced identical DNA fragments of predicted size (Figure 1(c), right panel). PCR analysis of total DNA extracted from virus-infected A549 cells also revealed the same results (data not shown).
Figure 1.
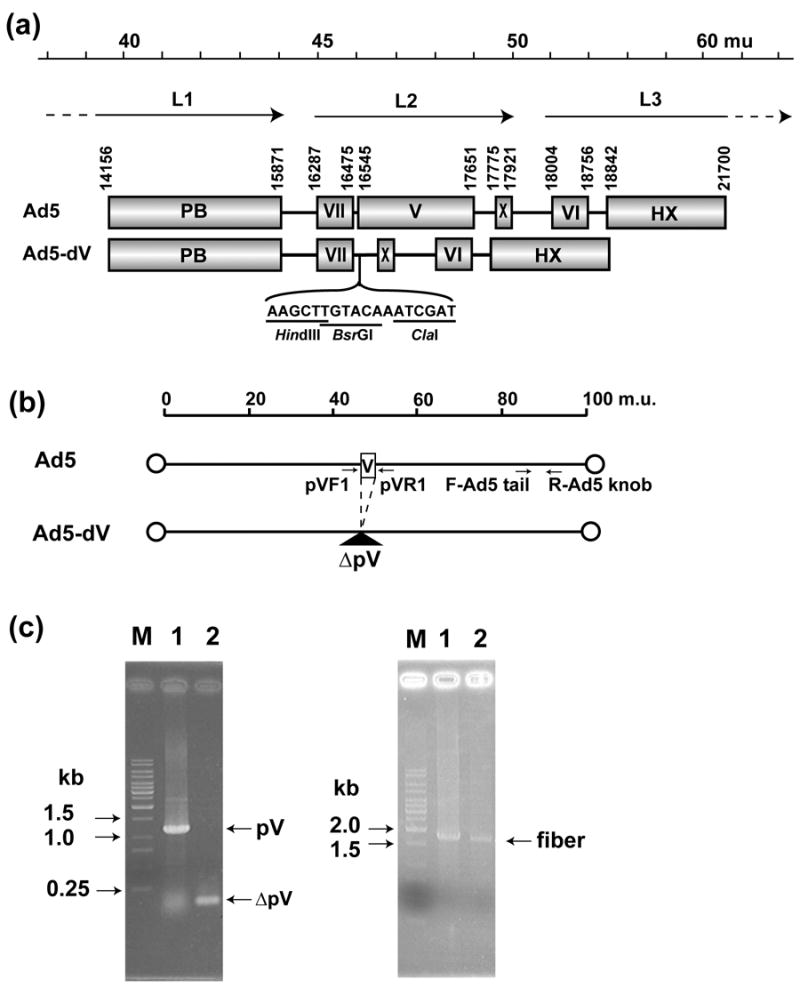
Genomic structure of the Ad5-dV and its PCR validation scheme. (a) Genomic structures of the wild type Ad5 and the pV-deletion mutant (Ad5-dV). The genome of Ad5-dV is carrying a deletion of nucleotides at position 16,544-17,650 of the wild type Ad5, thus this deletion removes the entire coding sequence of the pV gene. mu, map units; PB; penton base, VII; polypeptide VII, V; polypeptide V, X; polypeptide X (Mu), VI; polypeptide VII, HX; hexon, L1–L3; major late transcript units. (b) Schematic representation of the Ad5-dV genome PCR validation strategy. PCR primer pairs flanking pV and fiber gene sequences are indicated by arrows. pVF1, pV forward primer; pVR1, reverse pV primer; F-Ad5 tail, Ad5 tail sequence-specific forward primer; R-Ad5 knob, Ad5 knob sequence-specific reverse primer. (c) Validation of a pV deletion in the Ad5-dV genome by PCR. Left panel, PCR with pV flanking primers (pVF1 and pVR1); Right panel, PCR with fiber-specific primers (F-Ad5tail and R-Ad5knob); M, DNA size markers; Lane 1, PCR using total DNAs from wild type Ad5-infected cells as a template; Lane 2, PCR using total DNA from the Ad5-dV-infected cells as a template.
The infectious titers of the Ad5 and the Ad5-dV were 1.6 × 1010 PFU/ml and 2.5 × 106 PFU/ml, respectively, while the physical titers of viruses harvested from an equal number of infected A549 cells were 4.9 × 1011 VP/ml and 4.5 × 1011 VP/ml, respectively (Table 1). The ratios of physical to infectious titers for Ad5 and Ad5-dV were 31 and 1.8 × 105, respectively (Table 1). Thus, given almost identical physical titers, the infectious titer of the Ad5-dV was approximately 6,400-fold lower than that of Ad5 and, therefore, analysis of the growth kinetics and virus-specific gene expression of the mutant in culture was not feasible (Table 1). This suggests that deletion of gene V from Ad5 genome could dramatically reduce production of infectious virus in the infected cells.
Table 1.
Physical and infectious titers of Ad5 and pV-deleted Ad5
| Virus | VP/ml | PFU/ml a | VP/PFU |
|---|---|---|---|
| Ad5 | 4.9×1011 | 1.6×1010±7.64×108 | 31 |
| Ad5-dV | 4.5×1011 | 2.5×106±1.61×105 | 1.8×105 |
| Ad5-dV/TSB | 1.0×1012 | 1.6×109±1.44×108 | 625 |
Values are means ± SEM of results from triplicate experiments.
Plaque morphology and thermostability of Ad5-dV
As a part of virus characterization, we compared plaque morphology of Ad5 and Ad5-dV viruses propagated on confluent monolayers of A549 cells. The plaques formed from Ad5-dV had “unclear” morphology and were less than 1 mm in diameter at 12 days postinfection (Figure 2(a)) and merely 2 mm in size at 21 days postinfection (data not shown). The Ad5-dV generated plaques with similar morphology on 911 cells (data not shown). In contrast, plaques formed by Ad5 on A549 cells had a clear shape and were approximately 2 mm in diameter at 5 days postinfection (Figure 2(a)) and 3 to 5 mm at 12 days postinfection (data not shown). These observations suggested that viral spread and infectious progeny production were compromised as a result of the pV-deletion.
Figure 2.
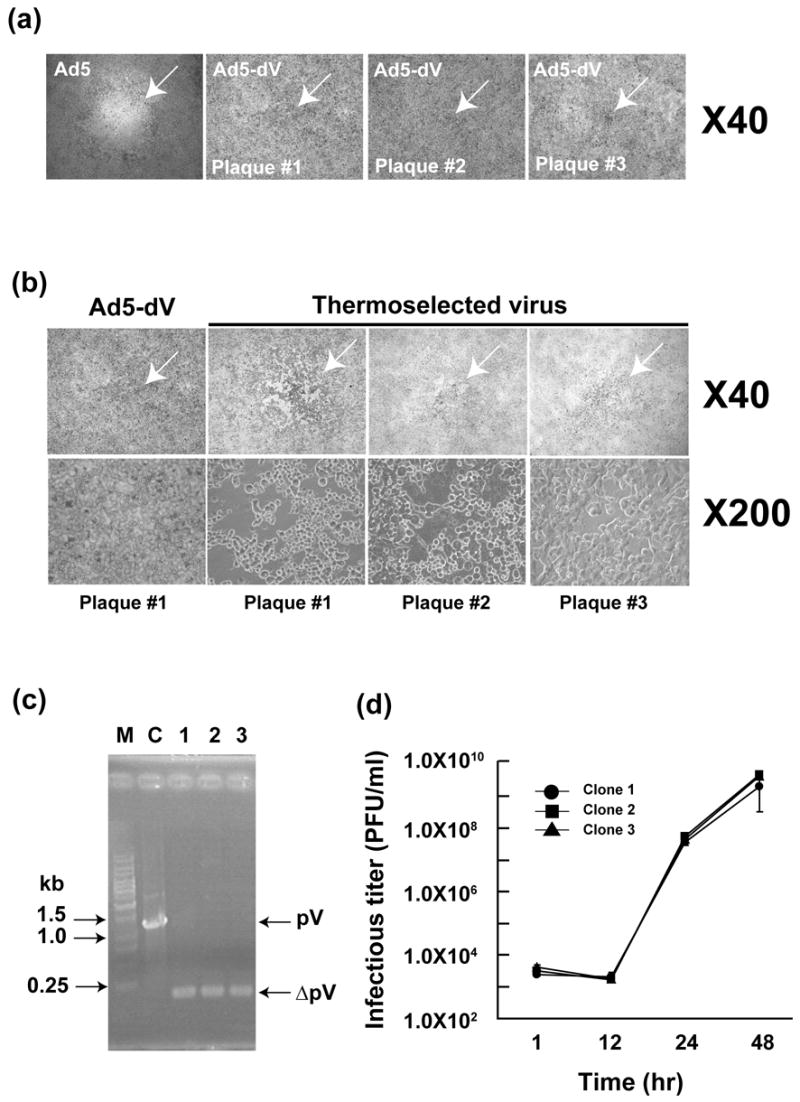
Comparative analysis of plaque morphology of Ad5, pV-deletion mutant (Ad5-dV) and the thermoselected Ad5-dV (Ad5-dV/TSB). (a) A light microscopic view (40 × magnification) of individual plaques formed by purified Ad5 and Ad5-dV on infected A549 cells after 10 days postinfection. (b) A comparative analysis of plaque morphology by 2 consecutive plaque purification and cytopathic effect (CPE) of the Ad5-dV and the thermoselected (Ad5-dV/TSB) viruses on A549 cells by light microscopy, at a 40 × magnification (upper panels) and 200 × magnification (lower panels). (c) PCR analysis of the region of gene V in the genomes of three independently isolated clones of Ad5-dV/TSB virus, same clones (plaque #1–#3) as shown in Figure 2(b). Lane 1; plaque #1, lane 2; plaque #2, lane 3; plaque #3. M, molecular size markers; C, control PCR using DNA from Ad5 with intact pV as the template. Total DNA was extracted from infected A549 cells and the region of gene V was amplified by PCR using the pV sequence-flanking primers. Others detail in Figure 1. (d) One step growth curve analysis of the three clones (same as shown in panels (b) and (c)) of the Ad5-dV/TSB virus.
Deletions and point mutations in the Ad5 genome are often associated with viral thermolability.13,27,29–34 Therefore we assessed whether the lack of pV from the virion affects its thermostability characteristics. To this end, we first incubated an equal number of particles (1010 VP) of Ad5 and of Ad5-dV, calculated on the basis of their physical titers (Table 1), at various temperatures for 3 days. The remaining infectivity of the heat-treated viruses was determined by titration using triplicate plaque assays on A549 cells. These experiments demonstrated that Ad5-dV lost infectivity much faster than Ad5 at each incubation temperature, except for −80ºC (Figure 3(a)). On the basis of these results we concluded that deletion of the pV gene from Ad5 genome reduces the thermostability of the virus.
Figure 3.
