

Abstract
Treatment of advanced lung cancer is one of the major challenges in current medicine because of the high morbidity and mortality of the disease. Advanced stage lung cancer is refractory to conventional therapies and has an extremely poor prognosis. Thus, new therapeutic approaches are needed. Lung tumor formation depends on angiogenesis in which the vascular endothelial growth factor (VEGF) produced by cancer cells plays a pivotal role. Neutralizing VEGF with a soluble VEGF receptor suppresses tumor growth; however, the anticancer effect with this therapy is weakened after the intratumoral vascular network is completed. In this study, we turned the expression of VEGF by tumors to therapeutic advantage using a conditionally replication-competent adenovirus (CRAd) in which the expression of E1 is controlled by the human VEGF promoter. This virus achieved good levels of viral replication in lung cancer cells and induced a substantial anticancer effect in vitro and in vivo. As a further enhancement, the cancer cell killing effect was improved with tropism modification of the virus to express the knob domain of Ad3, which improved infectivity for cancer cells. These VEGF promoter-based CRAds also showed a significant cell killing effect for various types of cancer lines other than lung cancer. Conversely, the VEGF promoter has low activity in normal tissues, and the CRAd caused no damage to normal bronchial epithelial cells. Since tumor-associated angiogenesis via VEGF signalling is common in many types of cancers, these CRAds may be applicable to a wide range of tumors. We concluded that VEGF promoter-based CRAds have the potential to be an effective strategy for cancer treatment.
Keywords: VEGF, replicative adenovirus, angiogenesis, chimeric vector
Introduction
Lung cancer, one of the most common malignant tumors in the world, is the leading cause of cancer deaths in many countries.1 Surgical resection remains the standard of care of patients with early-stage disease. Unfortunately, over 70% of all patients with non-small cell lung cancer (NSCLC) have inoperable disease at the time of diagnosis. Despite the use of optimal chemotherapy and radiotherapy regimens, the outcome of patients with advanced disease is extremely poor, and little progress has occurred in the last decade. This points to a need for developing more effective treatments.
Antiangiogenesis approaches to therapy are promising treatment alternatives, since several studies have shown that angiogenesis is one of the important factors in the growth, progression and metastasis of solid tumors.2,3 Among the many known angiogenic factors, such as basic fibroblast growth factor, angiogenin, interleukin-8, platelet-derived endothelial cell growth factor and vascular endothelial growth factor (VEGF), VEGF is now believed to play a pivotal role in tumor-associated angiogenesis in lung cancer as well as other solid tumors. In fact, several papers have reported that the resected lung cancer tissue express the VEGF mRNA and protein abundantly based on immunohistochemical and reverse transcription-polymerase chain reaction (RT-PCR) analysis.4,5 Moreover, the high expression levels of VEGF mRNA and protein are correlated with poor prognosis in lung cancer patients,4,5 indicating that advanced lung cancer tissues tend to express VEGF. We also reported the negative correlation between VEGF expression and dendritic cell infiltration in the tumor, suggesting an immunosuppressive function of VEGF, which may promote the tumor growth.6 These clinical findings show that VEGF is a possible target molecule for new lung cancer treatments. In fact, we reported that soluble VEGF receptor gene transfer was able to neutralize the angiogenic effect of VEGF and suppress tumorigenesis in vivo.7,8 However, the effect of anti-VEGF treatment is diminished in the well-established tumors that already have a developed blood vessel network. Nevertheless, the fact that established tumors still express higher levels of VEGF than normal tissues suggested that there might still be a way to exploit this differential for therapeutic advantage.
An emerging strategy for cancer therapy is the use of conditionally replicative adenoviruses (CRAds) that are designed to exploit key differences between tumor cells and normal cells to allow viral replication only in the former. Two basic strategies have been used to generate CRAds.9 A type I approach, such as Ad-dl1520 (ONYX-015) or AdΔ24, involves directly mutating a key Ad gene such as E1 to take advantage of the disordered cell cycle regulation in tumor cells with, for example, functionally deficient p53 or RB signalling.10,11 The type II approach involves replacement of wild-type Ad promoters with tumor-specific promoters to drive the expression of genes essential for Ad replication, including E1. The promoters of alpha-fetoprotein,12 prostate specific antigen,13 osteocalcin14 and MUC115 have already been used to generate type II CRAds to treat hepatocellular carcinoma, prostate cancer and breast cancer, respectively. We also reported that the midkine promoter and tyrosinase promoter based CRAds are useful for the treatment of pediatric solid tumors and melanoma, respectively.16,17 ONYX-015 and CV706 (PSA promoter-based) have already been used and shown to be safe in clinical trials.18,19
Based on these findings, we utilized the VEGF promoter, which is activated in advanced lung cancer to generate a type II CRAd, to achieve selective replication and oncolysis in VEGF positive tumors. To test our hypothesis, we replaced the native E1A promoter with the human 2.6 kb VEGF promoter and evaluated the oncolytic effect in various cell lines and in vivo. Furthermore, in view of the increasing evidence that levels of the coxsackie and adenoviral receptor (CAR) are low on many tumor types, we also generated a tropism-modified virus containing the receptor binding knob domain of Ad serotype 3 in place of the Ad5 knob.
Materials and methods
Cell culture
The NCI-H82, NCI-H460, NCI-H157, NCI-H322, NCI-H522, NCI-H1299, NCI-H358, NCI-N417, A427, A549, lung cancer cell lines, BEAS-2B, normal human bronchial epithelial cell line, Panc-I, pancreas cancer cell line and HEK293 adenoviral transformed human embryonic kidney cell line were obtained from ATCC (American Type Culture Collection, Manassas, VA). QG56 and QG90 were provided by National Kyushu Cancer Center, Fukuoka, Japan). Human ovarian adenocarcinoma cell line SKOV3.ip1 was obtained from Dr Janet Price (MD Anderson Cancer Center, Houston, TX). The MeWo cell line was a gift of Dr Ian R Hart (St Thomas Hospital, London, UK). Cells were cultured in the media recommended by each provider and incubated at 37°C and 5% CO2.
Adenovirus vectors
The recombinant adenoviral vectors that express firefly luciferase were constructed through homologous recombination in Esherichia coli using the AdEasy system.20 The 2.6 kb human VEGF promoter region derived from pVEGF-kpnI (provided from Dr Semenza at the Johns Hopkins University, Baltimore, MD)21 was placed in front of the firefly luciferase gene in an Ad E1 shuttle vector, recombined with the E1- and E3-deleted adenoviral backbone vector pAdEasy 1, then transfected into 293 cells by standard techniques to form Ad5VEGFLuc. The luciferase gene and simian virus 40 polyadenylation signal were derived from pGL3 Basic (Promega, Madison, WI). As a control, a vector that containing the ubiquitously active cytomegalovirus (CMV) immediate early promoter (derived from plasmid pCEP4; Invitrogen, Carlsbad, CA) instead of the VEGF promoter was also constructed and named Ad5CMVLuc. The replication competent adenovirus (Ad), Ad5VEGFE1, was also generated from the same E1- and E3-deleted adenoviral backbone vector. Briefly, the fragment corresponding 489–3533 bp from the left end of the type 5 adenoviral genome was amplified by PCR and inserted in the E1 deleted region of the backbone vector. This fragment contains the transcriptional start site of the E1A gene but not the native E1A promoter. The 2.6 kb VEGF promoter region was placed upstream of this fragment. A control vector was also constructed in which the CMV promoter was placed in the same position upstream of E1A. The strategy for these three constructs is summarized in Figure 1. Fiber modified CRAd, Ad5/3VEGFE1 was generated in similar manner as Ad5VEGFE1, but using Ad5/3E1- E3-deleted backbone vector derived from Ad5/3luc1 containing Ad3 knob in place of Ad5 wild-type knob gene as described previously.22 To compare the difference in infectivity between the Ad5 and Ad5/3 chimeric vectors on our target cells, an Ad vector (Ad5/3luc1) that contains a CMV driven luciferase gene in E1 was compared to AdCMVLuc. Wild-type p53 protein expressing Ad, Ad5p53, which contains CMV driven p53 cDNA, was provided from Dr Ueno (University of Occupational and Environmental Health, Kitakyusyu, Japan)23
Figure 1.

