

Abstract
What is already known about this subject
Mometasone furoate (MF) is a new inhaled glucocorticoid for which the first reports suggested a low degree of systemic side-effects and low systemic availability.
Recent studies of Fardon and colleagues have shown that MF's cortisol suppression is similar to that of fluticasone.
Pharmacokinetic/dynamic evaluations of MF's systemic side-effects, probing whether systemic side-effects can be explained by systemic availability, plasma protein binding and receptor binding affinity, are lacking in the literature.
What this study adds
This study shows that the systemic availability of MF and fluticasone propionate (FP) are similar and that systemic availability is directly related to the dose.
It also shows that the metabolites of MF are present only in very low concentrations at most, contrary to results in rats.
The observed cortisol suppression of FP and MF is related to the trough plasma concentrations and seems to be in agreement with its observed systemic availability, plasma protein binding and receptor binding affinity.
Aim
Fluticasone propionate (FP) and mometasone furoate (MF) are inhaled corticosteroids that possess a high ratio of topical to systemic activity. The systemic bioavailability of MF has been claimed to be minimal (1%). FP has been shown to exhibit the same degree of systemic effects, but its systemic availability is between 13 and 17%. We hypothesize that FP and MF have comparable systemic availabilities that can explain their potential to cause systemic effects.
Methods
Steady-state FP and MF trough plasma samples were determined from a clinical study by Fardon et al. in patients with persistent asthma (forced expiratory volume in 1 s = 91%). The percent plasma protein binding of FP and MF was measured using ultracentrifugation. Free FP plasma concentrations were normalized for their differences in receptor binding affinity compared with MF and linked to overnight urinary cortisol/creatinine with an inhibitory Emax.
Results
A plot of steady-state FP and MF total trough plasma concentrations vs. dose showed that both drugs exhibit dose linearity. MF has comparable bioavailability to FP based on the steady-state concentrations observed for the different doses. The free plasma concentration producing 50% of urinary cortisol suppression (IC50) for MF was not statistically different from the free, normalized IC50 for FP.
Conclusion
FP and MF have similar pulmonary deposition and the same potential to cause systemic side-effects due to their similar IC50 values. The observed urinary cortisol suppression of FP and MF is in agreement with their systemic availability, their differences in plasma protein binding and receptor binding affinity.
Keywords: fluticasone propionate, mometasone furoate, plasma protein binding, pulmonary deposition, systemic side-effects
Introduction
Asthma is a chronic inflammatory disorder of the airways that affects nearly 150 million people worldwide [1]. Since their introduction nearly 30 years ago, inhaled corticosteroids have become the mainstay of treatment for chronic asthma [2–4]. Although given locally into the lung, inhaled glucocorticoids can induce systemic side-effects, which can include skin thinning, easy bruising, decreased bone density, growth retardation and adrenal suppression [5–7]. Due to these unwanted effects, there have been efforts to improve further the safety profile of inhaled glucocorticoids. One of the newest inhaled glucocorticoids is mometasone furoate (MF), a halogenated glucocorticoid with a high lipophilicity as well as a high affinity for the glucocorticoid receptor, low systemic absorption and high metabolic clearance [8–11]. MF is available for use in allergic rhinitis in the USA [12, 13] and is approved for asthma therapy in Europe. At the time of the submission of this paper, it is being investigated for asthma therapy in the USA.
The relative binding affinity (RBA) of MF to the glucocorticoid receptor is higher than that of fluticasone propionate (FP), while its clearance is similar to that of other inhaled glucocorticoids [12, 14, 15]. Given the higher affinity and higher potency of MF compared with FP, it would seem logical to assume that similar doses of MF would lead to a greater degree of unwanted side-effects. However, early reports [16] have indicated that cortisol suppression of MF is negligible and that the low systemic bioavailability of 1%, compared with 13–17% for FP [9, 17, 18] and/or the high plasma protein binding (99% for MF [19] and 90% for FP [9, 20, 21]), could be the reason.
