

Abstract
Aims
To evaluate the effects of three ABCG2 variants (Q141K, V12M and Q126X), which are known to have altered transport properties in vitro, on the disposition of lamivudine in healthy subjects.
Methods
To evaluate whether lamivudine is a substrate of ABCG2, intracellular accumulation and vectorial transport of 3H-lamivudine were determined in MDCK-ABCG2 cells. The pharmacokinetic parameters of lamivudine were compared among subjects with four different ABCG2 genotypes, including wild type (seven subjects), K141/K141 (six subjects), Q126/Stop126 (four subjects) and M12/M12 (five subjects) after a single oral dose of 100 mg lamivudine.
Results
The intracellular accumulation of lamivudine in MDCK-ABCG2 cells was significantly lower than that in MDCK-mock cells, but fumitremorgin C reversed the intracellular lamivudine concentration to that of MDCK-mock cells. The ABCG2-mediated transport of lamivudine was saturable and the values of Km and Vmax were 216.5 ± 58 µm and 20.42 ± 2.9 nmol h−1 per 106 cells, respectively. After lamivudine administration to healthy subjects, the AUC of lamivudine showed no difference among subjects with different ABCG2 genotypes; 2480 ± 502, 2207 ± 1019, 2422 ± 239, 2552 ± 698 ng h−1 ml−1 for wild type, K141/K141, Q126/Stop126 and M12/M12 genotype, respectively (P = 0.85). The estimated 95% confidence intervals for the mean difference between K141/K141, Q126/Stop126, M12/M12 and wild as reference were (−1053, 507), (−555, 439) and (−552, 696), respectively. No other pharmacokinetic parameters were estimated to be significantly different among four different ABCG2 genotypes tested.
Conclusions
Lamivudine appeared to be a substrate of ABCG2 in vitro, but the disposition of lamivudine was not significantly influenced by known in vitro functional variants of ABCG2, Q141K, V12M and Q126X in healthy subjects.
Keywords: ABCG2, lamivudine, pharmacokinetics, polymorphism
Introduction
A major challenge in cancer treatment is the resistance of tumour cells to anticancer drugs. A primary mechanism of this resistance is the enhanced ability of tumour cells to actively efflux drugs, reducing the cellular accumulation to subtoxic levels. Active drug efflux is mediated by several members of the ATP binding cassette (ABC) superfamily of membrane transporters [1–5].
The breast cancer resistance protein (BCRP), also known as ABCG2, MXR and ABCP, belongs to a subfamily of ABC transporters and is expressed in the placenta, liver, brain, small intestine and breast in humans [6, 7]. It is found predominantly in the apical membrane of polarized cells in the intestine and liver. ABCG2 transport activity could potentially affect oral absorption and biliary elimination of substrate drugs. ABCG2 has been shown to transport various anticancer drugs, including SN-38 (the active metabolite of irinotecan), mitoxantron, methotrexate, topotecan and doxorubicin [8–11]. In addition, several cytological dyes such as rhodamine 123 and the fluorescent conjugate BODIPY-prazosin, exhibit decreased accumulation in cells overexpressing ABCG2 [5]. It was recently proposed that human immunodeficiency virus type 1 nucleoside reverse transcriptase inhibitors (HIV-1 NRTIs), such as lamivudine and zidovudine, are transported by ABCG2 in vitro [12, 13].
More than 40 single nucleotide polymorphisms (SNPs) have been identified in ABCG2, several of which have been shown to result in proteins with functional differences [2]. The ABCG2 C421A allele, resulting in a Gln141Lys change, has been associated with low levels of ABCG2 expression and altered sensitivity to the anticancer drugs SN-38, mitoxantrone and topotecan in vitro, compared with the wild type [14]. The C376T polymorphism, substituting a stop codon for Gln126, has been shown to prevent the expression of active ABCG2 [4, 14]. It has also been reported that the ABCG2 G34A allele, resulting in a Val12Met substitution, causes the apical plasma membrane dislocalization of ABCG2 and produces a protein with significantly reduced ability to transport several drugs [15]. These findings suggest that carriers of these ABCG2 alleles may have decreased clearance and/or increased oral bioavailability of ABCG2 substrate drugs due to the expression of variant ABCG2 proteins in the liver and small intestine. Recently, it was reported that patients with the ABCG2 Gln141Lys variant had elevated plasma concentrations of diflomotecan, a substrate of ABCG2, compared with patients with two wild-type alleles, after intravenous drug administration [16]. Very recently, it has also been demonstrated that ABCG2 Gln141Lys polymorphism may play an important role in the pharmacokinetics of rosuvastatin in healthy Chinese men, after excluding the impact of OATP-C and CYP2C9 genetic polymorphisms [17].
The aims of this study were to identify an ABCG2 substrate that could be administered to normal healthy volunteers and to evaluate the clinical relevance of ABCG2 variants on the disposition of this substrate. To perform this study, lamivudine was assessed as a substrate of ABCG2 in vitro and in vivo. Lamivudine (3TC, [-]-2′-deoxy-3′-thiacytidine), a known ABCG2 substrate, is a dideoxynucleoside analogue that undergoes anabolic phosphorylation by intracellular kinases to form lamivudine 5′-triphosphate, the active anabolite that prevents HIV-1 and hepatitis B virus (HBV) replication by competitively inhibiting viral reverse transcriptase and terminating DNA chain extension. Lamivudine is used at a high dose (300 mg day−1) in combination with other anti-HIV-1 agents in the treatment of HIV-1 infection and at a low dose (100 mg day−1) as a monotherapy in the treatment of HBV infection [18, 19]. Considering possible adverse drug reactions [18–21], it was determined that a single oral dose of 100 mg lamivudine was safe to administer to healthy volunteers. In this study, we demonstrated that lamivudine was transported by ABCG2 in vitro and then evaluated the effects of functional polymorphisms of ABCG2 on the disposition of lamivudine in vivo.