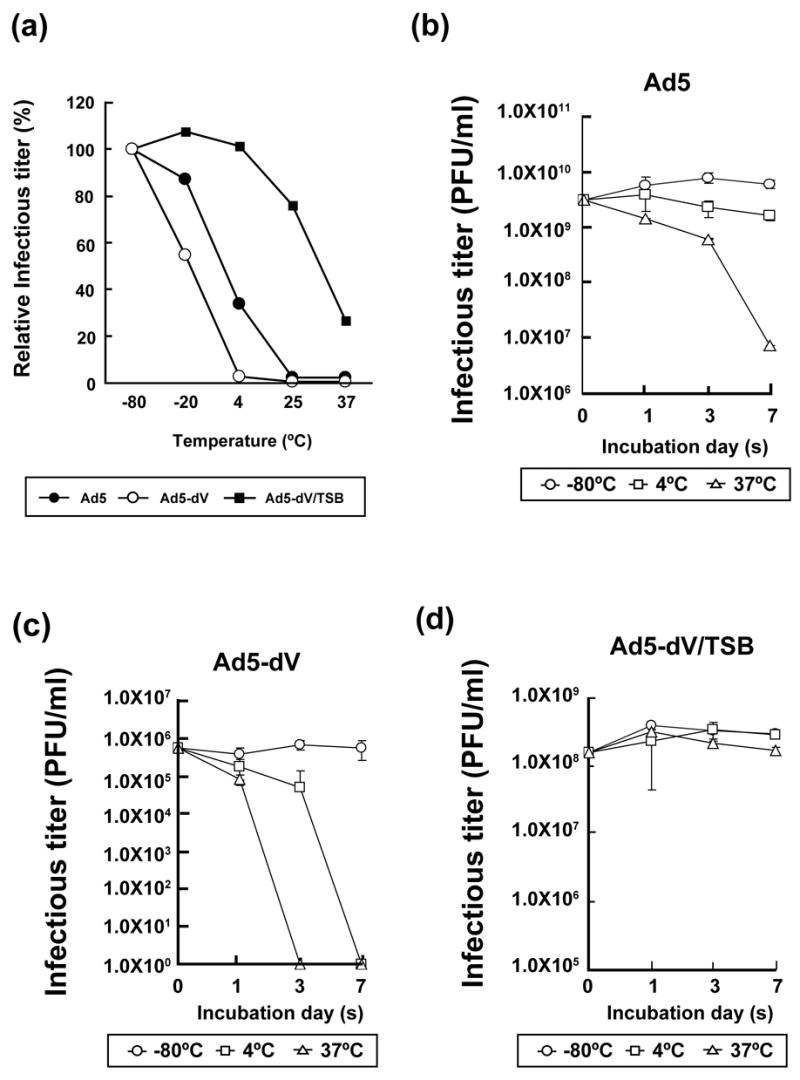
Thermostability of Ad5 and pV-deletion mutants (a) Thermostability analysis of Ad5, Ad5-dV and Ad5-dV/TSB particles. Purified viral particles (1010 VP) were incubated at various temperatures for 3 days and the resulting infectivity was determined by titration in a triplicate plaque assay on A549 cells. The residual (as % of the infectious titer at −80ºC after 3 days) infectious titers following incubation of the viruses at various temperatures for 3 days were plotted. (b–d) The kinetics of virus thermo-inactivation. 1010 VP of Ad5 (b), Ad5-dV (c) and Ad5-dV/TSB (d) viruses were incubated at various temperatures for 1, 3 and 7 days and the resulting infectious titers were determined by titration and plotted as an average in triplicate plaque assay on A549 cells.
Thermoselection of the pV-deleted virus
In the course of our thermostability experiments we noticed that following incubation at 37ºC for 3 days, the Ad5-dV generated larger plaques (approximately 2 mm in diameter) on the infected A549 cells than in prior experiments with freshly thawed virus preparations. This observation prompted us to suggest that during propagation of the rescued virus in A549 cells some virions in the intracellular pool of Ad5-dV acquired spontaneous mutations which improved their spread and thermostability. On this basis, we utilized a modified version of virus thermoselection approach described previously26,27 in order to improve growth and thermostability characteristics of the generated Ad5-dV. Following a 3-day incubation of the purified Ad5-dV at 37ºC (1010 VP), we were able to isolate single plaques approximately 2 mm in diameter by consecutive plaque assays on A549 cells (Figure 2(b), upper panels). The resulting thermoselected Ad5-dV, designated Ad5-dV/TSB, formed larger plaques on A549 cells and demonstrated a strong cytocidal effect in contrast to the original Ad5-dV virus (Figure 2(b), lower panels). As expected, all thermoselected viruses isolated from individual single plaques, shown in Figure 2(b), possessed the pV-deleted genomes as confirmed by PCR analysis of the total DNAs isolated from those individual plaques (Figure 2(c), lanes 1–3). A one-step growth curve analysis of three Ad5-dV/TSB clones independently isolated from the same individual plaques described above (Figure 2(b)), demonstrated similar kinetics of viral replication in culture (Figure 2(d)). We randomly chose one out of the three mentioned Ad5-dV/TSB clones (same as shown in Figure 2(b), plaque number 1, and Figure 2(c), lane 1) for further characterization. We assessed physical and infectious titers of each clone after purification. As shown in Table 1, the physical titer of the Ad5-dV/TSB was 1.0 × 1012 VP/ml, while the infectious titer was 1.6 × 109 PFU/ml (Table 1). The ratio of VP/ml to PFU/ml for this virus was 625:1 and, thus, the infectious titer of Ad5-dV/TSB was about 640-fold higher as compared to the Ad5-dV (Table 1). Furthermore, we found that thermostability of Ad5-dV/TSB was drastically improved (Figure 3(a)). In contrast to the Ad5-dV virus, the relative infectivity of the thermoselected mutant remained essentially unchanged during its 3-day storage at 4ºC and decreased by only 24% after storage at 25ºC (Figure 3(a)). Moreover, the relative thermostability of Ad5-dV/TSB appeared to be even higher than that of the Ad5 over a range of temperatures (Figure 3(a)). In addition to comparing the relative inactivation of each virus after storage at each given temperature for 3 days, we also analyzed the dynamics of each virus inactivation after its storage at each given temperature for 1, 3 and 7 days (Figure 3(b–d)). The remaining absolute infectious titers were determined as described in Materials and Methods. The results shown in Figure 3(b) and 3(c) demonstrate that inactivation of the Ad5-dV occurred much faster as compared to the Ad5. While the latter retained partial (~1000 fold reduced) infectivity after 7 days of incubation at 37ºC (Figure 3(b)), infectivity of the Ad5-dV was dramatically reduced already after 3 days of storage at 37ºC and even after 7 days of storage at 4ºC (Figure 3(c)). On the other hand, thermo-inactivation of the Ad5-dV/TSB was significantly slower at 37ºC and at 4ºC as compared to the Ad5-dV (Figures 3(a) and 3(d)). Comparison with the corresponding profile of the Ad5 again revealed a significantly slower inactivation, especially at 37ºC. Thus, the thermostability of the Ad5-dV/TSB appears to be significantly improved as compared to the Ad5-dV.
Validation of genomic structure of the thermoselected pV-deletion mutant
To confirm the genomic integrity of Ad5-dV/TSB, we subjected total DNA isolated from virus infected cells to restriction analysis with six different restriction endonucleases. This analysis confirmed the presence of additional sites for restriction endonucleases BsrGI, ClaI and HindIII created in place of the pV-deleted nucleotide sequence (Supplementary Figure 1(a–c), note asterisks). The absence of a single NotI site removed by the gene V sequence deletion was also confirmed in the Ad5-dV/TSB genome by a restriction test (Supplementary Figure 1(c), note asterisk). The above changes were additionally confirmed by sequencing of the viral genome (data not shown). Importantly, we observed no evidence for any random genomic rearrangements in the Ad5-dV/TSB which could theoretically confound viral stability and growth characteristics. In addition, we confirmed the lack of the pV gene in the genome of Ad5-dV/TSB by PCR assay (Supplementary Figure 1(d)).
Analysis of protein incorporation in the viral particles
To determine if the viral assembly is compromised by the absence of protein V, we analyzed protein composition of the double CsCl-purified Ad5-dV and Ad5-dV/TSB viral particles. Since the remaining amount of the original Ad5-dV preparation used for thermostability/infectivity characterization of the virus was not sufficient to complete this analysis, we re-propagated the parental Ad5-dV and assumed that the up-scaled Ad5-dV was genetically identical to the one used in our previous experiments. New preparations were also obtained for Ad5-dV/TSB as well as Ad5 in parallel. Both pV-deleted viruses fractionated on the CsCl gradients appeared as two bands (data not shown). We presumed that the lower band contained the “infectious” viral particles, whereas the upper band corresponded to “defective” viral particles as typically seen for Ad5 preparation. The purified viruses from both bands of the Ad5 and the pV-deleted viruses were then subjected to molecular and structural characterization.
First, to characterize protein composition of the pV-deleted viruses we compared protein patterns of the particles, purified from both lower and upper bands, respectively, and resolved by SDS-PAGE and the silver staining gel (Figure 4(a)) and GELCODE Blue (Supplementary Figure 2(a)). The observed protein patterns appear to be similar with respect to overall structural protein content as well as relative incorporation of the individual components of the viral particles (Figure 4(a)). Of note, the protein patterns observed for the upper band of all viruses (Figure 4(a), lanes 4–6) were clearly distinct from the ones of the lower band (Figure 4(a), lanes 1–3).
Figure 4.
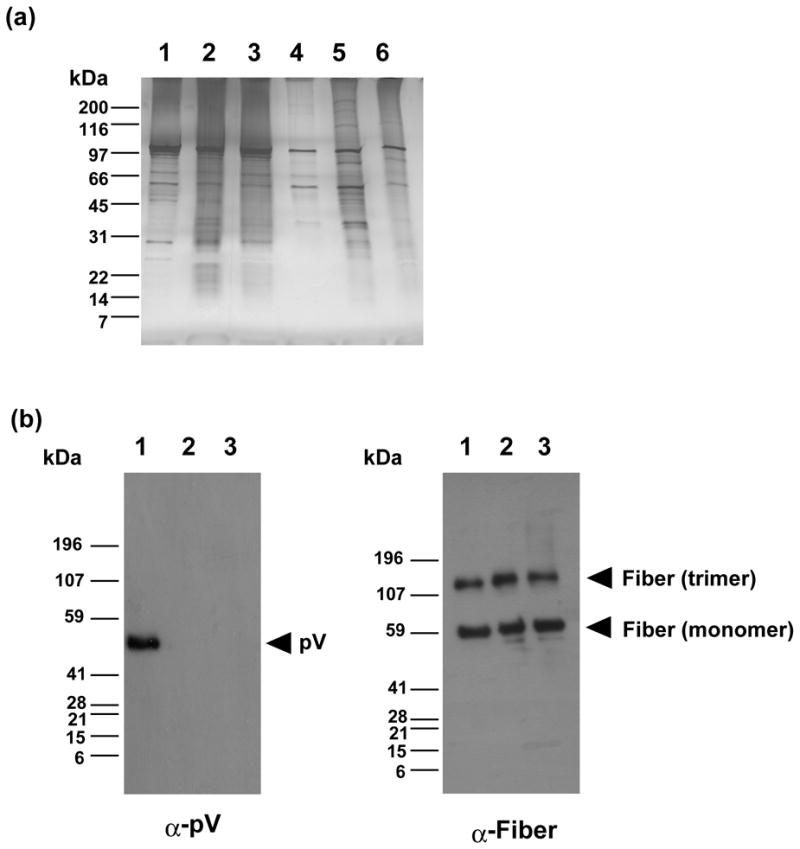
Analysis of protein composition of viral particles by gel-staining and Western blot. (a) Detection of adenoviral proteins incorporated in the particles. 1010 VP and 109 VP equivalents of purified infectious and defective viruses, respectively, were run on a 4–15% (w/v) gradient SDS-PAGE and the gel was stained with silver staining kit. Lanes 1–3; infectious particles (lower band), Lanes 4–6; defective particles (upper band), Lane 1; Ad5, lane 2; Ad5-dV; lane 3; Ad5-dV/TSB. (b) Comparison of the relative incorporation of pV and fiber proteins in purified infectious Ad5 (lane 1), Ad5-dV (lane 2) and Ad5-dV/TSB (lane 3) particles by Western blot with antibodies against pV and fiber. 1010 VP equivalents of the purified viruses were run on a 4–15% (w/v) gradient SDS-PAGE, electro-transferred onto a PVDF membrane, and probed by antibodies against pV and fiber. Protein molecular mass makers (in kilodaltons) are indicated on the left.
The lack of pV in the viral particles of Ad5-dV and Ad5-dV/TSB preparations in addition to gel staining was verified by Western blot analyses with anti-pV antibody (Figure 4(b) and Supplementary Figure 2(c)) as well as anti-serum against Ad5, (Supplementary Figure 2(b)). In sharp contrast to pV, we found no significant difference in the incorporation of minor capsid protein IX and fiber between Ad5 and Ad5-dV/TSB particles (Supplementary Figure 2(c)). This was consistent with the results of gel staining and further supported our conclusion that incorporation of most structural components of the capsid are not affected by the lack of pV in the particles of pV-deleted viruses.
Remarkably, among subtle changes seen in protein composition of the pV-less particles of Ad5-dV/TSB, we observed a discernible increase in incorporation of another core protein X/Mu that could be only identified by its size and relative mobility in SDS-PAGE gel (as pX/Mu-specific antibodies were not available) (Supplementary Figure 2(a), lane 2). In fact, the above results were consistently reproducible by different gel staining methods (data not shown).
Analysis of viral particle morphology by transmission electron microscopy
In order to provide a direct demonstration of Ad5-dV viral particle formation, infectious (lower band) and defective (upper band) adenoviral particles were also analyzed by transmission electron microscopy (TEM) (Figure 5). We found that the infectious particle fraction of each virus was represented by uniform particles of 70–80 nm in diameter (Figure 5(a), (c), (e) and (g–i)) most of which formed aggregation clusters. In sharp contrast, the viral particles from the upper band of all three viruses contained debris and partially degraded capsids (Figure 5(b), (d) and (f), see arrows), some of which appeared to be empty (Figure 5(b) and (f), see arrowheads).
