

Abstract
The emergence of multiresistant bacterial strains and the continuing burden of infectious disease globally point to the urgent need for novel affordable antimicrobial drugs. Thioridazine is a phenothiazine antipsychotic drug with well-recognized antimicrobial activity, but this property has not been harnessed for clinical use as a result of its central nervous system and cardiac side-effects. The cardiotoxicity of thioridazine has recently been shown to be structurally specific at a molecular level, whereas its antimicrobial properties are shared by a number of phenothiazine analogues. This raises the possibility that its enantiomers or its inactive metabolite, the ring sulphoxide, may act as a lead compound in the future development of antimicrobial drugs to face the new challenges in infectious disease.
Keywords: antimicrobial, drug development, drug resistance, QT prolongation, thioridazine, tuberculosis
Introduction
Phenothiazine drugs, in addition to their antipsychotic properties, have significant antimicrobial activity against a wide variety of intracellular microorganisms [1], as they are concentrated almost 100-fold in macrophages [2] and lung [3].
The prototypical phenothiazine drug, methylene blue (methylthioninium chloride), was shown to be active against Plasmodium falciparum by Ehrlich in 1891 [4]. Subsequently developed phenothiazines, such as chlorpromazine, have proven in vitro bacteriostatic and bactericidal activity against several microorganisms, including Mycobacterium tuberculosis. This antimicrobial potential has not been harnessed to date due to concern over the sedative and extrapyramidal side-effects and the cardiotoxicity of phenothiazine drugs at the plasma concentrations required to achieve bactericidal effects.
Thioridazine (an alkylpiperadine phenothiazine), previously used extensively for its antipsychotic properties, has recently attracted interest as a potential candidate for development as an antimicrobial drug, as it is associated with the lowest risk of extrapyramidal side-effects of the phenothiazine drugs [5]. However, thioridazine can cause cardiac repolarization abnormalities and QTc prolongation at therapeutic doses [6–8] and reports of torsade de pointes [9] are well documented. A series of studies showing increased risk of QTc prolongation [6, 10] and sudden death [11–13] in patients treated with thioridazine has led to a re-evaluation of its use by drug regulatory agencies worldwide, culminating in the voluntary withdrawal of branded versions of the drug by Novartis in June 2005. Although it is still available in generic form, there has been a marked decline in prescription of thioridazine in developed countries.
The antimicrobial properties of phenothiazine drugs (with particular reference to thioridazine) and recent developments in our understanding of thioridazine cardiotoxicity are reviewed in this article, followed by a discussion of their implications for potential clinical use and further drug development.
The need for new antimicrobial agents
The development of antibiotic therapy has led to a massive reduction in the morbidity and mortality associated with infectious diseases in the developed world. Nevertheless, the incidence of serious nosocomial infections resulting from the emergence of bacterial strains resistant to conventional antibiotics, in particular, methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococci, has significantly increased over the past decade [14]. Although glycopeptide (vancomycin and teicoplanin) and oxazolidinone antibiotics (linezolid) are available for the treatment of MRSA-associated infections, vancomycin-resistant strains are emerging and the cost of these drugs is prohibitive in developing countries.
Mycobacterium tuberculosis currently infects over 2 billion people worldwide and accounts for >1.5 million deaths annually. The global proportion of multidrug resistant (MDR) TB is estimated to be around 1–2% of all cases [15]. The resurgence of tuberculosis amidst the global acquired immunodeficiency syndrome epidemic and the increasing frequency of drug-resistant strains are matters of public health concern worldwide.
Resource-poor developing countries continue to suffer the socioeconomic and health consequences of endemic diseases such as malaria, leishmaniasis and Chagas disease. Over 3 billion people live in regions where malaria is endemic. Malaria is a devastating disease with an annual morbidity of 300–500 million people and annual mortality of over one million [16]. Chloroquine-resistant strains of P. falciparum, responsible for the most lethal form of human malaria, are now common in most malaria-endemic regions where artemisinin-based therapies are often unaffordable. Twelve million people are estimated to be infected and 2 million new cases of leishmaniasis occur annually worldwide [17]. Of the population of Latin America, 25% is at risk of acquiring Chagas' disease. Current antitrypanosomal drugs such as nifurtimox are highly toxic, resulting in poor patient compliance.
The emergence of multidrug-resistant bacteria has led to revived interest in the search and development of new antibiotics to add to our existing armamentarium. However, with the spiralling cost of new drug discovery estimated to exceed $750M per new chemical entity [18], there is insufficient economic incentive for the pharmaceutical industry to develop novel drugs to tackle infectious diseases endemic in developing countries. In this context, the development of existing chemical entities with documented antimicrobial activity must be explored in an attempt to bring affordable drugs to the billions of people worldwide afflicted by common endemic infectious diseases.
Antimicrobial activity of thioridazine
MRSA
Thioridazine, in addition to its activity against intracellular methicillin-susceptible S. aureus (MSSA) [19], has demonstrable activity against MRSA with minimum inhibitory concentrations (MIC) ranging between 16 and 50 mg l−1 [20–22]. Addition of thioridazine at concentrations of 25–50% of its MIC to conventional antibiotics has led to a two-to-eightfold reduction in the MIC of norfloxacin [22] and a reduction in the MIC of oxacillin from >500 mg l−1 to 10 mg l−1 against some MRSA strains [23]. This is due to inhibition by thioridazine of bacterial efflux pumps which confer antibiotic resistance [22–24]. In addition, at subinhibitory concentrations, thioridazine inhibits the replication of phagocytosed MRSA and causes ultrastructural changes in the cell envelope structure, resulting in bacterial lysis after phagocytosis [21]. The mechanism of action of thioridazine is not fully understood, but the ultrastructural changes are similar to those produced by β-lactam antibiotics, suggesting that inhibition of membrane-bound enzymes may partly be responsible.