Methods
Cellular accumulation assay
MDCK-mock cells and ABCG2-overexpressing MDCK cells (MDCK-ABCG2) were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air and were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 2 mmL-glutamine and 100 U ml−1 penicillin–streptomycin. For experiments, 3 × 105 cells were seeded on 12-well plastic cell culture clusters. After 95% confluence was achieved, the growth media were discarded and the attached cells washed with Eagle's media and preincubated for 1 h in serum-free DMEM at 37°C. The uptake of radiolabelled drug was initiated by the addition of 1 ml of medium containing 100 nm3H-lamivudine (Moraveck Biochemicals Inc., Brea, CA, USA), which was added to the cells in the absence or presence of 10 µm fumitremorgin C (FTC) at 37°C. The uptake was stopped by placing the plates on ice. The cells were washed three times with 2 ml of ice-cold phosphate-buffered saline (PBS). The radioactivity of the 3H-lamivudine in the cells was measured by liquid scintillation counting after lysing the cells with 100 µlof Cell Culture Lysis Reagent [CCLR buffer: 100 mm potassium phosphate (pH 7.8), 1 mm ethylenediamine tetraacetic acid, 7 mm 2-mercaptoethanol, 1% (v/v) Triton X-100, 10% (v/v) glycerol; Promega, Madison, WI, USA]. MDCK-ABCG2 cells and FTC were kindly provided by Dr A. H. Schinkel at NKI (Division of Experimental Therapy, the Netherlands Cancer Institute, Amsterdam, the Netherlands) and Dr S. E. Bates at NIH (Molecular Therapeutics Branch, NCI, NIH, Bethesda, USA), respectively.
Transport study
MDCK-ABCG2 cells were grown on a permeable polycarbonate insert (1 cm2, 0.2 µm pore size; Corning Costar Co., Cambridge, MA, USA) in 12-Transwell plates (4 cm2; Corning Costar Co.) at a density of 1 × 106 cells cm−2, and the medium was changed at 2-day intervals. After reaching the transepithelial electrical resistance value of the seeded cells of 250–300 Ω cm−2, vectorial transport study was performed.
For measurement of the apical to basal (A–B) transport of lamivudine, 0.5 ml of Dulbecco's modified PBS, pH 7.4 (DPBS) containing 3H-lamivudine (1 µm, 0.5 µCi) was added on the apical side and 1.5 ml of DPBS without lamivudine was added on the basal side of the insert. The insert was transferred to a well containing fresh DPBS medium every 15 min for 1 h. The radioactivity of an aliquot (50 µl) of the basal side of each well was determined by liquid scintillation counter. For measurement of the basal to apical (B–A) transport, 1.5 ml of DPBS containing 3H-lamivudine (1 µm, 1.5 µCi) was added on the basal side and 0.5 ml of DPBS without lamivudine was added on the apical side. The transport medium in the apical side was replaced with 0.35 ml of fresh incubation medium every 15 min for 1 h. The radioactivity of an aliquot (50 µl) of the apical side after each replacement was measured. To examine the concentration dependency of the B–A transport of lamivudine, 1.5 ml of DPBS containing 3H-lamivudine of a given concentration (1–1000 µm) was added on the basal side of the insert and 0.5 ml of DPBS without lamivudine was added on the apical side.
The total amount of lamivudine transported across the cell monolayer was plotted against time and the transport rate (nmol cm−2 h−1) was calculated from the linear portion of the curve (slope of the plot). The transport rate was then plotted against the initial concentration of lamivudine and the resulting profile was fitted to the Michaelis–Menten equation to estimate the kinetic parameters such as Vmax and Km. The intrinsic clearance for the transport (CLint) was obtained from Vmax/Km. Weighted nonlinear regression analysis was performed in the fitting using Sigma plot (version 9.0; Systat Software Inc., Richmond, CA, USA)
Subjects
Unrelated Korean subjects (n = 183) had been genotyped for the ABCG2 variants G34A (Val12Met), C376T (Gln126stop) and C421A (Gln141Lys). Of these, 22 healthy male subjects were enrolled in this trial. The genotypes of the subjects were wild type (n = 7), Lys141/Lys141 (n = 6), Gln126/Stop126 (n = 4) and Met12/Met12 (n = 5). All subjects were determined to be healthy by medical history, physical examination, vital signs and clinical laboratory tests performed within 2 weeks before the start of the study. Demographic data for the subjects are listed in Table 1. Subjects were not allowed to take any drugs, use alcohol or caffeine or drink any fruit juice during the entire study period. All subjects provided written informed consent before participating in the study. The study protocol was approved by the Institutional Review Board of Inje University Busan Paik Hospital, Busan, Korea.
Table 1.
Demographic data of subjects
| Wild (n = 7) | Met12/Met12 (n = 5) | Gln126/stop126 (n = 4) | Lys141/Lys141 (n = 6) | P-value | |
|---|---|---|---|---|---|
| Age (years) | 22.4 ± 1.0 | 22.8 ± 0.8 | 23 ± 1.1 | 22.3 ± 1.0 | 0.69 |
| Heigt (cm) | 176 ± 3 | 177 ± 6 | 180 ± 3 | 181 ± 4 | 0.12 |
| Weight (kg) | 70 ± 8 | 70 ± 8 | 77 ± 10 | 81 ± 9 | 0.08 |
Data are presented as mean ± SD.