Figure 5.
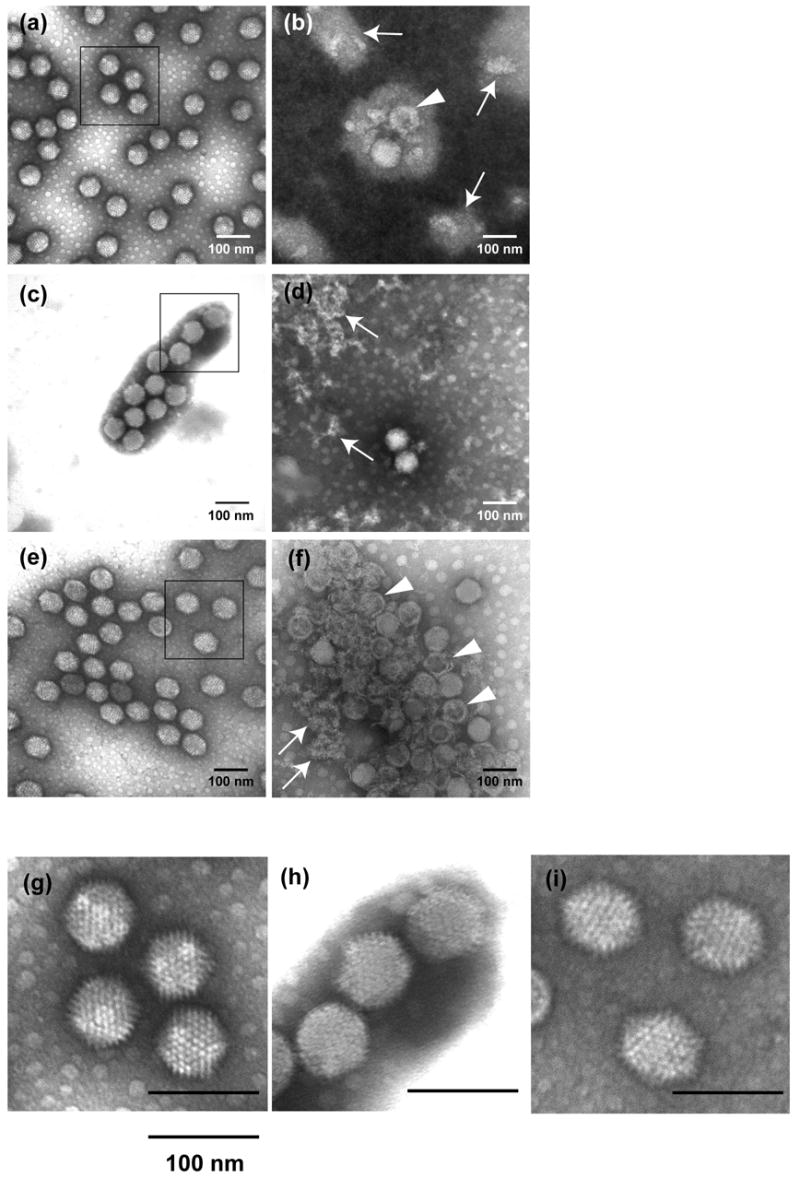
Negatively stained electron micrographs of purified Ad5 ((a) and (b)), Ad5-dV ((c) and (d)) and Ad5-dV/TSB ((e) and (f)) viral particles included in lower (a, c and e) and upper (b, d and f) bands after CsCl-ultracentrifugation. Arrows show debris and partially degraded capsids and arrowheads show empty particles. (g–i) Enlargement of highlighted areas in (a, c, e), respectively. Magnification: 200,000 ×.
Analysis of selected enlargement areas within the TEM images of the infectious particles revealed that the Ad5-dV particles have less clear morphology and appear to have more sphere-like shape as opposed to the icosahedron-like shape of both Ad5 and Ad5-dV/TSB particles (Figure 5(g–i)). Although the level of image resolution does not allow definitively determining the precise nature of the observed differences, the tentative changes in the Ad5-dV particle morphology might account for the dramatic loss of their thermostability and infectivity observed in our experiments. Based on the TEM structural analysis we suggest that deletion of pV could somewhat disturb viral assembly and cause detectable changes in capsid morphology, which are likely to explain the thermostability and infectivity defects. Conversely, the lack of discernible changes in the capsid structure of Ad5-dV/TSB, as compared to Ad5, correlates with the improved thermostability and infectivity phenotypes of Ad5-dV/TSB. Furthermore, it appears that the tentative defects in the viral structure caused by the lack of pV can be suppressed in the Ad5-dV/TSB, apparently, by some genetic changes acquired as a result of the selection procedure.
Analysis of viral replication and protein expression in infected cells
In order to characterize viral replication and production of Ad5-dV/TSB in infected cells, we performed a one-step growth curve analysis in A549 cells as described in the Materials and Methods section. Although the Ad5-dV/TSB-infected cells produced infectious progeny with kinetics similar to that of Ad5, the yield of infectious viral progeny between 24 and 144 hours postinfection was in the range from one to two orders of magnitude (10–100 fold) lower to that of Ad5 (Figure 6(a)).
Figure 6.
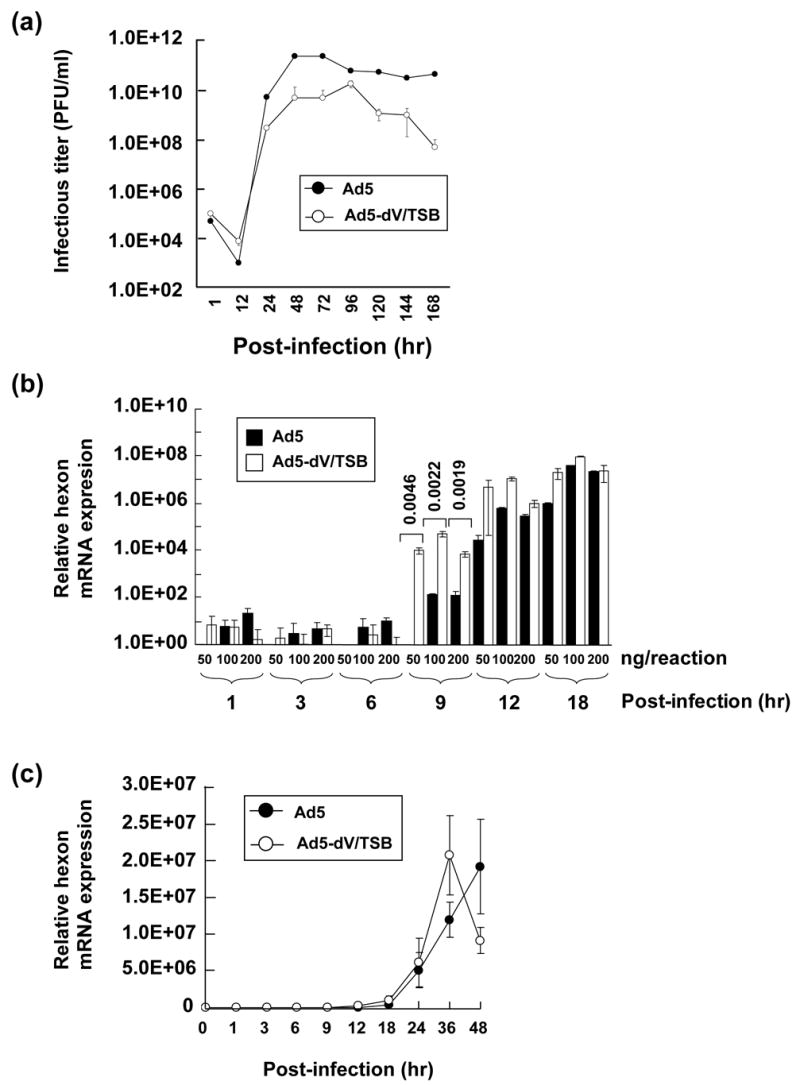
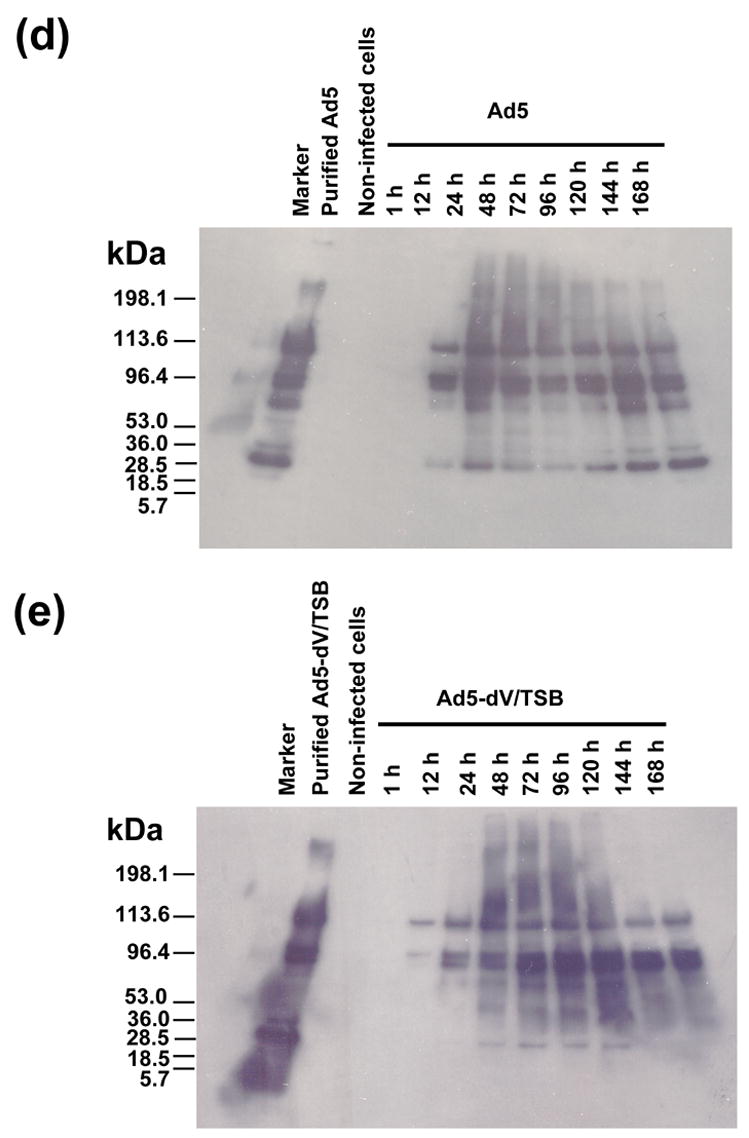
Comparative study of viral replication kinetics. (a). Comparison of one-step growth curves obtained for Ad5 (black circle) and Ad5-dV/TSB (open circle) in A549 cells infected at an MOI of 10 PFU/cell. (b) Quantification of hexon mRNA of Ad5 (black bar) and Ad5-dV/TSB (white bar) in A549 cells infected at an MOI 10 of PFU/cell followed by cell harvesting at various times postinfection. Three replicates were performed with various concentration of total RNA extracted from viral infected cells. P values calculated by two-tailed unpaired Student’s t tests are shown for comparison of the two groups indicated by the square brackets. (c) Quantification of hexon mRNA of Ad5 (black circle) and Ad5-dV/TSB (white circle) in A549 cells infected at an MOI 10 of PFU/cell followed by cell harvesting at various times postinfection. Three replicates were performed with 50 ng of total RNA extracted from viral infected cells. ((d) and (e)) Comparison of adenoviral late protein expression profiles. A549 cells were infected with Ad5 (d) and Ad5-dV/TSB (e) at an MOI of 10 PFU/cell followed by cell harvesting at various times postinfection. Five μg of total protein equivalents of cell lysates prepared from infected cells at each time point were analyzed on 4–20% (w/v) gradient SDS-PAGE, and detected with rabbit polyclonal anti-serum against Ad5.
