

Abstract
The utility of conditionally replicative adenoviruses (CRAds) in cancer gene therapy is based on their ability to destroy tumor cells by oncolysis. However, in order to achieve adequate therapeutic response, CRAds have to spread through the tumor tissue by replication. Thus, to study the potency of these viruses to replicate and penetrate in tumors, we created a herpes simplex virus type 1 thymidine kinase-green fluorescent protein fusion gene (TK-GFP) encompassing CRAd (Ad5 Δ24TK-GFP), whose tumor selectivity is mediated by a retinoblastoma (Rb)-binding site mutation. In addition, we evaluated the oncolytic efficacy of Ad5 Δ24TK-GFP in combination with the TK/ganciclovir (GCV) system. Based on our results, Ad5 Δ24TK-GFP replicates in cancer cells resulting in oncolysis and can efficiently penetrate into tumors. Additionally, the combination of GCV with Ad5 Δ24TK-GFP augmented cell death in vitro but this was not observed in vivo: tumor growth was significantly reduced by oncolysis when compared to non-replicative virus (p<0.001), but administration of GCV did not significantly enhance oncolysis. This suggests that in certain conditions, TK/GCV-mediated cell killing may be counterproductive to replication and oncolysis, which on the other hand might be useful feature for clinical trials in case of replication-associated toxicity.
Keywords: oncolytic adenovirus, cancer gene therapy, prodrug/suicide gene therapy, HSV-TK, viral spreading, green fluorescent protein
Introduction
Replication deficient adenoviruses have been widely used as gene transfer vectors, but there are several obstacles which might limit their efficient use in cancer gene therapy. One major challenge caused by low expression levels of the coxsackie-adenovirus receptor (CAR), is to achieve adequate and specific gene transfer into target tissue using reasonable and safe viral dose (1).
Secondly, expression of therapeutic gene has to be sufficient to elicit therapeutic response. When using non-integrating viruses such as adenoviruses, transgenes will remain extrachromosomal and are rapidly eliminated from proliferating cells diminishing the therapeutic effect (2). Nonetheless, these problems can be circumvented by using conditionally replicative adenoviruses (CRAds). Due to replication, virus multiplies itself in tumor tissue, kills the host cell and spreads throughout the tumor theoretically as long as tumor cells persist. Therefore, relatively low viral doses are needed for therapeutic response. The oncolytic potency of the viruses was first discovered in the early 1900’s, when occasional tumor regression was observed in cancer patients suffering from virus infections (3). Since then, several types of viruses, including adenoviruses and herpes viruses have been employed as anti-cancer agents due to their ability to spread through tumor tissue by virtue of viral replication and concomitant cell lysis (4–6). In addition, to minimize adverse side effects and increase the safety of these anti-cancer agents, replication can be limited to tumor tissue by genetically modifying the CRAd genome (i.e. partial deletions in the E1 region or use of tissue-specific promoters to drive genes responsible for viral replication) (7).
Another beneficial feature of CRAds compared to replication deficient viral vectors is their ability to multiply and amplify inserted therapeutic genes while replicating. Several studies suggest that combining therapeutic genes, such as p53, cytosine deaminase and HSV thymidine kinase (TK) with replicative viruses might improve their oncolytic potency and increase the anti-tumoral effect (8–12). However, in the case of TK, there have been also controversial findings suggesting that addition of the TK/GCV system does not augment the antineoplastic activity of oncolytic agents (13–17).
After infection of the target cells, the anti-tumoral efficiency of CRAds is mainly dependent on their ability to penetrate tumor tissue. Although this is a crucial feature, only a few studies have been able to evaluate this, due to difficulty in getting data on intratumoral replication dynamics. Most available data on viral spreading and distribution is based on histological staining, which is useful in endpoint studies but does not allow real-time imaging of living organisms. Due to the replicative nature of the virus, marking of viral particles with fluorescent tags has to be done by genetic modification to prevent loss of the labeling moiety. The major challenge of this approach is to fuse fluorescent proteins to viral structural proteins without disturbing the packing of the viral particles. Some promising results have been achieved by fusing enhanced green fluorescent protein (GFP) into adenovirus capsid protein IX (18). Instead of genetic labeling of the virus, another possibility is to image viral spreading non-invasively with the aid of inserted transgene. However, in contrast to fluorescent marker proteins, monitoring of the frequently used transgene products (e.g. thymidine kinase or luciferase) requires systemic injections of radiolabelled molecules or substrate (19,20).
The objective of this study was to create a novel, tumor selective CRAd, whose intratumoral spreading could be directly detected and that would allow investigation of the utility of CRAds with suicide/prodrug therapy. We created Ad5 Δ24TK-GFP, which encompasses 24-base pair deletion in the retinoblastoma (Rb) protein binding region in E1A (5) and a herpes simplex virus thymidine kinase-green fluorescent protein fusion gene (TK-GFP) (21) under a CMV promoter replacing E3. Due to the 24-base pair deletion, this virus is unable to bind to the Rb protein which facilitates selective replication in cells with a dysfunctional Rb-p16 pathway, a frequent phenomenon in most tumor cells (22). The efficacy of Ad5 Δ24TK-GFP was tested in vitro on several ovarian cancer cell lines and in vivo in subcutaneous ovarian tumors.
Materials and methods
Cell lines
Hey, SKOV3.ip1, and OV-4 human ovarian adenocarcinoma cell lines were kind gifts from Dr Judy Wolf, Dr Janet Price (both M.D. Andersson Cancer Center, Houston, TX) and Dr Timothy J. Eberlein (Harvard Medical School, Boston, MA), respectively. Human ovarian adenocarcinoma OV-3 and epithelial lung carcinoma A549 cell lines were obtained from the American Type Culture Collection (Manassas, VA) and human embryonic kidney epithelial 293 cells from Microbix (Toronto, Canada). Cell line 911 was obtained from Dr Alex J. van der Eb (University of Leiden, The Netherlands). All cell lines were cultured as recommended.