ABCG2 genotyping
ABCG2 genotyping was performed by pyrosequencing of polymerase chain reaction (PCR) products from the ABCG2 gene to determine the presence of the Val12Met, Gln126Stop and Gln141Lys alleles. PCR was performed in a reaction volume of 30 µl containing genomic DNA (150 ng), 1× PCR buffer, dNTPs (0.2 mm), primers (0.2 µm each), MgCl2 (1.5 mm) and rTaq polymerase (1 U; Takara, Tokyo, Japan). PCR was performed using a GeneAmp PCR 9700 (Applied Biosystems, Foster City, CA, USA) with an initial denaturation step of 95°C for 5 min, followed by 35 cycles of denaturation at 95°C for 30 s, annealing at 54–55°C for 35 s and extension at 72°C for 30 s. A final termination of elongation step was performed at 72°C for 5 min. The sequences of all primers and details of the annealing temperatures are listed in Table 2.
Table 2.
Sequences of primers used for the amplification and sequencing analysis of ABCG2 genotype and the denaturation temperatures used in the PCR
| Name | Primer sequence (5′→3′) | Size | PCR (Tm; °C) |
|---|---|---|---|
| ABCG2 V12M | F: Biotin-CTCTCCAGATGTCTTCCAGTAATG | 278 | 54 |
| R: GCCAAAACCTGTGAGGTTCA | |||
| S: CATTGGTGTTTCCTTGTGA | |||
| ABCG2 Q126X | F: GTCTTAGCTGCAAGGAAAGATCCA | 174 | 54.5 |
| R: Biotin-ACTATCAGCCAAAGCACTTACCC | |||
| S: AATGTAATTCAGGTTACGTG | |||
| ABCG2 Q141K | F: TGATGTTGTGATGGGCACTC | 69 | 54 |
| R: Biotin-GTTGCAAGCCGAAGAGCTG | |||
| S: GACGGTGAGAGAAAACTT |
F, forward primer; R, reverse primer; S, sequencing primer; Tm, melting temperature; PCR, polymerase chain reaction.
Biotinylated PCR products were immobilized on streptavidin-coated beads (Streptavidin Sepharose High Performance; Amersham Biosciences, Little Chalfont, UK), followed by strand separation and sample preparation, using a PSQ 96 sample preparation kit (Pyrosequencing; Amersham Biosciences). The Sepharose bead slurry (3 µl) was mixed with binding buffer (37 µl), PCR product (20 µl) and distilled water (20 µl) and the mixture was incubated at room temperature for 10 min at 160 g. The beads were then transferred to a filter plate and the liquid was removed by vacuum filtration (Multiscreen Resist Vacuum Manifold; Millipore, Bedford, MA, USA). The DNA strands were separated in denaturation solution (0.5 m sodium hydroxide) for 5 s. The immobilized template was washed in washing buffer (10 mm Tris–acetate, pH 7.6), transferred to a PSQ 96 plate and resuspended in annealing buffer (20 mm Tris–acetate, pH 7.6) containing each sequencing primer. The sequencing primer was annealed at 80–90°C for 3 min. The SNPs were analysed using a PSQ 96 system with a SNP reagent kit.
Study design
The trial was an open-label, parallel-group, single-dose study. All subjects received a single oral dose of 100 mg lamivudine (Zeffix®; GlaxoSmithKline Korea, Seoul, Korea) after fasting overnight. The subjects were asked to remain in a seated position for 4 h after taking lamivudine. They were then allowed to perform usual daily activities in the clinical trial room; however, strenuous activity and exercise were not allowed. Standardized meals were provided at 4 and 10 h after the administration of lamivudine. Blood samples were drawn immediately before and at 0.33, 0.67, 1, 1.5, 2, 3, 4, 6, 8, 10, 12 and 24 h after lamivudine administration. Urine samples were collected during time intervals of 0–12 h and 12–24 h after dosing. The blood samples were centrifuged and the separated plasma and the urine samples were stored at −80°C until assayed.
Assay of lamivudine concentrations
Lamivudine concentrations in plasma and urine samples were determined by liquid chromatography with ultraviolet (UV) detection, as described by Zheng et al. [22], with some modification. In brief, the Waters C18 Sepak cartridge column (100 mg), set on a Visiprep SPE vacuum manifold (Supelco, St Louis, MO, USA) connected to a vacuum pump, was successively washed with 1 ml of methanol and 1 ml of water. Then, 0.5 ml of plasma or urine sample, 0.5 ml of water and 25 µl of an internal standard (3-isobutyl-methylaxinthine, 1 mg ml−1) were applied to the column. After washing with 1 ml of water, lamivudine was eluted with 1 ml of acetonitrile. The eluate was evaporated to dryness at ambient temperature using a Speed-Vac (Savant, Holbrook, NY, USA). The residue was dissolved in 0.2 ml mobile phase and injected into the HPLC system. The mobile phase was a mixture of acetonitrile and 0.085% phosphoric acid (19 : 81, v/v) containing 10 mm octanesulphonate. The high-performance liquid chromatography (HPLC) system consisted of a Gilson model 307 pump (Gilson, Villiers Le Bel, France) with a Zorbax Eclipse XDB-C8 column (4.6 × 150 mm, 5 µm; Agilent, Waldbronn, Germany) and a Gilson model 118 UV-visible detector. Peaks were detected by absorbance at 270 nm at a flow rate of 1.0 ml min−1. The retention times for lamivudine and 2-isobutyl-methylaxinthine were 3.56 and 4.78 min, respectively. The lower limit of quantification for lamivudine was 10 ng ml−1. The interassay precision values for all of the samples were <15.0%.
Pharmacokinetic analysis
The pharmacokinetic parameters of lamivudine were calculated by noncompartmental analysis techniques using WinNonlin software (version 4.1; Pharsight, Mountain View, CA, USA). The peak plasma concentration (Cmax) values and time to reach Cmax (tmax) were estimated directly from the observed plasma concentration–time data. The elimination rate constant (Ke) was estimated from the least-squares regression slope of terminal plasma concentrations. The area under the plasma concentration–time curve (AUC0−24) from time zero to the last measurement was calculated according to the linear trapezoidal rule. The AUC from time zero to infinity (AUC0–∞) was calculated as AUC0–∞ = AUC0−24 + C24/Ke, where C24 is the plasma concentration measured 24 h after drug administration. The half-life (t1/2) of lamivudine was calculated as 0.693/Ke. The oral clearance of lamivudine (CL/F) was calculated as CL/F = Dose/AUC0−24. The renal clearance (CLR) was calculated as the ratio of Ae to AUC0−24, where Ae is the amount of lamivudine recovered in the urine collected for 24 h after oral administration.