Fardon et al. [10], compared the overnight urinary cortisol/creatinine concentrations after administration of low, medium and high doses of MF and FP at steady state. The authors found that at medium and high doses of FP and MF there was significant suppression of overnight urinary cortisol concentrations corrected for creatinine clearance [10]. The goal of this study was to evaluate whether systemic side-effects observed in the Fardon study are in agreement with observed plasma concentrations, receptor binding affinity and plasma protein binding of FP and MF.
Methods
Study design
Parts of this study, namely the effects of MF and FP on urinary cortisol, have been previously published by Fardon et al. [10]. The present study extends the results by evaluating the plasma concentrations of MF and FP and correlating these with the observed effects on urinary cortisol. In the original study by Fardon et al. [10], 22 patients with mild to moderate asthma [age range 23–67 years (mean 48 ± 13)] were assigned to two treatment groups. The first group received FP with an Accuhaler dry powder device at 250 µg twice a day for 2 weeks. This would be followed by 500 µg twice a day for 2 weeks and then 1000 µg twice daily for 2 weeks. After each 2-week treatment period, a urinary cortisol/creatinine measurement would be taken along with a corresponding plasma sample (for MF and FP measurements), 12 h after the last dose. The patients would then be subjected to a 1-week wash-out period where they were given 50 µg salmeterol twice a day and 10 mg montelukast once a day. After the wash-out period, the patients were given MF with a Twisthaler dry powder device at 200 µg twice a day for 2 weeks. This was followed by 400 µg twice a day for 2 weeks and then 800 µg twice a day for 2 weeks. A plasma sample and corresponding urinary cortisol measurement were taken after each 2-week treatment period. The other group of patients were given the same doses, but in the reverse order.
Analysis of FP and MF plasma samples
FP was purchased from Sigma-Aldrich (St Louis, MO, USA). MF was obtained from USP (Rockville, MD, USA), 6β-hydroxy MF was a gift from Professor P. Högger (Institut für Pharmazie und Lebensmittelchemie, Würzburg, Germany) and 13C3-fluticasone propionate was provided by GSK R&D (Ware, UK). High-performance liquid chromatography grade solvents were purchased from Fisher chemicals (Springfield, NJ, USA) and solid-phase LC18 cartridges for solid-phase extraction were acquired from Supelco (Bellafonte, PA, USA). Drug-free human plasma was purchased from the Civitan regional blood system (Gainesville, FL, USA).
FP and MF trough plasma samples from the clinical study conducted by Fardon and coworkers [10] were quantified with liquid chromatography tandem mass spectrometry (LC-MS-MS) after solid-phase extraction as previously described [22]. Surprisingly, low plasma concentrations were found in placebo samples. This might be due to patients being exposed to FP- or MF-contaminated air in the clinic, as such levels were not determined in relevant calibration and quality control samples.
For 6β-hydroxy MF, the MF method was adapted by using a transition m /z 537–501. The limit of quantification for FP was 15 pg ml−1, 45 pg ml−1 for MF and 200 pg ml−1 for 6β-hydroxy MF.
Plasma protein binding determination
Samples of FP and MF were prepared in three different batches of human drug-free plasma by spiking in a stock solution of drug in methanol into plasma to concentrations of 10, 50 and 150 ng ml−1 to a total volume of 5 ml. The concentration of methanol in plasma was <1% v/v. The samples were vortexed and then allowed to equilibrate for 20 min in a 37°C water batch. After equilibration, 3.5 ml of the sample was removed and put into polycarbonate centrifuge tubes. The tubes were then placed into a SW 55Ti swinging bucket rotor (Beckman Coulter, Fullerton, CA, USA) that had been incubated for 3 h at 37°C. The samples were spun at 300 000 g for 30 h at 37°C in an ultracentrifuge (Model L8-70M; Beckman Coulter). Under these conditions, but not after run times of 16 and 24 h, <0.5% of protein was remaining in the supernatant. The remaining 1.5-ml plasma samples were incubated at 37°C for 30 h followed by measuring MF and FP total plasma concentrations. This was done to adjust for the slight metabolism of MF in plasma at 37°C [23].
After ultracentrifugation of the samples, the tubes were taken out and the lipoprotein layer on top was carefully removed. One millilitre of the supernatant (plasma water) was extracted and stored at −70°C until analysis along with the plasma samples.
