

Abstract
What is already known about this subject
There are few data about the safety of paclitaxel in patients with clinically significant liver impairment. A study by Venook and colleagues (J Clin Oncol 1998; 16: 1811–19) studied paclitaxel pharmacokinetics (PK) and pharmacodynamics (PD) in patients with liver impairment. The results were mainly descriptive, as detailed PK–PD data were available for only a subgroup of patients.
Another study by Wilson and colleagues found a correlation between tumour involvement of the liver, aspartate aminotransferase and total bilirubin concentrations and reduced paclitaxel clearance in 48 patients with advanced breast cancer in an early combined Phase I/II study (J Clin Oncol 1994; 12: 1621–9).
Finally, the study by Huizing and colleagues (Ann Oncol 1995; 6: 699–704) described two advanced breast cancer patients with liver impairment who experienced higher paclitaxel AUC concentrations and more severe neuropathywhen exposed to paclitaxel 250 mg m−2 as a 3-h infusion.
Liver impairment has been studied as a covariate within population models of paclitaxel in patients with normal or mildly impaired liver function (Henningsson et al. Eur JCancer 2003; 39: 1105–14; Joerger et al. Clin Cancer Res 2006; 12: 2150–7). Both studies found a negative correlation between total bilirubin concentrations and paclitaxel elimination.
What this study adds
A direct relationship between liver impairment, paclitaxel elimination and susceptibility to neutropenia/thrombopenia.
As a result of PK–PD simulations, suggestions could be made for (further) dose adaptations for patients with more severe liver impairment.
Aims
To assess quantitatively the safety and pharmacology of paclitaxel in patients with moderate to severe hepatic impairment.
Methods
Solid tumour patients were enrolled into five liver function cohorts as defined by liver transaminase and total bilirubin concentrations. Paclitaxel was administered as a 3-h intravenous infusion at doses ranging from 110 to 175 mg m−2, depending on liver impairment. Covariate and semimechanistic pharmacokinetic–pharmacodynamic (PK–PD) population modelling was used to describe the impact of liver impairment on the pharmacology and safety of paclitaxel.
Results
Thirty-five patients were included in the study, and PK data were assessed for 59 treatment courses. Most patients had advanced breast cancer (n = 22). Objective responses to paclitaxel were seen in four patients (11%). Patients in higher categories of liver impairment had a significantly lower paclitaxel elimination capacity (R2 = −0.38, P = 0.05), and total bilirubin was a significant covariate to predict decreased elimination capacity with population modelling (P = 0.002). Total bilirubin was also a significant predictor of increased haematological toxicity within the integrated population PK–PD model (P < 10−4). Data simulations were used to calculate safe initial paclitaxel doses, which were lower than the administered doses for liver impairment cohorts III–V.
Conclusions
Total bilirubin is a good predictor of paclitaxel elimination capacity and of individual susceptibility to paclitaxel-related myelosuppression in cancer patients with moderate to severe liver impairment. The proposed, adapted paclitaxel doses need validation in prospective trials.
Keywords: drug safety, liver impairment, paclitaxel, pharmacokinetics, population analysis
Introduction
Paclitaxel formulated in Cremophor EL (CrEL)/ethanol is regularly administered intravenously to patients with non-small cell lung cancer (NSCLC), ovarian and breast cancer. The pharmacokinetics (PK) of paclitaxel are best described by saturable elimination and saturable distribution to the tissues [1, 2]. The nonlinear PK are related to the formulation vehicle of paclitaxel, CrEL, probably by entrapment of paclitaxel into micelles [3–7]. Generally, substantial interpatient variability of paclitaxel PK parameters has been noted [8]. 6-α-hydroxypaclitaxel, 3′-p-hydroxypaclitaxel and 6-α,3′-p-dihydroxypaclitaxel are the major metabolites of paclitaxel found in humans [9]. Hepatic metabolism and biliary excretion play pivotal roles in the elimination and distribution of paclitaxel and its metabolites [10]. Accordingly, patients with biochemical evidence of (cholestatic) liver impairment [11, 12] or significant tumour involvement of the liver [12, 13] experience more haematological and nonhaematological toxicity from paclitaxel. However, patients with severe hepatic impairment have usually not been included in clinical studies with paclitaxel and, as a consequence, our knowledge of paclitaxel safety and pharmacology in patients with moderate to severe hepatic impairment is very limited at best.
A population approach has previously been used for the analysis of clinical and biochemical covariates in the pharmacology of paclitaxel in cancer patients [14, 15] and both studies found a decrease of paclitaxel elimination capacity with increasing total bilirubin concentration. Subsequently, with the development of threshold models [1, 8, 13, 16] to correlate paclitaxel PK with drug toxicity, steady-state concentrations or total drug exposure (AUC) were found to be less relevant for paclitaxel-related myelosuppression. Instead, these models used the time above specific paclitaxel plasma concentrations as predictors of haematological toxicity [1, 8, 13, 16] or clinical outcome [17]. In this regard, patients with advanced NSCLC receiving paclitaxel/carboplatin combination chemotherapy, who had a time above paclitaxel plasma concentration of 0.1 µmol l−1 for >15 h, were found to have a superior clinical outcome [17]. Besides these threshold models, more physiology-based modelling approaches have recently been adapted. Friberg and colleagues have described a semimechanistic [PK–pharmacodynamic (PD)] model of paclitaxel-induced myelosuppression by using nonlinear mixed-effects modelling [18]. The semimechanistic modelling approach typically uses drug-related and system (patient)-related parameters and is capable of explaining and predicting both the degree and duration of haematological toxicity after various drug administration schedules, even with sparse PD data. Such more physiology-based models are often preferred because they are more predictive, with parameters that may reflect actual processes and conditions [18].
The aim of this study was to define quantitatively the impact of moderate to severe liver impairment on paclitaxel pharmacology and safety in solid tumour patients. Population PK–PD modelling was chosen as a modern tool to analyse the relationship between liver impairment, paclitaxel pharmacology and drug toxicity.