Statistical analysis
After assessing the normal distribution of the data using the Shapiro–Wilk test for normality, comparisons of the accumulation of lamivudine in the cells in vitro and of the pharmacokinetic parameters for the different genotype groups in vivo were made using the nonparametric Kruskal–Wallis test and parametric anova test for multigroup comparisons. SAS® (version 9.1.3, SAS Institute, Cary, NC, USA) software was used for the statistical analysis. P-values < 0.05 were considered to be statistically significant.
Results
Cellular accumulation and vectorial transport of lamivudine
To examine whether lamivudine is a substrate for ABCG2, uptake of lamivudine into MDCK-ABCG2 cells was investigated. When MDCK cells were incubated with 3H-lamivudine for 30 min, intracellular accumulation of the compound was 10.6 ± 1.9 pmol µg−1 protein. To evaluate the possible contribution of basal expression of ABCG2 in the MDCK cells, FTC, a specific inhibitor of ABCG2 [5], was added together with 3H-lamivudine, which revealed that the cellular accumulation was 10.3 ± 2.0 pmol µg−1 protein (Figure 1A). The cellular uptake of 3H-lamivudine was significantly reduced to 5.9 ± 1.5 pmol µg−1 protein in MDCK-ABCG2 cells. Furthermore, this decrease was reversed by treatment with FTC to 9.6 ± 0.8 pmol µg−1 protein.
Figure 1.
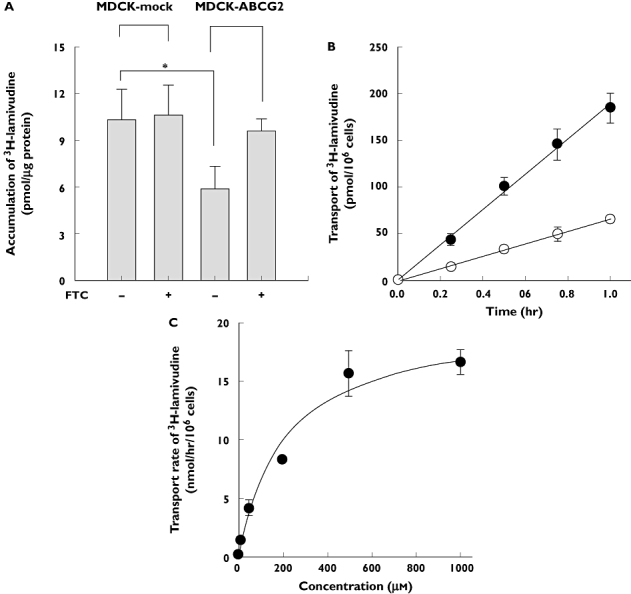
(A) Comparison of intracellular accumulation of 3H-lamivudine in the MCDK-mock cells and ABCG2-overexpressing MDCK (MDCK-ABCG2) cells. Cells were incubated for 30 min at 37°C with 3H-lamivudine applied from the apical sides of MDCK-mock cells or MDCK-ABCG2 cell monolayers in the absence or presence of 10 µm fumitremorgin C (FTC), the selective inhibitor of ABCG2. Data are presented as mean ± SD of three preparations. *P < 0.01. (B) Time course of apical to basal (○) and basal to apical (•) transport of 1 µm3H-lamivudine across MDCK-ABCG2 cell monolayer. Each data point represents the mean ± SD of four independent experiments. (C) Concentration-dependent basal to apical transport of lamivudine (1–1000 µm) across the MDCK-ABCG2 cell monolayer. Each data point represents the mean ± SD of four independent experiments
The B–A transport of 1 µm3H-methotrexate, which is a well-known substrate of ABCG2 [11], was threefold higher than that of the opposite direction, indicating the functional activity of ABCG2 in the MDCK-ABCG2 cell monolayer. The B–A transport of lamivudine was threefold greater than the corresponding A–B transport (Figure 1B), consistent with the above result that cellular accumulation of lamivudine was decreased in the ABCG2-overexpressed cell line. Since the involvement of ABCG2 was observed for the B–A transport of lamivudine, the characteristics of the transport were investigated further. A concentration dependency was found for the transport of lamivudine (Figure 1C) and the values of Km, Vmax and intrinsic clearance (CLint) obtained by the Michaelis–Menten equation were 216.5 ± 58 µm, 20.42 ± 2.9 nmol h−1 per 106 cells and 101.5 ± 38 µl h−1 per 106 cells, respectively. These results, taken together, suggested that lamivudine is a substrate for ABCG2.
Effect of ABCG2 variants on the pharmacokinetics of lamivudine
To study the influence of ABCG2 variants on lamivudine pharmacokinetics, we included subjects with the Val12Met, Gln126Stop or Gln141Lys ABCG2 variants.