Adenoviral vector construction and production
To create conditionally replicative Ad5 Δ24TK-GFP, XbaI-digested and blunted TK-GFP fragment (21) was inserted into previously constructed BglII-digested and blunted pKOE3ACMV to obtain pKOE3ACMVTK-GFP. FspI-linearized pKOE3AC MVTK-GFP and PacI-linearized Δ24 viral DNA were cotransfected into 911 cells with Effectine (Qiagen, Valencia, CA) followed by homologous recombination and formation of viral colonies. Both Ad5 Δ24 (5) and Ad5 Δ24TK-GFP viruses were propagated in A549 cells and purified with standard techniques. Non-replicative first generation Ad5TK-GFP vector was constructed earlier in our laboratory (23). Briefly, TK-GFP fragment was cloned into pShuttle CMV (AdEasy-kit™, Qbiogene, Carlsbad, CA) followed by homologous recombination in BJ5183 cells in order to obtain pAdTK-GFP. Adenoviral plasmid was linearized and transfected into 293 cells with Superfect (Qiagen) following the manufacturer’s instructions. Adenovirus colonies were picked and virus was propagated in 293 cells and purified using standard techniques. Viral particles (VP) were determined with spectrophotometry and plaque forming units (pfu) with plaque assay. Titers were Ad5TK-GFP: 4.2×1011 VP/ml; 2.2×1010 pfu/ml, Ad5 Δ24TK-GFP: 5.4×1011 VP/ml; 1.1×1010 pfu/ml and Ad5 Δ24: 8.4×1011 VP/ml; 1.8×1010 pfu/ml.
Verification of viral replication and oncolysis
Human ovarian cancer cells were plated onto a 24-well plate at the density of 15–30 000 cells/well (MTT-assay) and onto a 12-well plate at the density of 50–100 000 cells/well (crystal violet assay). The following day cells were infected with Ad5TK-GFP, Ad5 Δ24TK-GFP and Ad5 Δ24 overnight at 37°C using 1, 10 and 100 pfu/cell. After infection fresh growth media was added and replaced every other day. OV-3, OV-4, SKOV3.ip1 and Hey cells were analyzed with MTT-assay according to manufacturer’s instructions (Cell proliferation kit II, Roche Diagnostics, Indianapolis, IN) and stained with crystal violet 8, 12, 14 and 18 days post-infection, respectively.
Evaluation of viral spreading
For flow cytometry analysis, infections were carried out on 12-well plates using 50–100 000 cells per well. Cells were infected either with Ad5TK-GFP or Ad5 Δ24TK-GFP for 3 h at 37°C using 5 pfu/cell. After trypsination, cells were fixed with 4% paraformaldehyde (PFA) for 15 min at 4°C and resuspended in PBS. At each time point (2–10 days post-infection) percentage of GFP-expressing cells was determined by flow cytometry (FACSCalibur, BecktonDickinson).
For fluorescence microscopy, OV-4 cells were seeded onto a 6-well plate (2×105 cells per well) and infected either with Ad5TK-GFP or Ad5 Δ24TK-GFP overnight at 37°C. Cells were overlayed with 1:2 mixture of 2X Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% fetal bovine serum (FBS), 4 mM glutamine and gentamycin (100 mg/l) (Qibco BRL, Life Technologies, UK) and 1.8% seaplaque agarose (BioWhittaker Molecular Applications, Rockland, ME). Cells were examined under a fluorescence microscope (Axiovert 135M, Zeiss, Göttingen, Germany) 3, 5, 7 and 9 days after infection.
Ganciclovir sensitivity assays
Ovarian cancer cells were seeded in a 24-well plate (30–50 000 per well) day before infection. Cells were infected either with non-replicative Ad5TK-GFP or replicative Ad5 Δ24TK-GFP overnight at 37°C using 10 pfu/cell. Ganciclovir containing media was added 1 or 4 days post-infection and cells were grown in the presence of ganciclovir (10 μg/ml) for 5 days. Cell viability was analyzed with MTT-assay as above. The percentage of viable cells was determined by comparing values to non-infected control cells.
In vivo studies
Female NRMI nu/nu mice (age, 10 weeks) were purchased from Harlan, The Netherlands. Mice were anesthetized with phenyl-fluanisone-midazolam and human SKOV3.ip1 ovarian cancer cells (3×106 cells/tumor) were inoculated subcutaneously to two sites on the back of each animal (9 groups, n=4 in each group). A week after inoculation Ad5TK-GFP, Ad5 Δ24TK-GFP and Ad5 Δ24 viruses were injected intratumorally (3×108 pfu/tumor). To study the therapeutic efficiency of these viruses, seven groups of mice received 50 mg/kg/day ganciclovir for 12 days (Ad5TK-GFP + GCV, Ad5 Δ24TK-GFP + GCV and Ad5 Δ24 + GCV) or saline (control, Ad5TK-GFP, Ad5 Δ24TK-GFP, Ad5 Δ24) divided into two daily i.p. injections. After the treatment period, mice were euthanized and tumors were weighed. To monitor the spreading of the GFP-containing viruses, two groups of mice which received either Ad5TK-GFP or Ad5 Δ24TK-GFP were euthanized two animals at the time 2, 5, 9 and 12 days after infection. Tumors were harvested and minced with a scalpel and digested for 2 h in digestive enzyme solution [0.002% w/v Dnase I (type IV, Sigma) 0.1% w/v collagenase (type IV, Sigma) and 0.01% w/v hyaluronase (type V, Sigma) in OptiMEM]. Single cell suspensions were created by washing the digested cells twice with PBS and filtering with Falcon cell-strainer cap tubes (35-μm mesh, Beckton Dickinson Labware). The cell suspensions were analyzed using flow cytometry (FACSCalibur, Beckton Dickinson). This study was reviewed and approved by the Animal Care and Use Committee of the University of Kuopio.
Statistical analysis
One-way analysis of variance with Bonferroni’s post hoc test for multiple comparisons was used for statistical analysis in evaluation of viral spreading and in vivo studies, and two-way analysis of variance with Bonferroni’s post hoc test for multiple comparisons was used for GCV sensitivity studies (GraphPad Prism 3.0, GraphPad Software, Inc., San Diego, CA). p<0.05 was considered statistically significant.