Enterococcus species
Multiresistant enterococci have emerged as a cause of serious nosocomial infections over the past decade. These strains produce β-lactamase enzymes conferring resistance to multiple antibiotics, including penicillins, carbapenems and glycopeptides, and also possess multidrug resistance efflux pumps. The finding that methylene blue and two methylated derivatives have bactericidal activity against vancomycin-resistant pathogenic strains of Enteroccus species [25] has led to the investigation of thioridazine as a potential antienterococcal antibiotic. Thioridazine inhibits E. faecalis and E. faecium strains (originating from human infections and animal faecal flora) at a concentration of 16–32 mg l−1, regardless of their antibiotic sensitivity. At subinhibitory concentrations, thioridazine has shown synergistic effects when combined with vancomycin or ampicillin, by a mechanism unrelated to P-glycoprotein-mediated multidrug resistance [26].
Mycobacterium tuberculosis
Thioridazine has significant in vitro activity against susceptible and multidrug-resistant strains of M. tuberculosis, with reported MIC varying from 6 to 32 mg l−1 [27–29]. It also acts synergistically with some first-line antituberculous drugs [30] and may allow reduced doses of these drugs to be used. In vitro experiments using THP-1 macrophage cell lines and human peripheral monocyte-derived macrophages infected with M. tuberculosis have shown that the minimum bactericidal concentration of thioridazine is as low as 0.1 mg l−1, with complete killing occurring within 3 days of infection [29]. Phenothiazines affect a number of key mycobacterial targets [31–33]. They bind to and inactivate calmodulin, a calcium transport protein which is a vital constituent of the cell wall envelope of mycobacteria [34–36]. Development of resistance to thioridazine is unlikely, as mutations affecting mycobacterial calcium flux would affect the viability of the organism. Genomic analysis of M. tuberculosis led to identification of type II nicotinamide adenine dinucleotide (NADH) dehydrogenase as a key enzyme for bacterial growth under aerobic conditions and a specific target for drug action, as human mitochondria only use type I NADH dehydrogenase. The antituberculous activity of phenothiazines appears to be partially due to specific inhibition of type II NADH dehydrogenase, as determined by NADH:menaquinone oxidoreductase activity [37].
Plasmodium falciparum
Since the initial description of its activity against P. falciparum by Ehrlich [38], methylene blue has been shown to inhibit P. falciparum glutathione reductase. Evaluation of methylene blue in combination with chloroquine in children with uncomplicated P. falciparum malaria in sub-Saharan Africa has confirmed its antimalarial effects [39]. Newer phenothiazines have also been shown to have in vitro activity against P. falciparum [40–42]. In a preliminary screening study of existing chemical entities against two strains of P. falciparum, thioridazine inhibited growth of P. falciparum within clinically achievable therapeutic plasma concentrations of thioridazine (effective 50% inhibitory concentrations, EC50, of 1.9 and 2.6 μm) [43].
Trypanosoma cruzi
Trypathioninone plays a prominent role in the redox defences of pathogenic Trypanosoma species. Phenothiazines inhibit two key trypanosomal enzymes, trypanothione reductase [44] and dihydrolipoamide dehydrogenase [45, 46], and also induce mitochondrial disruption in epimastigote and tripomastigote forms by formation of cationic free radicals through the peroxidase/H2O2 sytem [47]. In mice with experimental Chagas' disease, thioridazine significantly improved survival and cardiac function in the acute phase [48, 49] and chronic phase of the disease [50].
Other parasites
Thioridazine has been shown to be the most active phenothiazine agent against Pseudomonas aeruginosa, Stenotrophomonas maltophilia [19] and M. avium in vitro [51]. Although thioridazine has not been tested specifically, other phenothiazines also appear to be active against Leishmania species [52, 53], Schistosoma mansoni and Trypanosoma brucei and gambiense [54].
Clinical pharmacology of thioridazine
Following oral administration, thioridazine is rapidly absorbed, with peak plasma concentrations occurring within 2–3 h [8]. Plasma concentrations of thioridazine and its metabolites achieved in clinical use show wide interindividual variability and are affected by factors such as age, smoking and genetic polymorphisms in drug-metabolizing enzymes [55–57]. Thioridazine is widely distributed to tissues throughout the body.
Thioridazine is metabolized to mesoridazine (thioridazine-2-sulphoxide), which undergoes further 2-oxidation to sulphoridazine (thioridazine-2-sulphone). Thioridazine also undergoes 5-oxidation to the ring sulphoxide (thioridazine-5-sulphoxide) (Figure 1) [8]. Studies using human liver microsomes suggest that the metabolism of thioridazine to mesoridazine and sulphoridazine is catalysed mainly by CYP2D6, but CYP1A2 and CYP3A4 are the main isoforms involved in the formation of the ring sulphoxide [58]. Drug–drug interactions may therefore occur when drugs which inhibit or induce CYP2D6, CYP1A2 or CYP3A4 are used concomitantly. Smokers have lower plasma concentrations of thioridazine, mesoridazine and sulphoridazine compared with nonsmokers [56, 57]. Patients who are genetically poor metabolizers of CYP2D6 or take drugs which are potent inhibitors of CYP2D6 have higher plasma concentrations of thioridazine and the ring sulphoxide due to inhibition of metabolism to mesoridazine [55, 57].
Figure 1.