Pharmacokinetic parameters for lamivudine were compared among the ABCG2 genotypes after a single oral dose of 100 mg lamivudine. The Cmax values (mean ± SD) for the wild-type, Lys141/Lys141, Gln126/Stop126 and Met12/Met12 genotypes were 742 ± 297, 655 ± 309, 840 ± 151 and 808 ± 193 ng ml−1 (P = 0.68), respectively, and the 95% confidence intervals (CIs) for the mean difference between Lys141/Lys141, Gln126/Stop126, Met12/Met12 and wild type as reference were (−389, 215), (−198, 394) and (−210, 342), respectively. The AUC values for the genotypes were 2480 ± 502, 2207 ± 1019, 2422 ± 239 and 2552 ± 698 ng h−1 ml−1 (P = 0.85), respectively, and the 95% CIs for the mean difference were (−1053, 507), (−555, 439) and (−552, 696), respectively. Thus, the pharmacokinetic parameters for lamivudine did not differ significantly among the genotypic groups. Pharmacokinetic data and statistical analyses are summarized in Table 3 and Figure 2. Large interindividual variations in Cmax and AUC values were observed in the wild-type, Lys141/Lys141 and Met12/Met12 groups, whereas interindividual variation of Cmax in the Gln126/Stop126 group was relatively small (Figure 3). As a whole, the renal clearances (CLR) showed large interindividual variations regardless of ABCG2 genotype. No significant differences were also observed among the parameters after normalization to 70 kg body weight to eliminate interindividual differences in weight of the subjects.
Table 3.
Pharmacokinetic parameters of lamivudine after single oral administration of 100 mg lamivudine in subjects with wild, Lys141/Lys141, Gln126/Stop126 and Met12/Met12 variant of ABCG2
| Mean difference between variants and wild | ||||||||
|---|---|---|---|---|---|---|---|---|
| Wild | Met12/Met12 | Gln126/Stop126 | Lys141/Lys141 | Met12/Met12 | Gln126/Stop126 | Lys141/Lys141 | P-value | |
| Cmax (ng ml−1) | 742 ± 297 | 808 ± 193 | 840 ± 151 | 655 ± 309 | 66 (−210, 342) | 98 (−198, 394) | −87 (−389, 215) | 0.68 |
| Cmax,normal (ng ml−1)* | 736 ± 303 | 850 ± 208 | 926 ± 230 | 746 ± 320 | 114 (−171, 399) | 190 (−133, 513) | 10 (−300, 320) | 0.57 |
| tmax (h) | 0.8 ± 0.4 | 1.0 ± 0.3 | 0.7 ± 0.3 | 0.8 ± 0.4 | 0.2 (−0.2, 0.6) | −0.1 (−0.5, 0.3) | 0 (−0.4, 0.4) | 0.63 |
| t1/2 (h) | 5.5 ± 2.4 | 7.1 ± 2.0 | 5.5 ± 2.1 | 7.7 ± 4.9 | 1.6 (−0.8, 4) | 0 (−2.6, 2.6) | 2.2 (−1.5, 5.9) | 0.59 |
| AUCinf (ng h−1 ml−1) | 2480 ± 502 | 2552 ± 698 | 2422 ± 239 | 2207 ± 1019 | 72 (−552, 696) | −58 (−555, 439) | −273 (−1053, 507) | 0.85 |
| AUCinf,normal (ng h−1 ml−1)* | 2440 ± 397 | 2604 ± 582 | 2654 ± 319 | 2573 ± 1326 | 164 (−345, 673) | 214 (−214, 642) | 133 (−807, 1073) | 0.69 |
| CLtotal/F (l kg−1 h−1) | 0.60 ± 0.12 | 0.59 ± 0.12 | 0.54 ± 0.07 | 0.66 ± 0.25 | −0.01 (−0.14, 0.12) | −0.06 (−0.18, 0.06) | 0.06 (−0.13, 0.25) | 0.76 |
| CLR (l kg−1 h−1) | 0.29 ± 0.05 | 0.26 ± 0.05 | 0.26 ± 0.05 | 0.28 ± 0.09 | −0.03 (−0.08, 0.02) | −0.03 (−0.09, 0.03) | −0.01 (−0.08, 0.06) | 0.85 |
| Vd/F (l kg−1) | 4.8 ± 2.6 | 5.9 ± 1.4 | 4.2 ± 1.2 | 4.7 ± 2.4 | 1.1 (−1.2, 3.4) | −0.6 (−3.2, 2.0) | −0.1 (−2.6, 2.4) | 0.66 |
Cmax,normal and AUCinf,normal: Cmax and AUCinf normalized to 70 kg body weight. Each value indicates mean ± SD (95% confidence interval).
Figure 2.
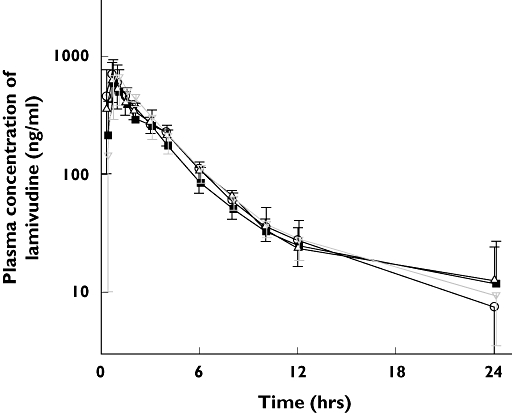
Mean concentration–time profiles of lamivudine after a single oral administration of 100 mg lamivudine in subjects with wild genotype (○), Lys141/Lys141 (▪), Gln126/Stop126 (▵), and Met12/Met12 ( ) variant of ABCG2
) variant of ABCG2
Figure 3.