Schematic diagram of vector construction. These vectors are constructed from an E3 region-deleted Ad5 backbone and do not contain the Ad E1A promoter region (from nucleotide 324 to 488 of the Ad genome). Deletion of the E3 region was necessary, because of the 2.6 kb VEGF promoter we chose was too long to insert into the Ad genome without deletion of adenoviral E3 region. AdCMVE1 and AdVEGFE1 differ in the promoter driving E1A expression. We also used wild-type Ad5 (Ad5wt) as a control.
The viruses were propagated in the Ad packaging cell line, 293HEK, and purified by double cesium chloride density gradient centrifugation, followed by dialysis against phosphate-buffered saline with 10% glycerol. The viral particle (VP) concentration was determined spectrophotometrically, using a conversion factor of 1.1 × 1012 VPs per absorbance unit at 260 nm,24 and standard plaque assays on 293 cells were performed to determine infectious particles.25
Analysis of VEGF RNA expression
The VEGF RNA status of cell lines was analyzed by RT-PCR as described previously.4 Total cellular RNA was extracted from 1 × 107 cells using the RNeasy kit (Qiagen, Valencia, CA) and analyzed for VEGF and glyceraldehydes-3-phosphate dehydrogenase (GAPDH) RNA with the GeneAmp RNA PCR core kit (Applied Biosystems), as described by the manufacturer. Briefly, 500 ng of total RNA was reverse-transcribed with the random hexamer and murine leukemia virus reverse transcriptase (50°C 30 min) and amplified by PCR with 50 nM of primer pairs described below using a cycling program (initial step of 95°C for 15 min, 27 cycles of 95°C for 1 min and 60°C for 1 min and 72°C for 1 min, final step of 72°C for 10 min). The primers used for the analyses were as follows; VEGF sense (5′-GAAGTGGTGAAGTTCATGGATGTC-3′), VEGF antisense (5′-CGATCGTTCTGTATCAGTCTTTCC-3′), GAPDH sense (5′-CCTTCATTGACCTCAACTA-3′), GAPDH antisense (5′-GGAAGGCCATGCCAGTGAGC-3′). The intensity of RT-PCR band corresponding to VEGF121 and VEGF165 isoform was quantified using image analyzer, NIH image.
Measurement of VEGF protein in culture media
The VEGF protein expression was evaluated as described previously.8 Briefly, 1 × 105 cancer cells were cultured for 24 h in the serum-free media, and then the medium was collected. After centrifugation, the supernatant was stored at −80°C until the assay. The VEGF protein in the culture medium was determined using an ELISA kit (Quantikine Human VEGF Immunoassay, R&D Systems, Minneapolis, MN), according to the manufacturer’s instructions. Each of the values given here is the mean of triplicate determination with respect to standardized cell numbers, 1 × 105 cells.
In vitro analysis of VEGF promoter activation
The activity of the VEGF promoter in an adenovirus context was analyzed by infection of cells with luciferase expression vectors as reported previously.16 Briefly, cells were plated in 12-well plates in triplicate at the density of 1 × 105 cells/well. The next day, the cells were infected with Ad5VEGFLuc or Ad5CMVLuc at the multiplicity of infection (MOI) of 10 plaque-forming unit (PFU)/cell in Dulbecco’s modified Eagle’s medium with 2% fetal calf serum (FCS) for 3 h and then maintained in complete medium. The infected cells were harvested and treated with 100 μl of lysis buffer (Promega, cat no. E153A) after 2 days culture. A luciferase assay (Luciferase Assay System; Promega) and an FB12 luminometer (Zyluc Corporation, Oak Ridge, TN) were used for the evaluation of luciferase activities of Ad-infected cells. Luciferase activities were normalized by the protein concentration in cell lysate (Bio-Rad DC Protein Assay kit).
In vivo analysis of VEGF promoter activation
For determination of luciferase gene expression in mouse organs, nude mice (Charles Rivers) received 1 × 109 PFU of Ad5CMVluc or Ad5VEGFLuc by tail vein injection as reported previously.16 After 2 days, mice were killed, and livers, kidneys, lungs and spleens were resected to measure the luciferase gene expression. The resected organs were placed in the polypropylene tubes, and immediately frozen in ethanol/dry ice. Frozen tissues ground to a fine powder was lysed using a tissue lysis buffer (Promega), and then luciferase activity was determined using a luciferase assay kit (Promega). The luciferase activity was normalized by protein concentration in the tissue lysate. Tumors were induced by injection of 1 × 107 H157 cells on nude mice subcutaneously (Charles Rivers). When tumor formation was confirmed (6–8 mm in diameter), Ad5VEGFLuc (1 × 108 PFU) or Ad5CMVLuc (1 × 108 PFU) were injected into the tumor. Measurement of luciferase activity in tumor was performed in same fashion with other organs described above.
Analysis of viral genome amplification
Viral DNA amplification was assessed as reported previously.16 Cells were plated in a 12-well culture plate in triplicate at the density of 1 × 105 cells/well. After overnight culture, cells were infected with replication-competent Ads (Ad5VEGFE1, Ad5CMVE1 or Ad5wt) or non-replicative Ad (Ad5CMVLuc) at the MOI of 10 for 3 h and then cultured for 24 h. The harvest of infected cells was followed by viral DNA isolation using Blood DNA kit (Qiagen, Valencia, CA). Viral DNA was eluted with 100 μl of elution buffer (10 mM Tris-Cl (pH 8.5)). Eluted samples (1 μl) were analyzed by real-time PCR analysis to evaluate Adenoviral E4 copy number using a LightCycler (Roche). Oligonucleotides corresponding to the sense strand of Ad E4 region (5′-TGACACGCATACTCGGAGCTA-3′: 34885–34905 nt), the antisense strand of E4 region (5′-TTTGAGCAGCACCTTGCATT-3′: 34977–34958 nt), and a probe (5′-CGCCGCCCATGCAACAAGCTT-3′: 34930–34951 nt) were synthesized, used as primers and probe for real-time PCR analysis. The PCR conditions were as follows: 35 cycles of denaturation (94°C, 20 s), annealing (55°C, 20 s), and extension (72°C, 30 s). Adenovirus backbone vector pTG3602 (Transgene, Strasbourg, France)26 was available for making a standard curve for Ad E4 DNA copy number. E4 copy numbers were normalized by the β-actin DNA copy number.