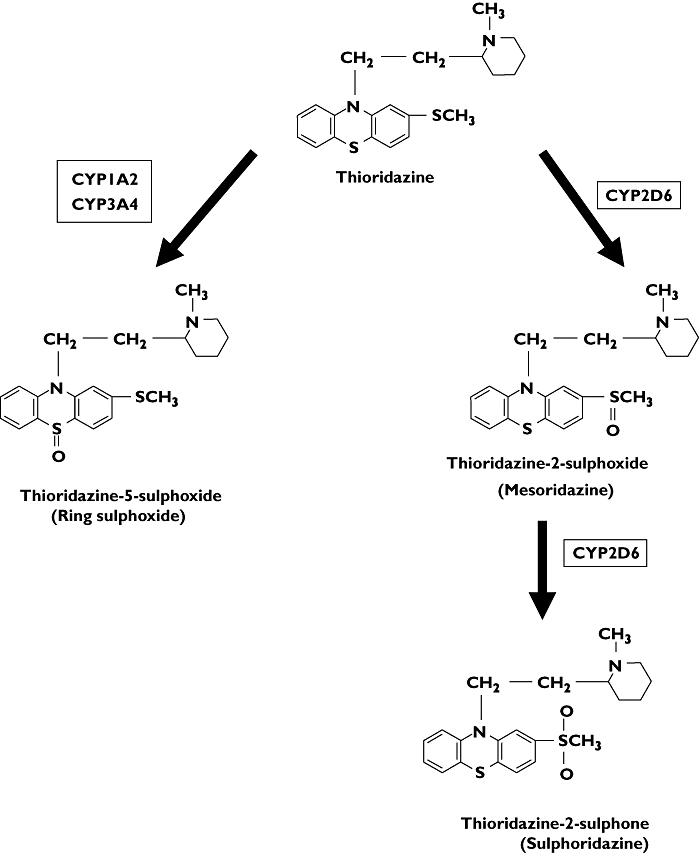
Metabolic pathway of thioridazine in man
The therapeutic thioridazine plasma concentration for its antipsychotic efficacy is 0.5–1.0 mg l−1. Thioridazine exhibits linear kinetics within the dose range used clinically. The plasma concentrations of thioridazine and its metabolites per mg of thioridazine given orally as a single dose and at steady state after chronic once-daily dosing are shown in Table 1. The mean half-life of thioridazine is 6.5 h in healthy volunteers after single dosing, but may be significantly prolonged to 40 h in the elderly. The ring sulphoxide is the main metabolite in chronically treated patients due to its longer elimination half-life (Table 1) [8].
Table 1.
Pharmacokinetic parameters of thioridazine and metabolites
| Thioridazine | Mesoridazine | Sulphoridazine | Ring sulphoxide | |
|---|---|---|---|---|
| Mean half-life after single 50-mg dose (h)* | 6.5 | 8.7 | 9.6 | 18.4 |
| Mean peak plasma concentration per mg thioridazine after single 50-mg dose (µg l−1 mg−1)* | 2.0 | 5.0 | 0.9 | 2.1 |
| Range of steady-state plasma concentration per mg thioridazine after once daily dosing (µg l−1 mg−1)† | 0.4–8.8 | 0.6–13.4 | 0.2–3.3 | 0.2–41.0 |
Thioridazine, mesoridazine and sulphoridazine are all potent D2 receptor blocking agents in rat and rabbit striatal membranes [59, 60], suggesting that they all contribute to the antipsychotic effects and the extrapyramidal side-effects of thioridazine. However, the ring sulphoxide metabolite (thioridazine-5-sulphoxide) is thought not to have antipsychotic activity [61].
Thioridazine is administered clinically as a 50 : 50 racemic mixture of its two enantiomers (R) and (S)-thioridazine. In isolated rat brain preparations (R)-thioridazine has 2.7 times higher affinity than (S)-thioridazine for D2 receptors, and acute administration of (R)-thioridazine to rats induced slightly more catalepsy than (S)-thioridazine and appeared to be more toxic at large doses [62].
Cardiotoxicity of thioridazine
The cardiac effects of thioridazine are related to the concentration-dependent blockade of the cardiac delayed inward rectifier potassium channel (Ikr) [63, 64]. Mesoridazine has similar concentration-dependent Ikr channel blocking activity to the parent drug [65]. Thioridazine and, more recently, mesoridazine have been shown to prolong the QTc interval following single-dose administration to healthy volunteers [8, 66]. It has been postulated that the cardiotoxic effects of thioridazine may be due to the ring sulphoxide as a result of the reported cardiotoxic effects in the isolated perfused rat heart [67] and in dogs [68]. Conflicting animal studies [68, 69] and findings of recent studies in patients treated with thioridazine and in healthy volunteers receiving mesoridazine have cast doubt on this hypothesis [57, 66]. The QTc interval at steady state was shown to be significantly correlated with the plasma concentrations of thioridazine and its 2-oxidation metabolites (mesoridazine and sulphoridazine), but not with the 5-oxidation metabolite (ring sulphoxide) [57]. The effect of the ring sulphoxide on Ikr channels has not been studied and these findings should be considered preliminary. Nevertheless, recent understanding of the mechanism of thioridazine Ikr blockade at the molecular level may explain why the ring sulphoxide does not appear to prolong the QTc interval in patients on thioridazine and may pave the way to derivatives of thioridazine which do not cause this side-effect.
Human ether-a-go-go-related gene (hERG) potassium (K+) channels mediate the rapidly activating delayed rectifier K+ current (IKr), which plays a key role in repolarization of the ventricular action potential. This is not involved in the electrophysiology of the rat and studies in the isolated perfused rat heart cannot therefore be extrapolated to man. In man, four hERG subunits assemble as a tetramer to form the ion channels that conduct the Ikr current. They consist of voltage sensor regions (S1–S4) and pore regions (S5–S6). The pore-forming unit of the Ikr channel contains a high-affinity drug binding site, of which aromatic amino acids present in the inner (S6) helices are key components [70]. It has been demonstrated that thioridazine causes Ikr blockade as a result of binding to the S6 helix amino acid residue F656 [71]. Single mutation F656A at this site leads to almost complete abolition of thioridazine-induced Ikr blockade [71].