Results
Replication of Ad5 Δ24TK-GFP kills ovarian cancer cells
To evaluate the replication potency of CRAds in tumor cells and the influence of TK/GCV system to oncolysis, we constructed a novel CRAd Ad5 Δ24TK-GFP by replacing E3 region with the TK-GFP fusion gene (Fig. 1). The functionality of Ad5 Δ24TK-GFP virus was compared to a replication deficient adenovirus containing the same fusion gene (Ad5TK-GFP) in the deleted E1 region and to an isogenic replicative E3-deleted virus without TK-GFP (Ad5 Δ24). In order to study the replication capacity of Ad5 Δ24TK-GFP, infected human ovarian cancer cells were analyzed both with crystal violet staining and with MTT-assay at given time points (Fig. 2a and b). Ad5 Δ24TK-GFP replicated and caused oncolysis in examined tumor cell lines (Fig. 2a). The rate of replication varied between cell lines showing the slowest replication in Hey cells (cell killing evident 18 days post-infection) and the most rapid replication in OV-3 cells (8 days post-infection). Compared to Ad5 Δ24, oncolysis was delayed in cells infected with Ad5 Δ24TK-GFP (Fig. 2b). When a dose of 10 pfu/cell was used, Ad5 Δ24 killed 60–80% of cells whereas cell killing rate of Ad5 Δ24TK-GFP varied between 10 and 70% in OV-3, OV-4 and SKOV3.ip1 cells. On the contrary, Hey cells seemed to be rather resistant to Ad5 Δ24TK-GFP replication even with the highest viral dose.
Figure 1.
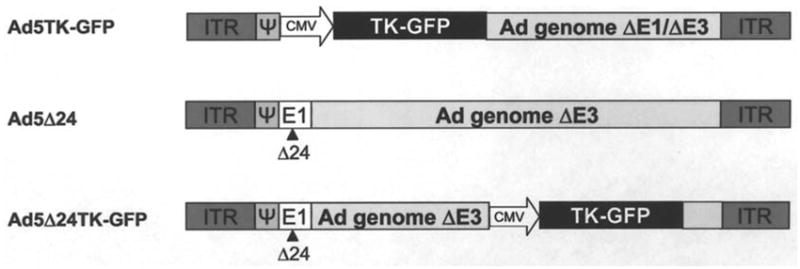
Schematic presentation of viral constructs. Non-replicative, E1/E3-deleted Ad5TK-GFP contains the TK-GFP fusion gene. Both replicative viruses (Ad5 Δ24TK-GFP and Ad5 Δ24) feature a 24-bp deletion in the Rb binding site of E1A. Additionally, in Ad5 Δ24TK-GFP E3 region is replaced with TK-GFP, which is driven by CMV promoter. Ψ, packaging signal; ITR, inverted terminal repeat; TK, thymidine kinase; CMV, cytomegalovirus promoter.
Figure 2.
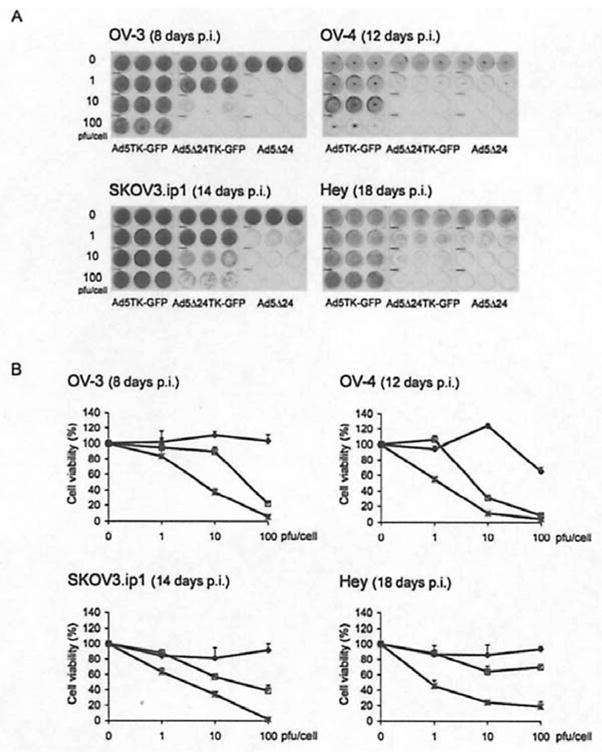
Ad5 Δ24TK-GFP replicates in cancer cells. Infected ovarian cancer cells were analyzed both with crystal violet staining (A) and with MTT-assay (B). Analyses were performed 8, 12, 14 and 18 days post-infection with OV-3, OV-4, SKOV3.ip1 and Hey cells, respectively. Error bars indicate standard error of the mean. Ad5TK-GFP (◆), Ad5 Δ24TK-GFP (□) and Ad5 Δ24 (
 ).
).
Ad5 Δ24TK-GFP spreads efficiently in cancer cells
To study the kinetics of viral spreading, Ad5TK-GFP and Ad5 Δ24TK-GFP-infected ovarian tumor cells were analyzed every 24 h for 10 days with flow cytometry to determine the percentage of GFP-expressing cells (Fig. 3a). When Ad5 Δ24TK-GFP was used, the amount of GFP-positive cells increased from an initial 10% up to 85% and from 3 to 38% in OV-4 and OV-3 cells, respectively. In Ad5TK-GFP-infected cells, the percentage of GFP-positive cells remained constant or showed a slight decrease. In SKOV3.ip1 cells, the proportion of GFP expressing cells increased from 12 to 28% and in Hey cells the percentage of infected cells remained nearly constant. When compared to Ad5TK-GFP, the increase in the proportion of GFP positive cells was significant after (p<0.001 or p<0.01) 7 days in OV-3, OV-4 and SKOV3.ip1 cells where as in Hey cells there was no significant difference between the two viruses. To visualize this effect, infected OV-4 cells were analyzed with fluorescence microscopy (Fig. 3b). After 9 days, the majority of Ad5 Δ24TK-GFP-treated cells exhibited strong fluorescence indicating spreading of the Ad5 Δ24TK-GFP virus throughout the cell layer. In contrast, the fluorescence of Ad5TK-GFP-infected cells faded and the number of GFP-expressing cells diminished.
Figure 3.