Materials and methods
Patient selection and study plan
Patients with histologically confirmed solid tumours were prospectively enrolled into five liver function cohorts as defined by liver transaminase [aspartateaminotransferase (ASAT), alanine aminotransferase (ALAT)] and total bilirubin concentrations (Table 1). The study was approved by the Medical Ethics Committees of the participating centres (Free University Hospital in Amsterdam, Academic Hospital Groningen and Academic Hospital Utrecht). Eligibility criteria included the following: patients with histologically proven nonhaematopoietic malignancy for which no curative therapy exists and paclitaxel has been shown to be effective or malignancy for which no standard therapy exists, at least a 4-week interval between the last dose of chemotherapy and registration (6 weeks in case of treatment with mitomycin C, carboplatin or nitrosoureas), hepatic impairment as defined by transaminase and bilirubin values (outlined in Table 1), age between 18 and 75 years, an Eastern Cooperative Oncology Group performance status = 2, a life expectancy of >12 weeks, adequate bone marrow function [absolute neutrophil count (ANC) = 1.5 × 109 l−1 and platelets = 100 × 109 l−1], serum creatinine = 1.5 times the upper limit of normal (ULN), patient accessible for treatment and follow-up and written informed consent. Exclusion criteria included the following: prior treatment with paclitaxel or other taxane analogues, pre-existing motor or sensory neurotoxicity = grade 2 according to World Health Organization (WHO) criteria, active infection or other serious underlying medical condition (including prior allergic reactions to drugs containing Cremophor), dementia or significantly altered mental status, symptomatic brain metastases, history of atrial or ventricular arrhythmias or congestive heart failure even if medically controlled, history of clinical and electrocardiographically documented myocardial infarction, increase of >50% of bilirubin and/or transaminase values compared with baseline studies when repeated within the 14 days prior to paclitaxel dosing. Tumour response rate was assessed according to the WHO criteria for solid tumours. Clinical (patient age, gender) and biochemical (liver impairment category) parameters were prospectively assessed to test their influence on paclitaxel safety and pharmacology. The presence of ascites was not assessed. For statistical purposes, at least three patients per liver impairment cohort were defined as adequate for assessing paclitaxel safety for each cohort, according to a common Phase I study design. More patients within lower liver impairment cohorts were anticipated.
Table 1.
Patient cohorts according to liver function
| Initial paclitaxel dose (mg m−2) | |||||
|---|---|---|---|---|---|
| Cohort | ALAT/ASAT | Bilirubin | Dose level | Predefined | Model-derived recommendation |
| I | <2.6 × ULN | = 1.25 × ULN | 0 | 175* | |
| II | 2.6–10 × ULN | = 1.25 × ULN | 0 | 175 | 175† |
| III | <10 × ULN | 1.26–2.0 × ULN | 0 | 175 | 115 |
| IV | <10 × ULN | 2.1–3.5 × ULN | −1 | 135 | 100 |
| V | <10 × ULN | = 3.6–10 × ULN | −2 | 110 | 70 |
ULN, Upper limit of normal. ULN for aspartate aminotransferase (ASAT) = 25 U l−1. ULN for alanine aminotransferase (ALAT) = 35 U l−1. ULN for total bilirubin = 20 µmol l−1.
ANC nadir from cohort I used as a base for dose recommendations cohorts II–V.
As cohort II is identical to cohort I with respect to total bilirubin concentration, the presented model does not predict increased toxicity in cohort II when administering the same dose of paclitaxel (175 mg m−2).
Dose reduction and dose modification
Initial paclitaxel doses for the five liver impairment cohorts are shown in Table 1. No dose escalations were performed in cohorts I, II or III, but dose escalation was attempted in cohorts IV and V in case no dose-limiting toxicities (DLTs) were observed. Dose reduction by one dose level was performed in case any of the following DLTs were experienced: ANC < 0.5 × 109 l−1 for >7 days, ANC < 0.1 × 109 l−1 for >3 days, WHO grade 3 or 4 thrombocytopenia for >7 days, increase of total bilirubin by ≥25% or transaminase by ≥50%, severe (WHO grade 3 or 4) mucositis, neuropathy, myalgia or arthralgia not responding to symptomatic treatment. Dose reduction by two dose levels was performed in case of febrile neutropenia (ANC < 0.5 × 109 l−1 and fever <38.2°C requiring intravenous antibiotics and hospitalization) or severe bleeding requiring platelet transfusion. No dose reductions were performed below dose level −3 (90 mg m−2). Patients went off-study if the following conditions were met: full haematological recovery was not achieved by day 42, increases of total bilirubin levels by >50% or transaminase by >100% and significant hypersensitivity reactions occurred (e.g. hypotension requiring vasopressor treatment, angio-oedema, acute respiratory distress syndrome requiring bronchodilation treatment and generalized urticaria). Haematological and nonhaematological toxicities were assessed at weekly intervals.
Drug administration
Paclitaxel (TaxolR; Bristol Myers Squibb, Syracuse, NY, USA) was administered as a 3-h continuous intravenous infusion. Adapted polyvinyl chloride-free administration equipment was used. Standard premedication with dexamethasone (20 mg orally at 12 and 6 h prior to paclitaxel administration), clemastine (2 mg intravenously 30 min prior to paclitaxel administration) and a H2-receptor antagonist was administered to prevent hypersensitivity reactions. A 3-h infusion was chosen, as this schedule is less myelosuppressive and well tolerated in heavily pretreated cancer patients [19].
Pharmacokinetic sampling and bioanalysis
Complete plasma concentration–time curves for paclitaxel were obtained by PK sampling during the first and second treatment cycles. The samples for paclitaxel analysis were collected in ethylenediamine tetraacetic acid tubes at 15 time points: prior to start, 10, 90 min after the start, at the end of infusion, and at 6, 15 min and 1, 2, 3, 4, 8, 12, 21, 30 and 48 h postinfusion. Whole blood was centrifuged immediately after venepuncture and the plasma fraction was stored at −20°C until analysis. The plasma concentrations of paclitaxel, 6-α-hydroxypaclitaxel and 3′-p-hydroxypaclitaxel were determined by a validated isocratic high-performance liquid chromatographic (HPLC) method with solid-phase extraction as the sample pretreatment procedure, as previously described [9, 20]. The quantification range of the HPLC method was 10–10 000 ng ml−1.