Samples made from a specific batch of plasma were quantified using standards and quality controls from the same batch of blank plasma in order to maintain uniformity. Drug-free plasma water was necessary in order to quantify the samples in plasma water. The standards and quality controls in plasma water were made up from the same batch of plasma in which the samples were made. The drug-free plasma was centrifuged in a similar manner to that described above and the resulting plasma water supernatant was used to make up the necessary standards and quality controls. The plasma samples, standards and quality controls for FP and MF were quantified in a manner similar to the samples from the clinical study (described above). The plasma water samples, standards and quality controls were also determined using the LC/MS/MS with the same conditions for the plasma samples. Equation 1 was used to calculate the percent plasma protein binding of FP and MF:
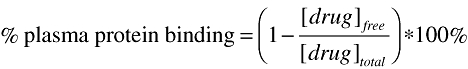 |
(1) |
Results for the plasma protein binding of FP and MF were obtained for three separate batches of human plasma in order to observe any differences.
The amount of nonspecific binding due to binding to the surface of the centrifuge tubes was determined by measuring FP and MF concentrations in phosphate-buffered saline (PBS) using the same ultracentrifugation technique described above.
Probing for pharmacokinetic/pharmacodynamic relationships
The total trough plasma concentrations (Cp) from the clinical study were linked to the urinary cortisol/creatinine concentrations (UC) using an inhibitory Emax model, Equation 2:
 |
(2) |
where UCo is the urinary baseline cortisol value and IC50 is the concentration that produces 50% of baseline urinary concentration. This model was the simplest model that gave the most adequate fit. The data were evaluated using the Nonlinear Mixed Effect Modelling (NONMEM) software program (version V, double precision) [24]. The fixed and random effect parameters were estimated using first-order conditional estimation. The intersubject variability was determined using an exponential error model and the intrasubject variability was described using a proportional error model.
Using the obtained values from the plasma protein binding experiments, the free MF and FP concentrations could then correlated to urinary cortisol concentrations using the model. The free FP concentrations were normalized for their differences in receptor binding affinity for the glucocorticoid receptor, in comparison with MF. The relative receptor binding affinity, relative to dexamethasone, is 1800 and 2938 for FP and MF, respectively [25] The calculation to determine the normalized free FP concentration was determined by Equation 3:
 |
(3) |
The normalized free FP concentrations and the free MF concentrations were then evaluated with the population pharmacodynamic (PD) model in order to compare the systemic exposure of FP and MF by modifying Equation 1:
 |
(4) |
The parameters generated from this analysis can be used to compare the systemic exposure and the potency of FP and MF to cause systemic effects.
Statistical analysis
All statistical tests were conducted using the GraphPad software program for Windows (version 4, InStat 4.0, San Diego, CA, USA). The population IC50 values for FP and MF were compared by using a t-test-based evaluation of their mean values and their standard deviation. A significance concentration of P < 0.05 was used to define statistically significant differences.
Results
FP and MF plasma samples
FP and MF steady-state trough plasma concentrations after low, medium and high doses in 22 patients under FP and MF treatment [10] were quantified by previously validated methods developed in our laboratory [22]. A plot of the total plasma concentrations observed for the different dosages of MF and FP vs. dose of FP or MF yielded a linear relationship between dose and steady-state plasma concentrations. The data for FP and MF were essentially superimposed (Figure 1; slope for FP was 4.05 × 10−5 and 4.22 × 10−5 for MF). This indicated that the systemic exposure for MF and FP would be identical if the same doses were given via dry powder inhalers. 6β-Hydroxy MF could not be quantified in any patient using the LC/MS assay and only two patients showed metabolite concentrations above the limit of detection. In these two patients, however, metabolite concentrations could be detected after low, medium and high doses.
Figure 1.