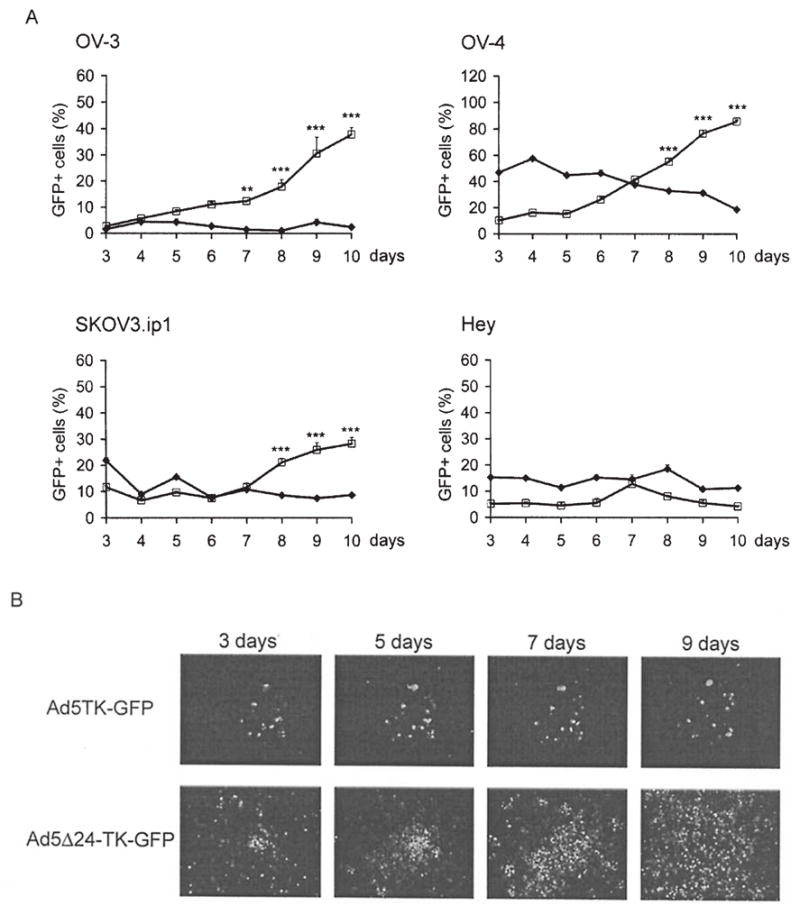
Viral spreading was directly detected by GFP fluorescence. (A) Ovarian cancer cells were infected either with Ad5TK-GFP (◆) or Ad5 Δ24TK-GFP (□) using 5 pfu/cell and analyzed with flow cytometry every 24 h for ten days. **p<0.01 and ***p<0.001, Ad5 Δ24TK-GFP vs. Ad5TK-GFP. Error bars indicate standard error of the mean. (B) Infected OV-4 cells were also analyzed with fluorescence microscopy at given time points.
Thymidine kinase/ganciclovir system enhances oncolysis in vitro
Combining a suicide/prodrug system with oncolytic viruses might offer one possibility to enhance cell death in tumor tissue. To examine this approach, Ad5TK-GFP- and Ad5 Δ24TK-GFP-infected cells were exposed to GCV followed by cell viability measurements (Fig. 4). To optimize the timing of the GCV treatment, GCV was added onto cells at various time points. Based on obtained results optimal time point was determined to be 1–4 days p.i. (results not shown).
Figure 4.
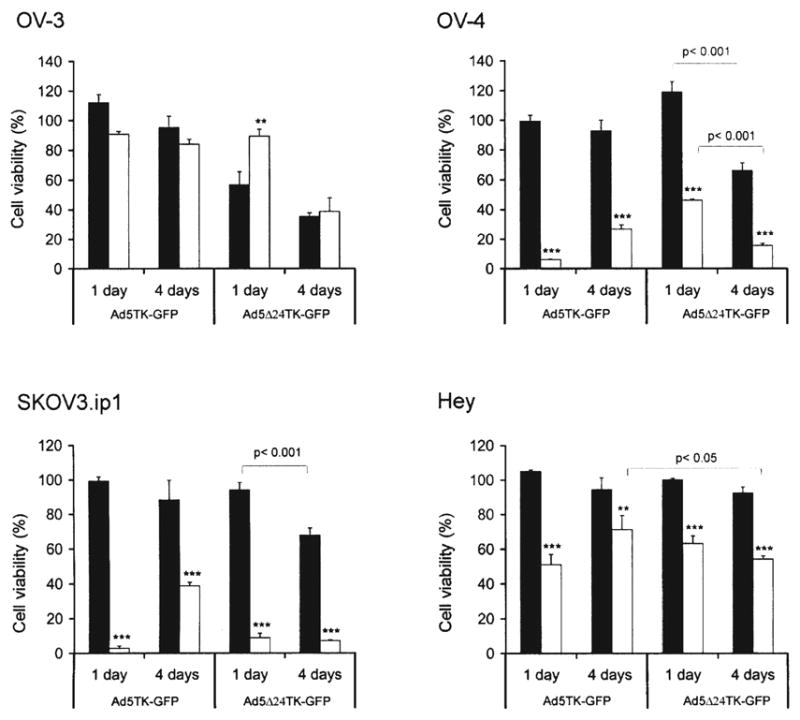
Ganciclovir improves the oncolytic potency of Ad5 Δ24TK-GFP in vitro. Ad5TK-GFP- and Ad5 Δ24TK-GFP-infected ovarian cancer cells were exposed to GCV 1 day or 4 days after infection and cultured in the absence (■) or presence (□) of GCV for 5 days followed by cell viability measurement. Cell viability was compared to non-infected control cells. **p< 0.01 and ***p< 0.001, no GCV vs. GCV. Error bars indicate standard error of the mean.
When Ad5 Δ24TK-GFP-infected OV-4 cells were exposed to GCV 1 day p.i., ~50% cell death was seen (p<0.001), although oncolytic cell death was not observed in Ad5 Δ24TK-GFP-infected cells. When cells were analyzed 3 days later, Ad5 Δ24TK-GFP alone killed ~34% of the cells due to significantly intensified replication (p<0.001, compared to 1 day p.i). Compared to virus alone, oncolysis was further enhanced with TK/GCV system; when GCV was added 4 days after infection, the combination treatment destroyed circa 85% of the cells (p<0.001). In addition, timing of the GCV treatment had significant effect on cell killing. Combination treatment was 1.7 times more efficient when GCV treatment was started 4 days after infection in comparison to 1 day p.i. (p<0.001).