Pharmacokinetic modelling
Population PK analysis of the concentration–time data of paclitaxel was performed using the nonlinear mixed-effect modelling program (NONMEM) version V (double precision, level 1.1) [21]. NONMEM uses a maximum likelihood criterion to estimate simultaneously population values of fixed-effects parameters (e.g. drug clearance) and values of the random effects parameters (e.g. interindividual and residual variability). Log-transformed plasma drug concentrations were used together with the first-order (FO) method throughout data analysis. Standard errors for all parameters were calculated using the COVARIANCE option of NONMEM and individual Bayesian PK parameters were obtained with the POSTHOC option [21]. The S-plus (MathSoft Inc., Seattle, WA, USA)-based model building aid Xpose 3.0 was used for graphical processing [22]. First, the three-compartment population PK and covariate model with Michaelis–Menten elimination and saturable distribution to one peripheral compartment, as described previously [15], was applied to the concentration–time data. The model used body surface area (BSA) as a covariate on the paclitaxel elimination capacity (VMEL), peripheral distribution volume (V3 in l) and intercompartmental clearance (Q in l min−1), patient gender as a covariate on VMEL, paclitaxel intercompartmental transport capacity (VMTR), total plasma concentration of paclitaxel at half VMTR (KMTR in µmol l−1) and K21, and total bilirubin and patient age as a covariate on VMEL. No additional covariate model building was performed except for the following parameters of liver impairment: total bilirubin, ASAT, ALAT. Except for the liver function parameters, the covariate to PK–parameter relationship was adapted from our previous paclitaxel population model [15]; however, the quantitative impact of the covariates on the respective PK–parameter was estimated for the current study population. To reduce potential bias from variability of variance of the residual error between individuals (σ2), two subgroups with different values for σ2 were assumed, with three estimated variables for the residual error model: the fractions of subpopulations, σ2 for subgroup 1 and σ2 for subgroup 2, according to our previous paclitaxel PK model [15]. The MIXTURE function of NONMEM was used for estimating the two subpopulations and their respective residual errors. Throughout data analysis, model selection was based on the minimum value of objective function (OFV), as calculated by NONMEM, the reliability of parameter estimates (according to the standard error values of the parameter estimates obtained by the COVARIANCE option of NONMEM) and the fit of the model to the data as approached by various graphical plots.
PK–PD modelling
For linking PK and PD data, sequential modelling was used, where individual Bayesian estimates of PK parameters were fixed from the prior PK analysis for subsequent PD modelling. This was separately done for neutrophils and thrombocytes. A semiphysiological model as introduced by Friberg et al. [18] was used in this study to describe paclitaxel-related neutropenia and thrombocytopenia. The model comprised a compartment representing the proliferating cells linked to a compartment representing the systemic circulation through three transit compartments, mimicking precursor cell maturation within the bone marrow. For graphical illustration of the PD model, see also the article of Friberg and colleagues [18]. A feedback mechanism (FB) imitated the effect of the release of endogenous growth factors as a response to the decrease of cells in the circulation pool. A linear slope function was used according to Equation 1, by which drug concentrations affect the proliferation rate of circulating blood cells:
| (1) |
where FB represents the feedback parameter (ANCbase/ANCt)γ, ktr the transition rate constant and kprol the cell proliferation constant. The following covariates were modelled on the slope values for neutrophils and thrombocytes: BSA, patient age, total bilirubin concentration, ASAT and performance status. Logarithmic transformation of both neutrophils and thrombocytes was used throughout model building together with the FO method. Neutrophil and thrombocyte counts, including nadir values, were assumed to be normally distributed.
Data simulation using the population PK–PD estimates was adopted to construct median values and distribution of absolute neutrophil and thrombocyte counts for patients within the various liver impairment cohorts. Simulation runs used both the final PK and PD models and a model for residual error that included two subpopulations of residual PK error to estimate neutrophil and thrombocyte toxicity. A uniform distribution of total bilirubin was used to simulate cohort III (bilirubin 20–32 µmol l−1), cohort IV (bilirubin 33–56 µmol l−1) and cohort V (bilirubin 57–160 µmol l−1). A normal distribution was used to simulate BSA (population mean 1.8 m2, SD 0.1 m2), baseline neutrophil (mean 5.6 × 109 l−1, SD 0.5 × 109 l−1) and thrombocyte count (mean 276 × 109 l−1, SD 70 × 109 l−1) and patient age (mean 50 years, SD 8 years). The distribution of gender was set equal to the original study. This procedure ensured that parameter distribution for the various simulation runs corresponded to the parameter distribution of the original study group. Ten thousand patients each were simulated for cohorts III–V to enable neutrophil and thrombocyte nadir estimates with reasonable confidence intervals. Median, 25% and 75% percentiles were calculated for absolute neutrophil and thrombocyte nadir, time to nadir, time <0.5 × 109 l−1 neutrophils and <50 × 109 l−1 thrombocytes and the frequency of dose-limiting neutrophil (<0.5 × 109 l−1 neutrophils for >7 days) and platelet toxicity (<50 × 109 l−1 thrombocytes for >7 days).
Results
Patient characteristics are shown in Table 2. Thirty-five patients were included into the study. Liver impairment cohort III accrued only two patients, whereas all other cohorts accrued at least three patients. PK data were assessed for 59 treatment courses. Twelve patients had PK data available from one treatment course, one patient had PK data available from three treatment courses and the remaining patients had PK data available from two treatment courses. In eight patients, treatment was stopped after a single treatment course, due to rapid disease progression (n = 4), severe hypersensitivity reaction (n = 1), prolonged thrombocytopenia requiring hospitalization and platelet transfusion (n = 1), early death due to pulmonary embolism (n = 1) and neutropenic infection (n = 1). There was no statistically significant trend across the study cohorts for performance status (P = 0.08), age (P = 0.46), gender (P = 0.10) and hepatic tumour involvement (P = 0.17) (all comparisons by two-sided Kruskal–Wallis test). Twenty-eight patients (80%) had hepatic tumour involvement as the most plausible cause of liver function impairment. Within cohorts III–V, only one patient in cohort IV had no hepatic tumour involvement. Additional hepatic morbidity as a potential cause of liver impairment were hepatitis C in one patient of cohort II and alcoholic liver cirrhosis in one patient each in cohorts IV and V. Most patients were diagnosed with advanced breast cancer (n = 22), of whom 17 had received previous hormonal treatment, nine previous adjuvant chemotherapy and 16 previous palliative chemotherapy. Objective responses to paclitaxel were partial remission in four patients (11%), stable disease in 12 (34%), progressive disease in 11 (31%) and not available in eight patients (23%). A partial response was found in one patient each in cohorts I, II, III and IV. Patient age, gender and liver impairment cohort were not correlated with treatment response (R2−0.02, −0.08, −0.01, respectively).
Table 2.