In vitro cytotoxicity assay
For determination of virus-mediated cytotoxicity, 5 × 103 cells were plated in 96-well plates in triplicate. After overnight culture, cells were infected with each Ads at various MOI for 3 h. The infection medium was then replaced with RPMI1640 containing 10% FCS. Viable cells using MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxy-methoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt) assay (CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay; Promega) were evaluated every 3 days. The MTS color development was quantified as optical density at 490 nm by an EL 800 Universal Microplate Reader (Biotec Instruments Inc.). To visualize the cytotoxic effect, crystal violet staining was also performed. 2 × 105 cells were plated in 12-well plates and infected with each Ad at various MOI for 3 h. The infection medium was replaced with growth medium the next day. When cell lysis was observed, cells were fixed and stained with 1% crystal violet in 70% ethanol for 45 min, followed by washing with tap water to remove excess color. The plates were dried, and images were captured with a Kodak DC260 digital camera (Eastman Kodak, Rochester, NY). All experiments were performed in duplicate wells.
In vivo studies – tumor formation in nude mice
All animals were used in accord with protocols approved by the animal care committees of Kyushu University. The experiment was carried out under both the Guidelines for Animal Experiments of Kyushu University and the Law (No. 105) and Notification (No. 6) of the Japanese government. Tumor suppressive effect in vivo was analyzed as described previously.8 Briefly, H157 cells (5 × 106) were injected s.c. into the dorsal skin of nude mice, and tumor growth was monitored for 25 days. Tumor volume was calculated according to the formula a2 × b, where a and b are the smallest and largest diameters, respectively, as described previously. When tumor formation was seen 10 days after inoculation, 1 × 108 PFU of each virus was injected into the tumor directly. One-way analysis of variance (ANOVA) was used to compare tumor volumes, with P<0.05 being considered significant.
Results
VEGF mRNA and protein expression in various cell lines
We wished to develop a strategy for the therapy of NSCLC based on the use of a CRAd in which the VEGF promoter controls the expression of E1 (Figure 1). In vitro, we first investigated a panel of 12 NSCLC cell lines, one bronchial epithelial cell line (BEAS-2B) as a normal cell control, one ovarian cancer cell line (SKOV3.ipl), one gastric cancer cell line (MKN28), and a pancreatic cancer cell line (Panc-I) for VEGF expression using a RT-PCR method as reported previously.4 There are four structural variants of VEGF (VEGF121, VEGF165, VEGF189 and VEGF206) resulting from alternative mRNA splicing in the regions encoding the cytoplasmic domains. Figure 2a shows amplification of a 408 bp fragment (representing VEGF121 cDNA) and a 541 bp fragment (representing VEGF165 cDNA) in all cell lines tested. The PCR bands corresponding to VEGF189 (615 bp) and VEGF206 (666 bp) were minimal or not detected, indicating VEGF121 and VEGF165 were dominant isoforms in these cell lines. The result is consistent with that of the previous similar analysis with primary lung cancer tissue.4 In all cells tested, H157, A427, N417, H358 and SKOV3.ipl showed relatively high expression of VEGF mRNA, while the BEAS-2B cells showed a less intense band than the cancer cell lines, although the band corresponding to VEGF121 was detected at very low levels. Furthermore, we investigated the correlation between mRNA expression and protein expression for VEGF. As shown in Figure 2b, the VEGF protein expression levels also varied in all cell lines. H157 secreted the highest concentration of VEGF protein into the culture media, and the concentration is over 100 times higher than that of BEAS-2B. Comparison between Figure 2a and b revealed that VEGF mRNA expression level correlated with VEGF protein expression level positively. Thus, these results suggested that we could predict the VEGF promoter activity from the VEGF protein concentration.
Figure 2.
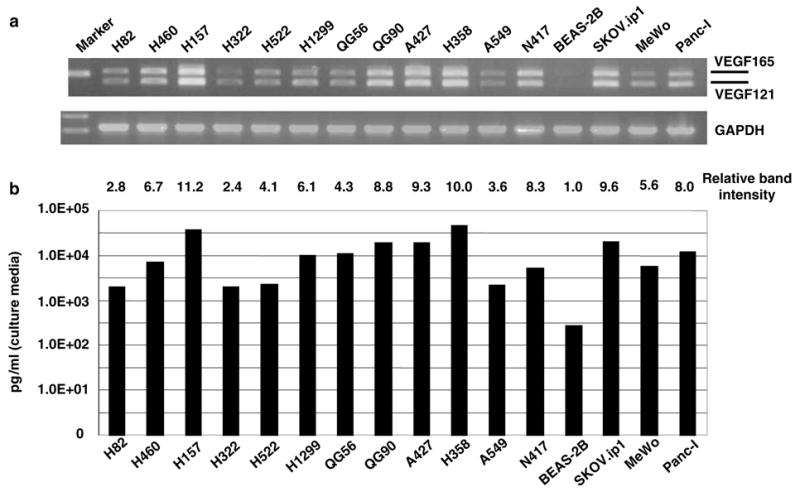
(a) Evaluation of VEGF mRNA expression of the each cell lines. Lane 1, H82; lane 2, H460; lane 3, H157; lane 4, H322; lane 5, H522; lane 6, H1299; lane 7, QG56; lane 8, QG90; lane 9, A427; lane 10, H358; lane 11, A549; lane 12, N417 (lanes 1–12 lung cancer cells); lane 13, BEAS-2B (normal bronchial epithelial cell line); lane 14, SKOV3.ipl (ovarian cancer cell line); lane 15, MeWo (melanoma cell); lane 16, Panc-I (pancreatic cancer cell line). The each RT-PCR product for VEGF121(408 bp), VEGF165 (541 bp) or GAPDH (574 bp) is shown in upper or lower panel, respectively. The intensity of RT-PCR band corresponding to VEGF121 and VEGF165 are quantitated by image analyzer, and expressed as relative intensity compared with no. 13. (b) Evaluation of VEGF protein expression of the each cell lines. 1 × 105 cancer cells were cultured for 24 h in the serum free media. Then the VEGF protein concentration in the media was measured by ELISA. Cell line in each lane is same as in (a).
Transgene expression by VEGF promoter in the Ad context in vitro
Putative tumor-specific promoters may lose their specificity when placed in the Ad genome. Thus, in the next step we assessed the VEGF promoter activity in an Ad vector (Ad5VEGFluc) containing the luciferase gene as a reporter in several cell lines, which spanned the range of VEGF levels we detected in Figure 2. In all of the lines tested, luciferase expression was achieved using the positive control Ad5CMVLuc, which contained luciferase gene driven by the non-selective CMV promoter. From the data with Ad5CMVLuc, A247 and H157 cells were most susceptible to Ad5 infection, which is over 100 times higher than that of H460 as shown in upper panel of Figure 3. These data were consistent with our previous report.8 To standardize the different susceptibility to Ad5 infection, VEGF promoter activity in each cell line was showed as the percentage of luciferase activity with Ad5VEGFLuc to Ad5CMVLuc. As shown in lower panel in Figure 3, H157 cells showed the strongest VEGF promoter activity, 28% of CMV promoter. While BEAS-2B cells presented the lowest VEGF promoter activity, less than 0.1% of CMV. The low transgene expression by the VEGF promoter in the adenoviral context with BEAS-2B was consistent with the results reported recently.27 Other cell lines showed various VEGF promoter activities, which correlate roughly to mRNA expression level of each cell line (cf Figure 2a). Based on these data, we concluded that the VEGF promoter was able to induce transgene expression in VEGF producing cells and importantly that the promoter retained its specificity when placed in the Ad genome.