We also analyzed expression of spliced hexon mRNA by quantitative reverse transcription-PCR (qRT-PCR) at various times postinfection. As can be seen from Figure 6(b), at 9 hours postinfection, the relative expression of spliced hexon mRNA shows a statistically significant increase in Ad5-dV/TSB- versus Ad5-infected cells at various concentrations of total RNA used for qRT-PCR analysis. On the other hand, the relative gene expression of hexon mRNA at later stage of infection (past 36 hrs postinfection) appears to decrease earlier in the Ad5-dV/TSB-infected cells (Figure 6(c)).
In order to validate that the observed phenomenon of early onset of gene expression for major late protein in Ad5-dV/TSB-infected cells, we performed Western blot analysis of the infected cell lysates using anti-serum against Ad5 to detect major structural proteins of Ad particles. As seen from Figure 6(d), most of the Ad5 structural proteins are expressed in the virus-infected cells starting at 24 hours postinfection. Comparison of the virus-specific protein expression in the Ad5-dV/TSB- versus the Ad5-infected cells revealed that it was essentially similar, except that expression of hexon protein (the uppermost protein band across the lanes) and other protein components of the capsid appears to start significantly earlier in the infection cycle, i.e. already at 12 hrs post infection (Figure 6(e)).
Another apparent difference in the gene expression profiles is down-regulation of some low molecular weight protein in the Ad5-dV/TSB-infected cells (Figure 6(d) and (e)). This protein is likely to be another core protein pVII, although the precise immunologic identification was not feasible in this study due to the lack of pVII-specific antibodies. The decrease of the protein expression (presumed to be the core protein pVII) observed during the infection, however, appears to have no significant effect on the incorporation in the viral particles (compare the “purified Ad5” and “purified Ad5-dV/TSB” control lanes in Figure 6(d) and (e); also Supplementary Figure 2(a)). These results strongly argue that the result obtained in the above Western blot experiment is unlikely to be explained by different quantity of structural proteins delivered to the cells with internalized viral capsids, which could potentially have occurred if unequal number of infectious particles were used in the experiment due to viral infectious titer miscalculation.
In order to rule out the possibility of more Ad5-dV/TSB genomic DNA reaching the nucleus of the infected cells due to the possible underestimation of the viral infectious titer, we also compared relative gene expression of E1A 12S mRNA by qRT-PCR in cells infected with Ad5 and Ad5-dV/TSB. As can be seen from Supplementary Figure 3, given the same infectious units (PFU) used for cell infection, the Ad5-dV/TSB-infected cells expressed significantly less E1A 12S mRNA at all time points as compared to Ad5. Thus, while gene expression of hexon mRNA at 9 hrs postinfection appeared to be statistically significant increased in Ad5-dV/TSB infected cells as compared to that in Ad5-infected cells, gene expression of E1A 12S mRNA in Ad5-dV/TSB-infected cells appears to be lower. This rules out the possibility of more Ad5-dV/TSB genomic DNA reaching the nucleus of infected cells as an explanation for the difference in the late gene expression profile observed in our Western blot experiment.
The improved biological characteristics of the Ad5-dV/TSB virus are associated with a cluster of three selectable mutations in the gene for the core protein Mu precursor
Although our results clearly demonstrate that Ad5 lacking the core protein V gene is viable and can be rescued, pV appears to play an important role in cytocidal effect, thermostability, viral assembly and expression of major late proteins (Figures 2, 3, 5 and 6, and Table 1). To get further insight into the possible functional roles of pV, we carried out mapping of the suppressor mutations that conferred thermostability and infectivity to the Ad5-dV/TSB particles, allowing their bioselection under restrictive conditions. The thermostability characteristic of Ad is apparently determined by physical stability of the viral capsid and the core. Stability of the core is likely to be determined by the efficiency of the association of the core components with one another and the genomic DNA. In this regard, we reasoned that the suppressor mutations, which rescue the thermostability and infectivity defects of Ad5-dV, are most likely to occur in Ad structural genes, particularly the ones that are functionally linked to the deleted gene V. Based on this assumption, we performed a judicious sequence analysis of selected areas of the Ad5-dV/TSB genomic DNA isolated from a single thermoselected clone after three rounds of its plaque-purification. We analyzed the mutation sites by comparison to database reference sequences (see Materials and Methods). Altogether about 91% (31,729 bp) of the entire genome from Ad5-dV/TSB has been sequenced. This sequence analysis did not cover inverted terminal repeats, E1 and E4 of Ad genome encoding non-structural genes, which are unlikely to affect viral assembly, thermostability and the thermoselection screening.
Examination of sequencing readouts obtained for the Ad5-dV/TSB genome did not reveal any mutations in the capsid protein-encoding genes. In contrast, we found three distinct but closely spaced nucleotide substitution mutations in the coding region of a gene for another component of Ad core, called protein X/Mu. One of those mutations generated a silent mutation changing guanine to adenine at nt position 17,714 (Figure 7), whereas nucleotide substitutions at nt positions 17,716 (guanine to adenine) and 17,728 (guanine to thymidine) of the Ad genome generated missense mutations that changed amino acids at positions 13 and 17 of the encoded polypeptide, respectively (Figure 7). Those amino acid substitutions included a change from neutral glycine to acidic glutamic acid (G13E), and basic arginine to hydrophobic isoleucine (R17I), respectively. Remarkably, all three mutations mapped to the N-terminal peptide of the protein X/Mu precursor, upstream from the known site of its proteolytic cleavage by Ad protease (Figure 8). Also, since the cluster of above mutations was not detected in gene for protein X/Mu precursor of Ad5-dV genome extracted from purified virus (Figure 7(e)), it appears that Ad5-dV/TSB acquired these mutations was isolated through thermoselection of a minor population of Ad particles with spontaneously occurred point mutations in the specified region present in the original Ad5-dV virus solution. Therefore, a cluster of selectable mutations in the gene for the core protein X/Mu precursor might account for the improved biological characteristics of Ad5-dV/TSB.
Figure 7.
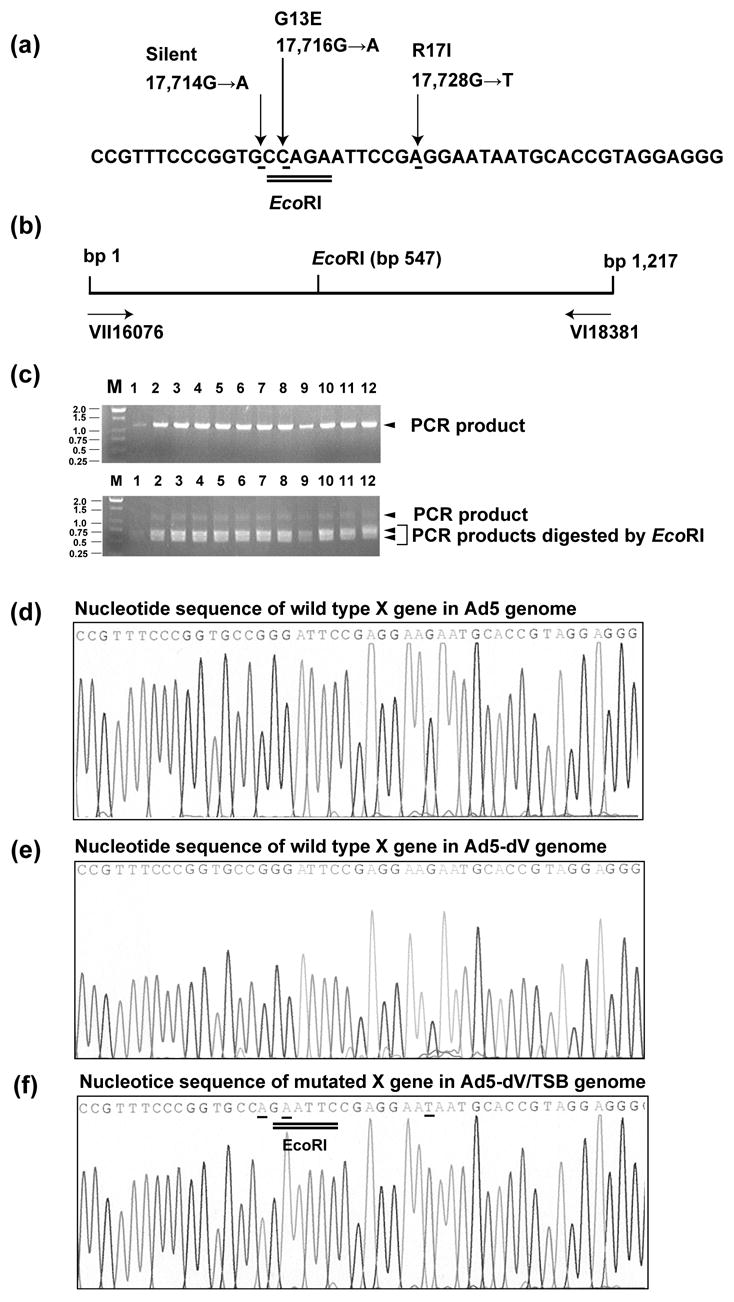
Detection of the suppressor mutation 17,716 in the Ad5-dV/TSB genome. (a) A sequence fragment of the gene X/Mu with three suppressor point mutations shown by arrows. The numbers designate nucleotide positions of the substitution mutations in the Ad5 genome and the amino acid changes generated by their nucleotide substitutions were described. Position 1 refers to the left end of wild type Ad5 genome (Accession number AY339865). Position 1 refers to the initiation site, methionine, of precursor Mu protein. (b) Schematic representation of the gene X/Mu containing region of the Ad5-dV/TSB genome amplified by PCR with VII16076 and VI18381 flanking primers. The PCR amplified approximately 1.22-kb fragment comprises the X/Mu gene mutation that creates a new EcoRI site at the genome nucleotide position 17,716. (c) Detection of the 17,716 suppression mutation by PCR-amplification of a specific gene X/Mu region and subsequent digestion of the PCR product with the EcoRI restriction enzyme. The cleavage generates approximately 0.55- and 0.67-kb fragments (d–f) A portion of the sequencing profiles obtained for the gene of X/Mu protein precursor in genomic DNAs of purified Ad5 (d) and Ad5-dV (e), and Ad5-dV/TSB (f). The mutated nucleotides are underlined. The EcoRI site created by the 17,716 mutation is double underlined.
Figure 8.
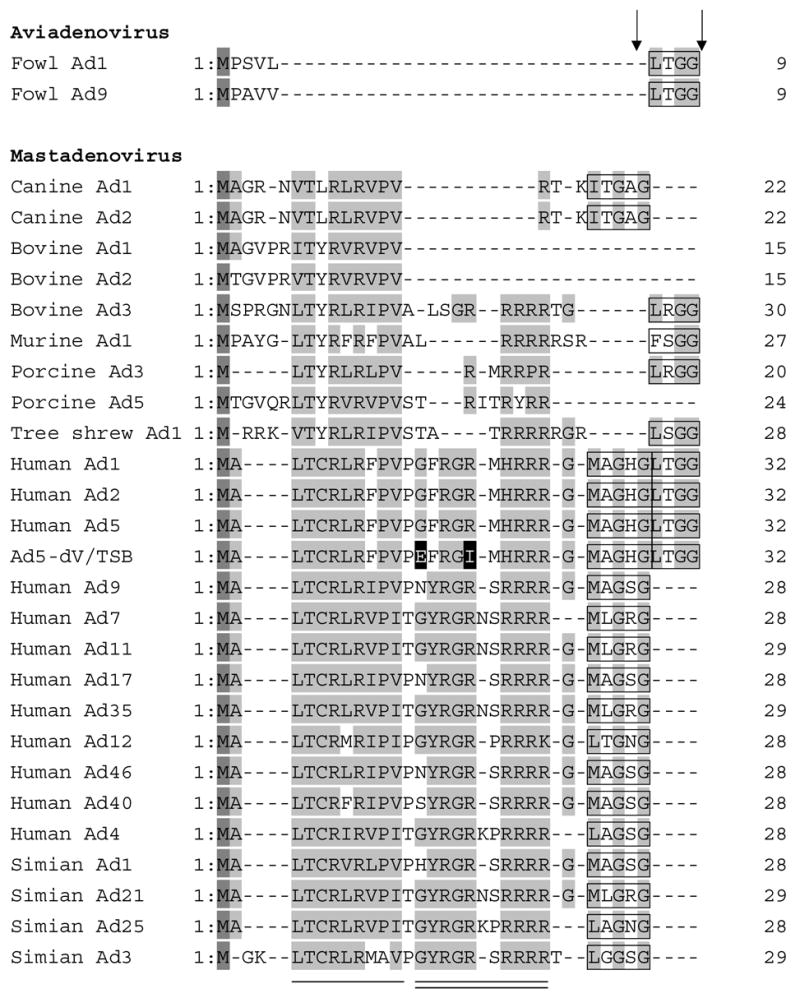
Alignment of the N-terminal domains of Mu protein in selected members of the Adenoviridae family. The amino acid sequences of the N-terminal domains of Mu protein of Adenoviridae were aligned using CLUSTALW software. The two mutated amino acid residues in Ad5-dV/TSB are shown with black background. Methionine residues are shown in a dark gray background and highly conserved residues are indicated by a gray background. The adenoviral protease recognition motif residues for cleavage are indicated by a black box and Ad protease cleavage sites are indicated by arrows. The amino acid sequences of the N-terminal domain of pre-Mu of Adenoviridae were retrieved from the sequence database accession numbers as shown below. fowl Ad1 (AP_000416), fowl Ad9 (AP_000382), canine Ad1 (AP_000057), bovine Ad1 (YP_094038), bovine Ad2 (AP_000012), bovine Ad3 (AAD09726), human Ad1 (AP_000510), human Ad2 (AP_000173), human Ad4 (YP_068030), human Ad5 (AP_000209), human Ad7 (AP_000546), human Ad9 (CAI05967), human Ad11 (AP_000450), human Ad12 (AP_000119), human Ad17 (AP_000148), human Ad35 (AP_000583), human Ad40 (NP_040860), humanAd46 (AAX70957), murine Ad1 (AP_000349), porcine Ad3 (YP_009208), porcine Ad5 (AP_000243), simian Ad1 (YP_213973), simian Ad3 (AAT84625), simian Ad21 (AP_000273), simian Ad25 (AP_000311). The conserved residues in Mastadenoviruses are shown by a single solid line and the conserved residues in human and simian Ads are indicated by a solid double line.