Patient characteristics (total patient population n = 35)
| Cohort I | Cohort II | Cohort III | Cohort IV | Cohort V | |
|---|---|---|---|---|---|
| No. of patients | 15 | 12 | 2 | 3 | 3 |
| Assessed treatment courses | 24 | 21 | 4 | 5 | 5 |
| Age (years) | |||||
| Median | 51.2 | 47.9 | 52.6 | 49.8 | 55.6 |
| Range | 25.2–69.7 | 38.3–72.2 | 47.2–57.9 | 41.9–68.3 | 49.0–64.6 |
| Sex | |||||
| Male | 4 | 0 | 0 | 2 | 2 |
| Female | 11 | 12 | 2 | 1 | 1 |
| BSA (m−2) | 1.81 | 1.82 | 1.81 | 1.82 | 1.83 |
| ECOG status | |||||
| 0 | 2 | 5 | 0 | 0 | 0 |
| 1 | 7 | 4 | 2 | 1 | 0 |
| 2 | 6 | 3 | 0 | 2 | 3 |
| Liver tumour involvement | 10 | 11 | 2 | 2 | 3 |
| Diagnosis | |||||
| Breast cancer | 7 | 9 | 2 | 2 | 2 |
| Ovarian cancer | 5 | 1 | 0 | 0 | 0 |
| Gastrointestinal cancer | 2 | 0 | 0 | 1 | 0 |
| Sarcoma | 1 | 1 | 0 | 0 | 0 |
| Gallbladder cancer | 0 | 0 | 0 | 0 | 1 |
| Endometrial cancer | 0 | 1 | 0 | 0 | 0 |
BSA, Body surface area; ECOG, Eastern Cooperative Oncology Group.
Toxicity
The following DLTs were seen: diarrhoea grade IV in one patient in cohort I, necessitating dose reduction to 135 mg m−2 paclitaxel in cycle 2, again followed by diarrhoea grade I. Another patient in cohort I experienced a >25% increase in total bilirubin requiring hospitalization. This DLT, however, was related to rapidly progressing pancreatic cancer and subsequent cholestatic icterus. One patient in cohort IV experienced febrile neutropenia requiring hospitalization and intravenous antibiotics, and the paclitaxel dose was accordingly reduced by two dose levels to 90 mg m−2. However, the patient experienced reversible hepatic coma shortly after the second treatment cycle. One patient from cohort V died 9 days after the first treatment course from complicated neutropenic infection.
Haematological toxicity is shown in Table 3. Of 35 patients, 32 had weekly haematological assessments available. Patients in higher categories of liver impairment experienced more severe anaemia (P = 0.02), leucocytopenia (P < 0.01), thrombocytopenia (P = 0.02) and neutropenia (P = 0.05) as measured by categorical (WHO) classification. Nonhaematological toxicity is detailed in Table 4. Nonhaematological toxicity was not significantly different across study groups with the exception of mucositis (P = 0.04) and neutropenic fever (P < 0.01). This is mainly due to two patients in cohorts IV and V experiencing both severe mucositis and neutropenic fever.
Table 3.
Haematological toxicity according to World Health Organization toxicity criteria
| Toxicity | Cohort I | Cohort II | Cohort III | Cohort IV | Cohort V | P-value for trend* |
|---|---|---|---|---|---|---|
| Anemia | 0.02 | |||||
| Grade I/II | 13 | 8 | 2 | 1 | ||
| Grade III/IV | 10 | 12 | 2 | 4 | 3 | |
| Leucocytopenia | <0.01 | |||||
| Grade I/II | 7 | 12 | 3 | 2 | 1 | |
| Grade III/IV | 4 | 8 | 1 | 2 | 3 | |
| Neutropenia | 0.06 | |||||
| Grade I/II | 4 | 9 | 2 | 1 | ||
| Grade III/IV | 9 | 11 | 2 | 4 | 3 | |
| Thrombocytopenia | 0.01 | |||||
| Grade I/II | 7 | 11 | 1 | 3 | ||
| Grade III/IV | 1 | 3 | 1 | 4 | ||
| Mean HB nadir (SE) | 8.5 (0.13) | 8.3 (0.46) | 8.4 (1.54) | 6.0 (0.31) | 7.0 (0.38) | 0.02 |
| Mean WBC nadir (SE) | 4.2 (0.45) | 2.2 (0.20) | 2.4 (0.37) | 1.5 (0.47) | 1.6 (0.65) | <0.01 |
| Mean ANC nadir (SE) | 1.9 (0.31) | 0.9 (0.15) | 0.9 (0.36) | 0.3 (0.19) | 0.5 (0.25) | 0.05 |
| Mean PLT nadir (SE) | 233 (29) | 145 (14) | 222 (38) | 85 (30) | 111 (17) | 0.02 |
HB, Haemoglobin (g dl−1); WBC, white blood cell count (103 µl−1); ANC, absolute neurophil count (103 µl−1); PLT, platelets (106 µl−1); SE, standard error.
Using the Kruskal–Wallis test.
Table 4.
Nonhaematological toxicity according to World Health Organization toxicity criteria (excluding fatigue, alopecia, nausea/vomiting)
| Toxicity | Cohort I | Cohort II | Cohort III | Cohort IV | Cohort V | P-value for trend* |
|---|---|---|---|---|---|---|
| Diarrhoea | 0.34 | |||||
| Grade I | 5 | 2 | ||||
| Grade III | 1 | 1 | ||||
| Mucositis | 0.04 | |||||
| Grade I/II | 2 | |||||
| Grade III | 1 | 1 | ||||
| Myalgia/arthralgia | 0.71 | |||||
| Grade I/II | 11 | 8 | 1 | |||
| Grade III | 1 | |||||
| Allergic reactions | 0.27 | |||||
| Grade I/II | 2 | 2 | 1 | |||
| Grade III | 1 | |||||
| Sensory polyneuropathy | 0.70 | |||||
| Grade I | 7 | 7 | 2 | |||
| Grade II | 3 | 2 | 1 | |||
| Neutropenic fever | <0.01 | |||||
| Grade III | 4 | 2 | 3 | |||
| Grade IV | 1 | 1 |
Using the Kruskal–Wallis test.