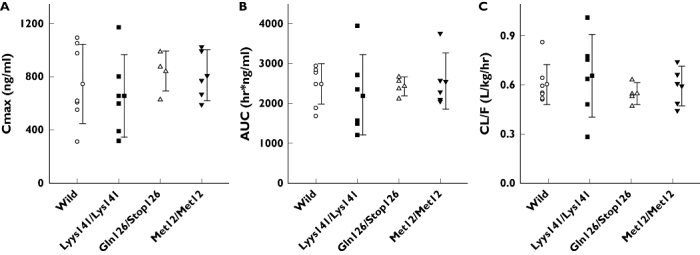
The peak plasma drug concentration (Cmax) (A), the area under the plasma drug concentration–time curve (AUC) (B) and the total clearance (CL/F) (C) of subjects with wild-type and three ABCG2 functional variants after oral administration of 100 mg lamivudine. Each symbol indicates the individual pharmacokinetic parameter of subjects with four different genotypes of ABCG2. Vertical lines with horizontal bar indicate mean ± SD
Discussion
It has been reported that the high level of ABCG2 expression in CD4+ T cells induces cellular resistance to HIV-1 NRTIs such as lamivudine and zidovudine in vitro [12, 13]. In our study, the decreased accumulation of lamivudine in MDCK-ABCG2 compared with that in MDCK-mock cells was reversed in the presence of FTC, which specifically inhibits ABCG2 activity. Moreover, concentration-dependent transport of lamivudine was saturable at a concentration range of 1–1000 µm. Thus, our results proved consistent with the hypothesis that lamivudine is transported by ABCG2 in vitro.
Among the known ABCG2 variants with altered functional properties in vitro, the ABCG2 Gln141Lys polymorphism has been reported to be involved in regulation of the level of protein expression [14, 23], drug resistance level and ATPase activity [15]. The Val12Met polymorphism has also been associated with change in membrane localization and produces a protein with significantly decreased activity in transporting several drugs [15]. The Gln126Stop polymorphism has been shown to prevent the expression of active ABCG2 [4, 14]. The ABCG2 Val12Met and Gln141Lys variants are common in Koreans, with allelic frequencies of 23 and 27.5%, respectively. Another variant, Gln126stop, has been found in Koreans at 1.8% (data not shown). Carriers of these ABCG2 variants may have decreased clearance and/or increased oral bioavailability of ABCG2 substrate drugs as a result of the polymorphic expression of ABCG2 in the liver and small intestine. This may be of particular importance in the Korean populations when administering substrate drugs of ABCG2 because of the high frequencies of functional ABCG2 variants.
We postulated that the pharmacokinetics and/or pharmacodynamics of lamivudine may be affected by functional variants of ABCG2. However, the pharmacokinetic parameters of lamivudine were not significantly different among the different ABCG2 genotypic groups in the healthy volunteers examined. These results suggest that the polymorphisms of ABCG2 did not affect the disposition of lamivudine in vivo, even though we confirmed that lamivudine is a substrate of ABCG2 in vitro.
There has been a previous demonstration that patients carrying the ABCG2 Gln141Lys variant have elevated plasma concentrations of diflomotecan, a substrate of ABCG2, compared with patients with two wild-type alleles, after intravenous drug administration [16]. In that report, however, differences between patients with wild-type and Lys141/Lys141 genotypes were not statistically significant after oral administration of diflomotecan. The authors stated that other anatomical and physiological factors might affect the overall rate and extent of intestinal transport of diflomotecan. Like diflomotecan, other physiological factors may influence the overall rate and extent of intestinal absorption of lamivudine, which might result in no effect of ABCG2 polymorphism being detected on lamivudine disposition.
Considering the complexity of lamivudine transport, it is also possible that the contribution of ABCG2 polymorphism is obscured by other proteins or mechanisms involved in lamivudine elimination. The disposition of lamivudine in human may be handled by several other transporters, such as the proton/organic cation antiporter [24, 25], SLC22A6 [26] and ABCC family-like ABCC4, 5, 11 [27–29], in addition to ABCG2. As renal excretion is a major factor in the disposition of lamivudine in humans [18, 19], the effects of proton/organic cation antiporter on renal secretion may be the greatest among effects of transporters on the disposition of lamivudine in vivo, and the contribution of ABCG2 to the disposition of lamivudine may be relatively small compared with proton/organic cation antiporter.
Another factor to be considered is the affinity of lamivudine to ABCG2. In our in vitro study, the Km value of lamivudine was 216.5 µm, which was larger than that of SN-38 (4 µm) [30] or estron-3-sulphate (17 µm) [31], although smaller than that of methotrexate (680 µm) [32]. Thus, the relatively low affinity of lamivudineto ABCG2 may also account for the lack of effect of ABCG2 polymorphism on the disposition of lamivudine.
Anderson et al. have recently shown that the intracellular concentration of lamivudine-triphosphate is not associated with ABCG2 Val12Met, Glu141Lys [33]. This is consistent with our data, with the difference that they analysed the intracellular concentration of lamivudine-triphosphate in peripheral blood mononuclear cells (PBMCs), whereas we obtained the plasma concentration of lamivudine. We have also tried to evaluate the intracellular concentration of lamivudine and lamivudine-triphosphate in PBMCs, but these were not detected at the maximum intracellular concentration time of lamivudine because a single low dose (100 mg) of lamivudine was administered.
In long-term treatment with nucleoside analogues, altered cellular factors such as increased efflux and decreased activation by cellular enzymes in host cells may contribute to inefficient activation of chemotherapeutic agents in HIV-1 patients [34]. It may also be meaningful to correlate functional polymorphisms of ABCG2 with the early or late failure of lamivudine treatment or failures unrelated to viral mutations. Further studies are needed to evaluate the clinical relevance of the genetic variants of ABCG2.
In conclusion, we sought to identify an ABCG2 substrate drug that could be administered to normal healthy volunteers and use it to evaluate the clinical relevance of ABCG2 polymorphisms on the disposition of the drug. We have confirmed that lamivudine is a substrate of ABCG2 in vitro. However, the Val12Met, Gln126stop and Gln141Lys polymorphisms of ABCG2 studied here, which have been previously shown to cause functional differences in the protein, did not alter the disposition of a single oral dose of 100 mg lamivudine in healthy volunteers. Thus, orally administered lamivudine may not be useful as a probe drug for ABCG2 activity in healthy subjects.