Total plasma concentration of mometasone furoate (□) and fluticasone propionate (•) vs. dose. Error bars represent the standard deviation about the mean
Plasma protein binding
Preliminary experiments performed in PBS showed that nonspecific binding to centrifugation tubes was minimal for FP (0.23%) and MF (0.22%). In addition, centrifugation using different centrifugation times showed that the percentage of plasma proteins in the supernatant did not decrease significantly after 30 h, so this time period was selected in the determination of the percent plasma protein binding of FP and MF. The stability of MF has been evaluated in a previous paper and shown to be relatively stable in plasma (degradation half-life of 85 h [23]). However, even a more pronounced degradation would not affect the results, as plasma samples used for the determination of the total plasma concentrations were incubated at 37°C for the time of centrifugation. A potential degradation of drugs during the centrifugation was therefore accounted for.
The method was validated by assessing the protein binding of budesonide to be in accordance with literature values (88%) [26].
Using ultracentrifugation, the average percent protein binding of three separate batches of human plasma was determined to be 94.8% [SD 1.06, 90% confidence interval (CI) 94.37, 95.24] for FP and 98.2% (SD 0.98, 90% CI 97.79, 98.61) for MF. There was no concentration dependency in plasma protein binding for either drug and no significant difference between batches of plasma. The differences between the two compounds were significant.
Concentration–effect relationship
Plasma concentrations of FP were converted to ‘MF’ equivalents by considering differences in receptor binding affinities. When urinary cortisol:creatinine ratios were plotted against these corticosteroid concentrations, the data could be described with a sigmoidal Emax model resulting in IC50 values of 0.06 ng ml−1 for FP and 0.10 ng ml−1 for MF (data not shown), which were statistically not significantly different. When free drug concentrations of MF and FP (expressed in MF equivalents) were linked to the urinary cortisol : creatinine ratios, a similar sigmoidal Imax relationship was indicated (Figure 2). The between-subject variability on IC50 of free concentrations of FP and MF was measured using an exponential error model and the residual error was best described with a proportional error model. The model was able to predict the urinary cortisol concentrations well and generate relevant parameters, as seen in Figure 2 and Table 1. The free, normalized IC50 for FP was 0.003 ng ml−1 and the free IC50 for MF was 0.002 ng ml−1. There was no statistical difference between these two values (P = 0.32) after performing a t-test-based evaluation of the mean population estimate and their SD.
Figure 2.

Urinary cortisol/creatinine vs. trough free corticosteroid concentrations. ○, Free mometasone furoate (MF) trough concentrations; •, free fluticasone propionate (FP) trough concentrations shown as free MF equivalents (free FP concentrations were normalized for differences in receptor binding affinity. Dashed line, model predictions for free MF concentrations; solid line, model prediction for free FP (shown as free MF equivalents). The corresponding IC50, with error bars representing the standard deviation, is also represented for MF and FP
Table 1.
Final parameter estimates for pharmacodynamic model
| Parameter | Population estimate | SE% | |
|---|---|---|---|
| FP | Free normalized IC50 (ng ml−1) | 0.003* | 34.2 |
| Between-subject variability on IC50 (%CV) | 42.3 | 92.2 | |
| Within-subject variability (%CV) | 44.3 | 25.1 | |
| UC0 | For FP data (nmol mmol−1) | 11.6 | 10.8 |
| Between-subject variability on UC0 (%CV) | 42.5 | 114.4 | |
| Within-subject variability (%CV) | 44.1 | 25.6 | |
| MF | Free IC50 (ng ml−1) | 0.002* | 38.2 |
| Between-subject variability on IC50 (%CV) | 77.9 | 73.5 | |
| Within-subject variability (%CV) | 45.3 | 20.4 | |
| UC0 | For MF data (nmol mmol−1) | 11.0 | 7.85 |
| Between-subject variability on UC0 (%CV) | 78.4 | 64.7 | |
| Within-subject variability (%CV) | 44.7 | 18.4 | |
P = 0.322, unpaired t-test.
Discussion
Systemic side-effects are an important factor when evaluating the safety profile of inhaled corticosteroids. The extent of systemic side-effects is thought to be determined by several factors, including the binding affinity of the drug to the glucocorticoid receptor, systemic clearance, plasma protein binding and the systemic bioavailability from oral and inhaled routes [27, 28].