Pharmacokinetic analysis
Population PK parameters are shown in Table 5. Dose adjustments according to liver impairment cohorts resulted in a lower mean paclitaxel AUC in study cohort IV (9.1 µmol h−1 l−1) and V (8.9 µmol h−1 l−1) compared with cohorts I–III (19 µmol h−1 l−1) (P < 0.01 for trend). Bayesian estimates for VMEL were lower in higher liver impairment cohorts (P = 0.05 for trend). In the final population PK model, as adopted from our previous paclitaxel population model [15], male gender and BSA were positively correlated with VMEL; patient age and total bilirubin were negatively correlated with VMEL (P < 0.01 for all correlations). With conventional correlation analysis, patients in higher liver impairment categories had a significantly lower paclitaxel elimination capacity (R2 = −0.38, P = 0.05). Typically, a 10-µmol increase of total bilirubin led to a 19% decrease in VMEL, translating into a higher paclitaxel AUC for patients with higher bilirubin concentrations. However, this higher AUC was offset by a lower paclitaxel dose administered in liver impairment cohorts IV and V. ASAT was not significantly correlated with paclitaxel VMEL (P > 0.1). The application of the previously reported covariate model [15] led to a significant improvement of the PK model (P < 10−5).
Table 5.
Population parameters of the final population pharmacokinetic model
| Pharmacokinetic parameters | Units | Estimate | RSE (%) |
|---|---|---|---|
| V1 | l | 10.2 | 15.3 |
| V3 | l | 642 | 19.7 |
| VMEL | µmol h−1 | 6.4 | 17.3 |
| KMEL | µmol l−1 | 0.06 | 35.0 |
| VMTR | µmol h−1 | 161 | 13.2 |
| KMTR | µmol l−1 | 0.55 | 13.4 |
| K21 | h−1 | 1.20 | 12.5 |
| Q | l h−1 | 16.1 | 8.82 |
| Interindividual variability | |||
| VMEL | % | 33.4 | 17.3 |
| VMTR | % | 22.2 | 17.9 |
| Residual variability* | |||
| Subpopulation 1 | % | 52.2 | 19.7 |
| Subpopulation 2 | % | 25.0 | 13.2 |
| Pharmacodynamic parameters | |||
| MTT (ANC) | h | 62.8 | NA |
| MTT (PLT) | h | 50.8 | 10.0 |
| γ (feedback ANC) | 0.11 | NA | |
| γ (feedback PLT) | 0.07 | 18.5 | |
| Slope (ANC) | l µmol−1 | 1.85 | NA |
| Slope (PLT) | l µmol−1 | 0.25 | 22.3 |
| Total bilirubin on slope (ANC) | 0.022 | NA | |
| Total bilirubin on slope (PLT) | 0.017 | 34.3 | |
| Interindividual variability | |||
| MTT (ANC) | % | 45.5 | NA |
| MTT (PLT) | % | 13.7 | 10.5 |
| Slope (ANC) | % | 34.7 | NA |
| Slope (PLT) | % | 57.0 | 37.9 |
| CV on baseline ANC | % | 67.0 | NA |
| CV on baseline PLT | % | 25.8 | 9.0 |
V1, Volume of the central compartment; V3, volume of the second peripheral compartment; VMEL, maximal elimination rate; KMEL, plasma concentration at half VMEL; VMTR, maximal transport rate from the central to the first peripheral compartment; KMTR, plasma concentration at half VMTR; K21, rate constant from the first peripheral compartment to the central compartment; Q, intercompartmental clearance between the central and second peripheral compartment; RSE, relative standard error (as obtained from the covariance step).
Fraction subgroup one 0.32 (RSE = 28.1%). MTT, Median transition time (h); γ, feedback, implemented in (ANCbase/ANCt)γ; slope, linear drug effect parameter; CV, coefficient of variation; NA, not available (resulting from failed variance–covariance step).
The single patient who died of complicated neutropenic infection had an estimated paclitaxel elimination capacity in the lower population range (4.5 µmol h−1, population range 2.5–15.2 µmol h−1), a paclitaxel AUC of 13 µmol h−1 l−1 (population range 5.9–29.9) and a Bayesian estimate for neutrophil nadir of 0.01 × 109 l−1.
PK–PD modelling
Population PD parameters are shown in Table 5. Total bilirubin was positively correlated with neutrophil and thrombocyte slopes (Figure 1) and the inclusion of total bilirubin significantly improved the PD model (P < 10−4). Patients with higher total bilirubin concentrations had higher slope values and were more susceptible to neutropenia and thrombocytopenia, independent of paclitaxel exposure. Typically, a 10-µmol l−1 increase of total bilirubin led to a 11% increase in the slope value for neutrophils and a 6.5% increase in the slope value for thrombocytes. The equations as outlined in Figure 1 are the mathematical (regression) functions explaining the higher susceptibility of patients with more advanced liver impairment to paclitaxel-related haematological toxicity. BSA, patient age, ASAT and performance status were not correlated with the slope values for neutrophils or thrombocytes, and did not significantly improve the PD model. As the variance–covariance step for the neutrophil PK–PD run failed in NONMEM, no estimates for relative standard errors are given in Table 5.
Figure 1.
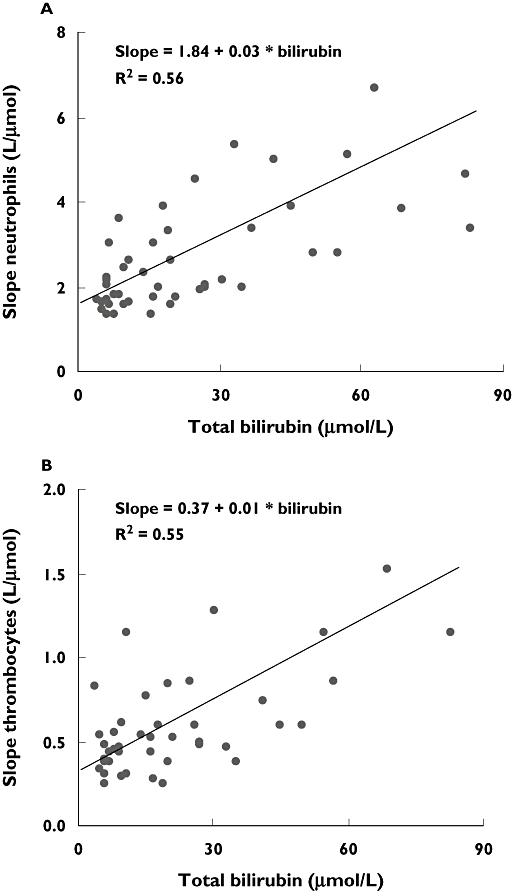
Linear regression of neutrophil (A), thrombocyte (B) slopes and bilirubin,with higher slopes indicating higher susceptibility to haematological toxicity
Population estimates for neutrophil and thrombocyte nadir are shown in Table 3. Patients in higher liver impairment cohorts had increased haematological toxicity, and this was supported by the Bayesian estimates for neutrophil and thrombocyte nadir. Results from data simulations showed a marked increase in haematological toxicity with liver impairment cohorts, and also a slight increase in time to nadir for higher paclitaxel doses compared with lower doses in every cohort (Table 6). This resulted in an increase of DLT for neutrophils from 14.7% in cohort I/II to 27.5% in cohort III, 18% in cohort IV and 30.8% in cohort V with the administered paclitaxel doses. Data simulations suggest a paclitaxel dose of 115 mg m−2, 100 mg m−2 and 70 mg m−2 for patients in cohorts III–V results in a comparable neutrophil nadir of a patient with normal liver function (total bilirubin 5 µmol l−1, typical neutrophil nadir 0.9 × 109 l−1). As liver impairment cohorts I and II were identical with respect to total bilirubin concentration, the PK–PD model does not predict increased haematological toxicity in a typical cohort II compared with a cohort I patient (Table 6).