In SKOV3.ip1 cells GCV-treatment significantly intensified oncolysis. One day after infection almost complete cell killing (93%) was observed, while virus alone did not cause significant cell death (p<0.001). When GCV was added 4 days after infection circa 85% of the cells were destroyed, whereas virus alone killed only 34% of the cells (p<0.001). In contrast to OV-4 cells, the timing of the GCV treatment did not have any significant influence to cell killing efficiency, although virus replication was more potent 4 days p.i. than 1 day after infection (p<0.001).
On the contrary, Hey cells seemed again to be rather resistant to viral replication since Ad5 Δ24TK-GFP alone did not cause any significant cell killing. Nonetheless, GCV treatment caused significant cell death at both time points when compared to Ad5 Δ24TK-GFP-infected cells (p<0.001). Although clear oncolysis was not observed due to inefficient viral replication, Ad5 Δ24TK-GFP caused significantly more efficient cell killing than replication deficient Ad5TK-GFP when GCV was added 4 days after infection (p<0.05).
Contrary to other cell lines, GCV significantly inhibited oncolysis in OV-3 cells when added 1 day after infection. While Ad5 Δ24TK-GFP alone killed almost half of the cell population, only 10% of GCV-treated cells were destroyed by oncolysis (p<0.01). However, the inhibitory effect of GCV was not observed when GCV was added 4 days after infection; Ad5 Δ24TK-GFP killed circa 65% of the cells with or without GCV treatment (no statistical significance).
Ad5 Δ24TK-GFP reduces tumor growth in vivo
In order to evaluate the functionality of the novel CRAd in vivo, viruses were injected intratumorally to subcutaneous SKOV3.ip1 tumors followed by GCV administration. Based on in vitro results, GCV treatment was started 1 day after virus injections. After 12 days of GCV treatment, tumor sizes were determined (Fig. 5a). Both replicative viruses (Ad5 Δ24TK-GFP and Ad5 Δ24) significantly reduced tumor growth in vivo when compared to the Ad5TK-GFP-treated group (p<0.001). When GCV was present, tumor size in each group decreased ~80% in comparison to Ad5TK-GFP alone (p<0.001). However, GCV did not reduce tumor size significantly in Ad5 Δ24TK-GFP-treated animals. In contrast, there seemed to be a trend suggesting inhibition of anti-tumor activity by GCV (no statistical significance).
Figure 5.
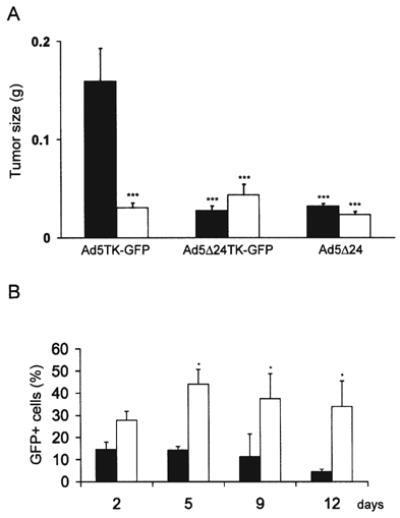
Efficacy of Ad5 Δ24TK-GFP in vivo. Ad5TK-GFP, Ad5 Δ24TK-GFP and Ad5 Δ24 viruses were injected intratumorally to subcutaneous SKOV3.ip1 tumors. (A) Animals received either saline (■) or GCV (□) intraperitoneally for 12 days starting 1 day after virus injections. After ganciclovir treatment animals were euthanized and tumors were weighted. Both replicative viruses significantly reduced tumor growth compared to Ad5TK-GFP treatment only, but GCV did not significantly increase the cell killing. ***p<0.001 compared to AdTK-GFP only treated cells. Error bars indicate standard error of the mean (B) For monitoring viral spreading, Ad5TK-GFP (■) and Ad5 Δ24TK-GFP (□) infected tumor cells were analyzed by flow cytometry at different time points. *p<0.05 Ad5TK-GFP vs. Ad5 Δ24TK-GFP. Error bars indicate standard error of the mean.
Tumor cells from Ad5TK-GFP and Ad5 Δ24TK-GFP-treated animals were analyzed with flow cytometry 2, 5, 9 and 12 days after infection to study the viral replication and its efficacy in vivo (Fig. 5b). The proportion of GFP-expressing cells decreased from the initial 15% to 4% in mice treated with the replication deficient virus Ad5TK-GFP. In contrast, the percentage of GFP-expressing cells remained relatively constant in Ad5 Δ24TK-GFP-treated animals (p<0.05 after 5 days when compared to Ad5TK-GFP). There was a minor increase in the proportion of GFP-expressing cells during the first 5 days (from 28 to 44%), but during the next days it stabilized to ~35%.
Discussion
Oncolytic viruses are studied as tools for cancer gene therapy. Although they have been demonstrated to destroy tumor cells via replication, clinical trials suggest that the ability of early generation agents to completely eradicate advanced tumor masses is limited. In order to achieve an increased antitumoral effect, combining enhancing elements to oncolytic agents might be useful. Consequently, several approaches to enhance the antitumor capabilities of CRAds have been evaluated. To improve the viral entry to tumor cells, several targeting approaches have been developed including transductional targeting of viruses to alternative cellular receptors, e.g. Ad 3 receptor and integrins (24,25). In addition, utilizing CRAds in conjunction with conventional therapy such as chemotherapy and radiotherapy has been found to augment antitumor activity (26–29). Another promising approach to enhance oncolytic potency of CRAds is to incorporate therapeutic genes expressing prodrug converting enzymes (8,9,12,30). To address whether the TK/GCV system can enhance the oncolytic potency of a CRAd and if a fluorescence transgene be utilized to visualize viral spreading in tumor cells, a Δ24-based CRAd expressing the TK-GFP fusion protein was created Ad5 Δ24TK-GFP).
The replication of Ad5 Δ24TK-GFP was slower when compared to the same virus without a transgene (Ad5 Δ24), presumably due to the activity of the CMV promoter and subsequent TK-GFP production, both of which might sap resources from virus replication and oncolysis (31–33). There was also variation in the replication rate between cell lines, which might be due in part to variable expression levels of CAR, as reported previously for ovarian cancer cells (34). However, despite the delayed replication kinetics and possible variability in CAR expression levels, Ad5 Δ24TK-GFP replicated efficiently in 3 out of 4 examined cell lines eventually resulting in almost complete oncolysis.