The study by Fardon et al. [10] compared the systemic side-effects of inhaled MF and FP at low, medium and high therapeutic doses by monitoring overnight urinary cortisol/creatinine as a surrogate marker of systemic effect. This study has demonstrated that FP and MF suppress cortisol production at medium and high doses of FP and MF to a similar extent. The present study aimed at evaluating whether the comparable cortisol suppression observed for MF and FP in Fardon's study, while in disagreement with earlier reports [8, 16, 29], was in agreement with MF's and FP's systemic exposure as it is related to the observed systemic steady-state plasma concentrations, the degree of plasma protein binding and receptor binding affinities.
Plasma trough concentrations were taken 12 h after the dose was administered. FP and MF both possess similar long half-lives (FP 7–14 h, MF 5.8 h) [9, 15, 18, 30]. This, and similar clearance values for FP and MF, close to the liver blood flow, ensured that trough values can be used as sufficiently good descriptor of the drug exposure at steady state. Considering that both drugs have an oral bioavailability of close to zero, the good agreement between FP and MF data in Figure 1 indicates that the pulmonary deposition and consequently the absorption into the systemic circulation are very similar for the FP and MF dry powder devices used in Fardon's study. These results are in agreement with a recent, more detailed pharmacokinetic (PK) study in asthmatics after single dosing of FP and MF dry powders [31]. Overall, these studies seem to indicate that the systemic spill-over is similar for FP and MF. In addition, linear PK are suggested from the low-, medium- and high-dose trough values for both MF and FP (Figure 1).
A possible explanation put forward by others for the systemic side-effects of MF is the presence of active metabolites [29]. The 6β-hydroxy metabolite and the free mometasone moiety may possess pharmacological activity [32]. We were able to detect 6β-hydroxy MF from 2/22 subjects with a limit of quantification of 0.2 ng ml−1 and detect metabolite concentrations in six of 87 samples. The low concentrations of 6β-hydroxy MF along with a lower receptor binding affinity when compared with the parent MF [23] would indicate that this metabolite does not play a significant role in producing systemic effects for MF. These results are contrary to previous results in rats [23], for which distinct concentrations of this active metabolite were identified.
It has been shown that the systemic activity of an inhaled glucocorticoid is determined by the steady-state plasma concentration, plasma protein binding and pharmacological activity at the site of action (generally described by the relative receptor binding affinity) [33]. In agreement with this relationship, it is now well accepted that a high degree of plasma protein is a means by which to improve the safety profile of inhaled corticosteroids by preventing the drug from interacting with systemic glucocorticoid receptors [34]. However, others have recently argued against the importance of plasma protein binding by stating that it is generally nonspecific and of low affinity, whereas systemic side-effects are mediated due to inhaled corticosteroids' high affinity for the glucocorticoid receptor, and protein binding is not important [35].
We chose to utilize ultracentrifugation as a method to determine the plasma protein binding of FP and MF, as it minimizes artefacts due to nonspecific binding of drugs to plastic and glass during the in vitro determinations. Protein binding was assessed in spiked blank plasma, as the limited volume of patient plasma did not allow a determination of both free and total concentrations in the actual patient samples. Although difficulties in determining protein binding estimates for high binding drugs have been pointed out [36], the MF estimate of 98.2% agreed well with previous estimates (MF 99%) [36]. We found the plasma protein binding of FP to be 94.8%, somewhat between the two estimates available from the literature (FP 90, 98.1%) [36], indicating some distinct differences in the free fraction of FP. The results of our and other studies reinforce that it is a challenge to provide high-quality protein binding data for highly protein-bound drugs.
In order to compare the systemic concentrations of MF and FP, total plasma concentrations and free concentrations calculated based on protein binding data were further adjusted for differences in biological activity. Receptor binding has been shown to be a valid biomarker for assessing differences of inhaled glucocorticoids at the site of action [27]. Differences in RBA have been reported for MF (RBA 2938) and FP ( RBA1800) [25].
However, if the free, normalized FP concentrations and the free MF concentrations were linked to overnight urinary cortisol/creatinine concentrations and analysed with the PDmodel described above, the results showed free IC50 values for MF and free, normalized FP resulted in IC50 values that were not statistically significantly different. While the lack of significant differences between free normalized FP and MF IC50 values might suggest that free concentrations are indeed relevant for the systemic effects, IC50 estimates for total normalized FP and MF values did not differ either. These findings argue for better designed studies with full PK and PD data collection to address the question conclusively.