Figure 3.

Upper panel: Luciferase activities in various cell lines infected by Ad5CMVLuc or Ad5VEGFLuc. 1 × 105 cells of each cell line were infected by Ad5CMVLuc or Ad5VEGFLuc for 3 h at MOI 10. At 48 h after infection cells were harvested and lysed in 100 μl of lysis buffer. Ten microliters of each lysate were used for luciferase assay. Mean + s.e. of triplicate determination is shown. Lower panel: The ratio of VEGF promoter activity to CMV promoter activity. To standardize the VEGF promoter activity in each cell line, the luciferase activity with Ad5VEGFLuc was expressed as the percentage of luciferase activity with Ad5CMVLuc.
Transgene expression by VEGF promoter in the Ad context in vivo
A key limitation of the adenovirus-mediated cancer gene therapy is the potential for toxicity to non-target organs. Because Ad has particular tropism for the liver, we were especially interested to determine whether the VEGF promoter would have low activity in the liver in vivo because the normal liver expresses minimal VEGF. Thus, Ad5VEGFLuc or Ad5CMVLuc (as a positive control) was injected i.v. via the tail vein into mice, and then the level of transgene expression at day 2 was determined (Figure 4). In this assay, transgene expression induced by the VEGF promoter was a mean 270-fold less than that seen with the CMV promoter in the liver. These results thus indicate the key property of VEGF promoter fidelity in the context of the Ad vector used in vivo. Additionally, we investigated the VEGF promoter activation in the tumor in vivo. As shown in Figure 4, the tumor injected by Ad5VEGFLuc showed the 20% of that of Ad5-CMVLuc. These studies confirmed the functionality of the VEGF promoter in VEGF-positive cells in vivo as well as in vitro.
Figure 4.

Tissue and tumor specificity of the VEGF promoter in the adenoviral context. Mice received 1 × × 109 PFU of Ad5VEGFLuc or Ad5CMVLuc via tail vein injection (three per group). At 2 days after virus injection, mice were killed to obtain the organ samples. Each organ lysate was asayed for luciferase activity and normalized for protein concentration. Mean + s.e. of triplicate determination is shown. Tumors were induced by injection of 1 × 107 H157 cells on nude mice subcutaneously. When tumor formation was confirmed (6–8 mm in diameter), Ad5VEGFLuc (1 × 108 PFU) or Ad5CMVLuc (1 × 108 PFU) were injected into the tumor. Measurement of luciferase activity in tumor was performed same as above.
VEGF promoter driving CRAd shows replication specificity
To exploit the cell specificity of the VEGF promoter in a CRAd context, we then constructed a recombinant Ad (Ad5VEGFE1) in which the native E1 promoter is replaced with the 2.6 kb human VEGF promoter. The genomic structures of replication-competent Ads used in this study are depicted in Figure 1. In addition to using the VEGF promoter to regulate E1 expression, we used an Ad in which E1 expression is controlled by the nonselective CMV (Ad5CMVE1) promoter as a control.16 These viruses are deleted in the E3 region and the E1A promoter region. The deleted E1A promoter region, containing native E1A TATA box, was replaced with either the VEGF promoter or CMV enhancer/promoter to produce the viruses Ad5VEGFE1 or Ad5CMVE1, respectively.
To determine the specificity of propagation of the AdVEGFE1, we infected the high VEGF expressing cell line (H157) and low expressing cell line (BEAS-2B), then used quantitative real-time PCR to determine the level of amplification of viral DNA. The non-replicative Ad5-CMVLuc and wild-type Ad5 virus (Ad5wt) were used as negative and positive controls, respectively. Since the all viruses tested here contain the Ad E4 region, viral DNA was quantified as E4 copy number by real-time PCR. As shown in upper panel in Figure 5, the Ad5VEGFE1 viral genome replicated in the high VEGF producing cancer cells, H157 to a similar extent as did the Ad5CMVE1 genome. The nonreplicative Ad5CMVLuc showed a background level of E4 signal indicating no replication in this cell line. Importantly, the replication ability of Ad5VEGFE1 decreased in the low VEGF expressing BEAS-2B cells, with values seen being is almost 3-logs lower than for Ad5CMVE1 (Figure 5, lower panel). These results indicate that theVEGF promoter retains fidelity even in the replication-competent adenoviral context.
Figure 5.
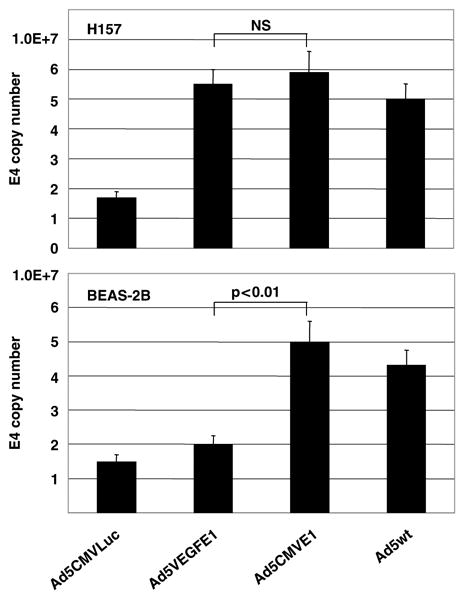
Assessment of viral DNA replication 24 h after infection. 1 × 105 cells were infected with replication-competent Ads (Ad5-VEGFE1, Ad5CMVE1 or Ad5wt) or nonreplicative Ad (Ad5VEGFLuc) at an MOI of 10 for 3 h and then cultured for 24 h. Viral DNA was isolated from the cells and analyzed by real-time PCR analysis to evaluate Adenoviral E4 copy number as described in Materials and methods. E4 copy numbers were normalized by the β-actin DNA copy number. Mean + s.e. of triplicate determination is shown.