Table 6.
Simulation analysis for haematological toxicity
| Simulated cohort | Paclitaxel dose, mg m−2 | Nadir, 103 cells µl−1 | Time to nadir, h | Time below threshold*, h | DLT†, % |
|---|---|---|---|---|---|
| III | |||||
| 175 | |||||
| ANC | 0.2 (0.04–0.5) | 170 (135–215) | 119 (0–173) | 27.5 | |
| Thrombocytes | 79 (41–123) | 124 (113–137) | 39 (0–65) | 6.5 | |
| 115 | |||||
| ANC | 0.92 (0.39–1.69) | 147 (113–193) | 35 (0–51) | 4.2 | |
| Thrombocytes | 139 (95–185) | 117 (105–132) | 0 | 0 | |
| IV | |||||
| 135 | |||||
| ANC | 0.32 (0.08–0.82) | 163 (128–208) | 91 (0–145) | 18.0 | |
| Thrombocytes | 75 (38–120) | 125 (113–138) | 42 (0–76) | 7.5 | |
| 100 | |||||
| ANC | 0.91 (0.38–1.68) | 148 (114–194) | 36 (0–55) | 4.9 | |
| Thrombocytes | 119 (76–166) | 120 (108–134) | 0 | 0 | |
| V | |||||
| 110 | |||||
| ANC | 0.18 (0.03–0.59) | 173 (136–220) | 125 (0–185) | 30.8 | |
| Thrombocytes | 33 (10–73) | 134 (121–150) | 102 (0–173) | 26.8 | |
| 70 | |||||
| ANC | 0.96 (0.39–1.81) | 149 (114–196) | 37 (0–52) | 4.4 | |
| Thrombocytes | 93 (50–142) | 124 (112–138) | 0 | 0 | |
Results given as median with 25% and 75% percentiles.
500 µl−1absolute neutrophil count (ANC) and 50 ×103 µl−1 thrombocytes.
Frequency of dose-limiting toxicity, defined as ANC <500 µl−1ANC or 50 × 103 µl−1thrombocytes for >7 days.
Discussion
This study was performed to assess quantitatively the safety and pharmacology of paclitaxel in patients with moderate to severe hepatic impairment. A previously published population model for paclitaxel in patients with solid tumours was applied to the concentration–time data [15]. The integrated PK–PD model was successfully applied to this group of 35 patients with increasing liver impairment. This approach allowed a more complete assessment of paclitaxel PK–PD in patients with liver impairment, as it was possible to include anthropometric and biochemical (liver function) covariates as well as haematological toxicity data into one population model. A population approach has previously been used for covariate testing in patients receiving intravenous paclitaxel [14, 15]. Both studies found a negative correlation between total bilirubin and paclitaxel elimination, which is confirmed by this study, in which a 10-µmol increase of total bilirubin led to a 19% decrease of paclitaxel elimination capacity. In the study by Henningsson and colleagues, an increase in total bilirubin of 10 µmol l−1 typically caused a decrease in paclitaxel clearance of 41 l h−1, which corresponds to roughly 12% of total drug clearance [14]. In our own previous study, a 10-µmol l−1 increase of total bilirubin typically led to a 14% decrease of paclitaxel elimination capacity [15]. While the former analysis included concentration–time data of total and unbound paclitaxel as well as CrEL, the latter included concentration–time data of total paclitaxel only. Despite these differences, the quantitative impact of total bilirubin as a surrogate for liver impairment on paclitaxel elimination capacity was similar. Most importantly, patients included in both studies had no severe liver impairment (total bilirubin range 3–41 µmol l−1 [14] and 2–24 µmol l−1 [15]), and extrapolation of the findings in patients with normal liver function or light liver impairment to patients with moderate to severe impairment is a concern.
The high incidence of hepatic tumour involvement, especially in cohorts III–V, suggests that liver impairment in these patients is probably due to hepatic tumour involvement. However, other causes of liver impairment, especially liver cirrhosis, might have contributed. The low incidence of known hepatic cirrhosis in this study (one patient each in cohorts IV and V) makes a separate analysis of paclitaxel PK–PD in patients with hepatic tumour involvement and liver cirrhosis impossible.