Conclusion
The data from this study have revealed that FP and MF, delivered via their corresponding dry powder inhalers, have comparable systemic availabilities and show linear PK over the dose range studied. The similar urinary cortisol suppression of FP and MF are in agreement with their systemic availability, plasma protein binding, and receptor binding affinity (although more studies are needed to confirm this), whereas MF metabolites do not seem to be involved in the induction of cortisol suppression.
Acknowledgments
Competing interests
G.H. has been reimbursed by Schering-Plough for attending a meeting and has received funds from AZ for performing research. G.H. has also received fees for consulting from Schering-Plough, Verus Pharmaceuticals and AZ. B.J.L. has spoken for Ivax, AstraZeneca, Sanofi-Aventis and Altana, who all make inhaled corticosteroids. B.J.L. has also received consulting fees from AstraZeneca, Neolab and Altana, who all make inhaled steroids.
We thank Professor Dr Petra Högger (Wuerzburg, Germany) for providing 6-β hydroxy mometasone furoate.
References
- 1.WHO. Geneva: World Health Organization; WHO fact sheet no. 206. [Google Scholar]
- 2.Barnes PJ, Adcock IM. Steroid resistance in asthma. QJM. 1995;88:455–68. [PubMed] [Google Scholar]
- 3.Barnes PJ, Pedersen S, Busse WW. Efficacy and safety of inhaled corticosteroids. New developments. Am J Respir Crit Care Med. 1998;157(3) Part 2:S1–53. doi: 10.1164/ajrccm.157.3.157315. [DOI] [PubMed] [Google Scholar]
- 4.Pedersen S, O'Byrne P. A comparison of the efficacy and safety of inhaled corticosteroids in asthma. Allergy. 1997;52(39 Suppl.):1–34. doi: 10.1111/j.1398-9995.1997.tb05047.x. [DOI] [PubMed] [Google Scholar]
- 5.Brown PH, Greening AP, Crompton GK. Large volume spacer devices and the influence of high dose beclomethasone dipropionate on hypothalamo–pituitary–adrenal axis function. Thorax. 1993;48:233–8. doi: 10.1136/thx.48.3.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lipworth BJ. Systemic adverse effects of inhaled corticosteroid therapy. A systematic review and meta-analysis. Arch Intern Med. 1999;159:941–55. doi: 10.1001/archinte.159.9.941. [DOI] [PubMed] [Google Scholar]
- 7.Mak VH, Melchor R, Spiro SG. Easy bruising as a side-effect of inhaled corticosteroids. Eur Respir J. 1992;5:1068–74. [PubMed] [Google Scholar]
- 8.Crim C, Pierre LN, Daley-Yates PT. A review of the pharmacology and pharmacokinetics of inhaled fluticasone propionate and mometasone furoate. Clin Ther. 2001;23:1339–54. doi: 10.1016/s0149-2918(01)80113-2. [DOI] [PubMed] [Google Scholar]
- 9.Derendorf H, Hochhaus G, Meibohm B, Mollmann H, Barth J. Pharmacokinetics and pharmacodynamics of inhaled corticosteroids. J Allergy Clin Immunol. 1998;101(4) Part 2:S440–6. doi: 10.1016/s0091-6749(98)70156-3. [DOI] [PubMed] [Google Scholar]
- 10.Fardon TC, Lee DK, Haggart K, McFarlane LC, Lipworth BJ. Adrenal suppression with dry powder formulations of fluticasone propionate and mometasone furoate. Am J Respir Crit Care Med. 2004;170:960–6. doi: 10.1164/rccm.200404-500OC. [DOI] [PubMed] [Google Scholar]