Specific cell killing efficacy of VEGF promoter driving CRAd
We next investigated the ability of Ad5VEGFE1 to achieve cell killing in the VEGF-positive cell lines using an MTS assay. The viability of the high VEGF H157 cells and the low VEGF BEAS-2B cells was quantified every 3 days after virus infection as shown in Figure 6a. For the H157 cells, Ad5VEGFE1 showed a strong cytotoxic effect as did the Ad5CMVE1, positive control virus. All cancer cells died at day 9 with infection at a low MOI of 0.1 although the MTS data did not go down to zero, the lack of any remaining viable cells was confirmed using a trypan blue exclusion test. The relatively steep fall in the survival curve after day 5 suggested that it took this long before sufficient replication occurred to begin to induce toxicity. To put these results in some context with another gene-based approach to cancer treatment, we also compared the cytotoxic effect of Ad5VEGFE1 with Ad5p53, which carries the wild-type p53 gene and has been used in clinical trials. We have previously shown that H157 cells, which have a mutated p53 gene, undergo apoptosis when infected with Ad5p53.23 Ad5p53 infection of H157 cells at MOI 0.1 showed a weak cytotoxic effect compared with Ad5VEGFE1. Similar results were obtained with A427 cells (data not shown). In contrast to the effect in the cancer cells, BEAS-2B cells were resistant to Ad5VEGFE1 toxicity even with infection at a high MOI of 10. These data were consistent with the crystal violet staining appearance as shown in Figure 5b. The cytotoxic effect of Ad5VEGFE1 to several other cancer cell lines was also evaluated in same manner with MTS assays. The data revealed that Ad5VEGFE1 had a significant cytotoxic effect to all cancer cells tested (see Discussion below).
Figure 6.
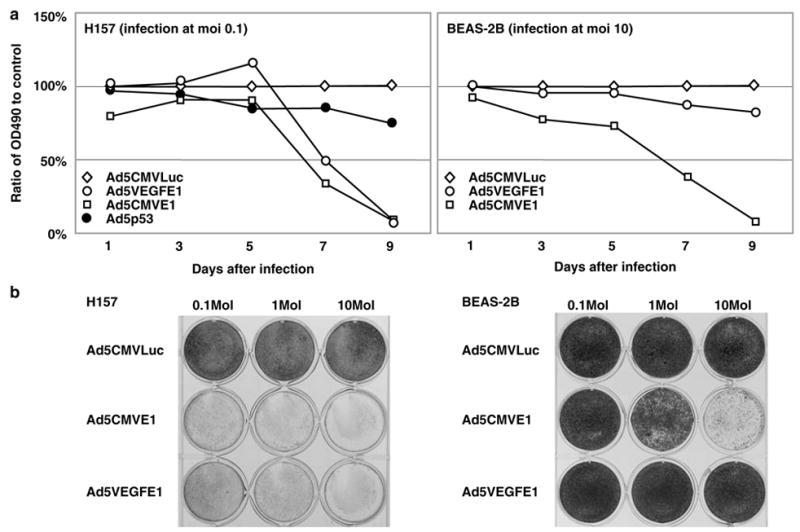
Cell killing effect of AdVEGFE1 and control. Upper panel, cell killing effect was evaluated by MTS assay. 5 × 103 H157 cells were infected with Ad5CMVLuc (negative control), Ad5CMVE1 (positive control), or Ad5VEGFE1 at MOI of 0.1. After infection cell viability in each well was quantified by MTS assay every 3 days. The cell viability of cells infected with Ad5VEGFE1 or Ad5CMVE1 is expressed as the percentage of the OD490 value to control cells infected with Ad5CMVLuc (100%). BEAS-2B cells were infected with each Ad at MOI 10 and evaluated by MTS assay in the same manner. Lower panel: 2 × 105 H157 cells and BEAS-2B cells were infected with each Ad at MOI 0.1, 1.0 or 10.9 days after infection all wells were stained by crystal violet to visualize the viable cells.
Tumor growth suppression by Ad5VEGFE1 in vivo
We next investigated whether Ad5VEGFE1 could suppress tumor growth in vivo. To this end, we established subcutaneous tumors in nude mice, then directly injected either Ad5CMVLuc, Ad5VEGFE1 or Ad5p53 into the tumor. Tumors become visible and injectable in size 10 days after subcutaneous inoculation. Our previous work revealed that the inoculated H157 cells completed angiogenesis at this time, and form the tumor with tumor stroma, resembling actual human tumors.8 1 × 108 PFU of each virus was injected into the tumor directly, and each tumor was observed for 2 weeks. As shown in Figure 7, the tumor injected with Ad5CMVLuc increased in size exponentially even after virus injection. Ad5p53 suppressed the tumor growth partially, but the suppressive effect was minimal. However, Ad5VEGFE1 suppressed the tumor growth significantly, but not completely at day 25 compared with Ad5p53. These findings suggested the CRAd may be a more efficacious agent than a non-replicative virus like Ad5p53 in vivo. Multiple intratumoral injection of Ad5VEGFE1 suppressed the tumor growth more strongly than single injection, and the tumor disappeared after five time injections every 5 days (data not shown).
Figure 7.

Ad5VEGFE1 suppressed tumor growth in vivo. Intact H157 cells (5 × 106) were injected s.c. into nude mice. When tumor formation was seen 10 days after inoculation, 1 × 108 PFU of each virus (diamond, Ad5CMVLuc; circle, Ad5VEGFE1; square, Ad5p53) was injected into the tumor directly. Three similar sized tumors were injected with each virus, and the mean volume + s.e. is shown. One-way ANOVA was used to compare the tumor volume.
Improvement of CRAd effect by fiber modification
The oncolytic effect of any CRAd is dependant on infectivity of the cancer cells as well as promoter activation intensity. The improvement of the Ad infectivity may lead directly to the enhancement of anticancer effect by the CRAd.28 In this study, we found Ad infectivity for H460 lung cancer cells and SKOV3.ipl ovarian cancer cells was almost 2 logs lower than H157 and A427 lung cancer cells (Figure 3), which may contribute to lower CRAd effect in these cell lines. We have previously reported that infectivity of serotype 5 adenovirus can be improved by fiber modifications. For example, a modified Ad with a chimeric fiber which express Ad3 knob instead of Ad5 knob, Ad5/3 showed enhanced infectivity to several kinds of cancer cells.28–30 Therefore, we analyzed the effect of infectivity enhancement with Ad5/3 for the cell lines tested in this study. As shown in Figure 8, the luciferase activities with Ad5/3 vector increased in all six cell lines tested. The increase rates are between 5.1 times in Panc I cells and 39.4 times in A549 cells. These findings led us to make a Ad5/3VEGFE1 in which the Ad5 knob is replaced with Ad3 knob. Ad5/3VEGFE1 was generated and propagated as described in Materials and methods. The oncolytic effect of Ad5/3VEGFE1 for the various cancer cells was evaluated using infection at an MOI 1 to highlight the difference from Ad5VEGFE1. Cytopathic effect with Ad5/3VEGFE1 infection was seen rapidly, almost 2 days earlier than with Ad5VEGFE1 in all cell lines. In this experiment, at day 9 after infection, complete cell death was seen for all lines infected with Ad5/3VEGFE1 while a significant number of cells survived with Ad5VEGFE1 infection. Moreover, Ad5/3VEGFE1 showed a stronger cell killing effect for H322 cells and SKOV3.ipl cells compared with Ad5CMVE1. These results suggested that the infectivity enhancement with modified adenovirus can improve the cell killing effect of the VEGF CRAd (Figure 9).
Figure 8.

Enhancement of infectivity to cancer cells with Ad5/3 chimeric vector. Luciferase activities in various cell lines infected by Ad5CMVLuc or Ad5/3luc1 containing CMV driven luciferase gene. 1 × 105 cells of each cell line were infected by Ad5CMVLuc or Ad5/3luc1 at MOI 10. At 48 h after infection, infected cells were harvested and lysed in 100 μl of lysis buffer. Ten microliters of each lysate were used for luciferase assay. Mean + s.e. of triplicate determination is shown.