To further verify homogeneity of the sequenced genomic DNA we analyzed the corresponding region of the gene X/Mu in twelve plaque-purified clones descending from the originally isolated clone of the thermoselected virus. Since one of the three mutations that occurred at nt position 17,716 was found to generate a new EcoRI site (Figure 7(a), double underline), we employed this finding to assess the presence of the restriction site in the gene X/Mu of each of the twelve clones. To this end, a region comprising the gene X/Mu sequence was amplified by PCR as illustrated in Figure 7(b) and the PCR product was digested with EcoRI restriction enzyme (Figure 7(c)). By using this analysis we found the missense mutation at nt position 17,716 in all 12 single plaque-derived genomes as the respective PCR products were fully cleaved by the restriction enzyme (Figure 7(c), lower panel). Since the genomes isolated from all 12 derivative clones bear the selectable amino acid alteration, we conclude that all the derivative clones were likely to be genetically identical. To validate this assumption we further confirmed the presence of all three mutations in all 12 clones by direct sequencing of the corresponding PCR products (Figure 7(f), underlines).
This provides a substantial evidence for genetic homogeneity of the originally sequenced genomic DNA and allows us to suggest that all three mutations identified in the X/Mu gene of Ad5-dV/TSB are indeed present in a single genomic DNA molecule.
The point mutations identified in a single clone of the thermoselected virus appear to be predominant suppressors of the Ad5-dV thermostability/infectivity defects
In order to verify that the discovered cluster of point mutations is the only type of spontaneous genetic alterations capable of rescuing thermostability of Ad5-dV, we utilized the above mentioned PCR-based mutation screening assay as well as a direct sequencing of the derived PCR products to analyze two more thermoselected clones isolated in two independent thermoselection experiments. Both clones were found to contain all three point mutations (data not shown). Thus, it appears that the cluster of two suppressor mutations is associated with the observed improvement in thermostability/infectivity of Ad5-dV/TSB, while the third silent point mutation has apparently no contribution to the Ad5-dV/TSB phenotype. We conclude that thermoselection of Ad5-dV yielded a genetically homogeneous suppressor mutant from a potentially heterogeneous population of viral particles. The cluster of two point mutations, thus, appears to be the sole or the predominant suppressor of the thermostability and infectivity defects associated with deletion of the pV gene.
The missense mutations mapped in the protein X/Mu precursor are located in the region of low homology among Adenoviridae family
Alignment of amino acid sequences of the protein X/Mu precursor (pre-Mu) from 27 different Ad species demonstrated some phylogenetic conservation in the N-terminal part of the protein (Figure 8). Two main clusters of conserved amino acid residues should be noted in the N-terminal part of pre-Mu. One cluster, proximal to the N-terminus is conserved among all the compared species of Mastadenovirus genus (Figure 8, underline), whereas the other cluster located closer to the center of the N-terminal portion of pre-Mu is conserved primarily among the analyzed human and simian Ad serotypes (Figure 8, double underline). It should be noted that the suppressor mutations which we mapped in the pre-Mu polypeptide of Ad5-dV/TSB change amino acids within the less conserved central cluster (Figure 8, double underline).1 This observation is consistent with the suggested role of these suppressor mutations as “adaptive” or “fitness” mutations which are less likely to occur in highly conserved regions of protein sequences.35 Furthermore, our phylogenetic comparison reveals that members of Atadenovirus and Siadenovirus genera that do not encode gene V orthologs in their genomes also lack most of the N-terminal domain in their pre-Mu proteins, including the above mentioned clusters of conservation. This observation indicates that the N-terminal domain of protein X/Mu may be functionally linked to the genus-specific protein V and, thus, the discovered mutations associated with the thermoselected virus phenotype could possibly account for the observed suppression effect.
Discussion
Both phylogenetic and experimental lines of evidence suggest that the genus-specific gene IX as well as E3 genes of the human adenovirus are not essential for the viral biology.12–16 Considering that the adenoviral gene V is found only in members of Mastadenovirus genus, we reasoned that the human adenovirus lacking the pV gene could be viable and, therefore, should be feasible to generate by genetic engineering. Validation of this hypothesis provided a rationale for this study. Moreover, generation and characterization of the pV-deletion mutant undertaken in this study was important for getting insight into the role of pV in viral assembly as well as its function in adenovirus-infected cells. Although we successfully rescued the first pV-deletion mutant (Ad5-dV), the originally generated and purified Ad5-dV demonstrated a strongly reduced thermostability (Figures 3 (a) and (c)) as well as an extremely low infectivity (Table 1), which appear to be associated with subtle changes in the particle morphology (Figure 5 (h)). Although the above characteristics of Ad5-dV prevented its further biological characterization, the obtained data indicated that protein V might be important for the correct assembly of the infectious viral particles.
Previous studies have demonstrated that a random mutagenesis and genetic engineering of human Ad, followed by multiple passages in cell culture under selective conditions, can be used to isolate temperature-sensitive13,27,29–34 as well as host range36–39 mutants. Furthermore, spontaneous mutations in modified regions of the Ad fiber genes could be generated as a result of replication errors during multiple passaging of fiber-modified recombinant Ads in certain cell lines40. These earlier studies prompted us to combine physical selection of viral thermostability characteristics with a bioselection screen in A549 cells in order to improve the thermostability and infectivity defects of Ad5-dV by a mechanism resembling natural selection. We also presumed that characteristics predicted to be acquired by Ad5-dV as a result of such thermoselection would be stably maintained during multiple rounds of viral propagation. The thermostable virus selected by this approach indeed showed a significantly improved thermostability characteristic associated with the improved viral infectivity (Figures 2(b), 3(d) and Table 1) and produced approximately 640-fold higher yield of infectious progeny in A549 cells as compared to the parental Ad5-dV (Table 1). In fact, the recovered virus was also capable of replicating in culture although the efficiency of replication at 48 hours postinfection was somewhat lower than that of Ad5 (Figure 6(a)). Since Ad5-dV subjected to the thermoselection screen gave rise to a new variant with strong cytocidal phenotype (Figure 2(b)) and improved thermostability (Figures 3(a) and (d)), we suggested that the originally rescued Ad5-dV arose from a heterogeneous population of variants, whose genomes carried various spontaneously generated point mutations. Those mutations could possibly emerge during the initial steps of viral propagation in culture due to random errors generated by Ad polymerase. Apparently, the thermoselection step physically inactivated the thermolabile virions and the subsequent bioselection screen allowed propagation of the variants, whose spontaneously acquired point mutations were capable of suppressing the thermostability/infectivity defects. Consequently, such mutations became selectable genetic markers.
We also found that the lack of core protein pV has no significant effect on incorporation of most structural proteins into the viral particles (Figure 4(a)) and does not prevent the assembly of viral particles per se, although it appears to somewhat alter their morphology (Figure 5(a), (c), (e) and (g–i)). These tentative changes in the Ad5-dV particle structure revealed by TEM studies are consistent with and could account for the dramatic loss of its stability and infectivity.
Interestingly, a detectable increase in incorporation of the structural protein, believed to be the core protein X/Mu, was consistently observed for the Ad5-dV/TSB particles isolated from the lower band in CsCl gradient (Supplementary Figure 2(a)). The excessive incorporation of this protein into the particles could occur by means of filling up the space inside the capsids that was freed up due to the lack of pV. Although the molecular mechanism for thermostability and infectivity improvement observed for the thermoselected virus remains elusive, the results of our further genetic analysis of the Ad5-dV/TSB suggest that it might involve changes in the structure of other core protein components. Importantly, since the originally rescued as well as the thermoselected viruses were both devoid of pV, our study provides the first experimental demonstration of viability of the pV-deleted Ads and validates a phylogenetic evidence-driven hypothesis that the adenoviral gene V is a non-essential, although gene V is a highly important adenoviral gene.
In addition to the role of pV in thermostability and infectivity of Ad, this study also provides the first evidence that pV might play also an important regulatory role in adenovirus-infected cells. This is supported by our observation that expression of major late structural proteins, including hexon, starts significantly earlier in Ad5-dV/TSB-infection cycle which correlates with hexon mRNA gene expression in the infected cells. Moreover, the hexon mRNA level appears to decline earlier at late phase of the viral replication as compared to those in Ad5-infected cells (Figure 6(b) and (c)). This phenomenon suggests that pV, just like pIX, might also have regulatory function and be required to directly or indirectly repress MLP activity and avoid premature transcription of major late structural genes. Since pV is a late gene, whose expression is driven by MLP as well, the tentative role of pV in MLP suppression at early stage of Ad infection can only be attributed to the virion-associated form of this core protein. In this regard, the lack of pV in the internalized Ad5-dV/TSB particles could result in premature activation of MLP and early onset of the late gene expression, observed in our experiments. Thus, another possible intracellular function of pV is to control the activity of MLP in the viral life cycle. The conspicuous downregulation of the protein, believed to be core protein VII, seen in the Ad5-dV/TSB virus-infected cells is not understood, but might indicate to the possible regulatory link between pV and the expression of pVII in the Ad5-infected cells.
Mapping of additional mutations, acquired by the Ad5-dV/TSB genome as a result of our thermoselection and biological screening procedures, has been achieved by employing a judicious genome sequencing approach. We hypothesized that the functional defects resulting from the absence of pV from the virion’s core could be most likely compensated by changes in other structural components of the virion. In this regard, we undertook sequencing of capsid and core protein coding genes in the first place. Although a careful sequence analysis of all capsid genes in the Ad5-dV/TSB genome revealed no changes relative to the counterpart sequences of Ad5, we found three single nucleotide substitution mutations in the sequence encoding precursor for the core protein X/Mu (Figure 7). Our discovery of suppressor mutations in another core protein was consistent with our theoretical prediction as X/Mu is known to be a functional partner of pV and pVII in the process of Ad genome condensation and packaging into the capsid. A detailed analysis of the genomic DNA sequence readouts argued for homogeneity of genomic DNA isolated from the purified Ad5-dV/TSB particles and, thus, suggested that all three mutations occurred within a single genomic DNA molecule. This conclusion was further reinforced by the fact that a single type of suppressor mutations was found in the genomic DNA derived from twelve distinct plaques descending from the original thermoselected clone after several rounds of its plaque purification. Furthermore, the cluster of exactly same suppressor mutations was also found in the genomic DNA from another two originally isolated thermoselected viral clones suggesting that it is the only or at least a predominant type of genetic alteration selected under the given experimental conditions. We could not isolate any of the individual point mutants and any other mutants as a result of our selection approach. The lack of heterogeneity among the isolated clones and the clustered nature of the selected mutations indicate that the acquired thermostability may in fact be a cumulative result of more than one mutation in the cluster. If an individual point mutation was sufficient to cause thermostability of Ad5-dV/TSB, we should also have been able to isolate variants with individual mutations contained in the cluster. In this basis, we suggest that a “cluster of suppressor mutations” in the precursor of the X/Mu protein gene rather than individual mutations, is associated with the thermostability/infectivity phenotype of the Ad5-dV/TSB. On the other hand, we suggest that the silent mutation cannot possibly have any effect on the phenotype and thus is unlikely to contribute to the restored thermostability/infectivity of the thermoselected virus.