- 11.Rago RP, Einstein A, Jr, Lush R, Beer TM, Ko YJ, Henner WD, Bubley G, Merica EA, Garg V, Ette E, Harding MW, Dalton WS. Safety and efficacy of the MDR inhibitor Incel (biricodar, VX-710) in combination with mitoxantrone and prednisone in hormone-refractory prostate cancer. Cancer Chemother Pharmacol. 2003;51:297–305. doi: 10.1007/s00280-003-0573-4. [DOI] [PubMed] [Google Scholar]
- 12.Onrustand SV, Lamb HM. Mometasone furoate. A review of its intranasal use in allergic rhinitis. Drugs. 1998;56:725–45. doi: 10.2165/00003495-199856040-00018. [DOI] [PubMed] [Google Scholar]
- 13.Storms WW. Treatment of seasonal allergic rhinitis with fluticasone propionate aqueous nasal spray: review of comparator studies. Allergy. 1995;50(23 Suppl.):25–9. doi: 10.1111/j.1398-9995.1995.tb02738.x. [DOI] [PubMed] [Google Scholar]
- 14.Issar M, Sahasranaman S, Buchwald P, Hochhaus G. Differences in the glucocorticoid to progesterone receptor selectivity of inhaled glucocorticoids. Eur Resp J. 2006;27:511–6. doi: 10.1183/09031936.06.00060005. [DOI] [PubMed] [Google Scholar]
- 15.Sharpe M, Jarvis B. Inhaled mometasone furoate: a review of its use in adults and adolescents with persistent asthma. Drugs. 2001;61:1325–50. doi: 10.2165/00003495-200161090-00011. [DOI] [PubMed] [Google Scholar]
- 16.Affrime MB, Cuss F, Padhi D, Wirth M, Pai S, Clement RP, Lim J, Kantesaria B, Alton K, Cayen MN. Bioavailability and metabolism of mometasone furoate following administration by metered-dose and dry-powder inhalers in healthy human volunteers. J Clin Pharmacol. 2000;40:1227–36. [PubMed] [Google Scholar]
- 17.Mackie AE, McDowall JE, Falcoz C, Ventresca P, Bye A, Daley-Yates PT. Pharmacokinetics of fluticasone propionate inhaled via the Diskhaler and Diskus powder devices in healthy volunteers. Clin Pharmacokinet. 2000;39(Suppl. 1):23–30. doi: 10.2165/00003088-200039001-00004. [DOI] [PubMed] [Google Scholar]
- 18.Thorsson L, Edsbacker S, Conradson TB. Lung deposition of budesonide from Turbuhaler (R) Is twice that from a pressurized metered-dose inhaler P-Mdi. Eur Resp J. 1994;7:1839–44. doi: 10.1183/09031936.94.07101839. [DOI] [PubMed] [Google Scholar]
- 19.Schering-Plough. Kenilwoth, NJ: ScheringPlough; 2004. Mometasone furoate package insert. [Google Scholar]
- 20.GlaxoSmithKline. Brentford: GlaxoSmithKline; 2004. Fluticasone propionate package insert. [Google Scholar]
- 21.Rohatagi S, Bye A, Falcoz C, Mackie AE, Meibohm B, Mollmann H, Derendorf H. Dynamic modeling of cortisol reduction after inhaled administration of fluticasone propionate. J Clin Pharmacol. 1996;36:938–41. doi: 10.1002/j.1552-4604.1996.tb04761.x. [DOI] [PubMed] [Google Scholar]
- 22.Rohatagi S, Krishnaswami S, Pfister M, Sahasranaman S. Model-based covariate pharmacokinetic analysis and lack of cortisol suppression by the new inhaled corticosteroid ciclesonide using a novel cortisol release model. Am J Ther. 2005;12:385–97. doi: 10.1097/01.mjt.0000155110.69831.75. [DOI] [PubMed] [Google Scholar]
- 23.Sahasranaman S, Issar M, Hochhaus G. Metabolism of mometasone furoate and biological activity of the metabolites. Drug Metab Dispos. 2006;34:225–33. doi: 10.1124/dmd.105.005702. [DOI] [PubMed] [Google Scholar]
- 24.Beal SL, Boeckman A, Sheiner LB. NONMEM Users' Guides. San Francisco: NONMEM Project Group, University of California; 1992. version IV. [Google Scholar]