Figure 9.

Enhancement of cell killing effect with Ad5/3 chimeric CRAd. Cell killing effect was evaluated by MTS assay. 5 × 103 cells of each cell line were infected with Ad5CMVLuc (negative control), Ad5CMVE1 (positive control), Ad5VEGFE1 or Ad5/3VEGFE1 at MOI of 1.0. After infection, cell viability in each well was quantified using OD490 by MTS assay every 3 days. The viability of cells infected with Ad5CMVE1, Ad5VEGFE1 or Ad5/3VEGFE1 was expressed as a percentage of cells infected with Ad5CMVLuc (100%).
Discussion
CRAds, which show tumor specific replication and oncolysis, are promising new therapies for malignancies resistant to conventional treatments. In the current report, we demonstrate a strategy based on the use of a replication-competent Ad controlled by a VEGF promoter. Furthermore, we demonstrated the use of Ad-VFEGFE1 applicable for the treatment of a wide spectrum of tumors. With regard to gene therapy of lung cancer, replication-incompetent Ad expressing wild-type p53 is already used in clinical trials. Whereas replication-incompetent viral vectors have demonstrated great promise as anticancer agents in preclinical studies, this has not been translated into patient benefit in the clinical setting.31 The poor anticancer effect with replication-incompetent Ad is partly depending on the weak penetration of the vector into the tumor mass. In this regard, a CRAd may achieve better intratumoral spread and penetration due to its replication ability.32
For clinical application, prevention of hepatic toxicity is also an important consideration. Tumor cells infected with replication-competent Ad may release new viruses in vivo. If this were to occur, there would be a potential for in vivo toxicity, especially in the liver, because this is the predominant site of Ad vector localization after systemic injection.33 In this regard, the VEGF promoter shows a low level of the promoter activity in the liver, and may avoid the hepatic injury. Since AdVEGFE1 showed good specificity in both replication rate and cytotoxicity for high versus low VEGF expressing cells, it can be expected to be less toxic to the liver compared with AdCMVE1 or wild-type Ad. Moreover, neurons express VEGF as a survival factor and cognitive damage might be expected if virus spreads to central nervous system. Unfortunately, at this time, no suitable animal models exist for the assessment of CRAd toxicity in vivo. To resolve this problem, we will investigate the CRAd toxicity using normal human hepatic cell culture in the next work. On the other hand, the results of a phase I clinical trial with VEGF inhibitors showed that these agents were well tolerated,34 indicating a marginal role for VEGF signalling in normal organs under physiological conditions except the ovary during the menstrual cycle.35
Another problem for the clinical use of a type II CRAd is that the relevant promoter activity in each tumor should be evaluated before treatment. From previous reports as well as our results, it is clear that tumors with low promoter activity are resistant to type II CRAds containing that promoter.14–17 Therefore, it is important to evaluate the promoter activity in advance to avoid potentially fruitless therapy. Analysis for RNA status needs some volume of live tissue obtained from the patient to prepare RNA samples for RT-PCR or Northern blotting. Precise evaluation of promoter activity with reporter gene like luciferase is more difficult in the clinical setting generally. In this regard, the VEGF promoter has an advantage for its activity evaluation. As shown in Figures 2 and 3, we revealed that there is a positive correlation between mRNA expression level, protein expression level and transgene activation for VEGF promoter, which is consistent with the previous report using clinical samples.36 Taken together, these data suggest that VEGF promoter activity can be predicted from VEGF protein expression levels. VEGF protein is easily detectable in clinical samples by ELISA evaluation of fluid samples37 and immunohistochemical staining of tissue samples.6 Thus, these tests could potentially be used to prospectively select the most appropriate patients for consideration of VEGF-CRAd therapy in the clinical setting.
VEGF production is an important mechanism for the development of tumor-associated angiogenesis in many types of tumors. In fact, many types of cancer are already known to express the VEGF protein at significant levels, and this VEGF expression is associated with poor prognosis in several disease contexts including leukemia, breast cancer, colorectal cancer, hepatocellular carcinoma, ovarian cancer and NSCLC.34 It seems that more advanced stage tumors actually express higher levels of VEGF protein. VEGF gene expression is known to be regulated transcriptionally. Although several transcription factors bind to the cis-elements on the promoter, hypoxia inducible factor (HIF) is the key factor to activate the promoter.21 Hypoxia induces the transcripitional activation of the VEGF gene via HIF. The central region of tumors are often hypoxic and necrotic due to decreased blood flow, and immunohistochemical analysis of primary tumor samples shows that VEGF protein expression is enhanced in the tumor tissue adjacent to the necrotic region.38 On the other hand, some kinds of cancer are known to express the HIF protein constitutively despite the oxygen tension, leading an increase VEGF promoter activation.39 Taken together these findings suggest that the antitumor effect of AdVEGFE1 may be even more efficacious in large in vivo tumors than under the normoxic conditions under which our in vitro experiments were performed.
Broadly speaking, the cell killing effect of a type II CRAd may be improved by several mechanisms such as promoter induction, infectivity enhancement or an armed CRAd strategy.40 A major obstacle to be overcome in Ad5-based cancer gene therapy has been the paucity of the primary receptor, CAR, on human primary tumor cells. Variable expression of CAR has been documented in many cancer types.29,41–43 Furthermore, downregulation of CAR may be associated with a more malignant phenotype.44 Due to variable expression of CAR on human primary cancer cells, the utility of Ad5 as a cancer gene therapy vector is compromised, limiting overall efficiency of cancer gene therapy including the use of CRAds. On this basis, systems to circumvent intervening tumor-associated CAR deficiency are required. In this regard, native Ad5 tropism can be modified to circumvent CAR deficiency and to enhance Ad infectivity. One approach is pseudotyping, that is, retargeting Ad by creating chimeric fibers possessing knob domains derived from alternate serotypes that bind to receptors other than CAR. To this end, we have constructed nonreplicating Ads containing chimeric fibers with the tail and shaft domains of Ad serotype 5 and the knob domain of serotype 3.22 Our previous work has revealed that a distinct Ad3 receptor exists in ovarian cancer cells based on a novel knob binding assay, and that the Ad5/3 chimeric vector is retargeted to the Ad3 receptor.29 Based on these findings, we constructed a CRAd based on the Ad5/3 chimeric approach in this study. As we hypothesized, Ad5/3VEGFE1 showed a stronger cell killing effect than that of the Ad5-based CRAd, probably due to infectivity enhancement. Thus, a chimeric vector-based CRAd is a promising way to improve the therapeutic effect. Recent data also show that Ad5/3VEGFE1 is effective to ovarian cancer in vivo.45
In conclusion, we believe that the data presented here provide a basis for the additional development of replication-competent adenovirus strategies based on the VEGF promoter for the therapy of various cancers. Furthermore, a CRAd based on the Ad5/3 chimeric vector is promising way to enhance the anticancer effect via infectivity improvement for cancer cells.