Although liver impairment was significantly correlated with lower drug elimination capacity in the present study, paclitaxel dose reductions in cohort IV (paclitaxel 135 mg m−2) and cohort V (paclitaxel 110 mg m−2) resulted in significantly lower drug exposure in these two cohorts. If one is looking exclusively to paclitaxel PK, one might expect patients in study cohorts IV and V to tolerate reduced paclitaxel doses without increased toxicity. However, toxicity assessment showed significantly increased haematological toxicity for patients with increasing liver impairment even with lower paclitaxel AUCs (Table 3), and PK–PD modelling suggested total bilirubin concentration to be a predictor for the susceptibility of individual patients to paclitaxel-related haematological toxicity. As anthropometric covariates such as patient age and performance status were not correlated with the susceptibility to haematological toxicity, total bilirubin as predictor of haematological toxicity seems not simply a surrogate for a poor health status. The pathophysiological mechanism of the correlation between total bilirubin concentration and increased susceptibility to haematological toxicity remains unclear, but the presented integrated PK–PD model suggests effects additional to a decrease in paclitaxel elimination in patients with increasing liver impairment. Although patients in higher liver impairment cohorts experienced more haematological toxicity, they usually did not experience more nonhaematological toxicity (Table 4), with the exception of mucositis and neutropenic fever. As a note of caution, the few events within these categories were highly influenced by two individuals in cohorts IV and V. The increase in time to nadir with increasing paclitaxel dose is suggested to be inherent to the model, as the feedback parameter was defined as a system parameter and not dependent on paclitaxel dose. Therefore, with increasing paclitaxel dose, a comparable feedback parameter leads to deeper nadir values and an increased time to nadir. As a general note, liver function parameters (total bilirubin, ASAT, ALAT) were modelled as quantitative covariates, whereas categorical liver impairment classification (e.g. Child–Pugh) has been proposed by the Food and Drug Administration for early pharmacological studies. On the one hand, quantitative covariates are known to be more informative for modelling purposes, on the other hand, ascites has not been assessed prospectively and had to be dropped as a potential covariate.
Although dose adaptations for patients in higher liver impairment cohorts, as suggested by this PK–PD model, may be efficient for equalizing haematological toxicity, our study does not clarify whether the proposed lower doses will not result in a loss of antitumour efficacy. This is of special interest, as the study by Huizing and colleagues has shown that exposure to paclitaxel as assessed by the time above threshold plasma concentration of 0.1 µmol l−1 for >15 h was correlated with superior clinical outcome in patients with NSCLC [17]. It may be argued that treating liver-impaired patients with effective paclitaxel doses is difficult without hitting a toxic ceiling, because their comorbidity hypersensitizes them to the DLT. Future studies should incorporate drug response into pharmacological models to address this question.
As patients with severe liver impairment were usually not eligible for clinical studies with paclitaxel, only limited information is available for paclitaxel PK–PD in patients with moderate to severe liver impairment. Wilson and colleagues have shown a significant correlation between tumour involvement of the liver, ASAT and total bilirubin levels and reduced paclitaxel clearance (P = 0.002, 0.009 and 0.035, respectively) in 48 patients with advanced breast cancer in an early combined Phase I/II study [12]. Our results revealed that ASAT concentrations were not a significant predictor of paclitaxel elimination capacity (P = 0.15), in contrast to total bilirubin (P = 0.002). We did not perform a formal analysis to correlate tumour involvement of the liver with paclitaxel elimination capacity, as only very few patients lacked hepatic tumour involvement. Venook and colleagues assessed the PK of paclitaxel in 81 patients with various solid tumours and liver impairment [11]. Their patients received paclitaxel at doses ranging between 50 and 125 mg m−2 as a 3-h infusion (n = 56), or at doses ranging between 50 and 200 mg m−2 as a 24-h infusion (n = 25). Patients were assigned to one of three cohorts: cohort I with bilirubin levels <1.5 mg dl−1 (<25.7 µmol l−1) and ASAT levels two times the ULN, cohort II with bilirubin levels 1.5–3 mg dl−1 (25.7–51.3 µmol l−1) with any ASAT level and cohort III with bilirubin levels >3 mg dl−1 (>51.3 µmol l−1). The analysis of potential correlations between liver impairment and paclitaxel pharmacology was mainly descriptive, as detailed PK–PD data were available for a subgroup of patients only. In a further study, Huizing and colleagues studied nine patients with advanced breast cancer receiving paclitaxel 250 mg m−2 as a 3-h infusion with subsequent granulocyte-colony stimulating factor support [13]. Two patients with liver impairment experienced higher paclitaxel and 6-OH AUC levels, as well as more severe neuropathy. No correlation between paclitaxel PK and sensory polyneuropathy was seen in the presented study (Table 5).
In conclusion, total bilirubin was shown to be a good predictor of paclitaxel elimination capacity and of individual susceptibility to paclitaxel-related myelosuppression in cancer patients with moderate to severe liver impairment. Dose adaptations were proposed based on data simulations, and these new adaptations need validation in prospective clinical trials.
Acknowledgments
M.J. is supported by a fellowship grant funded by the European Society of Medical Oncology and by a research grant from the Swiss National Science Foundation (PBBSB-102331). This study was sponsored by Bristol-Myers Squibb (Syracuse, NY, USA).
References
- 1.Gianni L, Kearns CM, Giani A, Capri G, Vigano L, Lacatelli A, Bonadonna G, Egorin MJ. Nonlinear pharmacokinetics and metabolism of paclitaxel and its pharmacokinetic/pharmacodynamic relationships in humans. J Clin Oncol. 1995;13:180–90. doi: 10.1200/JCO.1995.13.1.180. [DOI] [PubMed] [Google Scholar]