When tracking the viruses in vitro, the efficacy of viral spreading closely mirrored the oncolytic capacity of the virus resulting in the most efficient spreading in cell lines supporting rapid replication (OV-3 and OV-4). Within 10 days, almost 90% of OV-4 cells were expressing GFP while the initial proportion of GFP-positive cells was only 10%. Cells resistant to Ad5 Δ24TK-GFP replication (Hey) did not show any remarkable increase in the proportion of GFP-expressing cells. On the other hand, even though oncolysis was effective in OV-3 cells, the proportion of GFP-expressing cells was only 40% after 10 days. This may be influenced by the narrow window of GFP expression in CRAd-infected cells, which appear GFP positive only after completion of TK-GFP production but before lysis of the cell.
It has been suggested that GCV might inhibit viral replication and thus prevent viral spreading within tumor tissue (16,35). Since phosphorylated GCV-molecules can be transferred from one cell to another through gap-junctions, cells with insufficient gap-junctional communication possess a weak bystander effect, which attenuates the therapeutic outcome of TK/GCV gene therapy (36,37). The antagonistic effect of GCV to viral replication is observed especially in cells, which support rapid viral replication but lack efficient gap-junctional communication, where antiviral action of GCV predominates over its anticancer action (35). To evaluate the effect of suicide gene/prodrug therapy on oncolytic potency of Ad5 Δ24TK-GFP in vitro, infected cells were analyzed with a CGV-sensitivity assay. Based on our results, administration of GCV had clear additive effect on oncolysis in 2 out of 4 cell lines. The most dramatic increase in cell killing was observed in SKOV3.ip1 cell line, where cell death was ~10 times more efficient when compared to virus only-treated cells. In OV-4 cells oncolysis was 2-4 times more effective when GCV was present. In addition, TK/GCV system significantly enhanced cell killing in Hey cells even though clear oncolysis was not seen due to inefficient viral replication.
GCV inhibited oncolysis in OV-3 cells, supporting the theory of antagonistic effect of GCV to viral replication. However, this phenomenon was observed only when GCV was added before (1 day p.i.) but not after (4 days p.i.) the initiation of efficient viral replication. In addition, GCV treatment combined with the non-replicative virus (Ad5TK-GFP) caused only modest cell death despite the adequate gene transfer rate, suggesting weak bystander effect of these cells. Therefore, our results suggest that the relative efficacy of oncolysis vs. TK/GCV killing is determined by the relative sensitivity of the cells to virus replication and bystander effect, respectively. Obviously, increasing oncolytic potency by targeting to tumor-associated receptors (38) or increasing the efficacy of suicide gene/prodrug (39,40) will affect these parameters.
In order to study the antineoplastic activity of a CRAd combined with suicide/prodrug gene therapy and intratumoral penetration in vivo, mice bearing subcutaneous SKOV3.ip1 tumors were treated either with non-replicative or replicative viruses followed by GCV administration. Both replicative viruses showed similar significant antitumoral effects when compared to a non-replicative virus, indicating efficient replication and viral penetration of the tumor. When tumor cells were analyzed for GFP expression to monitor viral spreading, the percentage of GFP expressing cells was gradually decreased when a non-replicative virus was used. In contrast, the proportion of GFP expressing cells remained almost constant (~35%) after an initial slight increase, suggesting both viral replication and efficient transgene expression from the CRAd in tumor cells in vivo. Although high transgene expression levels were achieved in vivo, administration of GCV had no significant effect on tumor growth reduction. These results are in parallel with earlier studies, where GCV did not increase the antitumor efficiency of TK-expressing oncolytic viruses (13–16). On the other hand, there are several studies showing that oncolytic potency and antitumor effect can be enhanced with incorporated TK/GCV system (8,9,11,41). Therefore, the potential utility of the combination may be determined by the balance between the opposite effects of augmented GCV-induced cell killing mediated by phosphorylated GCV-metabolites on one hand and GCV-induced reduction of cell killing due to impaired viral replication on the other. It has been suggested that some cancer cells may feature abundant gap junctions (42). If confirmed, this might result in GCV-triphosphate rendering neighboring cells resistant to virus replication. Moreover, since TK/GCV killing is most effective in cycling cells, and one function of the viral proteins is to lock the cell in S-phase, cells allowing effective viral takeover might concomitantly become resistant to TK/GCV.
Many of the findings of this study suggest that the utility of TK/GCV in oncolytic viruses is determined by the relative anti-tumor effects of virus replication and GCV-triphosphate incorporation. In cells permissive for replication, TK/GCV might not be needed (e.g. OV-3), and in cells sensitive to TK/GCV killing (e.g. SKOV3.ip1), virus replication might not provide additional utility. Questions remaining include the importance of scheduling. Preliminary studies suggest that tumors may gain resistance to CRAd treatment (43). In such a situation, it might be useful to administer GCV in order to break resistance and perhaps kill remaining tumor cells. Further, it might be useful to administer GCV intermittently, to avoid abrogation of virus replication, while retaining gap junction-mediated killing of uninfected cells. Viruses such as Ad5 Δ24TK-GFP, which allow sensitive and non-invasive monitoring of replication and dissemination, might be useful tools in answering such questions.
In summary, Ad5 Δ24TK-GFP was capable of replication and spreading in ovarian tumor cells. Although transgene expression seemed to delay replication in vitro, it had no detectable influence on replication and antitumor effects in vivo. Although an efficacious therapeutic outcome was achieved for viral replication in vivo, enhancing effects attributable to the suicide gene/prodrug system were not observed. In some conditions, GCV seemed to even inhibit viral replication and oncolysis. If confirmed, GCV mediated impairment of oncolysis might be useful as a fail-safe mechanism to achieve virus inactivation in case of replication-associated toxicity.
Acknowledgments
We thank Tuula Salonen for help in preparing the adenoviral vectors. This study was supported by Finnish Cultural Foundation of Northern Savo and Cancer Society of Finland, the Academy of Finland, HUCH Research Funds, University of Helsinki Internal Funds, the Sohlberg Foundation, Biocentrum Helsinki, Sigrid Juselius Foundation, Instrumentarium Research Fund, National Institutes of Health (2 R01 CA083821-05A1, 5 R01 CA940484-03 and 1 R01 CA93796) and the Department of Defense (W81XWH-04-1-0025).