As a result of the specificity of thioridazine-induced blockade, it is possible that 5-sulphoxidation leads to molecular changes, which reduce the affinity of the ring sulphoxide for that binding site.
Potential antimicrobial use of thioridazine
Thioridazine shows most promise for the adjunctive treatment of infections caused by multiresistant strains of intracellular organisms such as M. tuberculosis and P. falciparum in resource-poor countries [40]. Although the in vitro MIC against M. tuberculosis is high, killing of phagocytosed mycobacteria in macrophages occurs at a thioridazine concentration of 0.1 mg l−1 (which is clinically achievable using oral doses as low as 10–20% of those used in psychiatry) because of the ability of macrophages to concentrate phenothiazines [29]. Plasma thioridazine concentrations corresponding to the in vitro inhibitory concentrations of 0.75–1.0 mg l−1 for two strains of P. falciparum [43] can be achieved using oral doses used in psychiatric practice. Further evaluation is required in animal models of infection to determine whether clinical efficacy can be achieved in vivo prior to the conduct of clinical trials in man. The potential use of low-dose thioridazine as antimycobacterial prophylaxis in HIV-infected patients in areas with a high prevalence of HIV seropositivity may also be explored.
With the advent of newer antipsychotic drugs with a better safety profile, the use of thioridazine in psychiatry has declined in developed countries as the risk–benefit ratio is considered to be unfavourable. However, large epidemiological studies have shown that the risk of cardiotoxicity is small, with <20 episodes of torsade de pointes or sudden death potentially attributable to use of thioridazine occurring per 10 000 patient-years of treatment [12, 72]. In areas with a high prevalence of drug-resistant malaria and TB, the potential benefits outweigh the risks and conduct of clinical trials and subsequent clinical use of thioridazine may be justified if clinical efficacy is demonstrated. Investigation of methylene blue in combination with chloroquine in children with malaria in an area of high chloroquine resistance demonstrates that such clinical trials are both ethically justifiable and feasible [39].
Although thioridazine is active against resistant strains of S. aureus and enterococci, the plasma concentrations required for bactericidal activity as a single agent are not clinically achievable without intolerable side-effects and therefore preclude its use for serious bacteraemic infections with these organisms. Nevertheless, the use of thioridazine in combination with other antibiotics for its synergistic effects deserves some further investigation, as higher concentrations of thioridazine may be achieved in infected tissues despite plasma concentrations lower than the MIC seen in vitro.
Potential avenues for drug development
Novel derivatives of thioridazine that have reduced affinity for the Ikr channel and dopamine receptors but have higher potency as an antimicrobial have potential to meet the new challenges in infectious disease.
This is theoretically possible since a number of phenothiazine agents and various derivatives of thioridazine exhibit antimicrobial activity in vitro, although the metabolites of thioridazine have not specifically been investigated. In preliminary studies, novel derivatives of thioridazine enhance the bactericidal activity of phagocytosed M. tuberculosis at concentrations of 0.1 mg l−1 [73]. Phenothiazine analogues with greater potency have also been characterized and shown to suppress M. tuberculosis growth in a mouse model of acute infection [37]. The S-enantiomer of thioridazine and other phenothiazine isomers has also recently been demonstrated to have antimicrobial properties with reduced central nervous system effects [74]. Different EC50 values were obtained for analogues of thioridazine when tested against two strains of P. falciparum, raising hope that structural optimization may be possible [43]. The antimicrobial properties of thioridazine therefore appear to lack the same structural specificity required for its cardiotoxic effects.
As thioridazine-5-sulphoxide does not appear to have antipsychotic effects which is the result of binding to dopaminergic receptors in the brain, it is unlikely to cause troublesome sedating and extrapyramidal side-effects and could therefore serve as a lead compound in the development of more potent and safer derivatives for clinical use. In addition, thioridazine-5-sulphoxide exist as two pairs of enantiomers in equal concentrations, known as thioridazine-5-sulphoxide fast-eluting (FE) and slow-eluting (SE), based on their separation properties by chromatography [75]. The characteristics of these enantiomers are unknown, but they are likely to have differing affinities for the D2 receptor and Ikr ion channels, offering another potential avenue for optimization of the structure–activity relationship.
Conclusions
Thioridazine may have lost its shine as an antipsychotic, but its potential as an antimicrobial can no longer be ignored, least of all in the vast areas of the world plagued by endemic infectious diseases. Further ‘proof of concept’ studies are required to investigate the binding of the ring sulphoxide and its enantiomers to the Ikr channel and dopamine receptors in vitro. Partnerships between academia and the pharmaceutical industry offer the best chance of developing a safe, effective and affordable drug using an existing chemical entity as a lead compound.
Acknowledgments
Competing interests: None to declare.