Acknowledgments
We thank Professor Semenza (John’s Hopkins University) for providing us with the human VEGF promoter. We also thank Dirk M Nettelbeck, Joel N Glasgow, Akiko Harada and Nobuyuki Hara (Kyushu university, Fukuoka, Japan) for their excellent technical support and expert advice. This work was supported by National Cancer Institute Grant CA83821, The CapCURE Foundation, The Lustgarten Foundation, United States Department of Defense Grant 991018 and the American Cancer Society.
Abbreviations
- NSCLC
non-small cell lung cancer
- IL-8
interleukin-8
- VEGF
vascular endothelial growth factor
- CAR
coxsackie and adenovirus receptor
- Ad5
serotype 5 adenovirus
- Ad3
serotype 3 adenovirus
- Ad5/3
Ad5 containing a chimeric fiber protein possessing the Ad3 knob
- CRAd
conditionally replicative adenovirus
- E1
early region 1
- E4
early region 4
- RT-PCR
revers transcription-PCR
- CMV
cytomegalovirus
- CsCl
cesium chloride
- VP
viral particle
- PFU
plaque forming unit
References
- 1.Levi F, Lucchini F, Negri E, La Vecchia C. Worldwide patterns of cancer mortality, 1990–1994. Eur J Cancer Prev. 1999;8:381–400. doi: 10.1097/00008469-199910000-00004. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J. What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst. 1990;82:4–6. doi: 10.1093/jnci/82.1.4. [DOI] [PubMed] [Google Scholar]
- 3.Liotta LA, Steeg PS, Stetler-Stevenson WG. Cancer metastasis and angiogenesis: an imbalance of positive and negative regulation. Cell. 1991;64:327–336. doi: 10.1016/0092-8674(91)90642-c. [DOI] [PubMed] [Google Scholar]
- 4.Ohta Y, Endo Y, Tanaka M, Shimizu J, Oda M, Hayashi Y, et al. Significance of vascular endothelial growth factor messenger RNA expression in primary lung cancer. Clin Cancer Res. 1996;2:1411–1416. [PubMed] [Google Scholar]
- 5.Fontanini G, Vignati S, Boldrini L, Chine S, Silvestri V, Lucchi M, et al. Vascular endothelial growth factor is associated with neovascularization and influences progression of non-small cell lung carcinoma. Clin Cancer Res. 1997;3:861–865. [PubMed] [Google Scholar]
- 6.Inoshima N, Nakanishi Y, Minami T, Izumi M, Takayama K, Yoshino I, et al. The influence of dendritic cell infiltration and vascular endothelial growth factor expression on the prognosis of non-small cell lung cancer. Clin Cancer Res. 8:3480–3486. [PubMed] [Google Scholar]
- 7.Goldman CK, Kendall RL, Cabrera G, Soroceanu L, Heike Y, Gillespie GY, et al. Paracrine expression of a native soluble vascular endothelial growth factor receptor inhibits tumor growth, metastasis, and mortality rate. Proc Natl Acad Sci USA. 1998;95:8795–8800. doi: 10.1073/pnas.95.15.8795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takayama K, Ueno H, Nakanishi Y, Sakamoto T, Inoue K, Shimizu K, et al. Suppression of tumor angiogenesis and growth by gene transfer of a soluble form of vascular endothelial growth factor receptor into a remote organ. Cancer Res. 2000;60:2169–2177. [PubMed] [Google Scholar]
- 9.Curiel DT. The development of conditionally replicative adenoviruses for cancer therapy. Clin Cancer Res. 2000;6:3395–3399. [PubMed] [Google Scholar]
- 10.Bischoff JR, Kirn DH, Williams A, Heise C, Horn S, Muna M, et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science. 1996;274:373–376. doi: 10.1126/science.274.5286.373. [DOI] [PubMed] [Google Scholar]
- 11.Fueyo J, Comez-Manzano C, Alemany R, Lee PSY, McDonell TJ, Mitlianga P, et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. 1999;19:1–11. doi: 10.1038/sj.onc.1203251. [DOI] [PubMed] [Google Scholar]
- 12.Hallenbeck PL, Chang Y-N, Hay C, Golightly D, Stewart D, Lin J, et al. A novel tumor-specific replication-restricted adenoviral vector for gene therapy of hepatocellular carcinoma. Hum Gene Ther. 1999;10:1721–1733. doi: 10.1089/10430349950017725. [DOI] [PubMed] [Google Scholar]
- 13.Rodriguez R, Schuur ER, Lim HY, Henderson GA, Simons JW, Henderson DR. Prostate attenuated replacation competent adenovirus (ARCA) CV706: a selective cytotoxic for prostate-specific antigen-positive prostate cancer cells. Cancer Res. 1997;57:2559–2563. [PubMed] [Google Scholar]
- 14.Matsubara S, Wada Y, Gardner TA, Egawa M, Park M-S, Hsieh C-L, et al. A conditional replication-competent adenoviral vector, Ad-OC-E1a, to cotarget prostate cancer and bone stroma in an experimental model of androgen-independent prostate cancer bone metastasis. Cancer Res. 2001;61:6012–6019. [PubMed] [Google Scholar]
- 15.Kurihara T, Brough DE, Kovesdi I, Kufe DW. Selectivity of a replication-competent adenovirus for human breast carcinoma cells expressing the MUC1 antigen. J Clin Invest. 2000;106:763–771. doi: 10.1172/JCI9180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adachi Y, Reynolds PN, Yamamoto M, Wang M, Takayama K, Matsubara S, et al. A midkine promoter-based conditionally replicative adenovirus for treatment of pediatric solid tumors and bone marrow tumor purging. Cancer Res. 2001;61:7882–7888. [PubMed] [Google Scholar]
- 17.Nettelbeck DM, Rivera AA, Balague C, Alemany R, Curiel DT. Novel oncolytic adenoviruses targeted to melanoma: specific viral replication and cytolysis by expression of E1A mutants from the tyrosinase enhancer/promoter. Cancer Res. 2002;62:4663–4670. [PubMed] [Google Scholar]
- 18.DeWeese TL, Van der Poel H, Li S, Mikhak B, Drew R, Goemann M, et al. A phase I trial of CV706, a replication-competent, PSA selective oncolytic adenovirus, for the treatment of locally recurrent prostate cancer following radiation therapy. Cancer Res. 2001;61:7464–7472. [PubMed] [Google Scholar]
- 19.Nemunaitis J, Ganly I, Khuri F, Arseneau J, Kuhn J, McCarty T, et al. Selective replication and oncolysis in p53 mutant tumors with ONYX-015, an E1B-55KD gene-deleted adenovirus, in patients with advanced head and neck cancer: a Phase II trial. Cancer Res. 2000;60:6359–6366. [PubMed] [Google Scholar]
- 20.He T-C, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating reombinant adenoviruses. Proc Natl Acad Sci USA. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forsythe JA, Jiang B-H, Iyer NV, Agani E, Leung SW, Koos RD, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor I. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krasnykh VN, Mikheeva GV, Douglas JT, Curiel DT. Generation of recombinant adenovirus vectors with modified fibers for altering viral tropism. J Virol. 1996;70:6839–6846. doi: 10.1128/jvi.70.10.6839-6846.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takayama K, Ueno H, Pei X-H, Nakanishi Y, Yatsunami J, Hara N. The levels of integrin αvβ5 may predict the susceptibility to adenovirus-mediated gene transfer in human lung cancers. Gene Therapy. 1998;5:361–368. doi: 10.1038/sj.gt.3300608. [DOI] [PubMed] [Google Scholar]