Adenoviral Mu protein is found in all members of the Adenoviridae family1 and is expressed as a precursor in Ad infected cells.41 The precursor form of Mu protein localizes to the nucleoli of the infected cells and modulates expression of the E2 and possibly also the late genes.42 It further undergoes processing by Ad protease24 into three fragments at two specific sites around amino acid positions 31 and 50. The central polypeptide fragment is subsequently incorporated in the virions,43 whereas the function(s) of the precursor’s N-terminal peptide in infected cells is unclear. Some recent evidence, however, suggests that the C-terminal peptide of pre-Mu is essential for its repression of the late genes (VI) in adenovirus-infected cells.42 Since the suppressor mutations of the Ad5-dV/TSB mapped in the pre-Mu coding sequence are distal to the precursor processing sites, they are unlikely to affect processing of the precursor.
Alignment of Mu protein sequences from different groups of adenoviruses shows that human, simian and some bovine adenovirus serotypes that possess the core protein V gene also have both clusters of conserved amino acid residues at the N-terminal peptide of the pre-Mu protein (Figure 8). On the contrary, members of Atadenovirus and Siadenovirus genera that do not encode gene V orthologs in their genomes also lack most of the N-terminal domain in their pre-Mu proteins, including the above mentioned clusters of conservation.1 Although C-terminal domain of pre-Mu is necessary for its trans-activating function,42 this activity cannot be carried out by the C-terminal domain alone and also requires the central portion (mature Mu) of the precursor.42 Since the pre-Mu appears to have function(s) distinct from the ones of the mature protein and is also capable of localizing to the nuclei of the infected cells,42 it can be hypothesized that the pre-Mu and/or its N-terminal peptide could play an important role in mediating viral assembly in Ad-infected cell. Elucidation of the mechanism whereby mutations in pre-Mu suppress the effect of pV deletion is currently underway.
Thus, our study implicates pV as a core component necessary for correct assembly of viral particles and provides an interesting biological insight into its possible functional relationships with the protein X/Mu precursor. Although the significance of the observed excessive incorporation of protein X/Mu into the Ad5-dV/TSB viral particles is yet unclear, this phenomenon might be a part of the molecular mechanism, whereby the absence of the core component pV is suppressed in the cells infected with thermoselected Ad5-dV/TSB.
The experimental approach used in this study has a potential utility for isolation and genetic characterization of other novel Ad mutants with suppressed thermostability defects generated in structural genes of recombinant Ad. Identification of such genetic suppressor mutations could contribute to understanding of molecular interactions between adenoviral proteins and their roles in the viral assembly and stabilization. The results of this study are consistent with our hypothesis that the process of losing or gaining of species-specific genes in the course of viral evolution should be accompanied by a “molecular adaptation” of other protein components to the new genetic environment. Such “adjustments” appear to be realized through accumulation of suppressor mutations inducing structural/functional changes in some other protein components of the virion that can compensate for the change at the molecular level.
Materials and Methods
Construction of recombinant plasmids
Deletion of the pV gene was generated by using a previously constructed shuttle vector pShuttle-V-EGFP-pA(−) (L.P. Le, unpublished data) as a template for PCR amplification of the Ad5 genome between the PmlI site located in the penton base gene and the sequence just upstream of the pV coding region. The following primers were used for PCR amplification: 5′-GTGTTCAATCGCTTTCCCGAG-3′ (forward primer; PmlI half site is shown in underlined.) and 5′-TTTGTACAAGCTTCGTTGCGCGCCG CCGC-3′ (reverse primer; BsrGI and HindIII sites are shown in italics and underlined, respectively.). The resulting PCR product was digested with BsrGI and cloned into the pShuttle-V-EGFP-pA(−), following digestion of the shuttle with PmlI and BsrGI, respectively, to replace the pV-EGFP sequence. The resulting pShuttle-dV-kan was used for homologous recombination with pTG3602 rescue vector28, harboring the wild type Ad5 genome, in Escherichia coli strain BJ518344 to introduce deletion of the gene V i.e. nucleotides 16,545-17,651 in the Ad5 genome (Genbank accession no. AY339865) (Figure 1(a)), resulting in pTG3602-dV-kan. Subsequently the kanamycin (kan) gene was removed by digestion of pTG3602-dV-kan with ClaI restriction enzyme and self-ligation of the digested vector to generate pTG3602-dV. The pV-deleted Ad5 genome, thus, contained a short heterologous sequence with artificially generated restriction sites, BsrGI, HindIII and ClaI. An intact pTG3602 was used to generate the wild type Ad5 virus.
Cells
Human embryonic retinoblast 911 cells45 and human lung epithelial A549 cells (ATCC CCL 185) were cultured in Dulbecco’s modified Eagle’s-Ham’s F12 50:50 medium (DMEM/F12) (Mediatech Inc., Herndon, VA) containing 10% fetal bovine serum (Hyclone, Logan, UT), 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. The cells were maintained as subconfluent adherent cultures in a 5% CO2 environment of 37°C. The Ad-infected cells were cultured in the same medium contained 2% FBS.
Generation and propagation of adenoviruses
Plasmids, pTG3602 and pTG3602-dV were linearized with PacI and transfected into 911 cells using UniFECTOR transfection reagent (B-Bridge International Inc., Sunnyvale, CA) to generate Ad5 and Ad5-dV viruses, respectively. Each generated Ad was plaque-purified on 911 cells by the method described elsewhere.40 All viruses were propagated in two 500-cm2 flasks of A549 cells and purified by a modified version of the CsCl-gradient ultracentrifugation method.46 Briefly, after visualization of the cytopathic effect (CPE) both infected cells and culture mediums were harvested and the cells were disrupted by three consecutive freeze-thaw cycles. The lysates were centrifuged at 3,000 × g for 10 min at 4°C to remove the cell debris and the crude supernatants were subsequently used for further virus purification. The gradient for the first ultracentrifugation was prepared with 5 ml of 1.50 g/ml CsCl in phosphate buffered saline (PBS, pH 7.4), 10 ml of 1.27 g/ml CsCl in PBS (pH 7.4) and 20 ml of the crude viral solution. Centrifugation was carried out in a SW28 rotor (Beckman Coulter Inc., Palo Alto, CA) at 112,700 × g for 1 h at 4°C. After centrifugation, the band of Ads was collected and mixed with an equal volume of a saturated solution of CsCl in PBS (pH 7.4). This mixture was transferred to a fresh tube that fitted an SW41 rotor (Beckman Coulter Inc.) and then 3 ml of 1.50 g/ml CsCl in PBS (pH 7.4) and 4 ml of 1.27 g/ml CsCl in PBS (pH 7.4) were overlaid on the mixture sequentially. After centrifugation at 209,900 × g for 1 h at 4°C, the band of Ad identified in the layer between 1.27 g/ml CsCl and 1.50 g/ml CsCl, was collected as a suspension of approximately 1 ml. The suspension was dialyzed twice for 2 hours and overnight each against 500 ml of PBS (pH7.4) contained 10% glycerol at 4 °C. The infectious titers (PFU/ml) of purified Ads were determined by triplicate plaque assays using A549 cells as described elsewhere40 and the physical titers (VP/ml) were determined by A260 absorbance of purified particles and assuming that 1.1 × 1012 VP/ml has an absorbance of 1.0 at 260 nm.47
Restriction and PCR analyses of adenoviral genome
For restriction and PCR analyses we used total DNA isolated from 6-cm dishes of Ad-infected A549 cells showing cytopathic effect (CPE) and Ad genomic DNA from CsCl-purified viruses. Both the total DNA and the Ad genomic DNA were prepared according to the method described by Saito et al..48 Analysis of the regions of pV and fiber genes was performed by PCR using TaqPCR Master Mix Kit (Qiagen Inc., Valencia, CA) and 1010 VP of viral genomic DNA as a template. Forward and reverse primers corresponding to nt positions 16,497-16,521 (pVF1) and 17,692-17,667 (pVR1) of the Ad5 genome were used to amplify the region containing gene V sequence. Forward and reverse primers corresponding to nt positions 30,943-30,964 (F-Ad5 tail) and 32,919-32,940 (R-Ad5 knob) of the Ad5 genome were used for PCR analysis of the fiber gene. The following PCR conditions applied: 20 sec at 96°C, 20 sec at 60°C and 72°C for 1 min when using the region of pV gene and 3 min, when using the region of fiber gene. All PCR primer sequences are available on request.
Sequence analysis
All plasmid constructs, PCR products and viral genomic DNAs of Ad5, Ad5-dV and Ad5-dV/TSB were validated by DNA sequencing. Sequencing was performed at the DNA sequencing core facility of the University of Alabama at Birmingham. Viral genomic DNA extracted from CsCl-purified viruses was completely sequenced between nt positions 1,239 and 32,967 using a set of forward and reverse primers (sequences available upon request). All mutation sites were validated by comparing the obtained genome sequencing data to the nucleotide sequence of Ad5 (Genebank accession no. AY339865) and the nucleotide sequences of the backbones (pTG3602 and pTG3602-dV) and the shuttle (pShuttle-dV-kan) vectors used in the vector construction.
Virus thermostability and thermoselection assays
Thermostability of Ads was assessed by using a modified method described previously.49,50 Briefly, 1010 VP of purified viral particles were incubated at various temperatures (−80, −20, +4, +25 and +37°C) for 3 days in PBS (pH 7.4) containing 10% glycerol. To assess dynamics of virus inactivation, 1010 VP of each purified Ad was incubated for 1, 3 and 7 days at each given temperature (−80, +4 and +37°C). The resulting infectious titers of the Ads were determined by triplicate plaque assays on A549 cells.40
To isolate thermoselected variants of the Ad5-dV which are more stable at 37ºC, we carried out a modified thermoselection screen26,27, which involved incubation of 1010 VP of purified Ad5-dV in PBS (pH 7.4) containing 10% (v/v) glycerol at 37ºC for 3 days. We performed two independent experiments and isolated thermostable variants of Ad5-dV by three rounds of plaque purification on A549 cells.
Screening of clones for the 17,716 suppressor mutation in the pre-Mu gene
The presence of the 17,716 suppressor mutation in genomes of plaque purified Ad5-dV/TSB clones was assessed by using EcoRI restriction analysis of the PCR products derived from amplification of a selected region of the Ad genome comprising gene for the protein X/Mu precursor with the following forward and reverse primers: 5′-GAGGCGCGCAACTACACGC-3′ (VII16076) and 5′-CCTCTGGAGACACTGT CTCCAC-3′ (VI18381), respectively. The following PCR conditions applied: 20 sec at 96°C, 20 sec at 55°C and 2 min at 72°C. The PCR products were purified with PCR purification kit (Qiagen Inc.) and digested with EcoRI restriction enzyme at 37°C for 1 hour. The digests were analyzed by electrophoresis in a 1% agarose gel to detect formation of a new EcoRI site generated as a result of the 17,716 suppressor mutation in the pre-Mu sequence. The undigested PCR products derived from twelve plaques in the same screening experiment were also sequenced with primer corresponding to nt positions 16,497–16,521 of the Ad5 genome (sequence available on request) to confirm the presence of all three suppressor mutations.
Viral replication
To assess viral replication efficiency A549 cells grown to 80% confluency in 6-cm dishes were infected at an MOI of 10 PFU/cell and each dish was maintained in 3 ml of 2% FBS containing medium. The infected cells were harvested with a cell scraper at various time points postinfection and pelleted down by centrifugation at 1,000 × g, 5 min, at 4 °C to remove culture medium. The cells were re-suspended in 100 μl of PBS (pH 7.4) containing 10% (v/v) glycerol and disrupted by three freeze-thaw cycles. The lysates were then centrifuged at 15,000 × g for 5 min at 4°C to remove cell debris. The supernatants prepared at various times postinfection were stored at −80°C prior to titration. Following determination of total protein concentration the lysates were analyzed by Western blot to assess adenovirus-specific protein expression as described below.