Acknowledgments
This study was supported in part by the Ministry of Science and Technology and in part by the Korea Health 21 R&D Project, Ministry of Health & Welfare, Korea (0412-CT02-0704-0006). We thank Dr A. H. Schinkel at NKI (Division of Experimental Therapy, the Netherlands Cancer Institute, Amsterdam, the Netherlands) for providing MDCK- ABCG2 cells and Dr S. E. Bates at NIH (Molecular Therapeutics Branch, NCI, NIH, Bethesda, MD, USA) for providing FTC. We also thank Dr Kwang-Hyeon Liu and Sang-Seop Lee for their helpful advice on the experiments and Mi-Gyung Go for her excellent technical assistance.
References
- 1.Haimeur A, Conseil G, Deeley RG, Cole SP. The MRP-related and BCRP/ABCG2 multidrug resistance proteins: biology, substrate specificity and regulation. Curr Drug Metab. 2004;5:21–53. doi: 10.2174/1389200043489199. [DOI] [PubMed] [Google Scholar]
- 2.Lockhart AC, Tirona RG, Kim RB. Pharmacogenetics of ATP-binding cassette transporters in cancer and chemotherapy. Mol Cancer Ther. 2003;2:685–98. [PubMed] [Google Scholar]
- 3.Sparreboom A, Danesi R, Ando Y, Chan J, Figg WD. Pharmacogenomics of ABC transporters and its role in cancer chemotherapy. Drug Resist Updat. 2003;6:71–84. doi: 10.1016/s1368-7646(03)00005-0. [DOI] [PubMed] [Google Scholar]
- 4.Sugimoto Y, Tsukahara S, Ishikawa E, Mitsuhashi J. Breast cancer resistance protein: molecular target for anticancer drug resistance and pharmacokinetics/pharmacodynamics. Cancer Sci. 2005;96:457–65. doi: 10.1111/j.1349-7006.2005.00081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allen JD, Schinkel AH. Multidrug resistance and pharmacological protection mediated by the breast cancer resistance protein (BCRP/ABCG2) Mol Cancer Ther. 2002;1:427–34. [PubMed] [Google Scholar]
- 6.Allikmets R, Schriml LM, Hutchinson A, Romano-Spica V, Dean M. A human placenta-specific ATP-binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res. 1998;58:5337–9. [PubMed] [Google Scholar]
- 7.Doyle LA, Yang W, Abruzzo LV, Krogmann T, Gao Y, Rishi AK, Ross DD. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci USA. 1998;95:15665–70. doi: 10.1073/pnas.95.26.15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Komatani H, Kotani H, Hara Y, Nakagawa R, Matsumoto M, Arakawa H, Nishimura S. Identification of breast cancer resistant protein/mitoxantrone resistance/placenta-specific, ATP-binding cassette transporter as a transporter of NB-506 and J-107088, topoisomerase I inhibitors with an indolocarbazole structure. Cancer Res. 2001;61:2827–32. [PubMed] [Google Scholar]
- 9.Kawabata S, Oka M, Shiozawa K, Tsukamoto K, Nakatomi K, Soda H, Fukuda M, Ikegami Y, Sugahara K, Yamada Y, Kamihira S, Doyle LA, Ross DD, Kohno S. Breast cancer resistance protein directly confers SN-38 resistance of lung cancer cells. Biochem Biophys Res Commun. 2001;280:1216–23. doi: 10.1006/bbrc.2001.4267. [DOI] [PubMed] [Google Scholar]
- 10.Nakagawa R, Hara Y, Arakawa H, Nishimura S, Komatani H. ABCG2 confers resistance to indolocarbazole compounds by ATP-dependent transport. Biochem Biophys Res Commun. 2002;299:669–75. doi: 10.1016/s0006-291x(02)02712-2. [DOI] [PubMed] [Google Scholar]
- 11.Volk EL, Farley KM, Wu Y, Li F, Robey RW, Schneider E. Overexpression of wild-type breast cancer resistance protein mediates methotrexate resistance. Cancer Res. 2002;62:5035–40. [PubMed] [Google Scholar]
- 12.Wang X, Furukawa T, Nitanda T, Okamoto M, Sugimoto Y, Akiyama S, Baba M. Breast cancer resistance protein (BCRP/ABCG2) induces cellular resistance to HIV-1 nucleoside reverse transcriptase inhibitors. Mol Pharmacol. 2003;63:65–72. doi: 10.1124/mol.63.1.65. [DOI] [PubMed] [Google Scholar]
- 13.Wang X, Nitanda T, Shi M, Okamoto M, Furukawa T, Sugimoto Y, Akiyama S, Baba M. Induction of cellular resistance to nucleoside reverse transcriptase inhibitors by the wild-type breast cancer resistance protein. Biochem Pharmacol. 2004;68:1363–70. doi: 10.1016/j.bcp.2004.05.052. [DOI] [PubMed] [Google Scholar]
- 14.Imai Y, Nakane M, Kage K, Tsukahara S, Ishikawa E, Tsuruo T, Miki Y, Sugimoto Y. C421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141K protein and low-level drug resistance. Mol Cancer Ther. 2002;1:611–6. [PubMed] [Google Scholar]
- 15.Mizuarai S, Aozasa N, Kotani H. Single nucleotide polymorphisms result in impaired membrane localization and reduced atpase activity in multidrug transporter ABCG2. Int J Cancer. 2004;109:238–46. doi: 10.1002/ijc.11669. [DOI] [PubMed] [Google Scholar]
- 16.Sparreboom A, Gelderblom H, Marsh S, Ahluwalia R, Obach R, Principe P, Twelves C, Verweij J, McLeod HL. Diflomotecan pharmacokinetics in relation to ABCG2 421C>A genotype. Clin Pharmacol Ther. 2004;76:38–44. doi: 10.1016/j.clpt.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 17.Zhang W, Yu BN, He YJ, Fan L, Li Q, Liu ZQ, Wang A, Liu YL, Tan ZR, Fen J, Huang YF, Zhou HH. Role of BCRP 421C>A polymorphism on rosuvastatin pharmacokinetics in healthy Chinese males. Clin Chim Acta. 2006;373:99–103. doi: 10.1016/j.cca.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 18.Johnson MA, Moore KH, Yuen GJ, Bye A, Pakes GE. Clinical pharmacokinetics of lamivudine. Clin Pharmacokinet. 1999;36:41–66. doi: 10.2165/00003088-199936010-00004. [DOI] [PubMed] [Google Scholar]
- 19.Epivir Prescribing Information. Available at URL. http://us.gsk.com/products/assets/us_epivir.pdf Last accessed 2 January 2005.