- 2.Sonnichsen DS, Hurwitz CA, Pratt CB, Shuster JJ, Relling MV. Saturable pharmacokinetics and paclitaxel pharmacodynamics in children with solid tumors. J Clin Oncol. 1994;12:532–8. doi: 10.1200/JCO.1994.12.3.532. [DOI] [PubMed] [Google Scholar]
- 3.Sparreboom A, van Tellingen O, Nooijen WJ, Beijnen JH. Nonlinear pharmacokinetics of paclitaxel in mice results from the pharmaceutical vehicle Cremophor EL. Cancer Res. 1996;56:2112–5. [PubMed] [Google Scholar]
- 4.Sparreboom A, van Zuylen L, Brouwer E, Loos WJ, de Bruijn P, Gelderblom H, Pillay M, Nooter K, Stoter G, Verweij J. Cremophor EL-mediated alteration of paclitaxel distribution in human blood: clinical pharmacokinetic implications. Cancer Res. 1999;59:1454–7. [PubMed] [Google Scholar]
- 5.van Tellingen O, Huizing MT, Panday VR, Schellens JH, Nooijen WJ, Beijnen JH. Cremophor EL causes (pseudo-) non-linear pharmacokinetics of paclitaxel in patients. Br J Cancer. 1999;81:330–5. doi: 10.1038/sj.bjc.6690696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Zuylen L, Gianni L, Verweij J, Mross K, Brouwer E, Loos WJ, Sparreboom A. Inter-relationships of paclitaxel disposition, infusion duration and cremophor EL kinetics in cancer patients. Anticancer Drugs. 2000;11:331–7. doi: 10.1097/00001813-200006000-00003. [DOI] [PubMed] [Google Scholar]
- 7.van Zuylen L, Karlsson MO, Verweij J, Brouwer E, de Bruijn P, Nooter K, Stoter G, Sparreboom A. Pharmacokinetic modeling of paclitaxel encapsulation in Cremophor EL micelles. Cancer Chemother Pharmacol. 2001;47:309–18. doi: 10.1007/s002800000215. [DOI] [PubMed] [Google Scholar]
- 8.Henningsson A, Karlsson MO, Vigano L, Gianni L, Verweij J, Sparreboom A. Mechanism-based pharmacokinetic model for paclitaxel. J Clin Oncol. 2001;19:4065–73. doi: 10.1200/JCO.2001.19.20.4065. [DOI] [PubMed] [Google Scholar]
- 9.Huizing MT, Sparreboom A, Rosing H, van Tellingen O, Pinedo HM, Beijnen JH. Quantification of paclitaxel metabolites in human plasma by high-performance liquid chromatography. J Chromatogr B Biomed Appl. 1995;674:261–8. doi: 10.1016/0378-4347(95)00308-8. [DOI] [PubMed] [Google Scholar]
- 10.Panday VR, Huizing MT, Willemse PH, De Graeff A, Bokkel Huinink WW, Vermorken JB, Beijnen JH. Hepatic metabolism of paclitaxel and its impact in patients with altered hepatic function. Semin Oncol. 1997;24:S11. [PubMed] [Google Scholar]
- 11.Venook AP, Egorin MJ, Rosner GL, Brown TD, Jahan TM, Batist G, Hohl R, Budman D, Ratain MJ, Kearns CM, Schilsky RL. Phase I and pharmacokinetic trial of paclitaxel in patients with hepatic dysfunction: Cancer and Leukemia Group B 9264. J Clin Oncol. 1998;16:1811–9. doi: 10.1200/JCO.1998.16.5.1811. [DOI] [PubMed] [Google Scholar]
- 12.Wilson WH, Berg SL, Bryant G, Wittes RE, Bates S, Fojo A, Steinberg SM, Goldspiel BR, Herdt J, O'Shaughnessy J. Paclitaxel in doxorubicin-refractory or mitoxantrone-refractory breast cancer: a phase I/II trial of 96-hour infusion. J Clin Oncol. 1994;12:1621–9. doi: 10.1200/JCO.1994.12.8.1621. [DOI] [PubMed] [Google Scholar]
- 13.Huizing MT, Vermorken JB, Rosing H, Bokkel Huinink WW, Mandjes I, Pinedo HM, Beijnen JH. Pharmacokinetics of paclitaxel and three major metabolites in patients with advanced breast carcinoma refractory to anthracycline therapy treated with a 3-hour paclitaxel infusion: a European Cancer Centre (ECC) trial. Ann Oncol. 1995;6:699–704. doi: 10.1093/oxfordjournals.annonc.a059287. [DOI] [PubMed] [Google Scholar]
- 14.Henningsson A, Sparreboom A, Sandstrom M, Freijs A, Larsson R, Bergh J, Nygren P, Karlsson MO. Population pharmacokinetic modelling of unbound and total plasma concentrations of paclitaxel in cancer patients. Eur J Cancer. 2003;39:1105–14. doi: 10.1016/s0959-8049(03)00126-6. [DOI] [PubMed] [Google Scholar]
- 15.Joerger M, Huitema AD, van den Bongard DH, Schellens JH, Beijnen JH. Quantitative effect of gender, age, liver function, and body size on the population pharmacokinetics of paclitaxel in patients with solid tumors. Clin Cancer Res. 2006;12:2150–7. doi: 10.1158/1078-0432.CCR-05-2069. [DOI] [PubMed] [Google Scholar]
- 16.Huizing MT, Keung AC, van Rosing H, van der Kuij V, Bokkel Huinink WW, Mandjes IM, Dubbelman AC, Pinedo HM, Beijnen JH. Pharmacokinetics of paclitaxel and metabolites in a randomized comparative study in platinum-pretreated ovarian cancer patients. J Clin Oncol. 1993;11:2127–35. doi: 10.1200/JCO.1993.11.11.2127. [DOI] [PubMed] [Google Scholar]
- 17.Huizing MT, Giaccone G, van Warmerdam LJ, Rosing H, Bakker PJ, Vermorken JB, Postmus PE, van Zandwijk N, Koolen MG, Bokkel Huinink WW, van der Vijgh WJ, Bierhorst FJ, Lai A, Dalesio O, Pinedo HM, Veenhof CH, Beijnen JH. Pharmacokinetics of paclitaxel and carboplatin in a dose-escalating and dose-sequencing study in patients with non-small-cell lung cancer. The European Cancer Centre. J Clin Oncol. 1997;15:317–29. doi: 10.1200/JCO.1997.15.1.317. [DOI] [PubMed] [Google Scholar]
- 18.Friberg LE, Henningsson A, Maas H, Nguyen L, Karlsson MO. Model of chemotherapy-induced myelosuppression with parameter consistency across drugs. J Clin Oncol. 2002;20:4713–21. doi: 10.1200/JCO.2002.02.140. [DOI] [PubMed] [Google Scholar]
- 19.Eisenhauer EA, Bokkel Huinink WW, Swenerton KD, Gianni L, Myles J, van der Burg ME, Kerr I, Vermorken JB, Buser K, Colombo N. European–Canadian randomized trial of paclitaxel in relapsed ovarian cancer: high-dose versus low-dose and long versus short infusion. J Clin Oncol. 1994;12:2654–66. doi: 10.1200/JCO.1994.12.12.2654. [DOI] [PubMed] [Google Scholar]
- 20.Willey TA, Bekos EJ, Gaver RC, Duncan GF, Tay LK, Beijnen JH, Farmen RH. High-performance liquid chromatographic procedure for the quantitative determination of paclitaxel (Taxol) in human plasma. J Chromatogr. 1993;621:231–8. doi: 10.1016/0378-4347(93)80100-i. [DOI] [PubMed] [Google Scholar]
- 21.Beal SL, Sheiner LB NONMEM User's Guide. University of California at San Francisco: NONMEM Project Group; 1998. [Google Scholar]
- 22.Jonsson EN, Karlsson MO. Xpose – an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Meth Programs Biomed. 1999;58:51–64. doi: 10.1016/s0169-2607(98)00067-4. [DOI] [PubMed] [Google Scholar]