- 24.Maizel JVJ, White O, Scharff MD. The polypeptides of adenovirus. I. Evidence for multiple protein components in the virion and a comparison of types 2, 7A, and 12. Virology. 1968;36:115–125. doi: 10.1016/0042-6822(68)90121-9. [DOI] [PubMed] [Google Scholar]
- 25.Mittereder N, March KL, Trapnell BC. Evaluation of the concentration and bioactivity of adenovirus vectors for gene therapy. J Virol. 1996;70:7498–7509. doi: 10.1128/jvi.70.11.7498-7509.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chartier C, Degryse E, Gantzer M, Dieterle A, Pavirani A, Mehtali M. Efficient generation of recombinant adenovirus vectors by homologous recombination in Escherichia coli. J Virol. 1996;70:4805–4810. doi: 10.1128/jvi.70.7.4805-4810.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaliberov S, Buchsbaum D, Gillespie G, Curiel DT, Arafat W, Carpenter M, et al. Adenovirus-mediated transfer of BAX driven by the vascular endothelial growth factor promoter induces apoptosis in lung cancer cells. Mol Ther. 2002;6:190–198. doi: 10.1006/mthe.2002.0648. [DOI] [PubMed] [Google Scholar]
- 28.Suzuki K, Fueyo J, Krasnykh V, Reynolds PN, Curiel DT, Alemany R. A conditionally replicative adenovirus with enhanced infectivity shows improved oncolytic potency. Clin Cancer Res. 2001;7:120–126. [PubMed] [Google Scholar]
- 29.Kanerva A, Mikheeva GV, Krasnykh V, Coolidge CJ, Lam JT, Mahasreshti PJ, et al. Targeting adenovirus to the serotype 3 receptor increases gene transfer efficiency to ovarian cancer cells. Clin Cancer Res. 2002;8:275–280. [PubMed] [Google Scholar]
- 30.Haviv YS, Blackwell JL, Kanerva A, Nagi P, Krasnykh V, Dmitriev I, et al. Adenoviral gene therapy for renal cancer requires retargeting to alternative cellular receptors. Cancer Res. 2002;62:4273–4281. [PubMed] [Google Scholar]
- 31.Hermiston T. Gene delivery from replication-selective viruses: arming guided missiles in the war against cancer. J Clin Invest. 2000;105:1169–1172. doi: 10.1172/JCI9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heise CC, Williams A, Olesch J, Kirn DH. Efficacy of a replication-competent adenovirus (ONYX-015) following intratumoral injection: intratumoral spread and distribution effects. Cancer Gene Ther. 1999;6:499–504. doi: 10.1038/sj.cgt.7700071. [DOI] [PubMed] [Google Scholar]
- 33.van der Eb MM, Cramer SJ, Vergouwe Y, Schagen FHE, van Krieken JHM, van der Eb AJ, et al. Severe hepatic dysfunction after adenovirus-mediated transfer of the herpes simplex virus thymidine kinase gene and ganciclovir administration. Gene Therapy. 1998;5:451–458. doi: 10.1038/sj.gt.3300637. [DOI] [PubMed] [Google Scholar]
- 34.Rosen LS. Clinical experience with angiogenesis signaling inhibitors: focus on vascular endothelial growth factor (VEGF) blockers. Cancer Control. 2002;9:36–44. doi: 10.1177/107327480200902S05. [DOI] [PubMed] [Google Scholar]
- 35.Ferrara N, Chen H, Davis-Smyth T, Gerber HP, Nguyen TN, Peers D, et al. Vascular endothelial growth factor is essential for corpus luteum angiogenesis. Nat Med. 1998;4:336–340. doi: 10.1038/nm0398-336. [DOI] [PubMed] [Google Scholar]
- 36.Yuan A, Yu C-J, Chen W-J, Lin F-Y, Kuo S-H, Luh K-T, et al. Correlation of total VEGF mRNA and protein expression with histologic type, tumor angiogenesis, patient survival and timing of relapse in non-small-cell lung cancer. Int J Cancer. 2000;89:475–483. doi: 10.1002/1097-0215(20001120)89:6<475::aid-ijc2>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 37.Mall JW, Schwenk W, Philipp AW, Meyer-Kipker C, Mall W, Muller J, et al. Serum vascular endothelial growth factor levels correlate better with tumor stage in small cell lung cancer than albumin, neuron-specific enolase or lactate dehydrogenase. Respirology. 2002;7:99–102. doi: 10.1046/j.1440-1843.2002.00386.x. [DOI] [PubMed] [Google Scholar]
- 38.Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- 39.Wiesener MS, Munchenhagen PM, Berger I, Morgan NV, Roigas J, Schwiertz A, et al. Constitutive activation of hypoxia-inducible genes related to overexpression of hypoxia-inducible factor-1 alpha in clear cell renal carcinoma. Cancer Res. 2001;61:5215–5222. [PubMed] [Google Scholar]
- 40.Haviv YS, Takayama K, Glasgow JN, Blackwell JL, Wang M, Lei X, et al. A model system for the design of armed replicating adenoviruses using p53 as a candidate transgene. Mol Cancer Ther. 2002;1:321–328. [PubMed] [Google Scholar]
- 41.Miller CR, Buchsbaum DJ, Reynolds PN, Douglas JT, Gillespie GY, Mayo MS, et al. Differential susceptibility of primary and established human glioma cells to adenovirus infection: targeting via the epidermal growth factor receptor achieves fiber receptor-independent gene transfer. Cancer Res. 1998;58:5738–5748. [PubMed] [Google Scholar]
- 42.Hemmi S, Geertsen R, Mezzacasa A, Peter I, Dummer R. The presence of human coxsackievirus and adenovirus receptor is associated with efficient adenovirus-mediated transgene expression in human melanoma cell cultures. Hum Gene Ther. 1998;9:2363–2373. doi: 10.1089/hum.1998.9.16-2363. [DOI] [PubMed] [Google Scholar]
- 43.Cripe TP, Dunphy EJ, Holub A, Vasi NH, Mahller YY, Collins MH, et al. Fiber knob modifications overcome low, heterogeneous expression of the coxsackievirus-adenovirus receptor that limits adenovirus gene transfer and oncolysis for human rhabdomyosarcoma cells. Cancer Res. 2001;61:2951–2960. [PubMed] [Google Scholar]
- 44.Okegawa T, Li Y, Pong RC, Bergelson JM, Zhou J, Hsieh JT. The dual impact of coxsackie and adenovirus receptor expression on human prostate cancer gene therapy. Cancer Res. 2000;60:5031–5036. [PubMed] [Google Scholar]
- 45.Lam JT, Kanerva A, Bauershcmitz GJ, Takayama K, Suzuki K, Yamamoto M, et al. Inter-patient variation in efficacy of five oncolytic adenovirus candidates for ovarian cancer therapy. J Gene Med. 2004;6:1333–1342. doi: 10.1002/jgm.635. [DOI] [PubMed] [Google Scholar]