References
- 1.Amaral L, Viveiros M, Kristiansen JE. ‘Non-Antibiotics’: alternative therapy for the management of MDRTB and MRSA in economically disadvantaged countries. Curr Drug Targets. 2006;7:887–91. doi: 10.2174/138945006777709539. [DOI] [PubMed] [Google Scholar]
- 2.Crowle AJ, Douvas GS, May MH. Chlorpromazine: a drug potentially useful for treating mycobacterial infections. Exp Chemother. 1992;38:410–9. doi: 10.1159/000239036. [DOI] [PubMed] [Google Scholar]
- 3.Forrest FM, Forrest IS, Roizin LE. Clinical, biochemical and post-mortem studies on a patient treated with chlorpromazine. Revue Agressologie. 1963;4:259–64. [PubMed] [Google Scholar]
- 4.Guttman P, Ehrlich P. Uber die Wirkung des Methylenblau bei Malaria. Berliner Klinika Wochenschrift. 1891;39:953–6. [Google Scholar]
- 5.Yang SY, Kao Yang YH, Chong MY, Yang YH, Chang WH, Lai CS. Risk of extrapyramidal syndrome in schizophrenic patients treated with antipsychotics: a population-based study. Clin Pharmacol Ther. 2007;81:586–94. doi: 10.1038/sj.clpt.6100069. [DOI] [PubMed] [Google Scholar]
- 6.Harrigan EP, Miceli J, Anziano R, Watsky E, Reeves KR, Cutler NR, Sramek J, Shiovitz T, Middle M. A randomized evaluation of the effects of six antipsychotic agents on QTc, in the absence and presence of metabolic inhibition. J Clin Psychopharmacol. 2004;24:62–9. doi: 10.1097/01.jcp.0000104913.75206.62. [DOI] [PubMed] [Google Scholar]
- 7.Thomas SHL. QT interval prolongation and ventricular arrhythmias. Adverse Drug React Toxicol Rev. 1994;13:77–102. [PubMed] [Google Scholar]
- 8.Hartigan-Go K, Bateman DN, Nyberg G, Martensson E, Thomas SHL. Concentration-related pharmacodynamic effects of thioridazine and its metabolites in humans. Clin Pharmacol Ther. 1996;60:543–53. doi: 10.1016/S0009-9236(96)90150-2. [DOI] [PubMed] [Google Scholar]
- 9.Kelly HQ, Fay JE, Laverty FG. Thioridazine hydrochloride (Mellaril): its effect on the electrocardiogram and a report of two fatalities with electrocardiographic abnormalities. CMAJ. 1963;89:546–54. [PMC free article] [PubMed] [Google Scholar]
- 10.Reilly JG, Ayis SA, Ferrier IN, Jones SJ, Thomas SH. QTc-interval abnormalities and psychotropic drug therapy in psychiatric patients. Lancet. 2000;355:1048–52. doi: 10.1016/s0140-6736(00)02035-3. [DOI] [PubMed] [Google Scholar]
- 11.Mehtonen OP, Aranko K, Malkonen L, Vapaatalo H. A survey of sudden death associated with the use of antipsychotic or antidepressant drugs: 49 cases in Finland. Acta Psychiatr Scand. 1991;84:58–64. doi: 10.1111/j.1600-0447.1991.tb01421.x. [DOI] [PubMed] [Google Scholar]
- 12.Ray WA, Meredith S, Thapa PB, Meador KG, Hall K, Murray KT. Antipsychotics and the risk of sudden cardiac death. Arch Gen Psychiatry. 2001;58:1161–7. doi: 10.1001/archpsyc.58.12.1161. [DOI] [PubMed] [Google Scholar]
- 13.Reilly JG, Ayis SA, Ferrier IN, Jones SJ, Thomas SH. Thioridazine and sudden unexplained death in psychiatric inpatients. Br J Psychiatry. 2002;180:515–22. doi: 10.1192/bjp.180.6.515. [DOI] [PubMed] [Google Scholar]
- 14.Klevens RM, Edwards JR, Richards CL, Jr, Horan TC, Gaynes RP, Pollock DA, Cardo DM. Estimating health care-associated infections and deaths in U.S. hospitals, 2002. Public Health Report. 2007;122:160–6. doi: 10.1177/003335490712200205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.World Health Organisation. WHO Report 2007. Geneva: WHO; 2007. Global Tuberculosis Control – Surveillance, Planning, Financing. WHO/HTM/TB/2007.376. [Google Scholar]
- 16.World Health Organisation. World Malaria Report 2005. Geneva: WHO; 2005. [Google Scholar]
- 17.World Health Organisation. Available at http://www.who.int/leishmaniasis/burden/magnitude/burden_magnitude/en/index.html (last accessed: 15 April 2007)
- 18.DiMasi JA, Hansen RW, Grabowski HG. The price of innovation: new estimates of drug development costs. J Health Econ. 2003;22:151–85. doi: 10.1016/S0167-6296(02)00126-1. [DOI] [PubMed] [Google Scholar]
- 19.Ordway D, Viveiros M, Leandro C, Arroz MJ, Amaral L. Intracellular activity of clinical concentrations of phenothiazines including thioridiazine against phagocytosed Staphylococcus aureus. Int J Antimicrob Agents. 2002;20:34–43. doi: 10.1016/s0924-8579(02)00110-3. [DOI] [PubMed] [Google Scholar]
- 20.Hendricks O, Butterworth TS, Kristiansen JE. The in-vitro antimicrobial effect of non-antibiotics and putative inhibitors of efflux pumps on Pseudomonas aeruginosa and Staphylococcus aureus. Int J Antimicrob Agents. 2003;22:262–4. doi: 10.1016/s0924-8579(03)00205-x. [DOI] [PubMed] [Google Scholar]
- 21.Martins M, Bleiss W, Marko A, Ordway D, Viveiros M, Leandro C, Pacheco T, Molnar J, Kristiansen JE, Amaral L. Clinical concentrations of thioridazine enhance the killing of intracellular methicillin-resistant Staphylococcus aureus: an in vivo, ex vivo and electron microscopy study. In Vivo. 2004;18:787–94. [PubMed] [Google Scholar]