- 20.Papatheodoridis GV, Hadziyannis SJ. Review article: current management of chronic hepatitis B. Aliment Pharmacol Ther. 2004;19:25–37. doi: 10.1046/j.1365-2036.2003.01810.x. [DOI] [PubMed] [Google Scholar]
- 21.Davis GL. Update on the management of chronic hepatitis B. Rev Gastroenterol Disord. 2002;2:106–15. [PubMed] [Google Scholar]
- 22.Zheng JJ, Wu ST, Emm TA. High-performance liquid chromatographic assay for the determination of 2′-deoxy-3′-thiacytidine (lamivudine) in human plasma. J Chromatogr B Biomed Sci Appl. 2001;761:195–201. doi: 10.1016/s0378-4347(01)00332-2. [DOI] [PubMed] [Google Scholar]
- 23.Kobayashi D, Ieiri I, Hirota T, Takane H, Maegawa S, Kigawa J, Suzuki H, Nanba E, Oshimura M, Terakawa N, Otsubo K, Mine K, Sugiyama Y. Functional assessment of ABCG2 (BCRP) gene polymorphisms to protein expression in human placenta. Drug Metab Dispos. 2005;33:94–101. doi: 10.1124/dmd.104.001628. [DOI] [PubMed] [Google Scholar]
- 24.Leung S, Bendayan R. Uptake properties of lamivudine (3TC) by a continuous renal epithelial cell line. Can J Physiol Pharmacol. 2001;79:59–66. [PubMed] [Google Scholar]
- 25.Takubo T, Kato T, Kinami J, Hanada K, Ogata H. Uptake of lamivudine by rat renal brush border membrane vesicles. J Pharm Pharmacol. 2002;54:111–7. doi: 10.1211/0022357021771814. [DOI] [PubMed] [Google Scholar]
- 26.Wada S, Tsuda M, Sekine T, Cha SH, Kimura M, Kanai Y, Endou H. Rat multispecific organic anion transporter 1 (rOAT1) transports zidovudine, acyclovir, and other antiviral nucleoside analogs. J Pharmacol Exp Ther. 2000;294:844–9. [PubMed] [Google Scholar]
- 27.Schuetz JD, Connelly MC, Sun D, Paibir SG, Flynn PM, Srinivas RV, Kumar A, Fridland A. MRP4: a previously unidentified factor in resistance to nucleoside-based antiviral drugs. Nat Med. 1999;5:1048–51. doi: 10.1038/12487. [DOI] [PubMed] [Google Scholar]
- 28.Turriziani O, Schuetz JD, Focher F, Scagnolari C, Sampath J, Adachi M, Bambacioni F, Riva E, Antonelli G. Impaired 2′,3′-dideoxy-3′-thiacytidine accumulation in T-lymphoblastoid cells as a mechanism of acquired resistance independent of multidrug resistant protein 4 with a possible role for ATP-binding cassette C11. Biochem J. 2002;368:325–32. doi: 10.1042/BJ20020494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wijnholds J, Mol CA, van Deemter L, de Haas M, Scheffer GL, Baas F, Beijnen JH, Scheper RJ, Hatse S, De Clercq E, Balzarini J, Borst P. Multidrug-resistance protein 5 is a multispecific organic anion transporter able to transport nucleotide analogs. Proc Natl Acad Sci USA. 2000;97:7476–81. doi: 10.1073/pnas.120159197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakatomi K, Yoshikawa M, Oka M, Ikegami Y, Hayasaka S, Sano K, Shiozawa K, Kawabata S, Soda H, Ishikawa T, Tanabe S, Kohno S. Transport of 7-ethyl-10-hydroxycamptothecin (SN-38) by breast cancer resistance protein ABCG2 in human lung cancer cells. Biochem Biophys Res Commun. 2001;288:827–32. doi: 10.1006/bbrc.2001.5850. [DOI] [PubMed] [Google Scholar]
- 31.Suzuki M, Suzuki H, Sugimoto Y, Sugiyama Y. ABCG2 transports sulfated conjugates of steroids and xenobiotics. J Biol Chem. 2003;278:22644–9. doi: 10.1074/jbc.M212399200. [DOI] [PubMed] [Google Scholar]
- 32.Volk EL, Schneider E. Wild-type breast cancer resistance protein (BCRP/ABCG2) is a methotrexate polyglutamate transporter. Cancer Res. 2003;63:5538–43. [PubMed] [Google Scholar]
- 33.Anderson PL, Lamba J, Aquilante CL, Schuetz E, Fletcher CV. Pharmacogenetic characteristics of indinavir, zidovudine, and lamivudine therapy in HIV-infected adults: a pilot study. J Acquir Immune Defic Syndr. 2006;42:441–9. doi: 10.1097/01.qai.0000225013.53568.69. [DOI] [PubMed] [Google Scholar]
- 34.Groschel B, Cinatl J, Cinatl J., Jr Viral and cellular factors for resistance against antiretroviral agents. Intervirology. 1997;40:400–7. doi: 10.1159/000150572. [DOI] [PubMed] [Google Scholar]