References
- 1.Volpers C, Kochanek S. Adenoviral vectors for gene transfer and therapy. J Gene Med. 2004;6 (Suppl 1):S164–S171. doi: 10.1002/jgm.496. [DOI] [PubMed] [Google Scholar]
- 2.Kootstra NA, Verma IM. Gene therapy with viral vectors. Annu Rev Pharmacol Toxicol. 2003;43:413–439. doi: 10.1146/annurev.pharmtox.43.100901.140257. [DOI] [PubMed] [Google Scholar]
- 3.Sinkovics J, Horvath J. New developments in the virus therapy of cancer: A historical review. Intervirology. 1993;36:193–214. doi: 10.1159/000150339. [DOI] [PubMed] [Google Scholar]
- 4.Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science. 1991;252:854–856. doi: 10.1126/science.1851332. [DOI] [PubMed] [Google Scholar]
- 5.Fueyo J, Gomez-Manzano C, Alemany R, et al. A mutant oncolytic adenovirus targeting the rb pathway produces anti-glioma effect in vivo. Oncogene. 2000;19:2–12. doi: 10.1038/sj.onc.1203251. [DOI] [PubMed] [Google Scholar]
- 6.Kirn D, Martuza RL, Zwiebel J. Replication-selective virotherapy for cancer: Biological principles, risk management and future directions. Nat Med. 2001;7:781–787. doi: 10.1038/89901. [DOI] [PubMed] [Google Scholar]
- 7.Bauerschmitz GJ, Barker SD, Hemminki A. Adenoviral gene therapy for cancer: from vectors to targeted and replication competent agents (review) Int J Oncol. 2002;21:1161–1174. [PubMed] [Google Scholar]
- 8.Wildner O, Blaese RM, Morris JC. Therapy of colon cancer with oncolytic adenovirus is enhanced by the addition of herpes simplex virus-thymidine kinase. Cancer Res. 1999;59:410–413. [PubMed] [Google Scholar]
- 9.Wildner O, Morris JC, Vahanian NN, Ford H, Jr, Ramsey WJ, Blaese RM. Adenoviral vectors capable of replication improve the efficacy of HSVtk/GCV suicide gene therapy of cancer. Gene Ther. 1999;6:57–62. doi: 10.1038/sj.gt.3300810. [DOI] [PubMed] [Google Scholar]
- 10.van Beusechem VW, van den Doel PB, Grill J, Pinedo HM, Gerritsen WR. Conditionally replicative adenovirus expressing p53 exhibits enhanced oncolytic potency. Cancer Res. 2002;62:6165–6171. [PubMed] [Google Scholar]
- 11.Freytag SO, Stricker H, Pegg J, et al. Phase I study of replication-competent adenovirus-mediated double-suicide gene therapy in combination with conventional-dose three-dimensional conformal radiation therapy for the treatment of newly diagnosed, intermediate- to high-risk prostate cancer. Cancer Res. 2003;63:7497–7506. [PubMed] [Google Scholar]
- 12.Fuerer C, Iggo R. 5-fluorocytosine increases the toxicity of wnt-targeting replicating adenoviruses that express cytosine deaminase as a late gene. Gene Ther. 2004;11:142–151. doi: 10.1038/sj.gt.3302148. [DOI] [PubMed] [Google Scholar]
- 13.Freytag SO, Rogulski KR, Paielli DL, Gilbert JD, Kim JH. A novel three-pronged approach to kill cancer cells selectively: concomitant viral, double suicide gene, and radiotherapy. Hum Gene Ther. 1998;9:1323–1333. doi: 10.1089/hum.1998.9.9-1323. [DOI] [PubMed] [Google Scholar]
- 14.Lambright ES, Amin K, Wiewrodt R, et al. Inclusion of the herpes simplex thymidine kinase gene in a replicating adenovirus does not augment antitumor efficacy. Gene Ther. 2001;8:946–953. doi: 10.1038/sj.gt.3301489. [DOI] [PubMed] [Google Scholar]
- 15.Morris JC, Wildner O. Therapy of head and neck squamous cell carcinoma with an oncolytic adenovirus expressing HSV-tk. Mol Ther. 2000;1:56–62. doi: 10.1006/mthe.1999.0014. [DOI] [PubMed] [Google Scholar]
- 16.Rogulski KR, Wing MS, Paielli DL, Gilbert JD, Kim JH, Freytag SO. Double suicide gene therapy augments the anti-tumor activity of a replication-competent lytic adenovirus through enhanced cytotoxicity and radiosensitization. Hum Gene Ther. 2000;11:67–76. doi: 10.1089/10430340050016166. [DOI] [PubMed] [Google Scholar]
- 17.Wildner O, Morris JC. The role of the E1B 55 kDa gene product in oncolytic adenoviral vectors expressing herpes simplex virus-tk: assessment of antitumor efficacy and toxicity. Cancer Res. 2000;60:4167–4174. [PubMed] [Google Scholar]
- 18.Le LP, Everts M, Dmitriev IP, Davydova JG, Yamamoto M, Curiel DT. Fluorescently labeled adenovirus with pIX-EGFP for vector detection. Mol Imaging. 2004;3:105–116. doi: 10.1162/15353500200404100. [DOI] [PubMed] [Google Scholar]
- 19.Contag CH, Spilman SD, Contag PR, et al. Visualizing gene expression in living mammals using a bioluminescent reporter. Photochem Photobiol. 1997;66:523–531. doi: 10.1111/j.1751-1097.1997.tb03184.x. [DOI] [PubMed] [Google Scholar]
- 20.Gambhir SS, Barrio JR, Wu L, et al. Imaging of adenoviral-directed herpes simplex virus type 1 thymidine kinase reporter gene expression in mice with radiolabeled ganciclovir. J Nucl Med. 1998;39:2003–2011. [PubMed] [Google Scholar]
- 21.Loimas S, Wahlfors J, Janne J. Herpes simplex virus thymidine kinase-green fluorescent protein fusion gene: new tool for gene transfer studies and gene therapy. Biotechniques. 1998;24:614–618. doi: 10.2144/98244st01. [DOI] [PubMed] [Google Scholar]