- 25.Issar M, Sahasranaman S, Buchwald P, Hochhaus G. Differences in the glucocorticoid to progesterone receptor selectivity of inhaled glucocorticoids. Eur Respir J. 2006;27:511–6. doi: 10.1183/09031936.06.00060005. [DOI] [PubMed] [Google Scholar]
- 26.Ryrfeldt A, Andersson P, Edsbaecker S, Tonnesson M, Davies D, Pauwels R. Pharmacokinetics and metabolism of budesonide, a selective glucocorticoid. Eur J Respir Dis. 1982;63(Suppl. 122):86–95. [PubMed] [Google Scholar]
- 27.Hochhaus G, Mollmann H, Derendorf H, Gonzalez-Rothi RJ. Pharmacokinetic/pharmacodynamic aspects of aerosol therapy using glucocorticoids as a model. J Clin Pharmacol. 1997;37:881–92. doi: 10.1002/j.1552-4604.1997.tb04262.x. [DOI] [PubMed] [Google Scholar]
- 28.Edsbacker S, Johansson CJ. Airway selectivity: an update of pharmacokinetic factors affecting local and systemic disposition of inhaled steroids. Basic Clin Pharmacol Toxicol. 2006;98:523–36. doi: 10.1111/j.1742-7843.2006.pto_355.x. [DOI] [PubMed] [Google Scholar]
- 29.Derendorf H, Daley-Yates PT, Pierre LN, Efthimiou J. Bioavailability and metabolism of mometasone furoate: pharmacology versus methodology. J Clin Pharmacol. 2002;42:383–7. doi: 10.1177/0091270002424003. [DOI] [PubMed] [Google Scholar]
- 30.Brutsche MH, Brutsche IC, Munawar M, Langley SJ, Masterson CM, Daley-Yates PT, Brown R, Custovic A, Woodcock A. Comparison of pharmacokinetics and systemic effects of inhaled fluticasone propionate in patients with asthma and healthy volunteers: a randomised crossover study. Lancet. 2000;356:556–61. doi: 10.1016/S0140-6736(00)02581-2. [DOI] [PubMed] [Google Scholar]
- 31.Mortimer KJ, Harrison TW, Tang Y, Wu K, Lewis S, Sahasranaman S, Hochhaus G, Tattersfield AE. Plasma concentrations of inhaled corticosteroids in relation to airflow obstruction in asthma. Br J Clin Pharmacol. 2006;62:412–9. doi: 10.1111/j.1365-2125.2006.02712.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Isogai M, Shimizu H, Esumi Y, Terasawa T, Okada T, Sugeno K. Binding affinities of mometasone furoate and related compounds including its metabolites for the glucocorticoid receptor of rat skin tissue. J Steroid Biochem Mol Biol. 1993;44:141–5. doi: 10.1016/0960-0760(93)90021-n. [DOI] [PubMed] [Google Scholar]
- 33.Derendorf H, Hochhaus G, Mollmann H, Barth J, Krieg M, Tunn S, Mollmann C. Receptor-based pharmacokinetic–pharmacodynamic analysis of corticosteroids. J Clin Pharmacol. 1993;33:115–23. doi: 10.1002/j.1552-4604.1993.tb03930.x. [DOI] [PubMed] [Google Scholar]
- 34.Rohatagi S, Luo Y, Shen L, Guo Z, Schemm C, Huang Y, Chen K, David M, Nave R, King SP. Protein binding and its potential for eliciting minimal systemic side effects with a novel inhaled corticosteroid, ciclesonide. Am J Ther. 2005;12:201–9. [PubMed] [Google Scholar]
- 35.Chen F, Kearney T, Robinson S, Daley-Yates PT, Waldron S, Churchill DR. Cushing's syndrome and severe adrenal suppression in patients treated with ritonavir and inhaled nasal fluticasone. Sex Transm Infect. 1999;75:274. [PubMed] [Google Scholar]
- 36.Taylor S, Harker A. Modification of the ultrafiltration technique to overcome solubility and non-specific binding challenges associated with the measurement of plasma protein binding of corticosteroids. J Pharm Biomed Anal. 2006;41:299–303. doi: 10.1016/j.jpba.2005.10.031. [DOI] [PubMed] [Google Scholar]