- 22.Kaatz GW, Moudgal VV, Seo SM, Kristiansen JE. Phenothiazines and thioxanthenes inhibit multidrug efflux pump activity in Staphylococcus aureus. Antimicrob Agents Chemother. 2003;47:719–26. doi: 10.1128/AAC.47.2.719-726.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kristiansen MM, Leandro C, Ordway D, Martins M, Viveiros M, Pacheco T, Kristiansen JE, Amaral L. Phenothiazines alter resistance of methicillin-resistant strains of Staphylococcus aureus (MRSA) to oxacillin in vitro. Int J Antimicrob Agents. 2003;22:250–3. doi: 10.1016/s0924-8579(03)00200-0. [DOI] [PubMed] [Google Scholar]
- 24.Kristiansen MM, Leandro C, Ordway D, Martins M, Viveiros M, Pacheco T, Molnar J, Kristiansen JE, Amaral L. Thioridazine reduces resistance of methicillin-resistant Staphylococcus aureus by inhibiting a reserpine-sensitive efflux pump. In Vivo. 2006;20:361–6. [PubMed] [Google Scholar]
- 25.Wainwright M, Phoenix DA, Gaskell M, Marshall B. Photobactericidal activity of methylene blue derivatives against vancomycin-resistant Enterococcus spp. J Antimicrob Chemother. 1999;44:823–5. doi: 10.1093/jac/44.6.823. [DOI] [PubMed] [Google Scholar]
- 26.Hendricks O, Molnar A, Butterworth TS, Butaye P, Kolmos HJ, Christensen JB, Kristiansen JE. In vitro activity of phenothiazine derivatives in Enterococcus faecalis and Enterococcus faecium. Basic Clin Pharmacol Toxicol. 2005;96:33–6. doi: 10.1111/j.1742-7843.2005.pto960105.x. [DOI] [PubMed] [Google Scholar]
- 27.Amaral L, Kristiansen JE, Abebe LS, Millett W. Inhibition of the respiration of multi-drug resistant clinical isolates of Mycobacterium tuberculosis by thioridazine: potential use for initial therapy of freshly diagnosed tuberculosis. J Antimicrob Chemother. 1996;38:1049–53. doi: 10.1093/jac/38.6.1049. [DOI] [PubMed] [Google Scholar]
- 28.Bettencourt MV, Bosne-David S, Amaral L. Comparative in vitro activity of phenothiazines against multidrug-resistant Mycobacterium tuberculosis. Int J Antimicrob Agents. 2000;16:69–71. doi: 10.1016/s0924-8579(00)00199-0. [DOI] [PubMed] [Google Scholar]
- 29.Ordway D, Viveiros M, Leandro C, Bettencourt R, Almeida J, Martins M, Kristiansen JE, Molnar J, Amaral L. Clinical concentrations of thioridazine kill intracellular multidrug-resistant Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2003;47:917–22. doi: 10.1128/AAC.47.3.917-922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Viveiros MB, Amaral L. Enhancement of antibiotic activity against polydrug-resistant Mycobacterium tuberculosis by phenothiazines. Int J Antimicrob Agents. 2001;17:225–8. doi: 10.1016/s0924-8579(00)00343-5. [DOI] [PubMed] [Google Scholar]
- 31.Ratnakar P, Murthy PS. Trifluoperazine inhibits the incorporation of labelled precursors into lipids, proteins and DNA of Mycobacterium tuberculosis H37Rv. FEMS Microbiol Lett. 1993;110:291–4. doi: 10.1111/j.1574-6968.1993.tb06337.x. [DOI] [PubMed] [Google Scholar]
- 32.Dhople AM. In vitro activities of phenothiazine-type calmodulin antagonists against Mycobacterium leprae. Microbios. 1999;98:113–21. [PubMed] [Google Scholar]
- 33.Katoch VM, Saxena N, Shivannavar CT, Sharma VD, Katoch K, Sharma RK, Murthy PS. Effect of trifluoperazine on in vitro ATP synthesis by Mycobacterium leprae. FEMS Immunol Med Microbiol. 1998;20:99–102. doi: 10.1111/j.1574-695X.1998.tb01115.x. [DOI] [PubMed] [Google Scholar]
- 34.Salih FA, Kaushik NK, Sharma P, Choudary GV, Murthy PS, Venkitasubramanian TA. Calmodulin-like activity in mycobacteria. Indian J Biochem Biophys. 1991;28:491–5. [PubMed] [Google Scholar]
- 35.Ratnakar P, Murthy PS. Antitubercular activity of trifluoperazine, a calmodulin antagonist. FEMS Microbiol Lett. 1992;76:73–6. doi: 10.1016/0378-1097(92)90366-v. [DOI] [PubMed] [Google Scholar]
- 36.Reddy PH, Burra SS, Murthy PS. Correlation between calmodulin-like protein, phospholipids, and growth in glucose-grown Mycobacterium phlei. Can J Microbiol. 1992;38:339–42. doi: 10.1139/m92-057. [DOI] [PubMed] [Google Scholar]
- 37.Weinstein EA, Yano T, Li LS, Avarbock D, Avarbock A, Helm D, McColm AA, Duncan K, Lonsdale JT, Rubin H. Inhibitors of type II NADH:menaquinone oxidoreductase represent a class of antitubercular drugs. Proc Natl Acad Sci USA. 2005;102:4548–53. doi: 10.1073/pnas.0500469102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ehrlich P. Chemotherapeutics: scientific principles, methods, and results. Lancet. 1913;ii:445–51. [Google Scholar]