Quantitative reverse transcription-polymerase chain reaction
For quantification of adenoviral hexon mRNA, A549 cells grown to 80% confluency in 6-cm dishes were infected at an MOI of 10 PFU/cell and each dish was maintained in 3 ml of 2% FBS containing medium. The infected cells were harvested at various time points postinfection and total RNA was extracted with Trizol reagent according to manufacture’s instructions (Invitrogen). The total RNA prepared from infected cells at various times postinfection were stored at −80°C prior to quantify hexon mRNA. Primers are specifically designed to amplify hexon mRNA based on the spliced site. Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) was performed using forward and reverse primers corresponding to nt positions 9714–9723 (5′-TAACCAGTCACAGTCGCAAG-3′) and 18,862-18,842 (5′-CATCATCGAAGGGGTAGCCAT-3′). For real-time RT-PCR using a LightCycler™ (Roche Molecular Biochemicals, version 3.0) each reaction was performed with various amounts (50, 100 and 200 ng) of total RNA of the sample and control sample without template as no template control, and Brilliant SYBER Green QRT-PCR Master mix prepared according to the manufacturer’s protocol (Strategene, La Jolla, CA). The following quantitative RT-PCR conditions applied: 30 min at 50°C for reverse transcription, 1 min at 95°C for denaturation reaction, 30 sec at 95°C, and 1 min at 50°C and 30 sec at 72°C for PCR. Results are presented as relative quantification of hexon mRNA related the PCR signal of the target transcript in a treatment group to that of another sample prepared at 0 hour post-infection by 2−ΔΔCT method.51 Normalization of the relative expression levels of the genes towards the levels of house keeping genes, such as glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was not possible because of the observed decrease in the levels of GAPDH after adenoviral infection (data not shown).
Transmission electron microscopy
All viruses were cultured in A549 cells of twenty 175-cm2 flanks, and purified by discontinuous CsCl-ultracentrifugation to collect upper band contained infectious virus and lower band contained defective virus.52 The upper and lower bands of adenovirus particles were collected and each band was CsCl-untracentrifuged again to decrease contaminates of cellular proteins and each viral bands. Transmission electron microscopy (TEM) was performed at Microscopy Center of Louisiana State University.
Determination of protein concentration
Protein concentration of purified Ads and total protein concentration in the lysates of Ad infected cells was determined using DC protein assay (Bio-Rad laboratories Inc., Hercules, CA) according to the manufacturer’s instructions.
Statistical analysis
Statistical analysis was performed with two-tailed unpaired Student’s t tests among groups. P values <0.05 were considered statistically significant.
Analysis of adenoviral proteins by staining and Western blots
Rabbit polyclonal anti-pV antibodies and rabbit polyclonal anti-pIX antibodies were a kind gift of Dr. D. A. Matthews (School of Biochemistry and Molecular Biology, University of Leeds, UK) and from Dr. I. Dmitriev (Gene Therapy Center, University of Alabama at Birmingham), respectively. Mouse monoclonal anti-Ad5 fiber tail primary antibody (4D2) was purchased from Abcam Inc. (Cambridge MA). Virus-specific protein expression was analyzed with rabbit polyclonal anti-serum against Ad5. Five μg of total proteins from virus-infected cell lysates and purified infectious (1010 VP) and defective (109 VP) Ads were separated by 4–20% (w/v) and 4–15% (w/v) gradient sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (Bio-Rad Laboratories, Inc., CA), respectively. Western blot analysis was carried out as described elsewhere.53 Since pVI- and pVII-specific antibodies were not available,21 we could not verify identification of these Ad proteins by Western blots. Pre-stained protein ladder of Kaleidoscope Standards (Bio-Rad Laboratories, Inc.) and SDS-PAGE molecular weight standards (Bio-Rad Laboratories, Inc.) were used for identification of the virus-specific proteins in stained gels and Western blots.
Supplementary Material
Validation of the genomic structure of the thermoselected Ad5-dV/TSB by restriction analysis. Restriction maps for various enzymes of wild type Ad5 (a) and Ad5-dV/TSB (b). Deletion of the pV gene from the genome of Ad5 removes a single NotI site and generates extra BsrGI, ClaI and HindIII sites in place of the deleted pV gene. (c) Validation of the genomic structure of Ad5-dV/TSB by restriction analysis. Total DNA isolated from infected A549 cells was digested with several restriction enzymes and resolved by electrophoresis in a 1% agarose gel. Asterisks show the specific DNA fragment digested with restriction enzymes. (d) Validation of the pV-deletion in the Ad5-dV/TSB genome by PCR. Genomic DNA was extracted from purified viruses and analyzed for gene V (lane 1, 2) and the fiber gene (lane 3, 4) regions by PCR using the same primers and strategy as shown in Figure 1. M, molecular size markers; lanes 1 and 3, PCR on wild type Ad5 genomic DNA; lanes 2 and 4, PCR on Ad5-dV/TSB genomic DNA as a template.
Analysis of protein composition of viral particles by Western blot and gel-staining. (a) Detection of adenoviral proteins incorporated in the virions. 2 × 1010 VP equivalents of purified infectious Ads were run on a 4–15% (w/v) gradient SDS-PAGE and the gel was stained with GELCODE Blue reagent (Pierce Biotechnology Inc., Rockford IL). Lane 1; Ad5, lane 2; Ad5-dV/TSB. (b) Detection of various proteins incorporated in Ad5 (lane 1) and Ad5-dV/TSB (lane 2) particles by Western blot with an anti-serum against Ad5. 1010 VP equivalents of the purified viruses were run on a 7.5% (w/v) SDS-PAGE, electro-transferred onto a PVDF membrane, and probed by an antiserum against Ad5. Protein molecular mass makers (in kilodaltons) are indicated on the left. (c) Comparison of the relative incorporation of pV, pIX and fiber proteins in the purified Ad5 (lanes 1, 3 and 5) and Ad5-dV/TSB viral particles (lanes 2, 4 and 6). Detection of viral proteins was performed by the corresponding (pV-, pIX- and fiber-specific) antibodies. 1010 VP equivalents of each purified virus were loaded on each lane of the 10% SDS-PAGE.
Relative quantification of E1A 12S mRNA at various times postinfection. Ad5 (black bar) and Ad5-dV/TSB (white bar) were infected A549 cells at an MOI 10 of PFU/cell and 50 ng of total RNA extracted from the infected cells was used as template for quantitative RT-PCR. Three replicates were performed. Primers are specifically designed to amplify E1A 12S mRNA based on the spliced site. Quantitative RT-PCR was performed using forward and reverse primers corresponding to nt positions 840–855 (5′-ACCTTTVVVGGCAGCC-3′) and 1104–1112+1229–1237 (5′-ACACAGGACCCTCTTCATCCT-3′). The following quantitative RT-PCR conditions applied: 30 min at 50°C for reverse transcription, 1 min at 95°C for denaturation reaction, 30 sec at 95°C, and 1 min at 50°C and 30 sec at 72°C for PCR. Results are presented as relative quantification of E1A 12S mRNA related the PCR signal of the target transcript in a treatment group to that of another sample prepared at 0 hour post-infection by 2−ΔΔCT method.51
Acknowledgments
We thank Drs. Joel N. Glasgow, Igor P. Dmitriev, Masato Yamamoto and Angel A. Rivera for useful advice and fruitful discussions. We are grateful to Dr. Igor P. Dmitriev for providing a rabbit polyclonal pIX-specific antibody, Dr. D. A. Matthews for providing a rabbit polyclonal pV-specific antibody, and Drs. Gene P. Siegel, Justin Roth Qiana L Matthews and Anand C. Annan for critical reading of the manuscript. We gratefully thank Dr Lacey McNally for technical supports. This work was supported by grants from the National Institute of Health (RO1CA111569 and T32CA075930).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Davison AJ, Benko M, Harrach B. Genetic content and evolution of adenoviruses. J Gen Virol. 2003;84:2895–2908. doi: 10.1099/vir.0.19497-0. [DOI] [PubMed] [Google Scholar]
- 2.Parks RJ. Adenovirus protein IX: a new look at an old protein. Mol Ther. 2005;11:19–25. doi: 10.1016/j.ymthe.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 3.Bahr U, Schondorf E, Handermann M, Darai G. Molecular anatomy of Tupaia (tree shrew) adenovirus genome; evolution of viral genes and viral phylogeny. Virus Genes. 2003;27:29–48. doi: 10.1023/a:1025120418159. [DOI] [PubMed] [Google Scholar]
- 4.Vellinga J, Van der Heijdt S, Hoeben RC. The adenovirus capsid: major progress in minor proteins. J Gen Virol. 2005;86:1581–1588. doi: 10.1099/vir.0.80877-0. [DOI] [PubMed] [Google Scholar]
- 5.Rosa-Calatrava M, Grave L, Puvion-Dutilleul F, Chatton B, Kedinger C. Functional analysis of adenovirus protein IX identifies domains involved in capsid stability, transcriptional activity, and nuclear reorganization. J Virol. 2001;75:7131–7141. doi: 10.1128/JVI.75.15.7131-7141.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lutz P, Rosa-Calatrava M, Kedinger C. The product of the adenovirus intermediate gene IX is a transcriptional activator. J Virol. 1997;71:5102–5109. doi: 10.1128/jvi.71.7.5102-5109.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horwitz MS. Function of adenovirus E3 proteins and their interactions with immunoregulatory cell proteins. J Gene Med. 2004;6(Suppl 1):S172–183. doi: 10.1002/jgm.495. [DOI] [PubMed] [Google Scholar]
- 8.Horwitz MS. Adenovirus immunoregulatory genes and their cellular targets. Virology. 2001;279:1–8. doi: 10.1006/viro.2000.0738. [DOI] [PubMed] [Google Scholar]
- 9.Tollefson AE, Scaria A, Hermiston TW, Ryerse JS, Wold LJ, Wold WS. The adenovirus death protein (E3-11.6K) is required at very late stages of infection for efficient cell lysis and release of adenovirus from infected cells. J Virol. 1996;70:2296–2306. doi: 10.1128/jvi.70.4.2296-2306.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tollefson AE, Ryerse JS, Scaria A, Hermiston TW, Wold WS. The E3-11.6-kDa adenovirus death protein (ADP) is required for efficient cell death: characterization of cells infected with adp mutants. Virology. 1996;220:152–162. doi: 10.1006/viro.1996.0295. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki K, Alemany R, Yamamoto M, Curiel DT. The presence of the adenovirus E3 region improves the oncolytic potency of conditionally replicative adenoviruses. Clin Cancer Res. 2002;8:3348–3359. [PubMed] [Google Scholar]
- 12.Sargent KL, Meulenbroek RA, Parks RJ. Activation of adenoviral gene expression by protein IX is not required for efficient virus replication. J Virol. 2004;78:5032–5037. doi: 10.1128/JVI.78.10.5032-5037.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Colby WW, Shenk T. Adenovirus type 5 virions can be assembled in vivo in the absence of detectable polypeptide IX. J Virol. 1981;39:977–980. doi: 10.1128/jvi.39.3.977-980.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bett AJ, Krougliak V, Graham FL. DNA sequence of the deletion/insertion in early region 3 of Ad5 dl309. Virus Res. 1995;39:75–82. [PubMed] [Google Scholar]
- 15.Mittal SK, Bett AJ, Prevec L, Graham FL. Foreign gene expression by human adenovirus type 5-based vectors studied using firefly luciferase and bacterial beta-galactosidase genes as reporters. Virology. 1995;210:226–230. doi: 10.1006/viro.1995.1337. [DOI] [PubMed] [Google Scholar]
- 16.Bett AJ, Prevec L, Graham FL. Packaging capacity and stability of human adenovirus type 5 vectors. J Virol. 1993;67:5911–5921. doi: 10.1128/jvi.67.10.5911-5921.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghosh-Choudhury G, Haj-Ahmad Y, Brinkley P, Rudy J, Graham FL. Human adenovirus cloning vectors based on infectious bacterial plasmids. Gene. 1986;50:161–171. doi: 10.1016/0378-1119(86)90321-5. [DOI] [PubMed] [Google Scholar]