- 22.Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 23.Hakkarainen T, Wahlfors T, Merilainen O, Loimas S, Hemminki A, Wahlfors J. VP22 does not significantly enhance enzyme prodrug cancer gene therapy as a part of a VP22-HSVTk-GFP triple fusion construct. J Gene Med. 2005;7:898–907. doi: 10.1002/jgm.737. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki K, Fueyo J, Krasnykh V, Reynolds PN, Curiel DT, Alemany R. A conditionally replicative adenovirus with enhanced infectivity shows improved oncolytic potency. Clin Cancer Res. 2001;7:120–126. [PubMed] [Google Scholar]
- 25.Kanerva A, Zinn KR, Chaudhuri TR, et al. Enhanced therapeutic efficacy for ovarian cancer with a serotype 3 receptor-targeted oncolytic adenovirus. Mol Ther. 2003;8:449–458. doi: 10.1016/s1525-0016(03)00200-4. [DOI] [PubMed] [Google Scholar]
- 26.Khuri FR, Nemunaitis J, Ganly I, et al. a controlled trial of intratumoral ONYX-015, a selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in patients with recurrent head and neck cancer. Nat Med. 2000;6:879–885. doi: 10.1038/78638. [DOI] [PubMed] [Google Scholar]
- 27.Lamfers ML, Grill J, Dirven CM, et al. Potential of the conditionally replicative adenovirus Ad5-Delta24RGD in the treatment of malignant gliomas and its enhanced effect with radiotherapy. Cancer Res. 2002;62:5736–5742. [PubMed] [Google Scholar]
- 28.Reid T, Galanis E, Abbruzzese J, et al. Hepatic arterial infusion of a replication-selective oncolytic adenovirus (dl1520): Phase II viral, immunologic, and clinical endpoints. Cancer Res. 2002;62:6070–6079. [PubMed] [Google Scholar]
- 29.Rogulski KR, Freytag SO, Zhang K, et al. In vivo antitumor activity of ONYX-015 is influenced by p53 status and is augmented by radiotherapy. Cancer Res. 2000;60:1193–1196. [PubMed] [Google Scholar]
- 30.Chen MJ, Green NK, Reynolds GM, et al. Enhanced efficacy of Escherichia coli nitroreductase/CB1954 prodrug activation gene therapy using an E1B-55K-deleted oncolytic adenovirus vector. Gene Ther. 2004;11:1126–1136. doi: 10.1038/sj.gt.3302271. [DOI] [PubMed] [Google Scholar]
- 31.Haviv YS, Takayama K, Glasgow JN, Blackwell JL, Wang M, Lei X, Curiel DT. A model system for the design of armed replicating adenoviruses using p53 as a candidate transgene. Mol Cancer Ther. 2002;1:321–328. [PubMed] [Google Scholar]
- 32.Hemminki A, Wang M, Hakkarainen T, Desmond RA, Wahlfors J, Curiel DT. Production of an EGFR targeting molecule from a conditionally replicating adenovirus impairs its oncolytic potential. Cancer Gene Ther. 2003;10:583–588. doi: 10.1038/sj.cgt.7700606. [DOI] [PubMed] [Google Scholar]
- 33.Rivera AA, Wang M, Suzuki K, Uil TG, Krasnykh V, Curiel DT, Nettelbeck DM. Mode of transgene expression after fusion to early or late viral genes of a conditionally replicating adenovirus via an optimized internal ribosome entry site in vitro and in vivo. Virology. 2004;320:121–134. doi: 10.1016/j.virol.2003.11.028. [DOI] [PubMed] [Google Scholar]
- 34.Kanerva A, Mikheeva GV, Krasnykh V, et al. Targeting adenovirus to the serotype 3 receptor increases gene transfer efficiency to ovarian cancer cells. Clin Cancer Res. 2002;8:275–280. [PubMed] [Google Scholar]
- 35.Post DE, Van Meir EG. A novel hypoxia-inducible factor (HIF) activated oncolytic adenovirus for cancer therapy. Oncogene. 2003;22:2065–2072. doi: 10.1038/sj.onc.1206464. [DOI] [PubMed] [Google Scholar]
- 36.Bi WL, Parysek LM, Warnick R, Stambrook PJ. In vitro evidence that metabolic cooperation is responsible for the bystander effect observed with HSVtk retroviral gene therapy. Hum Gene Ther. 1993;4:725–731. doi: 10.1089/hum.1993.4.6-725. [DOI] [PubMed] [Google Scholar]
- 37.Mesnil M, Piccoli C, Tiraby G, Willecke K, Yamasaki H. Bystander killing of cancer cells by herpes simplex virus thymidine kinase gene is mediated by connexins. Proc Natl Acad Sci USA. 1996;93:1831–1835. doi: 10.1073/pnas.93.5.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kanerva A, Hemminki A. Modified adenoviruses for cancer gene therapy. Int J Cancer. 2004;110:475–480. doi: 10.1002/ijc.20129. [DOI] [PubMed] [Google Scholar]
- 39.Kokoris MS, Sabo P, Adman ET, Black ME. Enhancement of tumor ablation by a selected HSV-1 thymidine kinase mutant. Gene Ther. 1999;6:1415–1426. doi: 10.1038/sj.gt.3300966. [DOI] [PubMed] [Google Scholar]
- 40.Black ME, Kokoris MS, Sabo P. Herpes simplex virus-1 thymidine kinase mutants created by semi-random sequence mutagenesis improve prodrug-mediated tumor cell killing. Cancer Res. 2001;61:3022–3026. [PubMed] [Google Scholar]
- 41.Nanda D, Vogels R, Havenga M, Avezaat CJ, Bout A, Smitt PS. Treatment of malignant gliomas with a replicating adenoviral vector expressing herpes simplex virus-thymidine kinase. Cancer Res. 2001;61:8743–8750. [PubMed] [Google Scholar]
- 42.Chipman JK, Mally A, Edwards GO. Disruption of gap junctions in toxicity and carcinogenicity. Toxicol Sci. 2003;71:146–153. doi: 10.1093/toxsci/71.2.146. [DOI] [PubMed] [Google Scholar]
- 43.Kanerva A, Zinn KR, Peng KW, et al. Noninvasive dual modality in vivo monitoring of the persistence and potency of a tumor targeted conditionally replicating adenovirus. Gene Ther. 2005;12:87–94. doi: 10.1038/sj.gt.3302387. [DOI] [PubMed] [Google Scholar]