- 39.Meissner PE, Mandi G, Coulibaly B, Witte S, Tapsoba T, Mansmann U, Rengelshausen J, Schiek W, Jahn A, Walter-Sack I, Mikus G, Burhenne J, Riedel KD, Schirmer RH, Kouyate B, Muller O. Methylene blue for malaria in Africa: results from a dose-finding study in combination with chloroquine. Malar J. 2006;5:84. doi: 10.1186/1475-2875-5-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amaral L, Viveiros M, Kristiansen JE. Phenothiazines: potential alternatives for the management of antibiotic resistant infections of tuberculosis and malaria in developing countries. Trop Med Int Health. 2001;6:1016–22. doi: 10.1046/j.1365-3156.2001.00804.x. [DOI] [PubMed] [Google Scholar]
- 41.Kristiansen JE, Jepsen S. The susceptibility of Plasmodium falciparum in vitro to chlorpromazine and the stereo-isomeric compounds cis(Z)- and trans(E)-clopenthixol. Acta Pathol Microbiol Immunol Scand [B] 1985;93:249–51. doi: 10.1111/j.1699-0463.1985.tb02884.x. [DOI] [PubMed] [Google Scholar]
- 42.Basco LK, Le Bras J. In vitro activities of chloroquine in combination with chlorpromazine or prochlorperazine against isolates of Plasmodium falciparum. Antimicrob Agents Chemother. 1992;36:209–13. doi: 10.1128/aac.36.1.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weisman JL, Liou AP, Shelat AA, Cohen FE, Guy RK, DeRisi JL. Searching for new antimalarial therapeutics amongst known drugs. Chem Biol Drug Des. 2006;67:409–16. doi: 10.1111/j.1747-0285.2006.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gutierrez-Correa J, Fairlamb AH, Stoppani AO. Trypanosoma cruzi trypanothione reductase is inactivated by peroxidase-generated phenothiazine cationic radicals. Free Radic Res. 2001;34:363–78. doi: 10.1080/10715760100300311. [DOI] [PubMed] [Google Scholar]
- 45.Gutierrez-Correa J, Krauth-Siegel RL, Stoppani AO. Phenothiazine radicals inactivate Trypanosoma cruzi dihydrolipoamide dehydrogenase: enzyme protection by radical scavengers. Free Radic Res. 2003;37:281–91. doi: 10.1080/1071576021000046622. [DOI] [PubMed] [Google Scholar]
- 46.Gutierrez-Correa J. Trypanosoma cruzi dihydrolipoamide dehydrogenase as target for phenothiazine cationic radicals. Effect of antioxidants. Curr Drug Targets. 2006;7:1155–79. doi: 10.2174/138945006778226615. [DOI] [PubMed] [Google Scholar]
- 47.Rivarola HW, Paglini-Oliva PA. Trypanosoma cruzi trypanothione reductase inhibitors: phenothiazines and related compounds modify experimental Chagas' disease evolution. Curr Drug Targets Cardiovasc Haematol Disord. 2002;2:43–52. doi: 10.2174/1568006023337745. [DOI] [PubMed] [Google Scholar]
- 48.Rivarola HW, Fernandez AR, Enders JE, Fretes R, Gea S, Suligoy M, Palma JA, Paglini-Oliva P. Thioridazine treatment modifies the evolution of Trypanosoma cruzi infection in mice. Ann Trop Med Parasitol. 1999;93:695–702. [PubMed] [Google Scholar]
- 49.Lo Presti MS, Rivarola HW, Bustamante JM, Fernandez AR, Enders JE, Fretes R, Gea S, Paglini-Oliva PA. Thioridazine treatment prevents cardiopathy in Trypanosoma cruzi infected mice. Int J Antimicrob Agents. 2004;23:634–6. doi: 10.1016/j.ijantimicag.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 50.Bustamante JM, Presti MS, Rivarola HW, Fernandez AR, Enders JE, Fretes RE, Paglini-Oliva P. Treatment with benznidazole or thioridazine in the chronic phase of experimental Chagas disease improves cardiopathy. Int J Antimicrob Agents. 2007;29:733–7. doi: 10.1016/j.ijantimicag.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 51.Viveiros M, Martins M, Couto I, Kristiansen JE, Molnar J, Amaral L. The in vitro activity of phenothiazines against Mycobacterium avium: potential of thioridazine for therapy of the co-infected AIDS patient. In Vivo. 2005;19:733–6. [PubMed] [Google Scholar]
- 52.Pearson RD, Manian AA, Harcus JL, Hall D, Hewlett EL. Lethal effect of phenothiazine neuroleptics on the pathogenic protozoan Leishmania donovani. Science. 1982;217:369–71. doi: 10.1126/science.6124040. [DOI] [PubMed] [Google Scholar]
- 53.Pearson RD, Manian AA, Hall D, Harcus JL, Hewlett EL. Antileishmanial activity of chlorpromazine. Antimicrob Agents Chemother. 1984;25:571–4. doi: 10.1128/aac.25.5.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gracio MA, Gracio AJ, Viveiros M, Amaral L. Since phenothiazines alter antibiotic susceptibility of microorganisms by inhibiting efflux pumps, are these agents useful for evaluating similar pumps in phenothiazine-sensitive parasites? Int J Antimicrob Agents. 2003;22:347–51. doi: 10.1016/s0924-8579(03)00204-8. [DOI] [PubMed] [Google Scholar]
- 55.Von Bahr C, Movin G, Nordin C. Plasma levels of thioridazine and metabolites are influenced by the debrisoquine hydroxylation phenotype. Clin Pharmacol Ther. 1991;49:234–40. doi: 10.1038/clpt.1991.22. [DOI] [PubMed] [Google Scholar]
- 56.Berecz R, de la Rubia A, Dorado P, Fernandez-Salguero P, Dahl ML, Llerena A. Thioridazine steady-state plasma concentrations are influenced by tobacco smoking and CYP2D6, but not by the CYP2C9 genotype. Eur J Clin Pharmacol. 2003;59:45–50. doi: 10.1007/s00228-003-0576-4. [DOI] [PubMed] [Google Scholar]