- 18.Vayda ME, Rogers AE, Flint SJ. The structure of nucleoprotein cores released from adenovirions. Nucleic Acids Res. 1983;11:441–460. doi: 10.1093/nar/11.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matthews DA, Russell WC. Adenovirus core protein V is delivered by the invading virus to the nucleus of the infected cell and later in infection is associated with nucleoli. J Gen Virol. 1998;79 (Pt 7):1671–1675. doi: 10.1099/0022-1317-79-7-1671. [DOI] [PubMed] [Google Scholar]
- 20.Chatterjee PK, Vayda ME, Flint SJ. Interactions among the three adenovirus core proteins. J Virol. 1985;55:379–386. doi: 10.1128/jvi.55.2.379-386.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Oostrum J, Burnett RM. Molecular composition of the adenovirus type 2 virion. J Virol. 1985;56:439–448. doi: 10.1128/jvi.56.2.439-448.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lehmberg E, Traina JA, Chakel JA, Chang RJ, Parkman M, McCaman MT, Murakami PK, Lahidji V, Nelson JW, Hancock WS, Nestaas E, Pungor E., Jr Reversed-phase high-performance liquid chromatographic assay for the adenovirus type 5 proteome. J Chromatogr B Biomed Sci Appl. 1999;732:411–423. doi: 10.1016/s0378-4347(99)00316-3. [DOI] [PubMed] [Google Scholar]
- 23.Chatterjee PK, Vayda ME, Flint SJ. Identification of proteins and protein domains that contact DNA within adenovirus nucleoprotein cores by ultraviolet light crosslinking of oligonucleotides 32P-labelled in vivo. J Mol Biol. 1986;188:23–37. doi: 10.1016/0022-2836(86)90477-8. [DOI] [PubMed] [Google Scholar]
- 24.Anderson CW, Young ME, Flint SJ. Characterization of the adenovirus 2 virion protein, mu. Virology. 1989;172:506–512. doi: 10.1016/0042-6822(89)90193-1. [DOI] [PubMed] [Google Scholar]
- 25.Matthews DA. Adenovirus protein V induces redistribution of nucleolin and B23 from nucleolus to cytoplasm. J Virol. 2001;75:1031–1038. doi: 10.1128/JVI.75.2.1031-1038.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shiomi H, Urasawa T, Urasawa S, Kobayashi N, Abe S, Taniguchi K. Isolation and characterisation of poliovirus mutants resistant to heating at 50 degrees Celsius for 30 min. J Med Virol. 2004;74:484–491. doi: 10.1002/jmv.20202. [DOI] [PubMed] [Google Scholar]
- 27.Young CS. Heat-stable variant of human adenovirus type 5: characterization and use in three-factor crosses. J Virol. 1975;15:1168–1175. doi: 10.1128/jvi.15.5.1168-1175.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chartier C, Degryse E, Gantzer M, Dieterle A, Pavirani A, Mehtali M. Efficient generation of recombinant adenovirus vectors by homologous recombination in Escherichia coli. J Virol. 1996;70:4805–4810. doi: 10.1128/jvi.70.7.4805-4810.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ensinger MJ, Ginsberg HS. Selection and preliminary characterization of temperature-sensitive mutants of type 5 adenovirus. J Virol. 1972;10:328–339. doi: 10.1128/jvi.10.3.328-339.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fried M. Isolation of temperature-semsitive mutants of polyoma virus. Virology. 1965;25:669–671. doi: 10.1016/0042-6822(65)90098-x. [DOI] [PubMed] [Google Scholar]
- 31.Mofford LM, Marusyk RG. Isolation and partial characterization of a human adenovirus type 4 temperature-sensitive mutant (Mastadenovirus h 4 tsl) Can J Microbiol. 1984;30:135–141. doi: 10.1139/m84-022. [DOI] [PubMed] [Google Scholar]
- 32.Praszkier J, Ginsberg HS. Isolation and characterization of temperature-sensitive mutants of adenovirus type 7. J Virol. 1987;61:3089–3095. doi: 10.1128/jvi.61.10.3089-3095.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kauffman RS, Ginsberg HS. Characterization of a temperature-sensitive, hexon transport mutant of type 5 adenovirus. J Virol. 1976;19:643–658. doi: 10.1128/jvi.19.2.643-658.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chee-Sheung CC, Ginsberg HS. Characterization of a temperature-sensitive fiber mutant of type 5 adenovirus and effect of the mutation on virion assembly. J Virol. 1982;42:932–950. doi: 10.1128/jvi.42.3.932-950.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Counago R, Chen S, Shamoo Y. In vivo molecular evolution reveals biophysical origins of organismal fitness. Mol Cell. 2006;22:441–449. doi: 10.1016/j.molcel.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 36.Anderson CW, Hardy MM, Dunn JJ, Klessig DF. Independent, spontaneous mutants of adenovirus type 2-simian virus 40 hybrid Ad2+ND3 that grow efficiently in monkey cells possess indentical mutations in the adenovirus type 2 DNA-binding protein gene. J Virol. 1983;48:31–39. doi: 10.1128/jvi.48.1.31-39.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aneskievich BJ, Taichman LB. Evidence for two points of restriction in the expression of adenovirus type 2 in cultured epidermal keratinocytes. J Virol. 1988;62:4365–4368. doi: 10.1128/jvi.62.11.4365-4368.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klessig DF. Isolation of a variant of human adenovirus serotype 2 that multiplies efficiently on monkey cells. J Virol. 1977;21:1243–1246. doi: 10.1128/jvi.21.3.1243-1246.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yan W, Kitzes G, Dormishian F, Hawkins L, Sampson-Johannes A, Watanabe J, Holt J, Lee V, Dubensky T, Fattaey A, Hermiston T, Balmain A, Shen Y. Developing novel oncolytic adenoviruses through bioselection. J Virol. 2003;77:2640–2650. doi: 10.1128/JVI.77.4.2640-2650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ugai H, Inabe K, Yamasaki T, Murata T, Obata Y, Hamada H, Yokoyama KK. Accumulation of infectious mutants in stocks during the propagation of fiber-modified recombinant adenoviruses. Biochem Biophys Res Commun. 2005;337:806–814. doi: 10.1016/j.bbrc.2005.09.112. [DOI] [PubMed] [Google Scholar]
- 41.Weber JM, Anderson CW. Identification of the gene coding for the precursor of adenovirus core protein X. J Virol. 1988;62:1741–1745. doi: 10.1128/jvi.62.5.1741-1745.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee TW, Lawrence FJ, Dauksaite V, Akusjarvi G, Blair GE, Matthews DA. Precursor of human adenovirus core polypeptide Mu targets the nucleolus and modulates the expression of E2 proteins. J Gen Virol. 2004;85:185–196. doi: 10.1099/vir.0.19352-0. [DOI] [PubMed] [Google Scholar]
- 43.Hosokawa K, Sung MT. Isolation and characterization of an extremely basic protein from adenovirus type 5. J Virol. 1976;17:924–934. doi: 10.1128/jvi.17.3.924-934.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hanahan D. Studies on transformation of Escherichia coli with plasmids. J Mol Biol. 1983;166:557–580. doi: 10.1016/s0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 45.Fallaux FJ, Kranenburg O, Cramer SJ, Houweling A, Van Ormondt H, Hoeben RC, Van Der Eb AJ. Characterization of 911: a new helper cell line for the titration and propagation of early region 1-deleted adenoviral vectors. Hum Gene Ther. 1996;7:215–222. doi: 10.1089/hum.1996.7.2-215. [DOI] [PubMed] [Google Scholar]
- 46.Kanegae Y, Makimura M, Saito I. A simple and efficient method for purification of infectious recombinant adenovirus. Jpn J Med Sci Biol. 1994;47:157–166. doi: 10.7883/yoken1952.47.157. [DOI] [PubMed] [Google Scholar]
- 47.Maizel JV, Jr, White DO, Scharff MD. The polypeptides of adenovirus. I Evidence for multiple protein components in the virion and a comparison of types 2, 7A, and 12. Virology. 1968;36:115–125. doi: 10.1016/0042-6822(68)90121-9. [DOI] [PubMed] [Google Scholar]
- 48.Saito I, Oya Y, Yamamoto K, Yuasa T, Shimojo H. Construction of nondefective adenovirus type 5 bearing a 2.8-kilobase hepatitis B virus DNA near the right end of its genome. J Virol. 1985;54:711–719. doi: 10.1128/jvi.54.3.711-719.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ugai H, Watanabe S, Suzuki E, Tsutsui-Nakata H, Yokoyama KK, Murata T. Stability of a recombinant adenoviral vector: optimization of conditions for storage, transport and delivery. Jpn J Cancer Res. 2002;93:598–603. doi: 10.1111/j.1349-7006.2002.tb01296.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shabram P, Vellekamp G, Scandella C. Purification of adenovirus. In: Curiel DT, Douglas JT, editors. Adenoviral vectors for gene therapy. Academic Press; London, UK: 2002. pp. 167–203. [Google Scholar]
- 51.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 52.Tellefson AE, Hermiston WT, Wold WSM. Preparation and titration of CsCl-banded adenovirus stock. In: Wold WSM, editor. Adenovirus methods and protocols. Humana Press Inc; Totowa, NJ: 1999. pp. 1–9. [Google Scholar]
- 53.Ugai H, Yamasaki T, Hirose M, Inabe K, Kujime Y, Terashima M, Liu B, Tang H, Zhao M, Murata T, Kimura M, Pan J, Obata Y, Hamada H, Yokoyama KK. Purification of infectious adenovirus in two hours by ultracentrifugation and tangential flow filtration. Biochem Biophys Res Commun. 2005;331:1053–1060. doi: 10.1016/j.bbrc.2005.03.227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Validation of the genomic structure of the thermoselected Ad5-dV/TSB by restriction analysis. Restriction maps for various enzymes of wild type Ad5 (a) and Ad5-dV/TSB (b). Deletion of the pV gene from the genome of Ad5 removes a single NotI site and generates extra BsrGI, ClaI and HindIII sites in place of the deleted pV gene. (c) Validation of the genomic structure of Ad5-dV/TSB by restriction analysis. Total DNA isolated from infected A549 cells was digested with several restriction enzymes and resolved by electrophoresis in a 1% agarose gel. Asterisks show the specific DNA fragment digested with restriction enzymes. (d) Validation of the pV-deletion in the Ad5-dV/TSB genome by PCR. Genomic DNA was extracted from purified viruses and analyzed for gene V (lane 1, 2) and the fiber gene (lane 3, 4) regions by PCR using the same primers and strategy as shown in Figure 1. M, molecular size markers; lanes 1 and 3, PCR on wild type Ad5 genomic DNA; lanes 2 and 4, PCR on Ad5-dV/TSB genomic DNA as a template.
Analysis of protein composition of viral particles by Western blot and gel-staining. (a) Detection of adenoviral proteins incorporated in the virions. 2 × 1010 VP equivalents of purified infectious Ads were run on a 4–15% (w/v) gradient SDS-PAGE and the gel was stained with GELCODE Blue reagent (Pierce Biotechnology Inc., Rockford IL). Lane 1; Ad5, lane 2; Ad5-dV/TSB. (b) Detection of various proteins incorporated in Ad5 (lane 1) and Ad5-dV/TSB (lane 2) particles by Western blot with an anti-serum against Ad5. 1010 VP equivalents of the purified viruses were run on a 7.5% (w/v) SDS-PAGE, electro-transferred onto a PVDF membrane, and probed by an antiserum against Ad5. Protein molecular mass makers (in kilodaltons) are indicated on the left. (c) Comparison of the relative incorporation of pV, pIX and fiber proteins in the purified Ad5 (lanes 1, 3 and 5) and Ad5-dV/TSB viral particles (lanes 2, 4 and 6). Detection of viral proteins was performed by the corresponding (pV-, pIX- and fiber-specific) antibodies. 1010 VP equivalents of each purified virus were loaded on each lane of the 10% SDS-PAGE.
Relative quantification of E1A 12S mRNA at various times postinfection. Ad5 (black bar) and Ad5-dV/TSB (white bar) were infected A549 cells at an MOI 10 of PFU/cell and 50 ng of total RNA extracted from the infected cells was used as template for quantitative RT-PCR. Three replicates were performed. Primers are specifically designed to amplify E1A 12S mRNA based on the spliced site. Quantitative RT-PCR was performed using forward and reverse primers corresponding to nt positions 840–855 (5′-ACCTTTVVVGGCAGCC-3′) and 1104–1112+1229–1237 (5′-ACACAGGACCCTCTTCATCCT-3′). The following quantitative RT-PCR conditions applied: 30 min at 50°C for reverse transcription, 1 min at 95°C for denaturation reaction, 30 sec at 95°C, and 1 min at 50°C and 30 sec at 72°C for PCR. Results are presented as relative quantification of E1A 12S mRNA related the PCR signal of the target transcript in a treatment group to that of another sample prepared at 0 hour post-infection by 2−ΔΔCT method.51