- 57.Thanacoody RH, Daly AK, Reilly JG, Ferrier IN, Thomas SH. Factors affecting drug concentrations and QT interval during thioridazine therapy. Clin Pharmacol Ther. 2007 doi: 10.1038/sj.clpt.6100195. Epub ahead of print. doi: 10.1038/sj.clpt.6100190. [DOI] [PubMed] [Google Scholar]
- 58.Wojcikowski J, Maurel P, Daniel WA. Characterization of human cytochrome p450 enzymes involved in the metabolism of the piperidine-type phenothiazine neuroleptic thioridazine. Drug Metab Dispos. 2006;34:471–6. doi: 10.1124/dmd.105.006445. [DOI] [PubMed] [Google Scholar]
- 59.Kilts CD, Knight DL, Mailman RB, Widerlov E, Breese GR. Effects of thioridazine and its metabolites on dopaminergic function: drug metabolism as a determinant of the antidopaminergic actions of thioridazine. J Pharmacol Exp Ther. 1984;231:334–42. [PubMed] [Google Scholar]
- 60.Niedzwiecki DM, Cubeddu LX. Comparative antidopaminergic properties of thioridazine, mesoridazine and sulphoridazine on the corpus striatum. J Pharmacol Exp Ther. 1989;250:117–25. [PubMed] [Google Scholar]
- 61.Dahl SG. Active metabolites of neuroleptic drugs: possible contribution to therapeutic and toxic effects. Ther Drug Monit. 1982;4:33–40. doi: 10.1097/00007691-198204000-00005. [DOI] [PubMed] [Google Scholar]
- 62.Svendsen CN, Froimowitz M, Hrbek C, Campbell A, Kula N, Baldessarini RJ, Cohen BM, Babb S, Teicher MH, Bird ED. Receptor affinity, neurochemistry and behavioral characteristics of the enantiomers of thioridazine: evidence for different stereoselectivities at D1 and D2 receptors in rat brain. Neuropharmacology. 1988;27:1117–24. doi: 10.1016/0028-3908(88)90006-8. [DOI] [PubMed] [Google Scholar]
- 63.Drolet B, Vincent F, Rail J, Chahine M, Deschenes D, Nadeau S, Khalifa M, Hamelin BA, Turgeon J. Thioridazine lengthens repolarization of cardiac ventricular myocytes by blocking the delayed rectifier potassium current. J Pharmacol Exp Ther. 1999;288:1261–9. [PubMed] [Google Scholar]
- 64.Tie H, Walker BD, Valenzuela SM, Breit SN, Campbell TJ. The heart of psychotropic drug therapy. Lancet. 2001;355:1825. doi: 10.1016/s0140-6736(05)73083-x. [DOI] [PubMed] [Google Scholar]
- 65.Su Z, Martin R, Cox BF, Gintant G. Mesoridazine: an open-channel blocker of human ether-a-go-go-related gene K+ channel. J Mol Cell Cardiol. 2004;36:151–60. doi: 10.1016/j.yjmcc.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 66.Salih IS, Thanacoody RH, McKay GA, Thomas SH. Comparison of the effects of thioridazine and mesoridazine on the QT interval in healthy adults after single oral doses. Clin Pharmacol Ther. 2007 doi: 10.1038/sj.clpt.6100194. Epub ahead of print. doi: 10.1038/sj.clpt.6100181. [DOI] [PubMed] [Google Scholar]
- 67.Hale PW, Jr, Poklis A. Thioridazine-5-sulphoxide cardiotoxicity in the isolated, perfused rat heart. Toxicol Lett. 1984;21:1–8. doi: 10.1016/0378-4274(84)90215-7. [DOI] [PubMed] [Google Scholar]
- 68.Heath A, Svensson C, Martensson E. Thioridazine toxicity – an experimental cardiovascular study of thioridazine and its major metabolites in overdose. Vet Hum Toxicol. 1985;27:100–5. [PubMed] [Google Scholar]
- 69.Hine B, Traficante LJ, Sakalis G, Gershon S. Thioridazine and EKG anomalies. Experientia. 1979;35:1631–2. doi: 10.1007/BF01953235. [DOI] [PubMed] [Google Scholar]
- 70.Kamiya K, Niwa R, Mitcheson JS, Sanguinetti MC. Molecular determinants of HERG channel block. Mol Pharmacol. 2006;69:1709–16. doi: 10.1124/mol.105.020990. [DOI] [PubMed] [Google Scholar]
- 71.Milnes JT, Witchel HJ, Leaney JL, Leishman DJ, Hancox JC. hERG K+ channel blockade by the antipsychotic drug thioridazine. An obligatory role for the S6 helix residue F656. Biochem Biophys Res Commun. 2006;351:273–80. doi: 10.1016/j.bbrc.2006.10.039. [DOI] [PubMed] [Google Scholar]
- 72.Glassman AH, Bigger JT. Antipsychotic drugs: prolonged QTc interval, torsade de pointes and sudden death. Am J Psychiatry. 2001;158:1774–82. doi: 10.1176/appi.ajp.158.11.1774. [DOI] [PubMed] [Google Scholar]
- 73.Martins M, Schelz Z, Martins A, Molnar J, Hajos G, Riedl Z, Viveiros M, Yalcin I, Aki-Sener E, Amaral L. In vitro and ex vivo activity of thioridazine derivatives against Mycobacterium tuberculosis. Int J Antimicrob Agents. 2007;29:338–40. doi: 10.1016/j.ijantimicag.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 74.Kristiansen JE, Hendricks O, Delvin T, Butterworth TS, Aagaard L, Christensen JB, Flores VC, Keyzer H. Reversal of resistance in microorganisms by help of non-antibiotics. J Antimicrob Chemother. 2007;59:1271–9. doi: 10.1093/jac/dkm071. [DOI] [PubMed] [Google Scholar]
- 75.Hale PW, Jr, Poklis A. Thioridazine 5-sulphoxide diastereoisomers in serum and urine from rats and man after chronic thioridazine administration. J Anal Toxicol. 1985;9:19. doi: 10.1093/jat/9.5.197. [DOI] [PubMed] [Google Scholar]